 Makayla Cook
Makayla Cook Tiffany Hensley-McBain
Tiffany Hensley-McBain Andrea Grindeland
Andrea Grindeland- McLaughlin Research Institute, Great Falls, MT, United States
Animal models are essential tools for investigating and understanding complex prion diseases like chronic wasting disease (CWD), an infectious prion disease of cervids (elk, deer, moose, and reindeer). Over the past several decades, numerous mouse models have been generated to aid in the advancement of CWD knowledge and comprehension. These models have facilitated the investigation of pathogenesis, transmission, and potential therapies for CWD. Findings have impacted CWD management and disease outcomes, though much remains unknown, and a cure has yet to be discovered. Studying wildlife for CWD effects is singularly difficult due to the long incubation time, subtle clinical signs at early stages, lack of convenient in-the-field live testing methods, and lack of reproducibility of a controlled laboratory setting. Mouse models in many cases is the first step to understanding the mechanisms of disease in a shortened time frame. Here, we provide a comprehensive review of studies with mouse models in CWD research. We begin by reviewing studies that examined the use of mouse models for bioassays for tissues, bodily fluids, and excreta that spread disease, then address routes of infectivity and infectious load. Next, we delve into studies of genetic factors that influence protein structure. We then move on to immune factors, possible transmission through environmental contamination, and species barriers and differing prion strains. We conclude with studies that make use of cervidized mouse models in the search for therapies for CWD.
1 Introduction
Chronic wasting disease (CWD) is an infectious prion disease of cervids (elk, deer, red deer, moose, and reindeer), and is the only prion disease to spread within populations of wild animals (1, 2). CWD is included in the disease classification of transmissible spongiform encephalopathies (TSEs) that are caused by proteinaceous infectious agents called ‘prions’ (1, 3, 4). TSEs are slowly progressive, fatal neurodegenerative disorders for which no treatment or vaccine is currently available (5). Prion disease results from a misfolded form of the prion protein (PrP), in which the normal cellular form (PrPC) is converted to the pathological form (PrPSc) by the misfolded molecule (3, 5). Misfolding of the prion protein occurs through a seeding mechanism that forms an accumulation of large amyloid-like fibrillar aggregates leading to spongiform change. PrPC is located at the cell surface and although the physiological function of PrP remains elusive, studies on PrPC knockout mice (Prnp0/0) demonstrate mild behavioral abnormalities (6, 7). Specifically, it was observed that these mice had sleep and circadian rhythm alterations, significantly poorer behavioral parameters including nest-building abilities, memory performance, and associative learning, a decrease in locomotor activity, and increased basal anxiety with age (7, 8).
CWD is just one of a host of similar diseases found in humans and animals. Currently, no cure exists for any of the TSEs and the result of contracting a TSE disease is generally death within a few years after the individual shows signs of disease (3, 4, 9–12). Five recognized human prion diseases and seven recognized animal prion diseases exist (1, 11). Human prion diseases consist of Creutzfeldt-Jakob disease (CJD), of which there are four main types, Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, variably protease sensitive prionopathy, and kuru (1, 3, 11–16). Animal prion diseases include bovine spongiform encephalopathy (BSE), ungulate spongiform encephalopathy, scrapie, transmissible mink encephalopathy, camel prion disease, feline spongiform encephalopathy, and CWD (1, 3, 4, 9, 11, 14). Prion diseases in humans can present as genetic, infectious, or sporadic disorders (12, 13). While it is accepted that other mammals acquire prion diseases mainly through infection (3), spontaneous forms have been identified (9, 12, 15–22).
2 Chronic wasting disease
The incidence of CWD in cervids is rapidly expanding; it currently circulates in white-tailed deer (Odocoileus virginianus), black-tailed deer (Odocoileus hemionus columbianus), mule deer (Odocoileus hemionus), red deer (Cervus elaphus), sika deer (Cervus Nippon), Reeves’ muntjac deer (Muntiacus reevesi), North American elk (Cervus canadensis), Rocky Mountain elk (Cervus elaphus nelsoni), moose (Alces alces), and reindeer (Rangifer tarandus tarandus) (11, 23, 24). Although the origins of CWD are unknown, CWD was first observed at a captive mule deer facility in Colorado in 1967, diagnosed as a TSE in 1978, and named chronic wasting disease in 1980 (25). Since its identification in Colorado, it has been detected throughout many regions of the world. According to the Centers for Disease Control and Prevention (CDC), as of June 2022, six countries have reported CWD cases in free-ranging cervids and/or commercial captive cervid facilities: Canada, Finland, Norway, South Korea, Sweden, and the United States. It is important to note that geographically distinct prions exist; CWD prions found in Norway, Sweden, and Finland were distinct from those found in North America (26), indicating that they are spontaneous and transmissible. Cases from South Korea, are linked to Canada via imported animals (11, 27).
In North America, CWD is currently present in 29 states in the United States and 4 provinces in Canada, affecting both free-ranging wildlife and captive animals according to the CDC and United States Geological Survey bureau. Its spread historically has been unpredictable in the captive cervid industry due to the animal movement being commercial, random, and previously inadequately regulated in many locations (28). CWD distribution pattern within the United States appeared as hotspots of various sizes and were separated by large distances instead of a pattern consistent with the natural movement of free-ranging animals (29). The inconsistent pattern was a result of many factors, including relocation of CWD-infected animals and direct or indirect contact between farm animals and wildlife populations (11).
The rapid spread of CWD is due to both direct (animal to animal) and indirect transmission. Indirect transmission consists of environmental contamination from feces, saliva, urine, blood, antler velvet, and infected carcasses (29–33). CWD prions are shed into the environment from symptomatic and asymptomatic deer (33), which complicates effective elimination through management practices and highlights the need for a convenient antemortem and environmental testing method. CWD prions are known to remain infectious in the environment for years making CWD challenging to contain and control (34, 35). Its transmissibility and environmental persistence may increase the risk of transmission to other species (36). CWD infectious prions also persist through the digestive tract and have been found in the feces of carnivores, although at significantly reduced infectious load (37, 38).
Clinical signs of CWD include weight loss, polydipsia, polyuria, excessive salivation, grinding of teeth, flaccid hypotonia of facial muscles, lowering of the head, drooping of the ears, ataxia, and terminal anorexia (25). Some affected animals experience esophageal hypotonia and dilation, difficulty swallowing, regurgitation of ruminal fluid, and ingesta, which may lead to pneumonia (25). Behavioral changes include episodes of apparent lack of awareness, decreased interactions with unaffected deer in the herd, occasional abnormal response to restraint, and hyperexcitability (25).
Methods of CWD detection historically have involved immunohistochemistry or enzyme-linked immunosorbent (ELISA) assays and require invasive tissue samples such as lymph node biopsies or the obex portion of the brain stem (39–42). Newer methods of detection are currently being developed such as protein misfolding cyclic amplification (PMCA) and Real Time Quaking Induced Conversion assay (RT-QuIC). PMCA is a sensitive method that amplifies PrPSc in vivo and has been used to generate CWD seeds for inoculation into human PRNP mouse models (43–48). RT-QuIC is a highly sensitive test that amplifies misfolded PrP seeds using PrPC substrates (usually from Syrian hamster or bank vole) which form amyloid fibrils and are detectible using a microplate reader (44, 45).
CWD is nearly impossible to eradicate once established in wild cervid populations, further supporting the need for research into ways to reduce transmission (49). New York is the only state to have eliminated CWD, due to intensive regulations put into place (50). CWD management has been attempted through active surveillance and wildlife management tools such as planned selective culling, selected breeding, targeted hunting, mandatory testing of hunted deer and elk in endemic locations, and even leading to more extreme strategies such as complete eradication of infected wild cervid populations (51–60). Newer strategies under investigation include vaccination (61, 62) and selective breeding for disease-resistant animals (51).
3 Mouse models in prion disease research
Mice are the most widely used laboratory animals for studying diseases due to their manageability, short generation time, and ease of genome manipulation (8). Mouse models possess the ability to recapitulate aspects of the neuropathological and biochemical characteristics of several human and animal diseases in a shorter time period (8).
Transgenic mice can be generated to express a Prnp gene matched to the species and prion strain under investigation. Most often, the transgenes are expressed on a Prnp null background (Prnp0/0) in order to avoid partial or full suppression of disease caused by co-expression of wild type Prnp (8, 63). The expression levels of Prnp in the models may affect prion disease susceptibility and incubation periods (64). While transgenic models are extremely valuable for understanding aspects of the disease, the downsides to classic transgenic mouse models include the potential for variable copy numbers of the transgene at undefined genomic locations due to random integration of transgene insertion sites (65). This aspect inhibits standardization of experiments and may affect recapitulating animal disease (8). Gene targeting, which directly replaces the mouse Prnp gene with the species of interest Prnp gene, offers advantages over traditional transgenic mice and appears to have a more accurate disease representation by showing peripheral pathogenesis and mechanisms of horizontal transmission (8, 66, 67).
Another advantage of laboratory mouse models is the ability to inoculate them with prions made from the species of interest, sometimes mouse passaged prior to inoculation (8). The levels of infectivity can then be quantified using brain homogenates from inoculated mouse models (8). Mice can be inoculated by multiple routes, including intracerebrally (i.c.), intranasally, orally (p.o.), intraperitoneal (i.p.) route, and in CWD studies, by horizontal exposure using infected cage mates (66). Following an incubation period that is dependent on the prion type and strain used, the inoculated mice, if susceptible to disease, will begin to develop progressive clinical signs of neurological illness and will eventually succumb to prion disease (8). Neuropathological characteristics of prion disease in mouse models vary depending on species and strain but may include spongiform degeneration, deposition of misfolded PrP in various brain regions (diffuse or plaques), and prominent astrocytic gliosis (8, 46, 67). Peripheral pathogenic signs may develop, including spleen and lymph node deposition of PrPSc (67–69). In addition to the bioassay of brain homogenates, transgenic mouse models have made it possible to bioassay prions in tissues, body fluids, and secretions of donor species, providing information on the mode of transmission (63).
3.1 Making and characterizing models
In the following sections, we explore the main contributions mouse models have provided to CWD knowledge and comprehension. We have identified various mouse models for CWD and relevant characteristics for each line (Table 1), which differ in the type of genetic modification (for example, transgene, direct modification), the PrP sequence that is modified, and expression level.
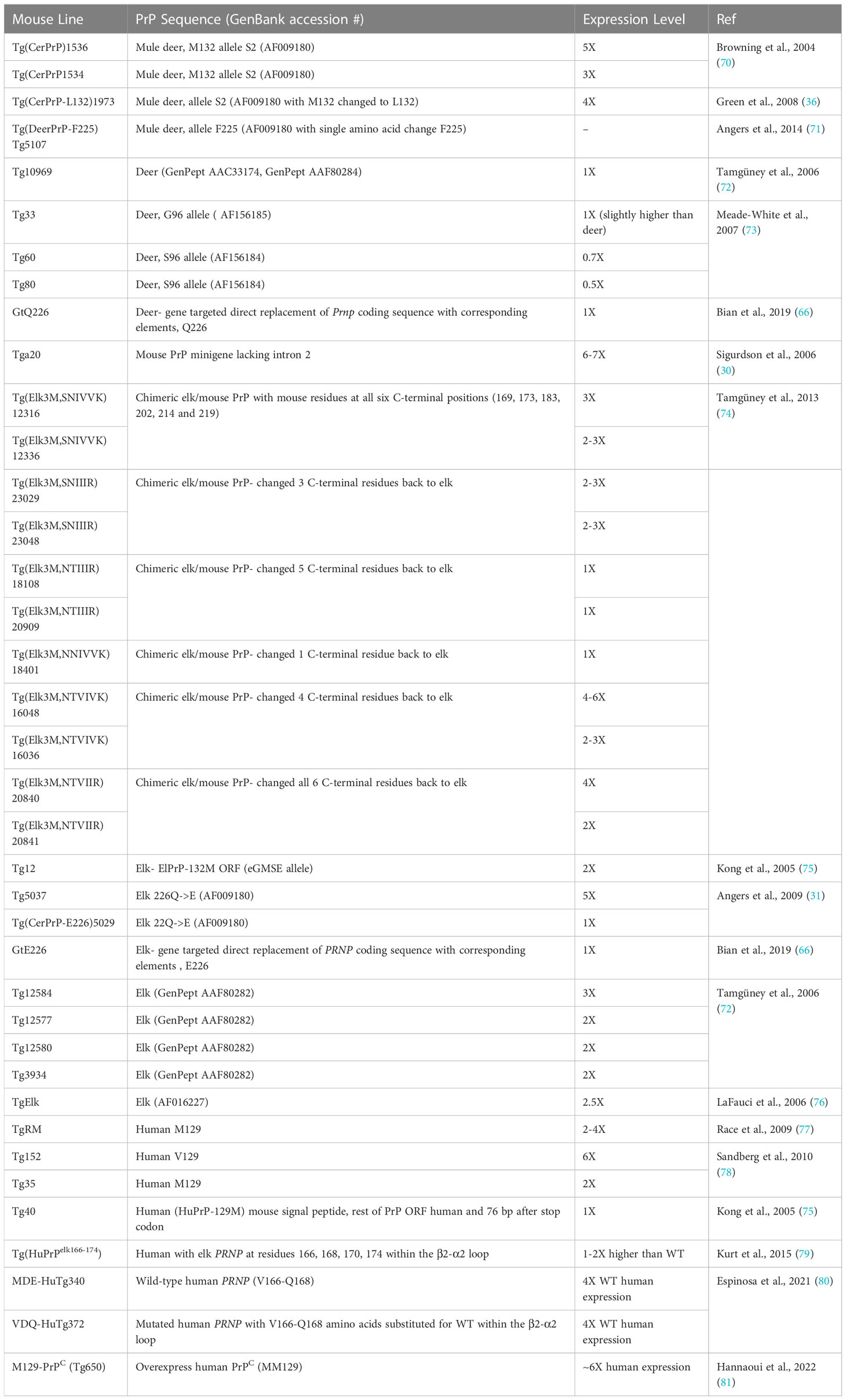
Table 1 Mouse models of CWD.
4 Transmission of CWD to cervidized mice
4.1 Investigation of infectious tissues
Cervid tissues can transmit disease through direct or indirect transmission routes as shown in studies highlighted in Table 2. Experimental direct inoculation is most commonly done by i.c. using brain tissue (8). Direct inoculation with deer feces into mouse models resulted in disease, suggesting that prolonged fecal prion excretion provides a plausible natural mechanism and the most likely contributing factor to the high incidence and efficient horizontal transmission of CWD within deer herds and between susceptible deer species (33).

Table 2 Investigations of infectious tissues.
Inoculation using antler velvet from two naturally infected elk to two transgenic mice models demonstrated the infectivity of low concentration of CWD prions in antler velvet by resulting in disease mouse models (31). Notably, not all source samples induced disease, and incubation periods were variable (31). The portion of the antler velvet processed and the age at harvest factored in the variable transmissibility and levels of infectivity (31). These results are significant, as antler velvet represented a previously unrecognized source of CWD prions in tissues and the environment. Infectious antler velvet may pose a seasonal risk factor for transmission in mule deer and elk during the rub and rut in early autumn (31).
Transmission has also been demonstrated via urine and saliva, particularly from experimentally infected white-tailed deer at the terminal state of disease (49). The rate of transmission was higher for mice inoculated with saliva (8/9 mice) compared to mice inoculated with urine (2/9 mice), suggesting a lower concentration or uneven PrP distribution of prion infectivity in urine (49). High infectivity of mouse saliva supports the hypothesis that it is a vehicle for CWD transmission (49). Potential vehicles of transmission based on transmissibility from mouse models include fat, whole blood, blood mononuclear cells, B cells, and platelets (77, 82). Cell-free plasma and monocytes did not transmit CWD in these studies (82).
In other studies, Seelig et al., assessed the longitudinal accumulation of protease-resistant prion protein (PrPRES) in tissues of inoculated mice. Tissues of the central nervous system, spleen, liver, mesenteric lymph nodes, bone marrow pancreatic islets, Peyer’s patches, tongue, salivary, adrenal, and pituitary glands, had detectable amounts of PrPRES (68). Horizontal transmission of CWD to naïve, cohabitating mice was confirmed by detectable PrPRES deposits in the obex, brainstem, cerebellum, hippocampus, hypothalamus, neocortex, spleen, Peyer’s patches, and pancreatic islets. PrPRES was not detected in additional peripheral tissues, including tissues from the gastrointestinal, urogenital, endocrine, and musculoskeletal systems (68).
4.2 Investigation of routes of transmission
Understanding the infectious routes of CWD provides insight into the management of the disease and transmission. As shown in Table 3, CWD routes of inoculation can result in transmission to cervidized mice by i.c., oral, aerosol, or intranasal pathways (29, 83). Parenteral route inoculation via intravenous and intraperitoneal were also shown to result in disease (68). These findings support further consideration for prion disease transmission and biosafety.

Table 3 Routes of transmission.
4.3 Investigation of transmission by passage through mice
Crossing species barriers in prion disease is hypothesized to be a two-step process involving normal host PrPC being recruited and misfolded by infective PrPSc. This conformational corruption may be an inefficient process depending on the compatibility of the protein structure of the donor and host (63). This initial conversion is thought to be followed by the PrPSc that is now structurally compatible with the host and can recruit PrPC more efficiently with increased misfolding recapitulating prion disease (63). This phenomenon results in prion-adapted conversion among species and is the basis for many studies in which mouse models can be used for studying prion diseases not acquired outside the laboratory environment (63, 84). LaFauci et al. and Lee et al. investigate how serial passage affects the transmission of CWD in the mouse models described in Table 4. Studies demonstrated that upon the second passage of CWD-positive elk brain homogenate there was a reduced incubation period for disease (76). The primary inoculation of CWD-positive elk brain homogenate caused an infection rate of 1/23 VM/Dk wild-type mice and the secondary passage had a 10/10 infection rate (85). Comparatively, the researchers observed a 100% infection rate (4/4 mice) for the primary passage in TgElk mice (85). Together these studies demonstrate the efficiency of infection, and that the disease increases with species specific adaptations of PrP in vivo.

Table 4 Transmission by passage through mice.
4.4 Investigation of transmission concerning genetics
Polymorphisms in the cervid Prnp may affect CWD susceptibility, progression of disease and incubation periods (2, 11, 51, 52, 86–93) several of which are shown in Figure 1. Sixteen amino acid polymorphisms exist within the 256 amino acid open reading frame in the third exon of the Prnp gene among the family of Cervidae (94). Codon 132 in elk corresponds to the human polymorphism 129 and results in longer incubation times with the leucine (LL) variants as opposed to the methionine (MM) variants (11, 86, 88, 95). White tailed-deer possess many Prnp variants, several have been shown to affect susceptibility (2, 51, 52, 89, 90). Mule deer have been documented to have a polymorphism at position 225 implicated in reduced disease prevalence, protracted time course, and even cause variations in disease pathology (96–98). Mouse models allow for a wide range of genomic manipulations (8) and facilitate CWD studies investigating the contribution of these genetic factors with considerable ease. The studies summarized in Table 5 have increased our understanding of how differing genetics (both donor and host) may affect disease phenotypes such as incubation time and neuropathological signs, and even susceptibility.
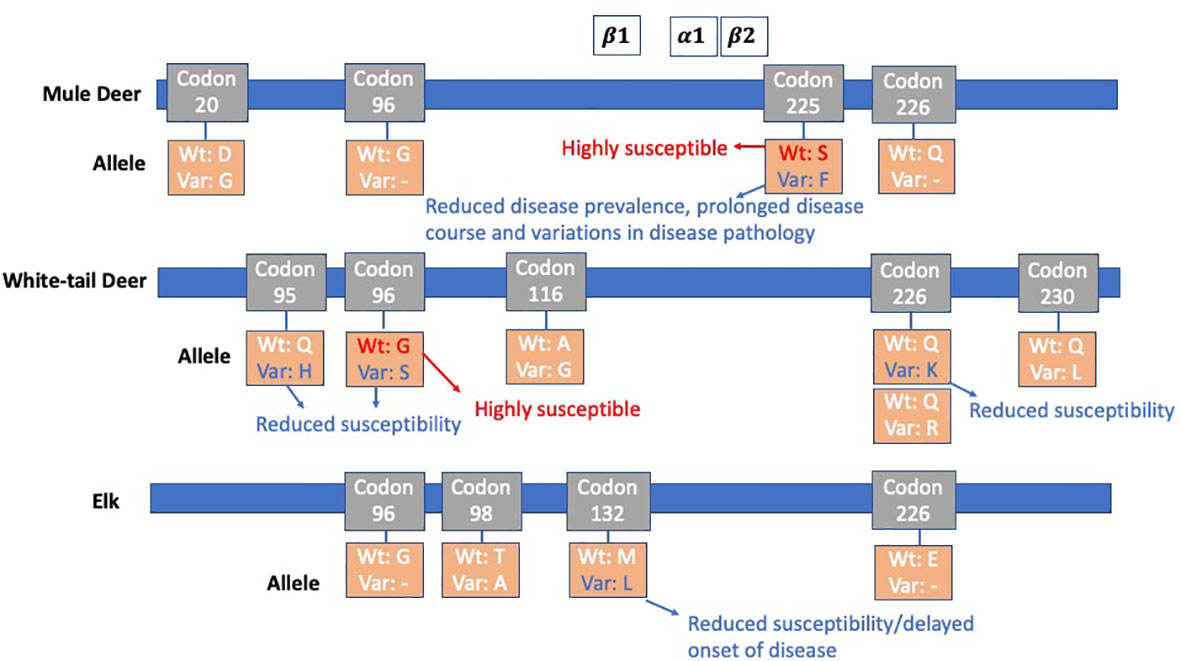
Figure 1 PrP Codon Polymorphisms among Mule Deer, White-tail Deer, and Elk: CWD susceptibility, disease onset, and pathology have been seen to be affected by polymorphisms in the cervid Prnp. β1, α1 and β2 and molecular structures are shown at their approximate location in reference to the codons (88). Mule deer have a polymorphism at position 225, referred to as 225F, which shows reduced disease prevalence, prolonged disease course, and variation in disease pathology. White-tailed deer contain many PRNP variants, several of which affect susceptibility. Codon 226 has a variant 226K, in which CWD susceptibility is reduced. In elk, codon 132 has a critical role in susceptibility and onset of disease. Animals expressing the 132M variant is overrepresented in both free-ranging and farmed elk positive for CWD. Those with 132L have a reduced rate of CWD and a delayed onset of disease progression.

Table 5 Transmission concerning genetics.
First, CWD susceptibility of transgenic mice expressing two naturally occurring allelic variants of deer PrP with either glycine (G) or serine (S) at residue 96 have been investigated (73). CWD positive brain tissue from elk and mule deer with the G96 PrP genotype and white-tailed deer expressing S96 PrP and/or G96 PrP genotypes were used to inoculate Tg mice expressing G96 deer PrP molecules and S96 deer PrP (73). Tg mice with S96 residue were found to be resistant to CWD infection past the 600-day observation period, unlike the mice with 96G residue where disease appeared as early as 160 days post inoculation (73). These results show allelic variation impacts incubation time regarding CWD in mouse models as well as wild cervids in vivo. Research with white-tailed deer populations also show that residue 96 has a great impact on CWD susceptibility, as deer carrying the 96S allele have reduced susceptibility and if infected with CWD deer have a 49% longer survival rate compared to deer with genotypes more susceptible to CWD (41, 52, 99).
Second, the influence of M or L at codon 132 on CWD pathogenesis was also investigated (36). Transgenic mice expressing cervid PrP with L or M at residue 132 were inoculated with CWD prions from mule deer and elk of various defined Prnp genotypes (M/M, L/L, or M/L) (36). It was found that mice expressing M at 132 (Tg(CerPrP)1536 and Tg(CerPrP)1534) were consistently susceptible to CWD prions from elk with all three genotypes. Mice expressing L (Tg(CerPrP)1973) failed to develop disease following the challenge with all CWD prions (36). These findings suggest that the elk 132 polymorphism controls prion susceptibility at the level of prion strain selection in transgenic mice and that the cervid PrP L132 variant restricts the propagation of CWD prions in these models (36). These findings are supported by cervid studies (100, 101) one of which involved orally administrating elk calves of three different genotypes (132MM, 132LM, and 132MM) with CWD-infected brain and comparing their susceptibility. At 23 months post inoculation (PI), elk with genotypes 132MM developed clinical signs of disease, elk with genotypes 132LM developed clinical signs during month 40 PI, and 132LL elk were still alive at 4 years (100).
Third, to explore the effects of Prnp residue 225 (71), Tg(DeerPrP-F225)5107+/- mice was produced. A phenylalanine (F) substitution for wild type serine (S) is a polymorphism found in cervids and has been implicated in reduced disease prevalence, protracted time course, and even cause variations in disease pathology. Two out of seven Tg(DeerPrP-F225)5107+/- mice developed CWD disease after inoculation with CWD prions after 582 and 609 days. Interestingly, histoblots showed different patterns of PrPSc deposition in the brains of Tg(DeerPrP-F225)5107+/- and Tg(DeerPrP)1536+/- (carrying the S225 allele). Additionally, impaired PMCA efficiency was discovered in the Tg(DeerPrP-F225)5107+/- compared to wild-type PrPC.
Fourth, the role of residue 226 on the selection and propagation of different CWD strains has been examined (66). Deer and elk PrP only differ only at codon 226, and gene-targeted (Gt) mice were developed for both (E226-cervid PrPC (elk) and Q226-cervid PrPC (deer), respectively). Both lines were i.c. challenged with prions isolated from CWD-affected North American deer and elk and the kinetics of disease onset was more rapid in GtE226+/+ mice. Additionally, these mice were inoculated with elk CWD prions that were previously passaged in Tg5037+/+ mice, resulting in disease onset that was 28% faster in GtE226 than in GtQ226 (66). The authors concluded that Gt mice expressing deer or elk PrP are highly susceptible to CWD prions and the effects of amino acid variation at residue 226 impact disease onset. The response of Gt mice to CWD prions introduced by the intraperitoneal (i.p.) and p.o. routes were further investigated (66). GtE226 mice had an incubation time that was 14% shorter compared to GtQ226 via i.p. inoculation (66). These findings concur that the p.o. route is less efficient than either the i.p. or i.c. routes. The incubation times of disease onset in GtE226 were 23% faster compared to GtQ226 mice (66). CWD transmission via cohousing inoculated with uninoculated Gt mice was observed (66). Five days after i.c. inoculation with CWD, GtE226 mice were transferred to new cages with uninoculated GtE226 mice. Following separation, the uninoculated cohoused mice were found to be infected and developed disease. In summary, Bian et al., concluded that GtE226 and GtQ226 mice had distinct kinetics of disease onset and distributions of prions in the brains of diseased mice following i.p., p.o., and i.c. inoculations (66).
4.5 Investigation of transmission concerning structure
Structure plays a significant role in the transmission of CWD, as seen in Tamgüney et al., in which studies demonstrate the C-terminal residue plays a role in the susceptibility and replication of prions (Table 6) (74). This group investigated this role by generating twelve new transgenic mouse lines with the wild-type mouse PrP gene (MoPRNP) removed and replaced with a series of chimeric elk/mouse PrP transgenes. They encoded the N terminus of ElkPrP up to residue Y168 and the C terminus of MoPrP beyond residue 169, designated Elk3M(SNIVVK). Between codons 169 and 219, six residues distinguish ElkPrP from MoPrP, including N169S, T173N, V183I, I202V, I214V, and R219K. The PrP C-terminal residues appear to affect susceptibility to CWD from one species to another and identified more complicated rules regarding prion recruitment (74).

Table 6 Transmission concerning structure.
4.6 Investigation of transmission in regard to immune system mediators
While prions have been found in the nervous tissue, muscles, blood, feces, urine, and saliva (Table 2), lymphoid tissue has been documented as the site of peripheral prion accumulation and formation (102). Michel et al., 2012 and 2013 investigated the immune system roles in CWD, specifically inhibition of prion accumulation, replication, and pathogenesis by CD21/35 complement receptors and complement activation (Table 7). Mice that lack CD21/35 expression (Tg5037;CD21/35-/-) were inoculated with CWD+ elk brain homogenate and showed complete resistance to CWD prions, while Tg5037 mice died of CWD (103). Despite the observation that mice lacking CD21/35 showed resistance to CWD disease manifestations, 3 of 11 brains displayed misfolded prion protein (PrPRES) from the Western blot and densitometric analysis (103). These results reveal the significance CD21/35 cells plays on prion pathogenesis and demonstrated that the host immune system mediators may be a possible target to slow the spread of prions due to its ability to delay peripheral prion accumulation further limiting replication and disease progression (103). Additionally, targeting CD21/35 may have benefits by eliminating the development of new prion strains with expanded host ranges and preventing transmission across species barriers. The impact of complement protein C3 on CWD infection has been studied in mice depleted in C3, Tg5037;C3-/- (102). These mice were inoculated with infected CWD+ brain homogenate and resulted in significant delays in disease development (102). Michel et al. (2012 and 2013) stated the roles of CD21/35 and C3 may further aid in possible therapeutic approaches utilizing complement proteins and their corresponding receptors (102, 103).

Table 7 Investigation of transmission in regard to immune system mediators.
4.7 Investigation of transmission through environmental contamination
Environmental contamination is a concern and complicates effective containment through management practices, as CWD prions are not only shed into the environment from symptomatic deer and asymptomatic deer (33). Environmental contamination may result in CWD prion transmission from contact with contaminated soil (Table 8). CWD transmissibility and environmental perseverance may increase the risk of transmission to other species in optimal conditions (36); therefore, environmental prion contamination is important to guide future management options. Chronic exposure to naturally contaminated soil from mule deer and elk is sufficient for CWD transmission in prion-susceptible mice (35). Environmental contamination was investigated by incubating CWD-infected brain homogenates in a variety of soils and inoculated into transgenic mice (34). The results demonstrated that long-term incubation of CWD prions with soils resulted in decreased recovery of PrPCWD that remained infectious (34). Overall, this study showed that although recovery of PrPCWD bound to soil mineral and whole soils became more difficult with time the prion infectivity remains stable. The detection of CWD prions in soil may be affected by soil type and by the length of time of the prion-soil interaction.

Table 8 Transmission through environmental contamination.
4.8 Investigating transmission in regard to species barriers
Cross-species transmission is influenced by at least two factors, including the sequence similarity between PrPC and PrPSc, and the PrPSc conformation (79). Among mammals, PrPC is a monomer; specific amino acids are hypothesized to impact the intermolecular binding of PrPC and PrPSc (79, 94). CWD is easily transmitted among the various cervid species as Prnp is highly conserved (88). Between residues 23 and 231 of mouse and mule deer Prnp amino acid sequences, there is a 90.2% similarity with the PrPC structures varying between amino acids 165 to 172 (30). It is hypothesized that this may contribute to the inefficient CWD prion conversion in wild-type mice and provide insight into prion species barriers (30).
History proves that disease research is crucial when looking back on the UK outbreak of BSE and the public health mistakes made at that time. Humans were diagnosed with variant CJD after consuming BSE-contaminated beef. Since the first report in 1996, there has been a total of 229 cases have been reported worldwide (104). Although this is quite a low number of cases when considering the high exposure rate, there may be longer incubation times. A clinical case for variant CJD has been reported 25 years after the estimated exposure in 1990 (105). The individual was heterozygous (129 M/V), which is equivalent to position 132 in elk (46, 105). Interestingly, delayed disease onset has been shown with heterozygous elk (132 M/L). Human susceptibility to CWD is unclear due to a lack of evidence of human prion infection linked to CWD-contaminated meat. Exposure is likely, as several million hunters consume venison from areas where the disease is endemic to the wildlife population (75). In multiple studies, CWD has failed to transmit clinical disease to transgenic mice expressing human PrP; however, a recent study has shown CWD infection in two lines of humanized transgenic mice (46). These animals expressed human prion protein (PrP) with amino acids valine or methionine at polymorphic codon 129, a known genetic risk factor for human CJD prion disease (46). When the mice were i.c. inoculated with infectious elk CWD- derived human seeds made using prion misfolding cyclic amplification (PMCA), they developed clinical CWD (46). Understanding what maintains and breaches species barriers provides a very valuable tool for preventing spread of CWD through management practices and avoiding another zoonotic disease crisis; mouse models designed to study species barriers are summarized in Table 9.
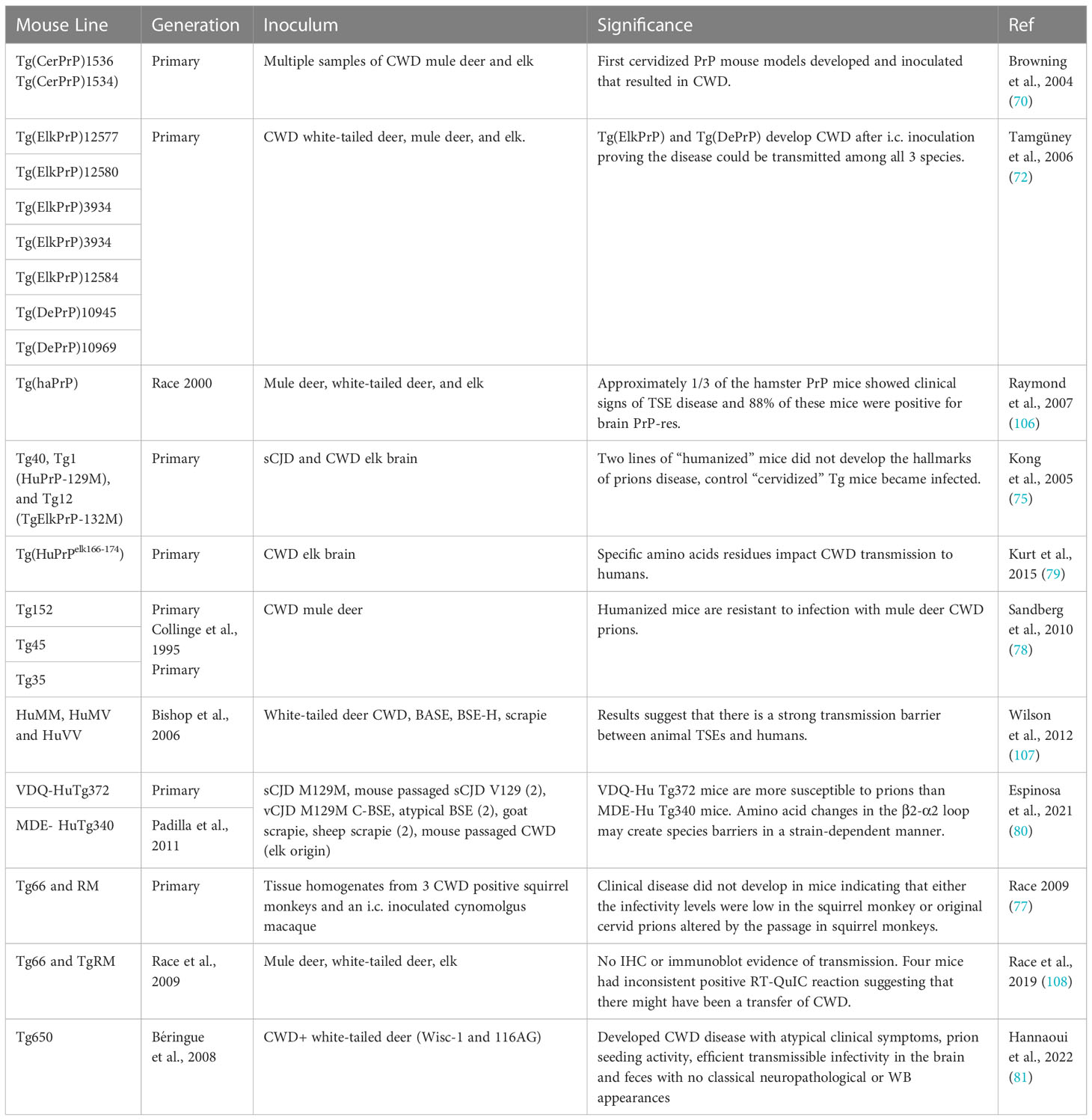
Table 9 Investigating transmission in regard to species barriers.
Browning et al. was the first group to develop and successfully inoculate the very popular transgenic cervidized mouse model lines Tg(CerPrP)1536 and Tg(CerPrP)1534 (70). Mice were i.c. inoculated with a variety of samples from CWD+ mule deer and elk which lead to the development of CWD. The disease presented in spongiform change in the brain and the presence of florid PrP plaques, recognized as key neuropathologic features in cervids with CWD (70). Seven lines of transgenic mice expressing elk and deer prion protein were created and inoculated with different species of CWD inoculum (elk, mule deer, and white-tailed deer), and the characteristics of disease were documented (72). All Tg(ElkPrP) lines developed CWD after inoculation as well as Tg(DePrP), confirming that the disease can be transmitted among all three species (72). Positive CWD isolates from mule deer, white-tailed deer, and elk have also been assessed and proved that CWD can be transmitted and adapted to some mouse models (Table 9) and provided some insight into CWD barriers between species (106). After the second and third serial passages, all Tg (HaPrP) mice showed clinical disease and reduced average incubation periods (106).
A specific area, the β2-α2 loop in PrP, has been implicated in modulating interspecies transmission (71, 109, 110). The transmissibility of CWD to humans was examined by i.c. inoculating humanized Tg mice (Table 9) with elk CWD+ materials (75). Results showed that while there is no species barrier for elk CWD transmission to the cervidized Tg12 mice, the same CWD inocula failed to cause disease in the humanized mice, indicating a species barrier for elk CWD transmission to humans (75). Specific amino acid changes present a substantial structural barrier to CWD zoonoses and provide a new determinant for cross-species prion transmission investigation (79). Transgenic mice were engineered to express human PrP modified with four elk amino acid substitutions at positions 166, 168, 170, and 174 within the beta2-alpha2 loop (79). These human-elk chimeric mice were susceptible to CWD from elk and deer inocula and had longer incubation times than for human CJD, indicating that the structural difference created by changes at residue 143 and 155 in the beta2-alpha2 loop affected cross-species prion transmission.
Sandberg et al. discovered methionine and valine 129 polymorphs of human PrP are resistant to pathological conversion by CWD prions (78). No clinical or subclinical prion infection was observed in i.c. inoculated lines of transgenic mice overexpressing human PrP two-to-six-fold with either methionine or valine at polymorphic residue 129 (78). Espinosa et al. also observed that CWD did not develop after i.c. inoculation in transgenic mice representing the genetic diversity of the PrP codon 129 M/V polymorphisms in the human population, HuMM, HuMV, and HuVV (107). CWD susceptibility and pathogenicity in mouse models consisting of changes in amino acids at positions 166 and 168 in human PrPC was investigated (80). It was discovered that these substitutions, M166V and E168Q affect the species barrier for CWD and other prion diseases in a strain-dependent manner.
TgDeer PrP mice and transgenic mice expressing human PrP were i.c. inoculated with tissue (brain) homogenates from infected squirrel monkeys with PrPres to determine whether the passage of CWD in squirrel monkeys altered the infectious agent (77). Clinical disease did not develop in the mouse lines, indicating that the infectivity levels were low or possible alteration of the original cervid CWD inocula by the passage in squirrel monkeys (77). The lack of transmission to humanized mouse lines indicates that passage through squirrel monkeys did not facilitate adaption to an agent with increased tropism for humans (77). CWD transmission into two human prion protein overexpressor transgenic mouse model lines (Tg66 and TgRM), was examined (108). Four out of 108 mice had inconsistent positive real-time quaking-induced conversion (RT-QuIC) reactions, indicating either detection of residual inoculum or CWD infection transfer (108). The remaining 104 inoculated mice did not have signs of CWD infection by RT-QuIC, immunohistochemistry (IHC), or immunoblot prion detection (108).
Transgenic mice overexpressing human M129-PrPC (tg650) appeared variably susceptible to CWD infection after inoculation with deer CWD isolates (81). Hannaoui et al., reported potential evidence of zoonoses from CWD by inoculating mice with two different CWD strains. The strains that were used were, Wisc-1, which is a strain derived from a white-tailed deer expressing wild type PrP, and 116AG, which is derived from a deer with a polymorphism in Prnp at position 116. Both groups of mice had variable results. A subset developed clinical signs, some of the mice progressed to terminal illness (81). RT-QuIC, western blot analysis, and neuropathological analyses were used to investigate CWD disease in the humanized mouse models. Additionally, they showed some evidence of positive seeding activity in CWD+ mouse feces. These results suggest that CWD may potentially cross the species barrier to humans.
4.9 Investigating transmission regarding strains
Prion strains are a heritable phenotype of disease thought to be determined by specific variations in the prion protein gene (PRNP) sequence and conformation of misfolded prion protein (PrP). They can affect the clinical presentation of disease, and incubation times, and result in differences in disease, neuropathology and biochemical profiles (3, 5, 26, 111, 112). New strains may emerge as a result of interactions of host polymorphisms in the prion protein gene and the invading prion agent, although host-specific pathways that are independent of Prnp may also alter strain phenotypes (113). Strain phenotypes may greatly influence the ability to cross species barriers (66, 114–116). Identification of the epidemiological origin of CWD in new locations may also be performed by comparing strain properties in newly emergent and endemic areas. Three examples of this were demonstrated in Korea, Norway, and Finland, from Jeon et al., Bian et al., and Sun et al. respectively. These, along with Sigurdson et al. and Duque Velasquez et al., utilized the mouse models in Table 10 to investigate transmission regarding strains.
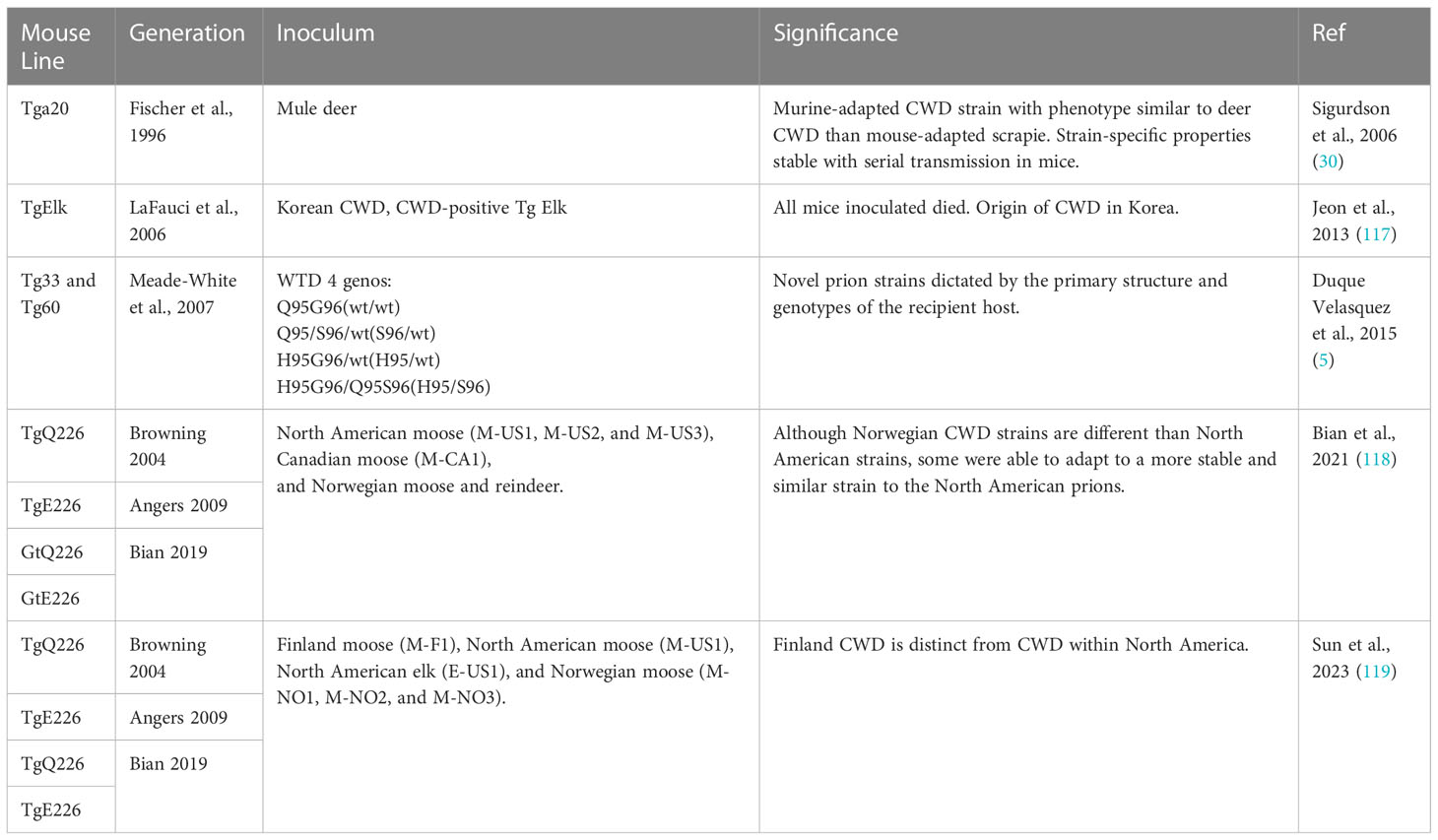
Table 10 Investigating transmission regarding strains.
A murine-adapted CWD strain of prion was generated using a transgenic mouse model that overexpresses murine PrP with a “half genomic” construct or mouse PrP “minigene” lacking intron 2 to aid in investigations studying strain properties (30). It was found that the C-terminal residues played a critical role in prion transmission between species (30). This prion strain displayed unique biochemical and biophysical properties more similar to deer CWD and distinct from the RML mouse-adapted scrapie prion (30).
The characteristics of CWD-associated prions isolated in Korea were investigated using TgElk (Prnp genotype 132 M/M) mice by inoculation of brain homogenate from an imported Canadian elk (117). Results showed incubation times, vacuolar degeneration, and PrPSc accumulation similar to what had previously been reported in the literature in North America. Data suggested that homozygous TgElk mice efficiently became infected and that this model is a valuable and reliable in vivo diagnostic tool (117).
CWD transmission between various genotypes of cervids may result in the generation of novel strains and an expanded host range for CWD (5). Investigation of CWD transmission properties from deer of four Prnp genotypes to transgenic mice expressing the wild-type allele G96 (Tg33) or S96 allele(Tg60) (Table 10) inoculated with four CWD agents from infected white-tailed deer based on their specific Prnp genotypes (5). There was an observed 100% attack rate for the Tg33 mice passaged with deer CWD prions, with significantly longer incubation periods with the CWD H95/S96 prions. The Tg60 mouse inoculated with CWD did not develop disease, however, in this study, the H95/wt and H95/S96 CWD allotypes did develop signs of disease. Serial passage in S96 Tg mice resulted in a new emergent CWD strain. When the first passage Tg60CWD-H95+ isolates were passaged into Tg33 mice, two prion disease presentations appeared to be a mixture of strains (5).
CWD cases in Europe were first discovered in Norway in 2016, approximately 50 years after its origin in North America. Gt mice (66, 118) were inoculated and utilized to determine characteristics of the surfacing Norwegian reindeer (NO) and moose prions and compare them with existing North American CWD prions. Their findings suggest that the emergent Norwegian strains were indeed different than North American strains, however, some isolated were able to adapt after propagation in the Gt mice to a more stable and similar strain to the North American prions. The Gt mice recapitulated the lymphotropic characteristics of naturally occurring CWD strains which improved the transmissibility of the unstable NO reindeer prions, demonstrating the advantages of Gt models in peripheral compartments as opposed to traditional overexpressing Tg mice (118). Similar studies were performed recently utilizing the Gt mouse models (66, 118) to investigate the etiology of CWD found in Finland. These experiments identified novel strain properties in a CWD positive moose, compared to Norweigan strain characteristics, suggesting it was not spread from North American wildlife (119).
4.10 Mouse models used for investigating CWD therapies
Cervidized mouse models have allowed investigation of potential CWD therapies, specifically vaccination (Table 11). To this point, Abdelaziz et al., 2017 and 2018 are the only studies that included cervidized mouse models. Active vaccination was tested to determine if it prevented peripheral CWD infection and prion shedding (61). Mice expressing cervid PrP revealed that all four immunogens (Mmo, Mdi, Dmo, and Ddi) effectively overcame self-tolerance against the prion protein, with high antibody titers and induced self-antibodies in Tg mice expressing deer PrP. Induced post-immune self-antibodies inhibited CWD seeding activity in RT-QuIC (61). This method was used as an in vitro screening platform for testing the potential of anti-PrP antibodies to prevent the formation of PrPSC and propagation of CWD. Active vaccination was also tested, and after mice were challenged with CWD brain homogenate (62). Vaccines included either the adjuvant CpG alone or one of four recombinant PrP immunogens: deer dimer;deer monomer; mouse dimer; and mouse monomer. All vaccinated mice developed ELISA-detectable antibody titers against PrP and lived longer than the unvaccinated control group (62). Overall, these studies provide possible models to test vaccines against CWD with subsequent studies in cervids to determine vaccine efficacy in the natural CWD hosts.

Table 11 Mouse models used for investigating CWD therapies.
5 Conclusion
In this comprehensive review, we have compiled tables and data from investigations that directly impacted our understanding of CWD. A subset of studies have been highlighted to illustrate the use of various mouse models contributing to our comprehension of prion diseases. Among these studies were investigations of infectious tissues and routes of transmission, mouse-adapted prion studies, genetics and structure of the protein, immune system mediators, environmental contamination, species barriers, prion strains, and investigating therapies. These factors are interrelated, and it is difficult to classify studies in just one category; however, we reasoned that it may be helpful for readers to have the clearly impactful findings organized under the main subheadings.
Although CWD has been studied in cervids and mouse models for almost 20 years (70), much remains unknown and unexplored. Critically, we need to identify and phenotype new emergent strains and their potential of transmission to humans and other species. vCJD, caused from ingesting BSE-contaminated meat is currently the only documented zoonotic prion disease, although it remains uncertain if CWD could be transmissible to humans (46, 107). Understanding and characterizing prion strains may lead to insight regarding the influence on species barriers and reduction of disease incidence. Duque Velasquez et al. and Bian et al., used gene-targeted models to investigate how prion strains affect disease pathology, transmissibility, and species barriers; much remains to be answered regarding strains and the effect they have on wildlife populations and epidemiologic factors (5, 66, 118). CRISPR Cas-9 “knock-in” models provide easily malleable representative models of the species of interest and utilizing this technology will be a useful addition for future model generations.
Convenient and early testing methods that could be used in the field would enable more effective management of this disease. Antemortem animal testing methods of easily obtained fluids or tissues using PMCA and RT-QuIC are in use and being improved upon continuously (38, 45, 120–122). Other creative testing methods could include biochemical signatures of disease using the microbiome or other biochemical markers. Biological interactions and the overall health status of the animal may also affect susceptibility and disease regarding CWD. Differential microbes in the gut have been reported based on disease status (123) and could potentially impact susceptibility and pathology of the disease. This “chicken or the egg” concept is intriguing; does the animal develop a differential microbiome due to alterations in the diet because of the disease or does an unhealthy animal become more susceptible?
Elegant experiments involving mouse models add information and provide tools for earlier diagnosis and management of this difficult disease. Investigation of CWD in the mouse model has led to the development of our understanding but needs to be applied in a practical manner regarding the management of wildlife populations, including studies in developing vaccines and investigating their efficacy (61, 62). However, one main concern with vaccines is the difficulty of distributing vaccines to uncontrolled wildlife populations. Disease interventions could include selective breeding for disease-resistant Prnp variants in farmed whitetail deer (51) and potentially apply to free-ranging populations. Perhaps expanding upon current phenotyping methods and development of more comprehensive phenotyping studies in mouse models would provide further insight into transmissibility, strain impact, and earlier diagnostic tools. By improving mouse model development, we may ultimately develop an earlier diagnosis of cervid or human disease and effective therapeutics for disease intervention in the future.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
McLaughlin Research Institute.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Escobar LE, Pritzkow S, Winter SN, Grear DA, Kirchgessner MS, Dominguez-Villegas E, et al. The ecology of chronic wasting disease in wildlife. Biol Rev Camb Philos Soc (2020) 95(2):393–408. doi: 10.1111/brv.12568
2. Ishida Y, Tian T, Brandt AL, Kelly AC, Shelton P, Roca AL, et al. Association of chronic wasting disease susceptibility with prion protein variation in white-tailed deer (Odocoileus virginianus). Prion (2020) 14(1):214–25. doi: 10.1080/19336896.2020.1805288
3. Prusiner SB. Prions. Proc Natl Acad Sci U S A. (1998) 95(23):13363–83. doi: 10.1073/pnas.95.23.13363
4. Saá P, Harris DA, Cervenakova L. Mechanisms of prion-induced neurodegeneration. Expert Rev Mol Med (2016) 18:e5. doi: 10.1017/erm.2016.8
5. Duque Velásquez C, Kim C, Herbst A, Daude N, Garza MC, Wille H, et al. Deer prion proteins modulate the emergence and adaptation of chronic wasting disease strains. J Virol (2015) 89(24):12362–73. doi: 10.1128/JVI.02010-15
6. Linden R. The biological function of the prion protein: A cell surface scaffold of signaling modules. Front Mol Neurosci (2017) 10:77. doi: 10.3389/fnmol.2017.00077
7. Schmitz M, Zafar S, Silva CJ, Zerr I. Behavioral abnormalities in prion protein knockout mice and the potential relevance of PrP(C) for the cytoskeleton. Prion (2014) 8(6):381–6. doi: 10.4161/19336896.2014.983746
8. Watts JC, Prusiner SB. Mouse models for studying the formation and propagation of prions. J Biol Chem (2014) 289(29):19841–9. doi: 10.1074/jbc.R114.550707
9. Imran M, Mahmood S. An overview of animal prion diseases. Virol J (2011) 8(1):493. doi: 10.1186/1743-422X-8-493
11. Moazami-Goudarzi K, Andréoletti O, Vilotte JL, Béringue V. Review on PRNP genetics and susceptibility to chronic wasting disease of cervidae. Vet Res (2021) 52(1):128. doi: 10.1186/s13567-021-00993-z
12. Prusiner SB. Inherited prion diseases. Proc Natl Acad Sci U S A. (1994) 91(11):4611–4. doi: 10.1073/pnas.91.11.4611
13. Wang H, Rhoads DD, Appleby BS. Human prion diseases. Curr Opin Infect Dis (2019) 32(3):272–6. doi: 10.1097/QCO.0000000000000552
14. Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. (2001) 24):519–50. doi: 10.1146/annurev.neuro.24.1.519
15. Kobayashi A, Teruya K, Matsuura Y, Shirai T, Nakamura Y, Yamada M, et al. The influence of PRNP polymorphisms on human prion disease susceptibility: an update. Acta Neuropathol (Berl). (2015) 130(2):159–70. doi: 10.1007/s00401-015-1447-7
16. Prusiner SB. Human prion diseases and neurodegeneration. Curr Top Microbiol Immunol (1996) 207:1–17. doi: 10.1007/978-3-642-60983-1_1
17. Bagyinszky E, Giau VV, Youn YC, An SSA, Kim S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat (2018) 14:2067–85. doi: 10.2147/NDT.S165445
18. Costassa EV, Iulini B, Mazza M, Acutis P, Maurella C, Meloni D, et al. Pathogenesis and transmission of classical and atypical BSE in cattle. Food Saf. (2016) 4(4):130–4. doi: 10.14252/foodsafetyfscj.2016018
19. Dudas S, Czub S. Atypical BSE: Current knowledge and knowledge gaps. Food Saf. (2017) 5(1):10–3. doi: 10.14252/foodsafetyfscj.2016028
20. Gurgul A, Polak MP, Larska M, Slota E. PRNP and SPRN genes polymorphism in atypical bovine spongiform encephalopathy cases diagnosed in polish cattle. J Appl Genet (2012) 53:337–42. doi: 10.1007/s13353-012-0102-4
21. Baylis M, Goldmann W. The genetics of scrapie in sheep and goats. Curr Mol Med (2004) 4:385–96. doi: 10.2174/1566524043360672
23. Rivera NA, Brandt AL, Novakofski JE, Mateus-Pinilla NE. Chronic wasting disease in cervids: Prevalence, impact and management strategies. Vet Med Res Rep (2019) 10:123–39. doi: 10.2147/VMRR.S197404
24. Nalls AV, McNulty E, Powers J, Seelig DM, Hoover C, Haley NJ, et al. Mother to offspring transmission of chronic wasting disease in reeves’ muntjac deer. PloS One (2013) 8(8):e71844. doi: 10.1371/journal.pone.0071844
25. Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis (1980) 16(1):89–98. doi: 10.7589/0090-3558-16.1.89
26. Tranulis MA, Gavier-Widén D, Våge J, Nöremark M, Korpenfelt SL, Hautaniemi M, et al. Chronic wasting disease in Europe: new strains on the horizon. Acta Vet Scand (2021) 63(1):48. doi: 10.1186/s13028-021-00606-x
27. Kim TY, Shon HJ, Joo YS, Mun UK, Kang KS, Lee YS. Additional cases of chronic wasting disease in imported deer in Korea. J Vet Med Sci (2005) 67(8):753–9. doi: 10.1292/jvms.67.753
28. Williams ES, Miller MW. Chronic wasting disease in deer and elk in north America. Rev Sci Tech Int Off Epizoot. (2002) 21(2):305–16. doi: 10.20506/rst.21.2.1340
29. Denkers ND, Seelig DM, Telling GC, Hoover EA. Aerosol and nasal transmission of chronic wasting disease in cervidized mice. J Gen Virol (2010) 91(Pt 6):1651–8. doi: 10.1099/vir.0.017335-0
30. Sigurdson CJ, Manco G, Schwarz P, Liberski P, Hoover EA, Hornemann S, et al. Strain fidelity of chronic wasting disease upon murine adaptation. J Virol (2006) 80(24):12303–11. doi: 10.1128/JVI.01120-06
31. Angers RC, Seward TS, Napier D, Green M, Hoover E, Spraker T, et al. Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis (2009) 15(5):696–703. doi: 10.3201/eid1505.081458
32. Waddell L, Greig J, Mascarenhas M, Otten A, Corrin T, Hierlihy K. Current evidence on the transmissibility of chronic wasting disease prions to humans-a systematic review. Transbound Emerg Dis (2018) 65(1):37–49. doi: 10.1111/tbed.12612
33. Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, et al. Asymptomatic deer excrete infectious prions in faeces. Nature (2009) 461(7263):529–32. doi: 10.1038/nature08289
34. Kuznetsova A, McKenzie D, Cullingham C, Aiken JM. Long-term incubation PrPCWD with soils affects prion recovery but not infectivity. Pathog Basel Switz. (2020) 9(4):E311. doi: 10.3390/pathogens9040311
35. Wyckoff AC, Kane S, Lockwood K, Seligman J, Michel B, Hill D, et al. Clay components in soil dictate environmental stability and bioavailability of cervid prions in mice. Front Microbiol (2016) 7:1885. doi: 10.3389/fmicb.2016.01885
36. Green KM, Browning SR, Seward TS, Jewell JE, Ross DL, Green MA, et al. The elk PRNP codon 132 polymorphism controls cervid and scrapie prion propagation. J Gen Virol (2008) 89(Pt 2):598–608. doi: 10.1099/vir.0.83168-0
37. Nichols TA, Fischer JW, Spraker TR, Kong Q, VerCauteren KC. CWD prions remain infectious after passage through the digestive system of coyotes (Canis latrans). Prion (2015) 9(5):367–75. doi: 10.1080/19336896.2015.1086061
38. Baune C, Wolfe LL, Schott KC, Griffin KA, Hughson AG, Miller MW, et al. Reduction of chronic wasting disease prion seeding activity following digestion by mountain lions. mSphere (2021) 6(6):e0081221. doi: 10.1128/msphere.00812-21
39. Holz CL, Darish JR, Straka K, Grosjean N, Bolin S, Kiupel M, et al. Evaluation of real-time quaking-induced conversion, ELISA, and immunohistochemistry for chronic wasting disease diagnosis. Front Vet Sci (2022) 8:824815. doi: 10.3389/fvets.2021.824815
40. Schwabenlander MD, Rowden GR, Li M, LaSharr K, Hildebrand EC, Stone S, et al. Comparison of chronic wasting disease detection methods and procedures: Implications for free-ranging white-tailed deer (Odocoileus virginianus) surveillance and management. J Wildl Dis (2022) 58(1):50–62. doi: 10.7589/JWD-D-21-00033
41. Thomsen BV, Schneider DA, O’Rourke KI, Gidlewski T, McLane J, Allen RW, et al. Diagnostic accuracy of rectal mucosa biopsy testing for chronic wasting disease within white-tailed deer (Odocoileus virginianus) herds in north America: Effects of age, sex, polymorphism at PRNP codon 96, and disease progression. J Vet Diagn Invest. (2012) 24(5):878–87. doi: 10.1177/1040638712453582
42. Bloodgood J, Kiupel M, Melotti J, Straka K. Chronic wasting disease diagnostic discrepancies: The importance of testing both medial retropharyngeal lymph nodes. J Wildl Dis (2021) 57(1):194–8. doi: 10.7589/JWD-D-20-00007
43. Bravo-Risi F, Soto P, Eckland T, Dittmar R, Ramírez S, Catumbela CSG, et al. Detection of CWD prions in naturally infected white-tailed deer fetuses and gestational tissues by PMCA. Sci Rep (2021) 11(1):18385. doi: 10.1038/s41598-021-97737-y
44. Makarava N. Savtchenko r, baskakov IV. methods of protein misfolding cyclic amplification. In: Lawson VA, editor. Prions. New York, NY: Springer New York (2017). p. 169–83. doi: 10.1007/978-1-4939-7244-9_13
45. Kramm C, Pritzkow S, Lyon A, Nichols T, Morales R, Soto C. Detection of prions in blood of cervids at the asymptomatic stage of chronic wasting disease. Sci Rep (2017) 7(1):17241. doi: 10.1038/s41598-017-17090-x
46. Wang Z, Qin K, Camacho MV, Cali I, Yuan J, Shen P, et al. Generation of human chronic wasting disease in transgenic mice. Acta Neuropathol Commun (2021) 9(1):158. doi: 10.1186/s40478-021-01262-y
47. Barria MA, Gonzalez-Romero D, Soto C. Cyclic amplification of prion protein misfolding. In: Sigurdsson EM, Calero M, Gasset M, editors. Amyloid proteins, vol. 849 . Totowa, NJ: Humana Press (2012). p. 199–212. doi: 10.1007/978-1-61779-551-0_14
48. Moda F. Protein misfolding cyclic amplification of infectious prions. Prog Mol Biol Transl Sci (2017) 150:361–74. doi: 10.1016/bs.pmbts.2017.06.016
49. Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PloS One (2009) 4(3):e4848. doi: 10.1371/journal.pone.0004848
51. Haley N, Donner R, Merrett K, Miller M, Senior K. Selective breeding for disease-resistant PRNP variants to manage chronic wasting disease in farmed whitetail deer. Genes (2021) 12(9):1396. doi: 10.3390/genes12091396
52. Haley NJ, Merrett K, Buros Stein A, Simpson D, Carlson A, Mitchell G, et al. Estimating relative CWD susceptibility and disease progression in farmed white-tailed deer with rare PRNP alleles. PloS One (2019) 14(12):e0224342. doi: 10.1371/journal.pone.0224342
53. Haley NJ, Henderson DM, Donner R, Wyckoff S, Merrett K, Tennant J, et al. Management of chronic wasting disease in ranched elk: conclusions from a longitudinal three-year study. Prion (2020) 14(1):76–87. doi: 10.1080/19336896.2020.1724754
54. Haley NJ, Henderson DM, Wyckoff S, Tennant J, Hoover EA, Love D, et al. Chronic wasting disease management in ranched elk using rectal biopsy testing. Prion (2018) 12(2):93–108. doi: 10.1080/19336896.2018.1436925
55. Conner MM, Miller MW, Ebinger MR, Burnham KP. A meta-BSCI approach for evaluating management intervention on chronic wasting disease in mule deer. Ecol Appl (2007) 1):140–53. doi: 10.1890/1051-0761(2007)017[0140:AMAFEM]2.0.CO;2
56. Manjerovic MB, Green ML, Mateus-Pinilla N, Novakofski J. The importance of localized culling in stabilizing chronic wasting disease prevalence in white-tailed deer populations. Prev Vet Med (2014) 113(1):139–45. doi: 10.1016/j.prevetmed.2013.09.011
57. Mysterud A, Rolandsen CM. A reindeer cull to prevent chronic wasting disease in Europe. Nat Ecol Evol (2018) 2:1343–5. doi: 10.1038/s41559-018-0616-1
58. Mysterud A, Hopp P, Alvseike KR, Benestad SL, Nilsen EB, Rolandsen CM, et al. Hunting strategies to increase detection of chronic wasting disease in cervids. Nat Commun (2020) 11(1):4392. doi: 10.1038/s41467-020-18229-7
59. Mateus-Pinilla N, Weng HY, Ruiz MO, Shelton P, Novakofski J. Evaluation of a wild white-tailed deer population management program for controlling chronic wasting disease in Illinois, 2003–2008. Prev Vet Med (2013) 110(3–4):541–8. doi: 10.1016/j.prevetmed.2013.03.002
60. Uehlinger FD, Johnston AC, Bollinger TK, Waldner CL. Systematic review of management strategies to control chronic wasting disease in wild deer populations in north America. BMC Vet Res (2016) 12(1):173. doi: 10.1186/s12917-016-0804-7
61. Abdelaziz DH, Thapa S, Abdulrahman B, Lu L, Jain S, Schatzl HM. Immunization of cervidized transgenic mice with multimeric deer prion protein induces self-antibodies that antagonize chronic wasting disease infectivity in vitro. Sci Rep (2017) 7(1):10538. doi: 10.1038/s41598-017-11235-8
62. Abdelaziz DH, Thapa S, Brandon J, Maybee J, Vankuppeveld L, McCorkell R, et al. Recombinant prion protein vaccination of transgenic elk PrP mice and reindeer overcomes self-tolerance and protects mice against chronic wasting disease. J Biol Chem (2018) 293(51):19812–22. doi: 10.1074/jbc.RA118.004810
63. Moreno JA, Telling GC. Insights into mechanisms of transmission and pathogenesis from transgenic mouse models of prion diseases. Methods Mol Biol Clifton NJ. (2017) 1658:219–52. doi: 10.1007/978-1-4939-7244-9_16
64. Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell (1990) 63(4):673–86. doi: 10.1016/0092-8674(90)90134-Z
65. Haruyama N, Cho A, Kulkarni AB. Overview: engineering transgenic constructs and mice. Curr Protoc Cell Biol (2009) 42:19.10.1-10.9. doi: 10.1002/0471143030.cb1910s42
66. Bian J, Christiansen JR, Moreno JA, Kane SJ, Khaychuk V, Gallegos J, et al. Primary structural differences at residue 226 of deer and elk PrP dictate selection of distinct CWD prion strains in gene-targeted mice. Proc Natl Acad Sci U S A. (2019) 116(25):12478–87. doi: 10.1073/pnas.1903947116
67. Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev (1996) 10(14):1736–50. doi: 10.1101/gad.10.14.1736
68. Seelig DM, Mason GL, Telling GC, Hoover EA. Pathogenesis of chronic wasting disease in cervidized transgenic mice. Am J Pathol (2010) 176(6):2785–97. doi: 10.2353/ajpath.2010.090710
69. Diack AB, Boyle A, Plinston C, Hunt E, Bishop MT, Will RG, et al. Variant Creutzfeldt-Jakob disease strain is identical in individuals of two PRNP codon 129 genotypes. Brain J Neurol (2019) 142(5):1416–28. doi: 10.1093/brain/awz076
70. Browning SR, Mason GL, Seward T, Green M, Eliason GAJ, Mathiason C, et al. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol (2004) 78(23):13345–50. doi: 10.1128/JVI.78.23.13345-13350.2004
71. Angers R, Christiansen J, Nalls AV, Kang HE, Hunter N, Hoover E, et al. Structural effects of PrP polymorphisms on intra- and interspecies prion transmission. Proc Natl Acad Sci (2014) 111(30):11169–74. doi: 10.1073/pnas.1404739111
72. Tamgüney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, et al. Transmission of elk and deer prions to transgenic mice. J Virol (2006) 80(18):9104–14. doi: 10.1128/JVI.00098-06
73. Meade-White K, Race B, Trifilo M, Bossers A, Favara C, Lacasse R, et al. Resistance to chronic wasting disease in transgenic mice expressing a naturally occurring allelic variant of deer prion protein. J Virol (2007) 81(9):4533–9. doi: 10.1128/JVI.02762-06
74. Tamgüney G, Giles K, Oehler A, Johnson NL, DeArmond SJ, Prusiner SB. Chimeric elk/mouse prion proteins in transgenic mice. J Gen Virol (2013) 94(Pt 2):443–52. doi: 10.1099/vir.0.045989-0
75. Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci Off J Soc Neurosci (2005) 25(35):7944–9. doi: 10.1523/JNEUROSCI.2467-05.2005
76. LaFauci G, Carp RI, Meeker HC, Ye X, Kim JI, Natelli M, et al. Passage of chronic wasting disease prion into transgenic mice expressing rocky mountain elk (Cervus elaphus nelsoni) PrPC. J Gen Virol (2006) 87(Pt 12):3773–80. doi: 10.1099/vir.0.82137-0
77. Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, et al. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis (2009) 15(9):1366–76. doi: 10.3201/eid1509.090253
78. Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O’Malley C, Powell C, et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol (2010) 91(Pt 10):2651–7. doi: 10.1099/vir.0.024380-0
79. Kurt TD, Jiang L, Fernández-Borges N, Bett C, Liu J, Yang T, et al. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest. (2015) 125(4):1485–96. doi: 10.1172/JCI79408
80. Espinosa JC, Marín-Moreno A, Aguilar-Calvo P, Torres JM. Met166 -Glu168 residues in human PrP β2-α2 loop account for evolutionary resistance to prion infection. Neuropathol Appl Neurobiol (2021) 47(4):506–18. doi: 10.1111/nan.12676
81. Hannaoui S, Zemlyankina I, Chang SC, Arifin MI, Béringue V, McKenzie D, et al. Transmission of cervid prions to humanized mice demonstrates the zoonotic potential of CWD. Acta Neuropathol (Berl). (2022) 144(4):767–84. doi: 10.1007/s00401-022-02482-9
82. Mathiason CK, Hayes-Klug J, Hays SA, Powers J, Osborn DA, Dahmes SJ, et al. B cells and platelets harbor prion infectivity in the blood of deer infected with chronic wasting disease. J Virol (2010) 84(10):5097–107. doi: 10.1128/JVI.02169-09
83. Trifilo MJ, Ying G, Teng C, Oldstone MBA. Chronic wasting disease of deer and elk in transgenic mice: oral transmission and pathobiology. Virology (2007) 365(1):136–43. doi: 10.1016/j.virol.2007.03.032
84. Barria MA, Ironside JW, Head MW. Exploring the zoonotic potential of animal prion diseases: in vivo and in vitro approaches. Prion (2014) 8(1):85–91. doi: 10.4161/pri.28124
85. Lee YH, Sohn HJ, Kim MJ, Kim HJ, Park KJ, Lee WY, et al. Experimental chronic wasting disease in wild type VM mice. J Vet Med Sci (2013) 75(8):1107–10. doi: 10.1292/jvms.13-0018
86. Monello RJ, Galloway NL, Powers JG, Madsen-Bouterse SA, Edwards WH, Wood ME, et al. Pathogen-mediated selection in free-ranging elk populations infected by chronic wasting disease. Proc Natl Acad Sci (2017) 114(46):12208–12. doi: 10.1073/pnas.1707807114
87. Johnson C, Johnson J, Vanderloo JP, Keane D, Aiken JM, McKenzie D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J Gen Virol (2006) 87(Pt 7):2109–14. doi: 10.1099/vir.0.81615-0
88. Arifin MI, Hannaoui S, Chang SC, Thapa S, Schatzl HM, Gilch S. Cervid prion protein polymorphisms: Role in chronic wasting disease pathogenesis. Int J Mol Sci (2021) 22(5):2271. doi: 10.3390/ijms22052271
89. O’Rourke KI, Spraker TR, Hamburg LK, Besser TE, Brayton KA, Knowles DP. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol (2004) 85(5):1339–46. doi: 10.1099/vir.0.79785-0
90. Robinson SJ, Samuel MD, O’Rourke KI, Johnson CJ. The role of genetics in chronic wasting disease of north American cervids. Prion (2012) 6(2):153–62. doi: 10.4161/pri.19640
91. Robinson AL, Williamson H, Güere ME, Tharaldsen H, Baker K, Smith SL, et al. Variation in the prion protein gene (PRNP) sequence of wild deer in great Britain and mainland Europe. Vet Res (2019) 50(1):59. doi: 10.1186/s13567-019-0675-6
92. Miller WL, Walter WD. Spatial heterogeneity of prion gene polymorphisms in an area recently infected by chronic wasting disease. Prion (2019) 13(1):65–76. doi: 10.1080/19336896.2019.1583042
93. LaCava MEF, Malmberg JL, Edwards WH, Johnson LNL, Allen SE, Ernest HB. Spatio-temporal analyses reveal infectious disease- driven selection in a free-ranging ungulate. R. Soc. Open Sci. (2021) 8:210802. doi: 10.1098/rsos.210802
94. Orge L, Lima C, Machado C, Tavares P, Mendonça P, Carvalho P, et al. Neuropathology of animal prion diseases. Biomolecules (2021) 11(3):466. doi: 10.3390/biom11030466
95. O’Rourke KI, Besser TE, Miller MW, Cline TF, Spraker TR, Jenny AL, et al. PrP genotypes of captive and free-ranging rocky mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol (1999) 80(10):2765–679. doi: 10.1099/0022-1317-80-10-2765
96. Fox KA, Jewell JE, Williams ES, Miller MW. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol (2006) 87(11):3451–61. doi: 10.1099/vir.0.81999-0
97. Wolfe LL, Fox KA, Miller MW. “Atypical” chronic wasting disease in PRNP genotype 225FF mule deer. J Wildlife Dis (2014) 50(3):660–5. doi: 10.7589/2013-10-274
98. Jewell JE, Conner MM, Wolfe LL, Miller MW, Williams ES. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J Gen Virol (2005) 86(8):2127–34. doi: 10.1099/vir.0.81077-0
99. Robinson SJ, Samuel MD, Johnson CJ, Adams M, McKenzie DI. Emerging prion disease drives host selection in a wildlife population. Ecol Appl (2012) 22(3):1050–9. doi: 10.1890/11-0907.1
100. Hamir AN, Gidlewski T, Spraker TR, Miller JM, Creekmore L, Crocheck M, et al. Preliminary observations of genetic susceptibility of elk (Cervus elaphus nelsoni) to chronic wasting disease by experimental oral inoculation. J Vet Diagn Invest. (2006) 18(1):110–4. doi: 10.1177/104063870601800118
101. O’Rourke KI, Spraker TR, Zhuang D, Greenlee JJ, Gidlewski TE, Hamir AN. Elk with a long incubation prion disease phenotype have a unique PrPd profile. NeuroReport (2007) 18(18):1935–8. doi: 10.1097/WNR.0b013e3282f1ca2f
102. Michel B, Ferguson A, Johnson T, Bender H, Meyerett-Reid C, Wyckoff AC, et al. Complement protein C3 exacerbates prion disease in a mouse model of chronic wasting disease. Int Immunol (2013) 25(12):697–702. doi: 10.1093/intimm/dxt034
103. Michel B, Ferguson A, Johnson T, Bender H, Meyerett-Reid C, Pulford B, et al. Genetic depletion of complement receptors CD21/35 prevents terminal prion disease in a mouse model of chronic wasting disease. J Immunol Baltim Md 1950 (2012) 189(9):4520–7. doi: 10.4049/jimmunol.1201579
104. Maheshwari A, Fischer M, Gambetti P, Parker A, Ram A, Soto C, et al. Recent US case of variant Creutzfeldt-Jakob disease-global implications. Emerg Infect Dis (2015) 21(5):750–9. doi: 10.3201/eid2105.142017
105. Mok T, Jaunmuktane Z, Joiner S, Campbell T, Morgan C, Wakerley B, et al. Variant Creutzfeldt-Jakob disease in a patient with heterozygosity at PRNP codon 129. N Engl J Med (2017) 376(3):292–4. doi: 10.1056/NEJMc1610003
106. Raymond GJ, Raymond LD, Meade-White KD, Hughson AG, Favara C, Gardner D, et al. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. J Virol (2007) 81(8):4305–14. doi: 10.1128/JVI.02474-06
107. Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J Gen Virol (2012) 93(Pt 7):1624–9. doi: 10.1099/vir.0.042507-0
108. Race B, Williams K, Chesebro B. Transmission studies of chronic wasting disease to transgenic mice overexpressing human prion protein using the RT-QuIC assay. Vet Res (2019) 50(1):6. doi: 10.1186/s13567-019-0626-2
109. Kurt TD, Sigurdson CJ. Cross-species transmission of CWD prions. Prion (2016) 10(1):83–91. doi: 10.1080/19336896.2015.1118603
110. Harrathi C, Fernández-Borges N, Eraña H, Elezgarai SR, Venegas V, Charco JM, et al. Insights into the bidirectional properties of the sheep–deer prion transmission barrier. Mol Neurobiol (2019) 56(8):5287–303. doi: 10.1007/s12035-018-1443-8
111. Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol (2007) 8:552–61. doi: 10.1038/nrm2204
112. Duque Velásquez C, Kim C, Haldiman T, Kim C, Herbst A, Aiken J, et al. Chronic wasting disease (CWD) prion strains evolve via adaptive diversification of conformers in hosts expressing prion protein polymorphisms. J Biol Chem (2020) 295(15):4985–5001. doi: 10.1074/jbc.RA120.012546
113. Crowell J, Hughson A, Caughey B, Bessen RA. Host determinants of prion strain diversity independent of prion protein genotype. J Virol (2015) 89(20):10427–41. doi: 10.1128/JVI.01586-15
114. Herbst A, Duque Velasquez C, Triscott E, Aiken JM, McKenzie D. Chronic wasting disease prion strain emergence and host range expansion. Emerg Infect Dis (2017) 23(9):1598-1600. doi: 10.3201/eid2309.161747
115. Otero A, Duque Velasquez C, McKenzie D, Aiken J. Emergence of CWD strains. Cell Tisue Res (2022). doi: 10.1007/s00441-022-03688-9
116. Hannaoui S, Triscott E, Duque Velásquez C, Chang SC, Arifin MI, Zemlyankina I, et al. New and distinct chronic wasting disease strains associated with cervid polymorphism at codon 116 of the prnp gene. PloS Pathog (2021) 17(7):e1009795. doi: 10.1371/journal.ppat.1009795
117. Jeon YC, Choi JK, Choi EK, Carp RI, Kim YS. Pathological characterization of TgElk mice injected with brain homogenate from elk with chronic wasting disease. J Vet Sci (2013) 14(1):21–6. doi: 10.4142/jvs.2013.14.1.21
118. Bian J, Kim S, Kane SJ, Crowell J, Sun JL, Christiansen J, et al. Adaptive selection of a prion strain conformer corresponding to established north American CWD during propagation of novel emergent Norwegian strains in mice expressing elk or deer prion protein. PloS Pathog (2021) 17(7):e1009748. doi: 10.1371/journal.ppat.1009748
119. Sun JL, Kim S, Crowell J, Webster BK, Raisley EK, Lowe DC, et al. Novel prion strain as cause of chronic wasting disease in a moose, Finland. Emerg Infect Dis (2023) 29(2):323–32. doi: 10.3201/eid2902.220882
120. Ferreira NC, Charco JM, Plagenz J, Orru CD, Denkers ND, Metrick MA, et al. Detection of chronic wasting disease in mule and white-tailed deer by RT-QuIC analysis of outer ear. Sci Rep (2021) 11(1):7702. doi: 10.1038/s41598-021-87295-8
121. Kramm C, Soto P, Nichols TA, Morales R. Chronic wasting disease (CWD) prion detection in blood from pre-symptomatic white-tailed deer harboring PRNP polymorphic variants. Sci Rep (2020) 10(1):19763. doi: 10.1038/s41598-020-75681-7
122. Hwang S, Greenlee JJ, Nicholson EM. Real-time quaking-induced conversion detection of PrPSc in fecal samples from chronic wasting disease infected white-tailed deer using bank vole substrate. Front Vet Sci (2021) 8:643754. doi: 10.3389/fvets.2021.643754
Keywords: chronic wasting disease, mouse models, transmission, prion, cervids
Citation: Cook M, Hensley-McBain T and Grindeland A (2023) Mouse models of chronic wasting disease: A review. Front. Virol. 3:1055487. doi: 10.3389/fviro.2023.1055487
Received: 28 September 2022; Accepted: 03 February 2023;
Published: 20 February 2023.
Edited by:
Pedro Martinez-Gomez, Center for Edaphology and Applied Biology of Segura (CSIC), SpainReviewed by:
Christopher J. Silva, United States Department of Agriculture (USDA), United StatesJulie Ann Moreno, Colorado State University, United States
Copyright © 2023 Cook, Hensley-McBain and Grindeland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Grindeland, YW5kcmVhQG1jbGF1Z2hsaW5yZXNlYXJjaC5vcmc=