
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Virol. , 23 September 2022
Sec. Modeling of Viral Replication and Pathogenesis
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.973997
This article is part of the Research Topic Kinase/phosphatase signaling and axonal function in health and disease View all 8 articles
 Alexsia Richards1*
Alexsia Richards1* Rudolf Jaenisch1,2
Rudolf Jaenisch1,2Coronaviruses have been responsible for severe outbreaks worldwide over the past 20 years. Most recently, the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) virus was responsible for a global pandemic leading to over 500 million documented cases and over 6 million deaths as of May 2022 (1). Although the primary symptoms of SARS-CoV-2 infection are associated with the respiratory system, there is increasing evidence that severe infection is also associated with neurological complications. Over one third of patients diagnosed with SAR-CoV-2 experience neurological symptoms including stroke, headache, fatigue, impairment of consciousness, myalgia, seizures, smell impairment and taste impairment (2–4). The cause of the neurological symptoms remains unknown.
Damage to neuronal axons in the central nervous system (CNS) can cause headaches, fatigue, nausea, and disorientation (5, 6). In addition, axonal damage is associated with the development of several neurodegenerative diseases including multiple sclerosis (MS), Amyotrophic Lateral Sclerosis (ALS), and Alzheimer’s disease (7–9). The overlap in the symptoms of axonal damage and the neurological symptoms associated with severe SARS-CoV-2 infection led to the hypothesis that SARS-CoV-2 infection may result in axonal damage (10, 11). In support of this hypothesis serum levels of neurofilament light chain (NfL), a highly specific biomarker of axonal damage, are elevated in in SARS-CoV-2 infected patients.
Coronaviruses, including Middle Eastern Respiratory (MERS), SARS-CoV and SARS-CoV-2, activate the cellular kinases p38-MAPK and Casein kinase 2 (CK2) (12–20). Inhibition of these kinases during infection reduces viral replication suggesting that these kinases are specifically targeted by the virus to promote infection (16, 19). Aberrant activation of both p38-MAPK and CK2 can induce axonal damage through the disruption of axonal transport (21–25). In this opinion piece we discuss a possible role for SARS-CoV-2 mediated activation of CK2 and p38-MAPK in inducing the axonal damage associated with infection. We also discuss how the release of pro-inflammatory cytokines by neighboring cells may trigger the initial phosphorylation events that ultimately result in axonal damage. Finally, we discuss how pharmaceutical interventions targeting aberrant kinase activation during SARS-CoV-2 infection could be used to reduce the axonal damage associated with infection.
Although they are not typically thought of as neuroinvasive viruses, there is ample evidence demonstrating that coronaviruses can infect neurons if given the opportunity. SARS-CoV can infect neurons within the central nervous system and cause neurological symptoms similar to what has been observed for SARS-CoV-2 (26). Another closely related coronavirus, HCoV-OC43, uses axonal transport to spread between neurons (27). The neurotropism of SARS-CoV-2 has been an area of intense research since the beginning of the pandemic. The majority of the early data on this topic came from autopsies on patients who succumbed to the infection. In support of direct neuronal infection by SARS-CoV-2, neurons stained positive for the SARS-CoV-2 spike protein in sections of fixed brain tissue (28). However, it should be noted that viral RNA levels in the brain appear significantly lower than in other organs such as the lung or kidneys (29). In addition, some studies have failed to find viral infection in neurons of patients that succumbed to infection (30). These data suggest that the CNS is likely not a primary target of SARS-CoV-2, however, even a low level of infection may be sufficient to cause significant damage.
While primary brain samples have provided valuable insights into the neurological pathology of SARS-CoV-2 infection, the interpretation of results is complicated by interpatient variability and the heterogenous nature of the clinical course of infection. Recent advances in the derivation of 3-D cerebral organoids from human pluripotent stem cells have facilitated the study of viral CNS pathology in a highly controlled system. Cerebral organoids contain the cell types, multi-cellular structures, and epigenomic signatures typically found in the developing human brain (31, 32). Immunofluorescence imaging of SARS-CoV-2 infected cerebral organoids showed co-localization of the SARS-CoV-2 nucleocapsid protein with both neurons and neuronal progenitor cells further demonstrating the capacity of SARS-CoV-2 to infect neural cells (28, 33). Additional data generated using single cell sequencing of infected brain organoids showed SARS-CoV-2 transcripts present in multiple CNS cell types including neurons, glia, and neural stem cells (28). Areas of the brain organoid with high levels of infection also had increased levels of TUNEL-positive cells suggesting that infection may lead to local toxicity within the brain (33). This result is consistent with reports on SARS-CoV which showed extensive neuronal loss at the sites of high levels of infection (34).
Axons are long projections in nerve cells that are responsible for transmitting information both within the CNS and from the CNS to peripheral organs. The severe neurological manifestations of SARS-CoV-2 infection prompted researchers to investigate if infection could cause damage to axons within the CNS. To determine if axonal damage occurred as a result of SARS-CoV-2 infection researchers measured the levels of neurofilament light chain (NfL) in the serum of patients hospitalized for SARS-CoV-2 infection. Neurofilaments (NFs) are highly abundant cytoskeletal proteins found primarily in axons. Damage to the axonal membrane leads to release of NFs into the cerebrospinal fluid (CSF) and blood. Serum and CSF levels of neurofilament light change (NfL), which has the highest solubility of the subunits, are used as a biomarker to detect axonal damage (35, 36). During the acute stage of infection serum NfL levels were significantly higher than in healthy controls (37–39). Higher serum NfL levels correlated with worse clinical outcomes including admittance to intensive care unit and the need for mechanical ventilation (39). Follow up examination on patients with elevated NfL levels showed that NfL levels decline rapidly after the acute stage of infection (37). These data suggest that the axonal damage caused be SARS-CoV-2 infection may be more temporary than what is observed in neurodegenerative diseases.
As research continues into the neurological manifestations of SARS-CoV-2 infection it will be important to quantify the extent of axonal damage using proton magnetic resonance spectroscopy to noninvasively measure axonal damage. This method has been successfully used to measure axonal damage in patients with MS (5). Analysis of magnetic resonance imaging (MRI) images from SARS-CoV-2 patients with neurological complications showed significant changes due to infection however a more targeted examination is needed to determine if these changes included axonal injury (40).
Healthy neurons require the translocation of a wide variety of cellular cargo to spatially discrete neuronal regions including the neuronal soma, axonal initial segment, pre-synaptic terminals, and nodes of Ranvier (24, 41). Within the axon cellular cargo is primarily transported by the molecular motor proteins conventional kinesin (kinesin-1) and cytoplasmic dynein (CDyn) which move along microtubules in a process referred to as fast axonal transport (FAT) (42). Conventional kinesin powers anterograde FAT which moves cargo away from their place of synthesis in the neuronal soma to distal regions of the axon. Conversely, retrograde FAT is driven by CDyn and involves the movement of degraded materials, defective organelles, or neurotrophic signals from axonal subdomains back to the neuronal soma. Inhibition or misregulation of FAT can directly lead to axonal damage and eventual neuronal death [reviewed in (43–45)]. More specifically, mutations that disrupt the activity of molecular motor proteins result in progressive axonal degeneration in a distal to proximal manner (22, 46).
The activity of both CDyn and conventional kinesin can be modulated through cellular kinases. These effects can either be direct, through the phosphorylation of specific subunits of the motor proteins [reviewed (47, 48)], or indirect by phosphorylation of the adaptor proteins that link cellular cargoes to the motor proteins. Activation of the cellular kinases casein kinase 2 (CK2) and p38-MAPK can inhibit retrograde FAT, however, it is not known whether this inhibitory effect involves direct phosphorylation of specific CDyn subunits (23–25). CK2 and p38-MAPK activation also has the potential to impact anterograde FAT. p38-MAPK directly phosphorylates the motor domain of kinesin-1, which reduces the interaction of kinesin-1 with axonal microtubules (Figure 1) (24). Aberrant CK2 activation leads to increased phosphorylation of both kinesin-1 heavy and light chains, the later resulting in the release of kinesin from its cargo (Figure 1) (25).
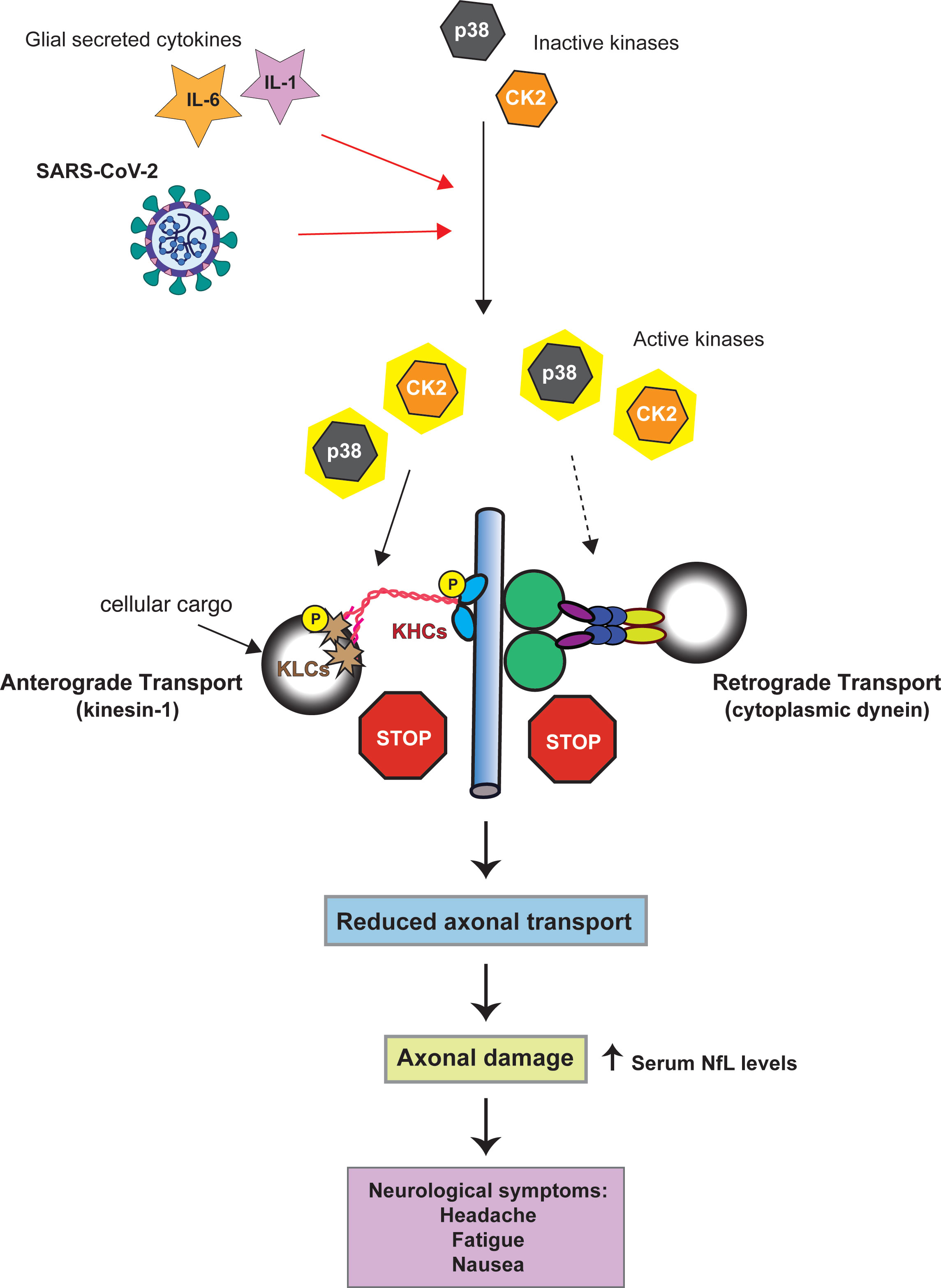
Figure 1 Model linking SARS-CoV-2 kinase activation to axonal damage. Both SARS-CoV-2 and the cytokines IL-1 and IL-6 can activate p38-MAPK and CK2 (red arrows). Activation of p38-MAPK and CK2 can impair the axonal transport of the molecular motor proteins cytoplasmic dynein and kinesin-1(conventional kinesin). Solid black arrows indicate phosphorylation of kinesin-1 subunits (KHC = kinesin heave chain, KLC = kinesin light chain). Whether p38-MAPK and CK2 inhibit retrograde axonal transport through dynein phosphorylation has not yet been established (dashed arrow). By altering the activity of kinases involved in the regulation of FAT SARS-CoV-2 may promote FAT abnormalities eventually triggering neuronal dysfunction and pathology.
Multiple respiratory viruses including respiratory syncytial virus, and influenza virus, have been shown to active p38 signaling (49, 50). Coronavirus infection can result in activation of both p38-MAPK and CK2 (12–14, 16–19). The spike protein of SARS-CoV was sufficient to activate CK2 whereas the 3a protein has been shown to activate p38-MAPK (12, 14). Bouhaddou et al. investigated overall changes in phosphorylation of cellular and viral proteins following SARS-CoV-2 infection (16). Their results showed that infection of Vero E6 cells, which are highly susceptible to SARS-CoV-2, resulted in dramatic increases in the activity of multiple cellular kinases including p38-MAPK and CK2. Expression of the SARS-CoV-2 N protein alone was sufficient to activate CK2, suggesting that this protein may be responsible for kinase activation during infection (16). Inhibitors of p38-MAPK reduce viral replication of both MERS and SARS-CoV-2 demonstrating that this kinase is likely specifically targeted by these viruses and increasing that likelihood of activation being conserved across multiple cell types (16, 19). In addition to activation of p38-MAPK through the expression of SARS-CoV-2 proteins, it is also possible that the generation of double stranded RNA (dsRNA), which occurs during viral genome replication, could result in p38-MAPK activation (51, 52).
Neurons express both p38 and CK2 (53, 54). Therefore, if neurons were to be infected with SARS-CoV-2 it is possible that these kinases could become activated. Consideration of the activation of p38-MAPK and CK2 during SARS-CoV-2 infection – both pathways with known connections to axonal dysfunction, and the observed axonal damage in SARS-CoV-2 infected patients results in a hypothesis that SARS-CoV-2 infects cortical neurons resulting in increased activity of p38-MAPK and CK2 which in turn causes deficits in FAT. This misregulation of axonal transport damages the axon which leads to the observed increase in serum NfL (Figure 1).
As noted in the previous section, SARS-CoV-2 infection of CNS neurons remains a rare outcome of infection (30). In addition, olfactory sensory neurons appear to be resistant to infection, with the virus instead infecting sustentacular cells in the olfactory epithelium (55). In mice ACE2 is primarily expressed in astrocytes around the microvasculature, radial glial cells, epithelial cells, as well as cerebral pericytes (56). A very recent publication showed that in the developing human cortex SARS-CoV-2 infection was limited to cortical astrocytes with minimal infection of other cortical populations (57). For this reason, one essential question to ask is if the virus could induce axonal damage through kinase activation in the absence of direct neuronal infection. SARS-CoV-2 infection induces a massive inflammatory cytokine response which includes IL-6 and IL-1β (58–60). Exposure to the SARS-CoV-2 spike protein alone is sufficient to induce secretion of IL-6 by epithelial cells (61). In addition, viral activation of IL-1β can promote the secretion of IL-6 demonstrating the feedback loops that promote the production of these cytokines from infected cells (62). Both IL-6 and IL-1β can activate CK2 and p38 respectively (63–66). p38 activation following viral infection has been shown to promote IL-1β expression which may in turn promote further kinase activation (50). For this reason, direct infection of neurons by SARS-CoV-2 may not be required for the induction of axonal damage. If the virus infects non-neuronal supporting cells, such as astrocytes, which are adjacent to neurons these cells may secret IL-1β and IL-6 which could activate p38-MAPK and CK2 within nearby neurons leading to misregulated axonal transport and subsequent axonal damage (Figure 1).
Critical to testing both of these hypotheses will be an examination of axonal FAT following SARS-CoV-2 infection. To determine if any observed defects in axonal transport are due to kinase activation, in vitro experiments should be performed with and without inhibitors of p38-MAPK and CK2.
SARS-CoV-2 activates cellular kinases as it proceeds through the viral life cycle. Activation of these kinases has the potential to impact a number of cellular processes. It has begun to be appreciated that one of the processes directly impacted by cellular kinases is axonal transport. Misregulation of axonal transport can cause axonal damage. Therefore, activation of cellular kinases may be one of the mechanisms by which SARS-CoV-2 induces the axonal damage that leads to impaired nerve function. The association between the acute and chronic neurological symptoms associated with SARS-CoV-2 infection is only beginning to be understood but it is possible that axonal damage contributes to both of these pathological conditions. It’s worth noting that related coronaviruses have been found in the brains of patients with MS and a link has been suggested between viral infection and development of the disease (67). The symptoms of MS differ widely from those associated with SARS-CoV-2 infection, however, this finding suggests the potential for long term neurological complications following viral neuroinvasion.
Viral activation of neuronal kinases may represent an essential event in the development of axonal damage, and could be a promising target of therapeutic intervention. Highly specific, brain-permeable kinase inhibitors have recently been developed (68). Inhibitors of p38 and CK2 activation show great potential as antivirals against SARS-CoV-2 (69–71). Treatment with these drugs may have a multifaceted effect on the development of neurological symptoms first by reducing the overall viral load, second by rescuing axonal transport and improving the neurological symptoms associated with SARS-CoV-2 infection.
AR and RJ contributed equally to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. WHO. WHO coronavirus (COVID-19) dashboard. Available at: https://covid19.who.int/ (Accessed July 10, 2022)
2. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol (2020) 19:767–83. doi: 10.1016/S1474-4422(20)30221-0
3. Misra S, Kolappa K, Prasad M, Radhakrishnan D, Thakur KT, Solomon T, et al. Frequency of neurologic manifestations in COVID-19: A systematic review and meta-analysis. Neurology (2021) 97:e2269–81. doi: 10.1212/WNL.0000000000012930
4. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in wuhan, China. JAMA Neurol (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
5. Tartaglia MC, Narayanan S, Francis SJ, Santos AC, De Stefano N, Lapierre Y, et al. The relationship between diffuse axonal damage and fatigue in multiple sclerosis. Arch Neurol (2004) 61:201–7. doi: 10.1001/archneur.61.2.201
6. Mesfin FB, Nishant G, Shapshak AH, Taylor RS. Diffuse axonal injury. StatPearls Publishing (2022). Available at: https://www.ncbi.nlm.nih.gov/books/NBK448102/.
7. Kanaan NM, Pigino GF, Brady ST, Lazarov O, Binder LI, Morfini GA. Axonal degeneration in alzheimer’s disease: when signaling abnormalities meet the axonal transport system. Exp Neurol (2013) 246:44–53. doi: 10.1016/j.expneurol.2012.06.003
8. Brettschneider J, Petzold A, Sussmuth SD, Ludolph AC, Tumani H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology (2006) 66:852–6. doi: 10.1212/01.wnl.0000203120.85850.54
9. Haines JD, Inglese M, Casaccia P. Axonal damage in multiple sclerosis. Mt Sinai J Med (2011) 78:231–43. doi: 10.1002/msj.20246
10. Virhammar J, Naas A, Fallmar D, Cunningham JL, Klang A, Ashton NJ, et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur J Neurol (2021) 28:3324–31. doi: 10.1111/ene.14703
11. Sutter R, Hert L, De Marchis GM, Twerenbold R, Kappos L, Naegelin Y, et al. Serum neurofilament light chain levels in the intensive care unit: Comparison between severely ill patients with and without coronavirus disease 2019. Ann Neurol (2021) 89:610–6. doi: 10.1002/ana.26004
12. Padhan K, Minakshi R, Towheed MAB, Jameel S. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J Gen Virol (2008) 89:1960–9. doi: 10.1099/vir.0.83665-0
13. Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem Biophys Res Commun (2004) 319:1228–34. doi: 10.1016/j.bbrc.2004.05.107
14. Chen IY, Chang SC, Wu HY, Yu TC, Wei WC, Lin S, et al. Upregulation of the chemokine (C-c motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway. J Virol (2010) 84:7703–12. doi: 10.1128/JVI.02560-09
15. Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol (2006) 80:785–93. doi: 10.1128/JVI.80.2.785-793.2006
16. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell (2020) 182:685–712 e19. doi: 10.1016/j.cell.2020.06.034
17. Banerjee S, Narayanan K, Mizutani T, Makino S. Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J Virol (2002) 76:5937–48. doi: 10.1128/JVI.76.12.5937-5948.2002
18. Li SW, Wang CY, Jou YJ, Yang TC, Huang SH, Wan L, et al. SARS coronavirus papain-like protease induces egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep (2016) 6:25754. doi: 10.1038/srep25754
19. Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother (2015) 59:1088–99. doi: 10.1128/AAC.03659-14
20. Lim YX, Ng YL, Tam JP, Liu DX. Human coronaviruses: A review of virus-host interactions. Diseases (2016) 4(3):26. doi: 10.3390/diseases4030026
21. Gibbs KL, Kalmar B, Rhymes ER, Fellows AD, Ahmed M, Whiting P, et al. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis (2018) 9:596. doi: 10.1038/s41419-018-0624-8
22. Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, et al. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase c. Proc Natl Acad Sci U.S.A. (2007) 104:2442–7. doi: 10.1073/pnas.0611231104
23. Morfini G, Szebenyi G, Richards B, Brady ST. Regulation of kinesin: implications for neuronal development. Dev Neurosci (2001) 23:364–76. doi: 10.1159/000048720
24. Morfini GA, Bosco DA, Brown H, Gatto R, Kaminska A, Song Y, et al. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PloS One (2013) 8:e65235. doi: 10.1371/journal.pone.0065235
25. Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, et al. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U.S.A. (2009) 106:5907–12. doi: 10.1073/pnas.0901229106
26. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med (2005) 202:415–24. doi: 10.1084/jem.20050828
27. Dube M, Le Coupanec A, Wong AHM, Rini JM, Desforges M, Talbot PJ. Axonal transport enables neuron-to-Neuron propagation of human coronavirus OC43. J Virol (2018) 92(17):e00404–18. doi: 10.1128/JVI.00404-18
28. Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med (2021) 218(3):e20202135. doi: 10.3892/etm.2021.10943
29. Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med (2020) 383:590–2. doi: 10.1056/NEJMc2011400
30. Solomon IH, Normandin E, Bhattacharyya S, Mukerji SS, Keller K, Ali AS, et al. Neuropathological features of covid-19. N Engl J Med (2020) 383:989–92. doi: 10.1056/NEJMc2019373
31. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501:373–9. doi: 10.1038/nature12517
32. Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, Ecker JR. Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep (2016) 17:3369–84. doi: 10.1016/j.celrep.2016.12.001
33. Ramani A, Muller L, Ostermann PN, Gabriel E, Abida-Islam P, Muller-Schiffmann A, et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J (2020) 39:e106230. doi: 10.15252/embj.2020106230
34. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol (2008) 82:7264–75. doi: 10.1128/JVI.00737-08
35. Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol (2018) 14:577–89. doi: 10.1038/s41582-018-0058-z
36. Barro C, Zetterberg H. Neurological symptoms and blood neurofilament light levels. Acta Neurol Scand (2021) 144:13–20. doi: 10.1111/ane.13415
37. Bozzetti S, Ferrari S, Zanzoni S, Alberti D, Braggio M, Carta S, et al. Neurological symptoms and axonal damage in COVID-19 survivors: are there sequelae? Immunol Res (2021) 69:553–7. doi: 10.1007/s12026-021-09220-5
38. Kanberg N, Ashton NJ, Andersson LM, Yilmaz A, Lindh M, Nilsson S, et al. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology (2020) 95:e1754–9. doi: 10.1212/WNL.0000000000010111
39. Prudencio M, Erben Y, Marquez CP, Jansen-West KR, Franco-Mesa C, Heckman MG, et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci Transl Med (2021) 3(602):eabi7643. doi: 10.1126/scitranslmed.abi7643
40. Yeahia R, Schefflein J, Chiarolanzio P, Rozenstein A, Gomes W, Ali S, et al. Brain MRI findings in COVID-19 patients with PRES: A systematic review. Clin Imaging (2022) 81:107–13. doi: 10.1016/j.clinimag.2021.10.003
41. Black MM. Axonal transport: The orderly motion of axonal structures. Methods Cell Biol (2016) 131:1–19. doi: 10.1016/bs.mcb.2015.06.001
42. Morfini G, Burns M, Stenoien D, Brady S. Axonal transport. In: Brady SS,R, Albers W, Price. ,D, editors. Basic neurochemistry: Principles of molecular, cellular and medical neurobiology, 8 ed, vol. p . Boston: Elsevier (2012). p. 146–64.
43. Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol (2005) 109:5–13. doi: 10.1007/s00401-004-0952-x
44. Brady ST, Morfini GA. Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol Dis (2017) 105:273–82. doi: 10.1016/j.nbd.2017.04.010
45. Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G. Axonal transport and neurological disease. Nat Rev Neurol (2019) 15:691–703. doi: 10.1038/s41582-019-0257-2
46. Griffin JW, Watson DF. Axonal transport in neurological disease. Ann Neurol (1988) 23:3–13. doi: 10.1002/ana.410230103
47. Gibbs KL, Greensmith L, Schiavo G. Regulation of axonal transport by protein kinases. Trends Biochem Sci (2015) 40:597–610. doi: 10.1016/j.tibs.2015.08.003
48. Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci (2009) 29:12776–86. doi: 10.1523/JNEUROSCI.3463-09.2009
49. Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J Immunol (2000) 164:3222–8. doi: 10.4049/jimmunol.164.6.3222
50. Meusel TR, Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-kappa b-independent pathway. J Immunol (2003) 171:3768–74. doi: 10.4049/jimmunol.171.7.3768
51. Steer SA, Moran JM, Christmann BS, Maggi LB Jr., Corbett JA. Role of MAPK in the regulation of double-stranded RNA- and encephalomyocarditis virus-induced cyclooxygenase-2 expression by macrophages. J Immunol (2006) 177:3413–20. doi: 10.4049/jimmunol.177.5.3413
52. Iordanov MS, Paranjape JM, Zhou A, Wong J, Williams BR, Meurs EF, et al. Activation of p38 mitogen-activated protein kinase and c-jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase l, protein kinase r, and alternative pathways. Mol Cell Biol (2000) 20:617–27. doi: 10.1128/MCB.20.2.617-627.2000
53. Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of p38 MAPK in synaptic function and dysfunction. Int J Mol Sci (2020) 21(16):5624. doi: 10.3390/ijms21165624
54. Castello J, Ragnauth A, Friedman E, Rebholz H. CK2-an emerging target for neurological and psychiatric disorders. Pharm (Basel) (2017) 10(1):7. doi: 10.3390/ph10010007
55. Khan M, Yoo SJ, Clijsters M, Backaert W, Vanstapel A, Speleman K, et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell (2021) 184:5932–5949 e15. doi: 10.1016/j.cell.2021.10.027
56. Zhang Y, Archie SR, Ghanwatkar Y, Sharma S, Nozohouri S, Burks E, et al. Potential role of astrocyte angiotensin converting enzyme 2 in the neural transmission of COVID-19 and a neuroinflammatory state induced by smoking and vaping. Fluids Barriers CNS (2022) 19:46. doi: 10.1186/s12987-022-00339-7
57. Andrews MG, Mukhtar T, Eze UC, Simoneau CR, Ross J, Parikshak N, et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc Natl Acad Sci U.S.A. (2022) 119:e2122236119. doi: 10.1073/pnas.2122236119
58. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
59. Hsu RJ, Yu WC, Peng GR, Ye CH, Hu S, Chong PCT, et al. The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front Immunol (2022) 13:832394. doi: 10.3389/fimmu.2022.832394
60. Chen R, Lan Z, Ye J, Pang L, Liu Y, Wu W, et al. Cytokine storm: The primary determinant for the pathophysiological evolution of COVID-19 deterioration. Front Immunol (2021) 12:589095. doi: 10.3389/fimmu.2021.589095
61. Patra T, Meyer K, Geerling L, Isbell TS, Hoft DF, Brien J, et al. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PloS Pathog (2020) 16:e1009128. doi: 10.1371/journal.ppat.1009128
62. Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PloS Pathog (2014) 10:e1004077. doi: 10.1371/journal.ppat.1004077
63. O’Neill LA, Greene C. Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects, and plants. J Leukoc Biol (1998) 63:650–7. doi: 10.1002/jlb.63.6.650
64. Palsson EM, Popoff M, Thelestam M, O’Neill LA. Divergent roles for ras and rap in the activation of p38 mitogen-activated protein kinase by interleukin-1. J Biol Chem (2000) 275:7818–25. doi: 10.1074/jbc.275.11.7818
65. Liu X, Ye F, Xiong H, Hu DN, Limb GA, Xie T, et al. IL-1beta induces IL-6 production in retinal Muller cells predominantly through the activation of p38 MAPK/NF-kappaB signaling pathway. Exp Cell Res (2015) 331:223–31. doi: 10.1016/j.yexcr.2014.08.040
66. Su YW, Xie TX, Sano D, Myers JN. IL-6 stabilizes twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PloS One (2011) 6:e19412. doi: 10.1371/journal.pone.0019412
67. Murray RS, Brown B, Brian D, Cabirac GF. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol (1992) 31:525–33. doi: 10.1002/ana.410310511
68. Maphis N, Jiang S, Xu G, Kokiko-Cochran ON, Roy SM, Van Eldik LJ, et al. Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res Ther (2016) 8:54. doi: 10.1186/s13195-016-0221-y
69. Grimes JM, Grimes KV. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J Mol Cell Cardiol (2020) 144:63–5. doi: 10.1016/j.yjmcc.2020.05.007
70. Cheng Y, Sun F, Wang L, Gao M, Xie Y, Sun Y, et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics (2020) 10:12223–40. doi: 10.7150/thno.50992
71. Cruz LR, Baladron I, Rittoles A, Diaz PA, Valenzuela C, Santana R, et al. Treatment with an anti-CK2 synthetic peptide improves clinical response in COVID-19 patients with pneumonia. a randomized and controlled clinical trial. ACS Pharmacol Transl Sci (2021) 4:206–12. doi: 10.1021/acsptsci.0c00175
Keywords: SARS-C0V-2, neuron, axon damage, kinase, axonal transport disruption
Citation: Richards A and Jaenisch R (2022) Starting signal: Aberrant kinase activation as a trigger for SARS-CoV-2 induced axonal damage. Front. Virol. 2:973997. doi: 10.3389/fviro.2022.973997
Received: 21 June 2022; Accepted: 05 September 2022;
Published: 23 September 2022.
Edited by:
Stefan Kins, University of Kaiserslautern, GermanyReviewed by:
Prasun K. Datta, Tulane University, United StatesCopyright © 2022 Richards and Jaenisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexsia Richards, YXJpY2hhcmRAd2kubWl0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.