 Dong-Qiang Cheng
Dong-Qiang Cheng Sandra Kolundžija2
Sandra Kolundžija2 Federico M. Lauro
Federico M. Lauro
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Virol. , 05 September 2022
Sec. Bioinformatic and Predictive Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.981177
This article is part of the Research Topic Methods in Bioinformatic and Predictive Virology View all 4 articles
Viruses of the kingdom Orthornavirae are the causative agents of many diseases in humans, animals and plants and play an important role in the ecology of the biosphere. Novel orthornaviral viral sequences are constantly being discovered from environmental datasets, but generating high-quality and comprehensive phylogenetic trees of Orthornavirae to resolve their taxonomic and phylogenetic relationships is still a challenge. To assist microbial ecologists and virologists with this task, we developed OrViT (OrthornaVirae Tree), a pipeline that integrates and updates published methods and bridges various public software to generate a global phylogenetic tree of the RNA-dependent RNA polymerase (RdRp) encoded by all orthornaviral genomes. The pipeline can infer the phylogenetic relationships between RdRp sequences extracted from the RefSeq viral database and the users’ own assembled contigs or protein datasets. The results from OrViT can be used for the taxonomic identification of novel viruses and suggest revisions of the existing phylogeny of RNA viruses. OrViT includes several Perl and Bash scripts assembled into a Makefile, making it portable between different Linux-based operating systems and easy to use. OrViT is freely available from https://github.com/chengdongqiang/OrViT.
RNA viruses are essential components of the earth ecosystem (1). All RNA viruses are a part of the realm, Riboviria (2) which includes two kingdoms. Kingdom Pararnavirae contains the RNA viruses encoding for a reverse transcriptase (RT). Kingdom Orthornavirae contains the RNA viruses that replicate with a RNA-dependent RNA polymerase (RdRp) and may have one of three genome types: positive-strand (+) ssRNA viruses, negative-strand (-) ssRNA viruses, and double-stranded dsRNA viruses. With the proliferation of massive environmental genome surveys, new Orthornavirae viruses are constantly being discovered (3–8). However, their evolutionary relationships and classification remains challenging. In fact, the only available tool to identify the DNA and RNA viruses is VirSorter2 (9), but the results are hampered by low sensitivity and high false-positive rate. Therefore, the most accurate identification method still relies on high-quality multiple-sequence alignment and phylogeny.
Here we report the development, validation and usage of OrViT, a pipeline that integrates and improves on two published approaches (the Starr method (5) and the Wolf method (4)) and bridges various public software (Prodigal (10), HMMER (hmmer.org), JAligner (11), Mafft (12), Usearch (13), MUSCLE (14), HHsuite (15), PHYLIP (16), Modeltest-ng (17), IQ-TREE 2 (18), and Taxonkit (19)) to generate a global phylogenetic tree of all Orthornavirae with minimal input from the user.
OrViT is based on the fact that all orthornaviral genomes encode a universal molecular marker RdRp. The issue is that the diversity and limited aminoacid conservation of RdRp sequences results in poor quality multiple sequences alignments and phylogenetic trees that are error-prone and subjective. Nevertheless, using sequence alignments, Kamer and Argos first reported a conserved Asp-Asp (DD) motif from 8 different plant and animal (+) ssRNA viruses in 1984 (20). Olivier et al. first reported the four conserved motifs A, B, C, and D among the RdRps in 1989 (21). In 1999, the first complete structure of an RdRp, the HCV RdRp, was reported (22–24). With the identification of these conserved features, a structure-based superposition of RdRps was developed to aid in their alignment, because distantly related RdRps are more structurally conserved than their sequence (25, 26). Yet, even with the structural conservation, it is still challenging to get a high quality sequence alignment of all orthornaviral sequences.
The OrViT pipeline overcomes these limitations by initially extracting the RdRp core domains from the RefSeq sequences and from the user-assembled contigs. Then, it employs a high-quality sequence alignment and tree reconstruction for accurate placement of the users’ sequences on the global RefSeq tree backbone.
OrViT performs the following steps (Figure 1):
1. Search for viral RdRp sequences. The pipeline automatically downloads the latest release of the RefSeq viral protein database from NCBI (ftp.ncbi.nih.gov/refseq/release/viral/). Genes from the user’s assembled contigs are predicted using Prodigal v2.6.3 (10) with default translation table and the “-p meta” option. The RdRp homologs are searched using hmmsearch in HMMER v3.3.2 (hmmer.org) as described by Starr et al. (5) with an e-value cutoff of 0.01. The available Hidden Markov Models (HMMs) of RdRp which Starr employed from PFAM database are downloaded. Those HMMs of RdRp include: Mononeg_RNA_pol [PF00946], RdRP_5 [PF07925], Flavi_NS5 [PF00972], Bunya_RdRp [PF04196], Mitovir_RNA_pol [PF05919], RdRP_1 [PF00680], RdRP_2 [PF00978], RdRP_3 [PF00998], RdRP_4 [PF02123], RVT_1 [PF00078], RVT_2 [PF07727], and Birna_RdRp [PF04197]. One additional HMM profile of Cystoviridae RdRp is downloaded from the figshare repository (5).
2. Excise the RdRp core domain sequences. The full length RdRp sequences may contain accessory domains in addition to the core domain, which must be isolated to produce accurate multiple sequence alignments (MSA). To achieve this, six distantly-related protein structures from PDB database are selected as reference templates (Figure 2). The PDB IDs are 1RA6 (Pisuviricota RdRp), 6V85 (Negarnaviricota RdRp), 4GZK (Duplornaviricota RdRp), 2XI2 (Kitrinoviricota RdRp), 4R71 (Lenarviricota RdRp) and 1MU2 (Reverse Transcriptase). The RdRp core domain of 1RA6 comprises residues 139 to 404 of chain A (1RA6:A:139-404) based on the GenBank annotation. Other core domains are with pairwise flexible structure alignments of each structure to 1RA6:A:139-404 using FATCAT methods v2.0 (Figure 2) (27) with the following resulting residue-ranges: 6V85:A:527-851, 4GZK:A:236-537, 2XI2:A:116-398, 4R71:B:202-422 and 1MU2:A:18-234. Each full length sequence is then aligned to the six reference templates with JAligner (11) and assigned to the reference template with the highest JAligner Smith-Waterman score. JAligner is an open-source Java implementation of the Smith-Waterman algorithm to perform local pairwise sequence alignment with default parameters (the scoring matrix used was BLOSUM62, with open gap penalty of 10, and extend gap penalty of 0.5). The core domain is finally excised after pairwise alignment against its best template using Mafft v7.490 (12), which improved the quality of the global pairwise alignment for latter core domain excision. Because the full length RdRp candidates are longer than their best template, if there are more than 30 gaps at both ends of the alignment, these sites are treated as the excision sites. When performing RdRp core domain excision, we did not consider the domain shuffling or sequence recombination. For example, the family birnaviridae has been reported to include permuted RdRp motifs in C-A-B pattern (28). Here we assumed that the most conserved single core domain, which are critical for the function of RdRp, must maintain the A-B-C order in motifs.
3. Calculate high-quality MSA using RdRps from the RefSeq database. OrViT employs two rounds of iterative hhalign with improved sensitivity and specificity compared to the original Wolf’s approach (4). The first round uses strict filtering parameters to produce a high-quality MSA used as anchor. The second round uses looser parameters that increase the sensitivity with a larger number of homologs. The empirically determined strict filtering parameters exclude sequences with JAligner Smith-Waterman score below 200 for RT (1MU2) templates and 100 for other RdRp templates. After that, the first round of iterative hhalign is performed. Clusters are obtained using Usearch v11.0 with a similarity threshold of 0.5 (13). Each cluster is aligned to the MSA using MUSCLE v5.1 (14). All pairs of clusters are aligned using HHsuite v3.3.0 hhalign command (15). The hhalign scores (SA,B) for a pair of clusters (A and B) are converted to distances as: dA,B=log[sA,B/min(sA,A, sB,B)]. The neighbor command in PHYLIP (Felsenstein, 2005) is used on the matrix of pairwise distance to generate a UPGMA (Unweighted Pair Group Method with Arithmetic mean) tree. Iterative hhalign is performed based on the UPGMA tree progressively replacing each pair of closest leaves with their parental node until one MSA is produced. Partial RdRp core domain sequences are omitted from the MSA if they contain gaps at first conserved aspartic acid (D) of motif A (DXXXXD) or at the last conserved aspartic acid (D) of motif C (GDD). Alignments with e-value less than 0.01 and spanning the full conserved motif A, B, and C are preserved for a less stringent and more sensitive second round of hhalign. The final MSA is checked for gaps at the two most conserved locations.
4. Align the user’s RdRps sequences to the RefSeq RdRps. Individual RdRp sequences extracted from the user’s contigs are aligned to the final reference MSA generated in (3) with hhalign scores sorted from high to low. The final MSA is checked to exclude incomplete sequences if gaps are present at the two most conserved sites.
5. Build the global phylogenetic tree. The evolutionary model is selected by modeltest-ng v0.2.0 (17). The Maximum Likelihood (ML) tree is computed using IQ-TREE 2 (18).
6. Annotate the tree leaves. Annotated sequences from the RefSeq database can be used as references in the global tree. Each RefSeq protein ID is assigned to its corresponding taxon ID. The taxon ID is assigned its taxonomic rank using Taxonkit v0.10.1 software (19).
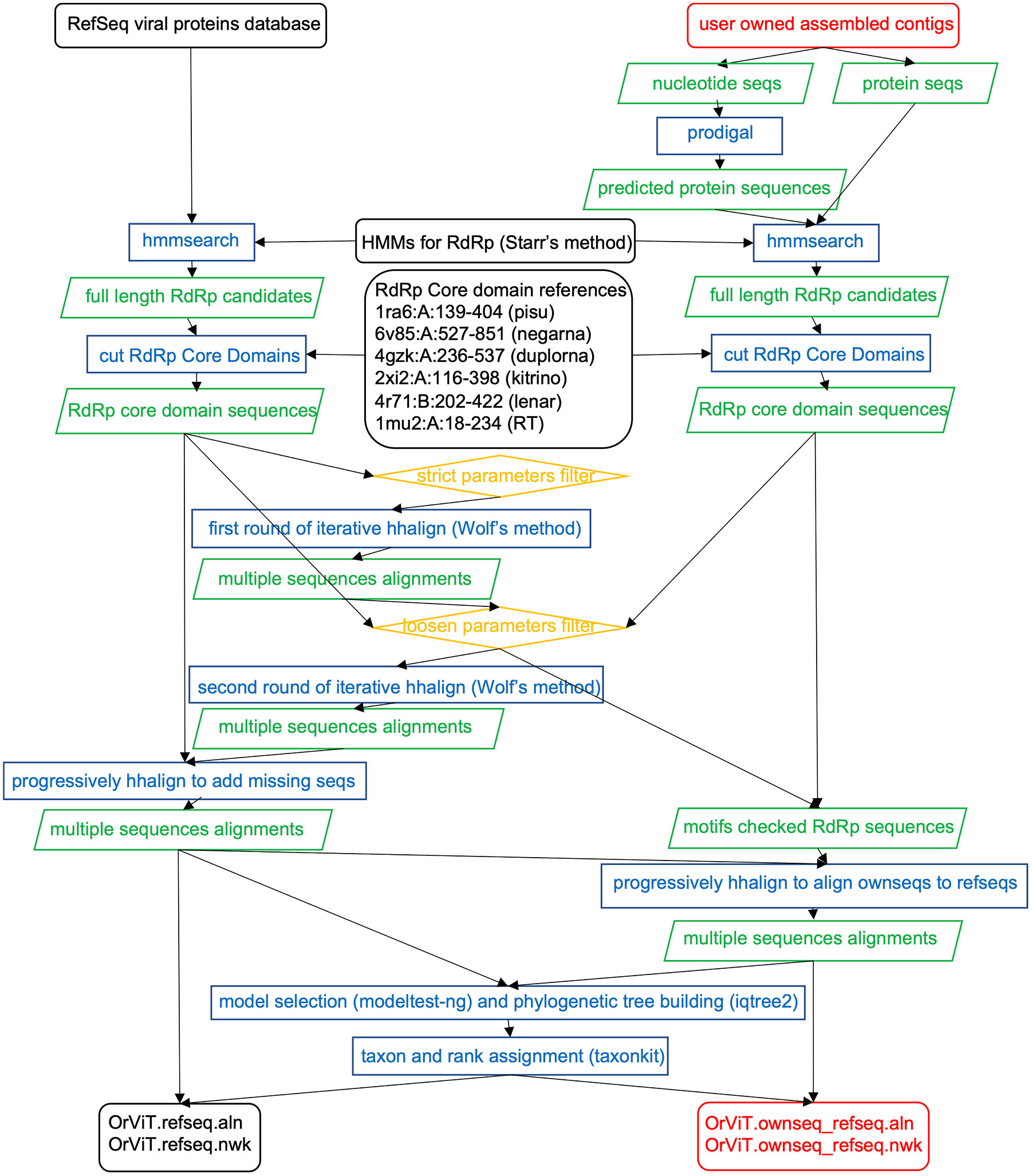
Figure 1 Flowchart of the OrViT pipeline.

Figure 2 Template references used to excise the RdRp core domain. (A) Pairwise flexible structure alignments showing the sequences alignments of the conserved motifs A, B and C. (B) Pairwise flexible structure alignments of the RdRp core domain using FATCAT.
Depending on the usage mode, OrViT generates two phylogenetic trees: OrViT.refseq.tre (Supplementary Figure S1 and Supplementary Figure S2) and OrViT.ownseq_refseq.tre (Supplementary Figure S3) that can be edited and collapsed (Figures 3, 4) using the iTOL webserver (29). In the phylogenetic tree, most of the major clades of Orthornavirae viruses were partitioned consistently with the manually-curated phylogeny of Wolf, etal. (4). After comparing the two separate trees, OrViT.refseq.tre and OrViT.refseq_ownseq.tre, it is clear that the placing of some clades remains uncertain: Duplornaviricota (Reoviridae), Duplornaviricota (Cystoviridae), and Negarnaviricota (Qinviridae). These topologically unstable clades and other unclassified clades are potential new orthornaviral groups. The motif logos of each major clade of the tree (i.e. the three conserved motifs of the RdRp) are consistently aligned as further validation of the robustness and accuracy of the pipeline (Figures 5, 6) (30, 31).

Figure 3 Collapsed phylogenetic tree (OrViT.refseq.tre) of the major groups of Orthornavirae viruses based on sequences from the RefSeq viral proteins database. The names in bracket imply the suffix -viridae.

Figure 4 The collapsed phylogenetic tree (OrViT.ownseq_refseq.tre) showing the Orthornavirae viruses classification of 9 samples (S1-S9) from the Tara Oceans (TO) and Tara Oceans Polar Circle (TOPC) expeditions. Additional information for each sample ID is reported in Supplementary Table S1 and the number of Identified Orthornavirae viruses for each sample is displayed in red.
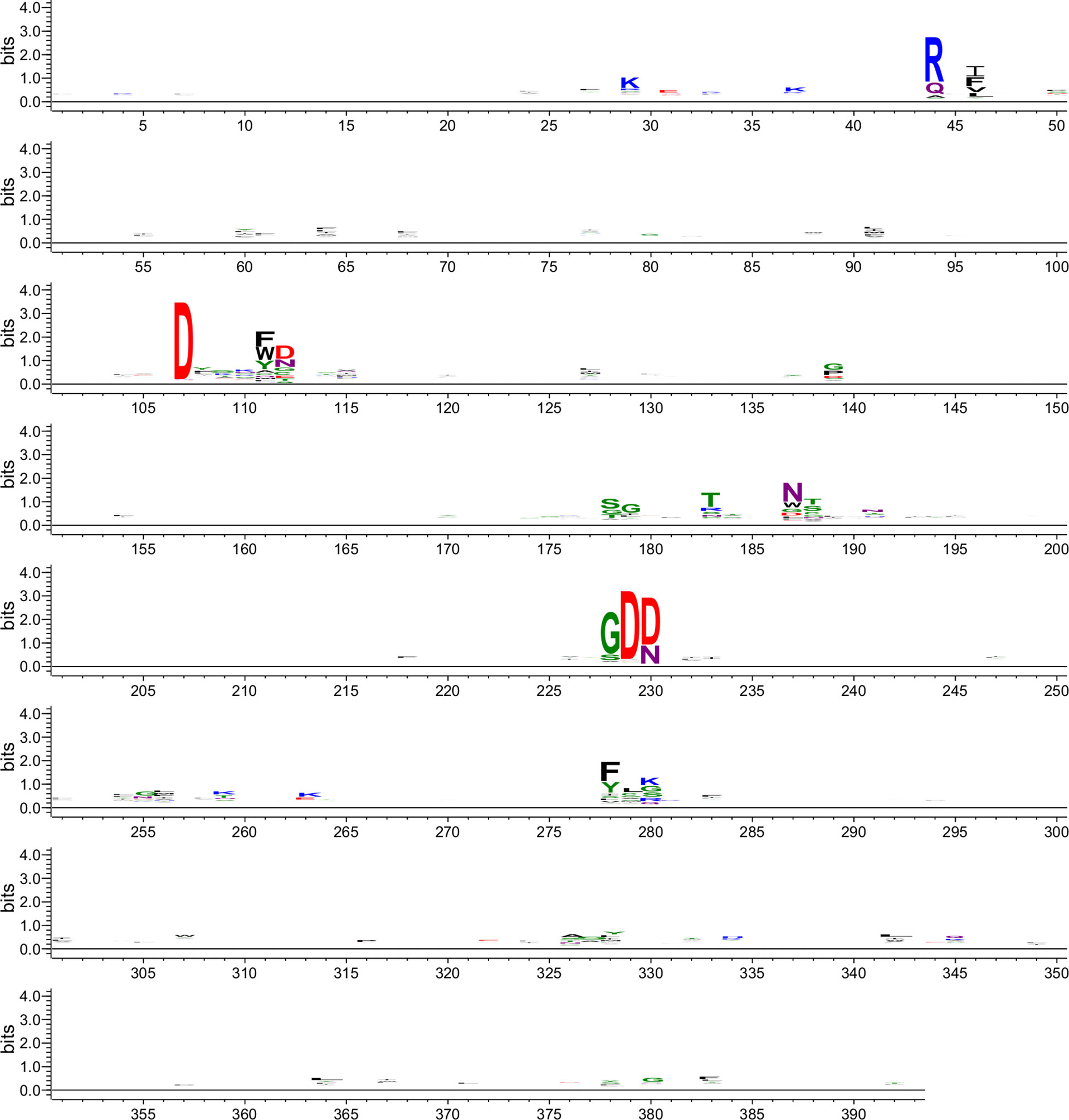
Figure 5 The motif logo generated for all the RdRp core domain sequences searched by OrViT from the RefSeq viral proteins database. Sites 104-115 are the conserved motif A. Sites 175-189 are the conserved motif B. Sites 223-234 are the conserved motif C.
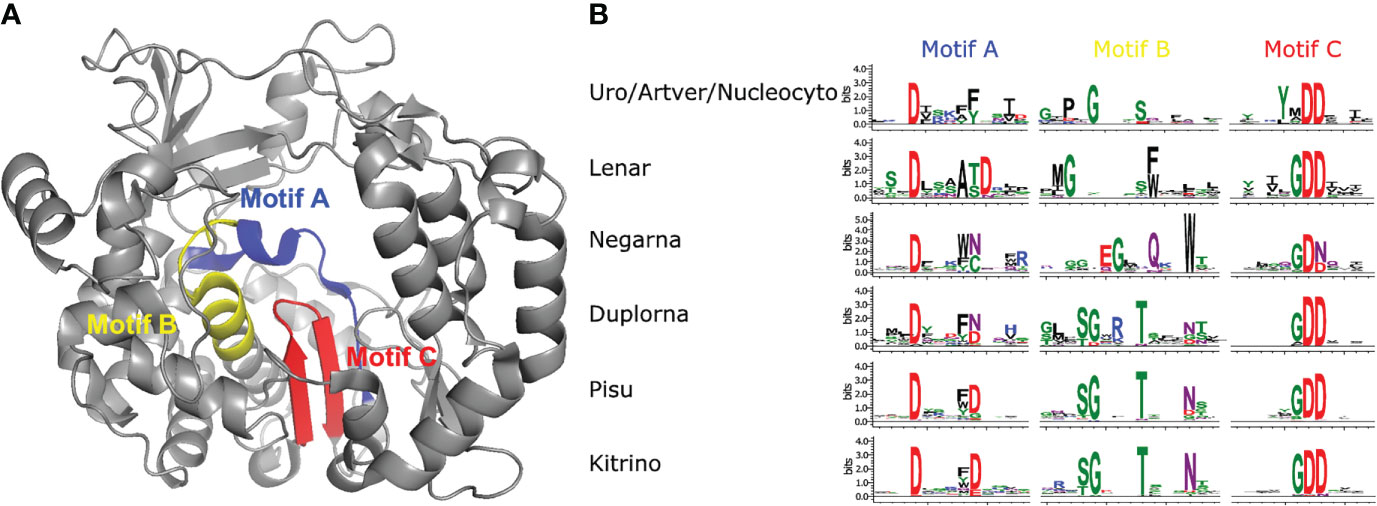
Figure 6 Palmprint motif sequence logos of the RdRp core domains based on the multiple alignments from OrViT.refseq.aln. (A) Motif location superimposed to the PDB 1RA6 structure. (B) Palmprint motif sequence logos. Uro/Artver/Nucleocyto refers to the outgroup Uroviricota/Artverviricota/Nucleocytoviricota. Lenar refers to Lenarviricota (botourmiaviridae/duinviridae/fiersviridae/leviviridae/mitoviridae/narnaviridae/steitzviridae). Negarna refers to Negarnaviricota (aliusviridae/artoviridae/bornaviridae/chuviridae/filoviridae/lispiviridae/mymonaviridae/myriaviridae/natareviridae/nyamiviridae/paramyxoviridae/pneumoviridae/rhabdoviridae/sunviridae/xinmoviridae). Duplorna refers to Duplornaviricota (chrysoviridae/megabirnaviridae/quadriviridae/totiviridae). Pisu refers to Pisuviricota (astroviridae/caliciviridae/dicistroviridae/fusariviridae/hypoviridae/iflaviridae/marnaviridae/picornaviridae/polycipiviridae/potyviridae/secoviridae/solinviviridae/amalgaviridae/curvulaviridae/partitiviridae/picobirnaviridae/arteriviridae/coronaviridae/euroniviridae/gresnaviridae/mesoniviridae/tobaniviridae). Kitrino refers to Kitrinoviricota (alphaflexiviridae/astroviridae/benyviridae/betaflexiviridae/bromoviridae/carmotetraviridae/closteroviridae/deltaflexiviridae/endornaviridae/flaviviridae/hepeviridae/kitaviridae/luteoviridae/matonaviridae/mayoviridae/nodaviridae/sinhaliviridae/togaviridae/tombusviridae/tymoviridae/virgaviridae).
The performance of the pipeline was tested using nine samples from the Tara Oceans (TO) and Tara Oceans Polar Circle (TOPC) expeditions (8, 32, 33). The test samples were chosen to encompass the widest geographic coverage (Supplementary Table S1), were assembled with MEGAHIT v1.2.9 (34) and analysed with the OrViT pipeline resulting in 48 RdRps that could be assigned to 5 different RNA groups (Table 1).

Table 1 Identified Orthornavirae viruses RdRps (n=48) from 9 samples of TO and TOPC expeditions.
The pipeline was further tested using the protein datasets from the assembly of 483 samples (more than half of the total samples) from the TO/TOPC expeditions. The RdRp protein candidates were de-replicated 90% sequence identity using CD-HIT (35). OrViT identified 1442 RdRps (Supplementary Figures S4, S5 and Supplementary Table S2).
The pipeline consists of Perl and Bash scripts, assembled into a Makefile. The GNU parallel shell was applied for executing multiple command jobs at the same time (36). It was tested on a HPE ProLiant DL380 Gen9 Server running Linux (Ubuntu 22.04 LTS) but should be portable with minimal modifications to any Linux-based operating system. The following software dependencies must be installed and made available in the path: curl, wget, hmmer, parallel, mafft, usearch, muscle, hhalign (with -all option), phylip (neighbor), modeltest-ng, iqtree2, taxonkit. The Java software JAligner is included in the src directory.
A global tree based on sequences from both user’s assembled contigs and the RefSeq viral proteins database can be obtained by:
~$git clone https://github.com/chengdongqiang/OrViT.git
~$cd OrViT/src
~$make CONTIGS=/path/to/your/contigs.fasta
Or user’s input contigs as protein sequences by:
~$make CONTIGS=/path/to/your/contigs.fasta SEQTYPE=pro
The global tree based on sequences only from the RefSeq viral proteins database can be obtained by:
~$git clone https://github.com/chengdongqiang/OrViT.git
~$cd OrViT/src
~$make refseq
The outputs will be available in OrViT/results directory. OrViT.refseq.aln and OrViT.ownseq_refseq.aln contain the multiple sequence alignments of the RdRp core domain. OrViT.refseq.tre and OrViT.ownseq_refseq.tre are the phylogenetic trees based on these alignments.
The execution time is approximately 4 days using 50 threads (Makefile with the default option CPU=50) with each thread running at 2.0 GHz. We recommend the use of a terminal multiplexer such as ‘screen’ to ensure the completion of the long-running task.
The novel aspect of OrViT is the ability to identify the RdRp core domain using sequences that are distantly related making use of high-quality pairwise structural alignments. By excising the RdRp core motifs, the pipeline successfully achieves high-quality sequence alignments and phylogenetic trees. Additionally, the integrated pipeline greatly simplifies the challenges in the analysis of orthornaviral taxonomy.
In 2018, Wolf et al. used an iterative hhalign method of 4617 RdRps to obtain a global orthornaviral phylogenetic tree for the first time that determined the identity of the Lenarviricota, Pisuviricota, Kitrinoviricota, Duplornaviricota and Negarnaviricota phyla (4). Subsequently, by mapping an aquatic virome to the backbone of this global tree, they identified more than 4500 distinct novel members of the Orthornavirae, doubling the previously known diversity (6). Edgar et al. performed a sequence alignment (singular on sequence) based data mining of 5.7 million biologically diverse samples (10.2 petabases) and discovered more than 100,000 novel RNA viruses (37). Neri et al. performed data-mining on 5150 metatranscriptomes and identified about 330,000 novel RdRps, their findings also identified two putative new phyla and numerous novel classes and orders (7). Finally, Zayed et al. analysed 771 Global Ocean metatranscriptome samples (28 terabases) and characterised novel phyla as the Taraviricota, Pomiviricota, Arctiviricota, Paraxenoviricota, Wamoviricota (8). The reanalysis of a subset of 483 samples of this data, corroborated the discovery of novel taxonomic groups of Orthornavirae (Supplementary Figures S3, S4). However, only three sequences after de-replication at 90% sequence identity (TARA_N000000269_k119_138062_2, TARA_N000001754_k119_849460_1, and TARA_N000001941_k119_504_1) could be assigned by OrViT to novel taxonomic groups of Orthornavirae. On the contrary, most of the novel TARA sequences grouped into clades located inside the closely-related outgroups of Artverviricota, Nucleocytoviricota, and Uroviricota.
This apparent discrepancy suggests that more research, targeted at the isolation, host-specificity and characterization of the whole genomes harboring environmental RdRps will be needed to establish stable phylogenetic relationships between newly discovered viral groups. Nevertheless, by automating the process of RdRp extraction, core domain alignment and phylogenetic construction, we envision that OrViT will help to expand our understanding of the diversity of Orthornavirae.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
D-QC and FL designed the experiments. D-QC and FL co-write the article. D-QC, SK, and FL revised and approved the submitted version.
This work was supported by the National Research Foundation, Prime’s Minister’s Office, Singapore under its Intra-CREATE Seed Collaboration Grant (Award NRF2018-ITS004-0001) and Competitive Research Programme (Award CRP21-2018-0005). SK is supported by a Singapore International Graduate Award (SINGA) from the Agency for Science, Technology & Research (A*STAR).
We thank Yuri I. Wolf (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health) for his insightful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.981177/full#supplementary-material
Supplementary Figure 1 | Complete phylogenetic tree (OrViT.refseq.tre) of the Orthornavirae based on sequences from the RefSeq viral proteins database.
Supplementary Figure 2 | The collapsed phylogenetic tree (OrViT.refseq.tre) with phylum and class rank labelled.
Supplementary Figure 3 | Complete phylogenetic tree (OrViT.ownseq_refseq.tre) showing the Orthornavirae identified from 9 samples of the TO and TOPC expeditions.
Supplementary Figure 4 | The collapsed phylogenetic tree showing potential novel clades of Orthornavirae based on the protein sequences after de-replication at 90% sequence identity. The de-replicated protein sequences were extracted from 483 sample assemblies from the TO and TOPC expeditions.
Supplementary Figure 5 | The full phylogenetic tree of the Orthornavirae viruses based on the protein sequences after de-replication at 90% sequence identity.
Supplementary Table 1 | List of 9 selected samples from the TO and TOPC expeditions. These 9 samples were used to test the OrViT pipeline with DNA sequences as input.
Supplementary Table 2 | List of the 438 samples from the TO and TOPC expeditions. These 438 samples were used to test the OrViT pipeline with protein sequences as input.
1. Kolundžija S, Cheng D-Q, Lauro FM. RNA Viruses in aquatic ecosystems through the lens of ecological genomics and transcriptomics. Viruses (2022) 14:702. doi: 10.3390/v14040702
2. Koonin EV, Dolja VV, Krupovic M, Varsani A, Wolf YI, Yutin N, et al. Global organization and proposed megataxonomy of the virus world. Microbiol Mol Biol Rev (2020) 84:e00061–19. doi: 10.1128/MMBR.00061-19
3. Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature (2016) 540:539–43. doi: 10.1038/nature20167
4. Wolf YI, Kazlauskas D, Iranzo J, Lucia-Sanz A, Kuhn JH, Krupovic M, et al. Origins and evolution of the global RNA virome. mBio (2018) 9:e02329–18. doi: 10.1128/mBio.02329-18
5. Starr EP, Nuccio EE, Pett-Ridge J, Banfield JF, Firestone MK. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc Natl Acad Sci USA (2019) 116:25900–8. doi: 10.1073/pnas.1908291116
6. Wolf YI, Silas S, Wang Y, Wu S, Bocek M, Kazlauskas D, et al. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat Microbiol (2020) 5:1262–70. doi: 10.1038/s41564-020-0755-4
7. Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, et al. A five-fold expansion of the global RNA virome reveals multiple new clades of RNA bacteriophages. bioRxiv (2022) 02.15.480533. doi: 10.2139/ssrn.4047248
8. Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of earth’s RNA virome. Science (2022) 376:156–62. doi: 10.1126/science.abm5847
9. Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome (2021) 9:37. doi: 10.1186/s40168-020-00990-y
10. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf (2010) 11:119. doi: 10.1186/1471-2105-11-119
11. Moustafa A. JAligner: Open source Java implementation of smith-waterman (2014). Available at: http://jaligner.sourceforge.net.
12. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol (2013) 30:772–80. doi: 10.1093/molbev/mst010
13. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics (2010) 26:2460–1. doi: 10.1093/bioinformatics/btq461
14. Edgar RC. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv (2021) 2021.03.02.433648. doi: 10.1101/2021.06.20.449169
15. Steinegger M, Meier M, Mirdita M, Vohringer H, Haunsberger SJ, Soding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinf (2019) 20:473. doi: 10.1186/s12859-019-3019-7
16. Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics (1989) 5:164–6. doi: 10.1111/j.1096-0031.1989.tb00562.x
17. Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol (2019) 37:291–4. doi: 10.1093/molbev/msz189
18. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
19. Shen W, Ren H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J Genet Genomics (2021) 48:844–50. doi: 10.1016/j.jgg.2021.03.006
20. Kamer G, Argos P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res (1984) 12:7269–82. doi: 10.1093/nar/12.18.7269
21. Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J (1989) 8:3867–74. doi: 10.1002/j.1460-2075.1989.tb08565.x
22. Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis c virus reveals a fully encircled active site. Nat Struct Biol (1999) 6:937–43. doi: 10.1038/13305
23. Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis c virus. Structure (1999) 7:1417–26. doi: 10.1016/S0969-2126(00)80031-3
24. Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis c virus. Proc Natl Acad Sci USA (1999) 96:13034–9. doi: 10.1016/s0969-2126(00)80031-3
25. Peersen OB. A comprehensive superposition of viral polymerase structures. Viruses (2019) 11:745. doi: 10.3390/v11080745
26. Jia H, Gong P. A structure-function diversity survey of the RNA-dependent RNA polymerases from the positive-strand RNA viruses. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.01945
27. Li Z, Jaroszewski L, Iyer M, Sedova M, Godzik A. FATCAT 2.0: towards a better understanding of the structural diversity of proteins. Nucleic Acids Res (2020) 48:W60–4. doi: 10.1093/nar/gkaa443
28. Gorbalenya AE, Pringle FM, Zeddam JL, Luke BT, Cameron CE, Kalmakoff J, et al. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J Mol Biol (2002) 324:47–62. doi: 10.1016/S0022-2836(02)01033-1
29. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res (2021) 49:W293–6. doi: 10.1093/nar/gkab301
30. Yu YK, Capra JA, Stojmirovic A, Landsman D, Altschul SF. Log-odds sequence logos. Bioinformatics (2015) 31:324–31. doi: 10.1093/bioinformatics/btu634Shirley Landicho Ocampo
31. Babaian A, Edgar RC. Ribovirus classification by a polymerase barcode sequence. bioRxiv (2021) 2021.03.02.433648. doi: 10.1101/2021.03.02.433648
32. Salazar G, Paoli L, Alberti A, Huerta-Cepas J, Ruscheweyh HJ, Cuenca M, et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell (2019) 179:1068–83.e21. doi: 10.1016/j.cell.2019.10.014
33. Carradec Q, Pelletier E, Da Silva C, Alberti A, Seeleuthner Y, Blanc-Mathieu R, et al. A global ocean atlas of eukaryotic genes. Nat Commun (2018) 9:373. doi: 10.1038/s41467-017-02342-1
34. Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
35. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
Keywords: orthornavirae, RdRp, global phylogeny, RNA viruses, pipeline
Citation: Cheng D-Q, Kolundžija S and Lauro FM (2022) Global phylogenetic analysis of the RNA-dependent RNA polymerase with OrViT (OrthornaVirae Tree). Front. Virol. 2:981177. doi: 10.3389/fviro.2022.981177
Received: 29 June 2022; Accepted: 15 August 2022;
Published: 05 September 2022.
Edited by:
Samuel Ken-En Gan, Kean University-Wenzhou, ChinaReviewed by:
Xiang Ji, Tulane University, United StatesCopyright © 2022 Cheng, Kolundžija and Lauro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federico M. Lauro, ZmxhdXJvQG50dS5lZHUuc2c=; Dong-Qiang Cheng, ZG9uZ3FpYW5nLmNoZW5nQG50dS5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.