 Roger Badia
Roger Badia Edurne Garcia-Vidal
Edurne Garcia-Vidal Ester Ballana
Ester Ballana- 1IrsiCaixa AIDS Research Institute, Hospital Germans Trias i Pujol, Universitat Autònoma de Barcelona, Badalona, Spain
- 2Health Research Institute Germans Trias i Pujol (IGTP), Hospital Germans Trias i Pujol, Universitat Autònoma de Barcelona, Badalona, Spain
The development of antiviral drugs, has provided enormous achievements in our recent history in the fight against viral infections. To date, most of the approved antiviral drugs target virus-encoded proteins to achieve direct antiviral activity. Nonetheless, the inherent idiosyncrasy of viral mutations during their replication cycle, enable many viruses to adapt to the new barriers, becoming resistant to therapies, therefore, representing an ever-present menace and prompting the scientific community towards the development of novel therapeutic strategies. Taking advantage of the increasing knowledge of virus-host cell interactions, the targeting of cellular factors or pathways essential for virus survival turns into an alternative strategy to intervene in almost every step of viral replication cycle. Since host factors are evolutionary conserved, viral evasion to host-directed therapies (HDT) would impose a higher genetic barrier to the emergence of resistant strains. Thus, targeting host factors has long been considered an alternative strategy to overcome viral resistance. Nevertheless, targeting host factors or pathways potentially hints undesired off targets effects, and therefore, a critical risk-benefit evaluation is required. The present review discusses the current state-of-the-art on the identification of viral host dependency factors (HDF) and the workflow required for the development of HDT as antivirals. Then, we focus on the feasibility of using a specific class of host factors, those involved in innate immune modulation, as broad-spectrum antiviral therapeutic strategies. Finally, a brief summary of major roadblocks derived from targeting host cellular proteins and putative future strategies to overcome its major limitations is proposed.
1 Targeting Host Factors as Antiviral Strategies
Viral diseases represent a major cause of mortality across the world and can devastate our global health systems, as illustrated by the current coronavirus disease 2019 (COVID-19) pandemic (1). COVID-19 also demonstrates the need to develop innovative tools that allow the rapid identification of potential therapies when new viral epidemics occur. While vaccines are often the most efficient approach to control viral infections in the long term, their success is somewhat limited to the huge efforts and time needed to administer to the general population together with the fact that they do not provide 100% antiviral efficiency (2). Therefore, it is critical to develop antiviral therapies to treat new emerging viruses or viral infections when prevention fails.
To date, most FDA-approved antiviral drugs target viral proteins involved in its replication cycle (3, 4). However, a major challenge of these antiviral compounds is that they facilitate the common emergence of drug-resistant viral strains (5) and are effective only against particular infections, hampering the possibility of presenting pan-viral efficacy and limiting putative repurposing strategies against emerging new pathogens. Thus, targeting the host proteins required for viral replication is a viable and innovative strategy that can avoid resistance and lead to potentially broad-spectrum therapeutics, as families of viruses often exploit common cellular pathways and processes.
One of the main roadblocks for the identification and development of HDTs is that they require in-depth knowledge of virus-host interactions and their biological significance to virus replication. Indeed, only host factors significantly contributing to viral replication and whose modulation might not alter critical endogenous molecular functions are suitable for pharmacological inhibition for HDT. Deciphering the complex network between the host and the virus is a challenging unsolved question. In recent years new approaches have been developed aimed at identifying these cellular host factors, providing a range of powerful tools to elucidate the direct and indirect interactions between host and pathogens, as well as perturbations to the host cell function, either caused by viral infection itself or by the targeted modulation of specific cellular proteins involved in viral replication.
The initial part of the present article reviews some of the experimental approaches currently in use for the identification of host factors required for viral replication. In the second part of the article, we review the modulation of the innate immune response as a potential therapeutic strategy for the management of virus associated diseases. We focus specifically on how the infections caused by emerging viruses or less studied pathogens, which have little or non-available approved pharmacological interventions, could benefit of host-directed enhancement of the innate sensing as prophylactic and/or therapeutic strategies.
2 Identification of Host Dependency Factors
Drug development is often a too slow process to be readily deployable to an emerging viral outbreak. It is therefore key to establish new methods that facilitate the rapid identification of antiviral compounds to treat viral infections (2). To date, several different approaches have been successfully implemented to identify HDF, although only a few of them have successfully been translated into effective HDT. Examples of successful HDT include the use of C-C chemokine receptor type 5 (CCR5) antagonists for the human immunodeficiency virus (HIV), cyclosporine for influenza A virus (IAV) and anti-claudin-1 and antioccludin monoclonal antibodies for hepatitis C virus (HCV). However, despite these examples, more efforts on translational research are needed to be able to develop effective therapies targeting the host to combat infectious disease.
Overall, current technologies can be classified depending on the type of technique used into:
- Functional genomics; including loss of function/gain of function screenings.
- Genomic-based approaches; including genome-wide association studies and whole transcriptome profiling.
- Proteomic-based approaches, including virus-host protein-protein interactions.
Each screening methodology has its own inherent technical characteristics, providing a picture of a specific moment in the vast context of a viral infection (summarized in Figure 1). Therefore, combined and/or integrative approaches together with functional characterization are necessary to validate the biological relevance of any potential novel HDF that might be putatively developed as HDT. This section provides a brief overview on the fundamentals of the distinct approaches (extensively reviewed elsewhere (6–13), with special focus on their key strengths and weaknesses, and comparing and contrasting the three screening approaches for elucidating host-virus interactions that may ultimately lead to HDT.
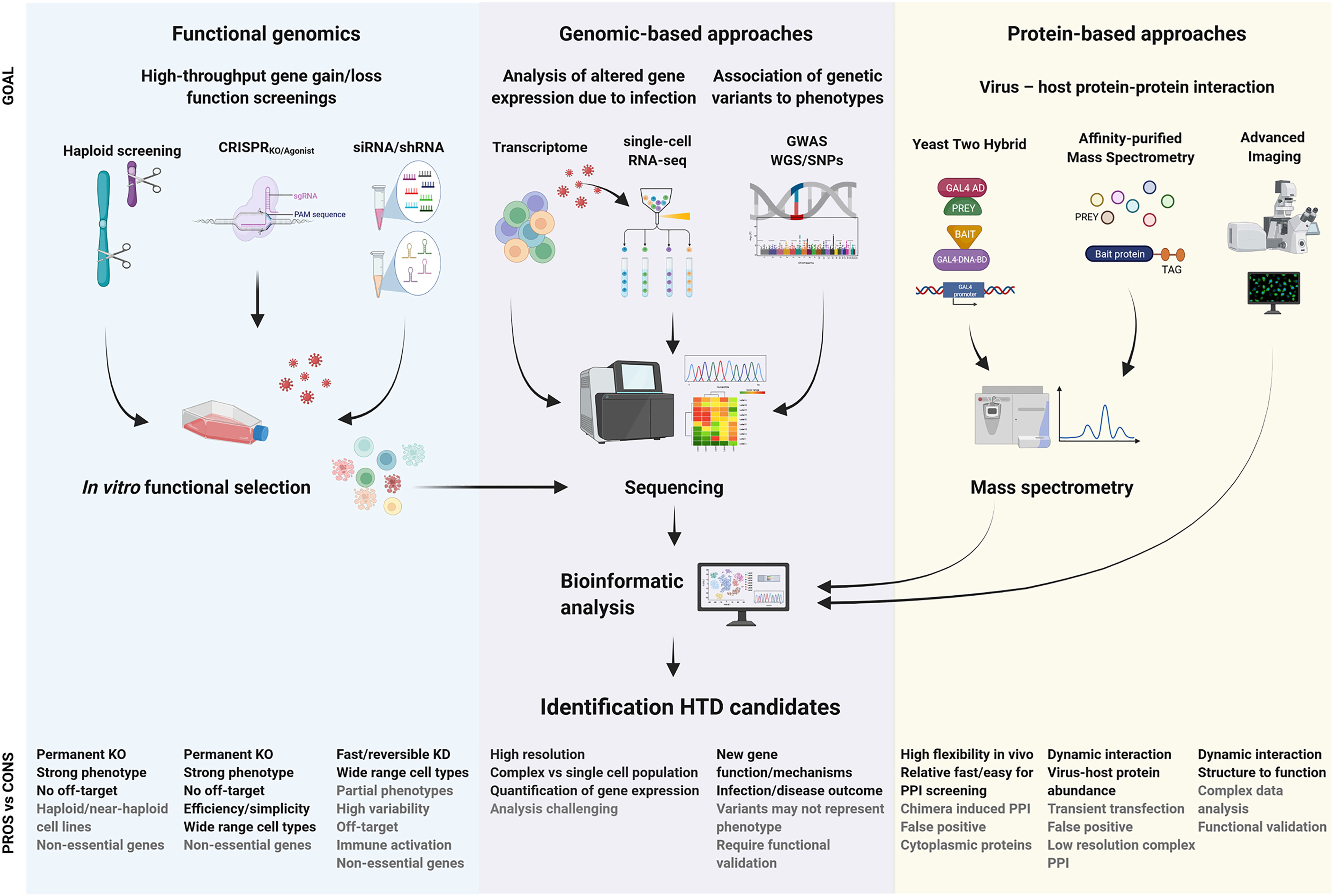
Figure 1 Summary of milestones for the identification of viral host dependency factors and development of host-based therapeutic intervention. The expected purpose of outcome information of each technique is described as “goal” in the figure. The advantages and disadvantages are indicated as “pros vs cons”. Whole-genome sequencing, WGO; single nucleotide polymorphism, SNP; genome-wide association studies, GWAS; knock-out, KO; knock-down, KD; protein-protein interaction, PPI.
2.1 Functional Genomics
Over the last several years a wealth of transformative human-virus interaction discoveries has been produced using loss-of-function functional genomics (9). These insights have greatly expanded our understanding of how human pathogenic viruses exploit our cells to replicate. Two technologies have been at the forefront of this genetic revolution, RNA interference (RNAi) and random retroviral insertional mutagenesis using haploid cell lines (haploid cell screening), with the former technology largely predominating. Currently, the cutting-edge gene editing of the CRISPR/Cas9 system has also been harnessed for large-scale functional genomics and is displacing the earlier methods.
2.1.1 RNA Interference
The RNA interference (RNAi) technology consists of the sequence-specific knockdown of host cell mRNA, resulting in loss-of-function phenotypes. RNAi can either achieve the transiently knock down expression of the gene of interest by the transfection of small interfering RNA (siRNA) or a long-term silencing through transfection of plasmids containing short hairpin RNAs (shRNAs) that integrate into the host genome (14). Although siRNA has identified many cellular components, including host kinases, cellular receptors, transcription factors and transporter proteins as essential HDF factors for viral replication, RNAi screenings have several limitations. First, limited overlap of top hits has been observed in RNAi screening of independent studies, thus, candidate genes found in one screening are rarely confirmed by independent RNAi screenings. This can be attributed to differences in the RNAi sequences for each gene, RNAi reagents, hit selection criteria and the cellular model. Secondly, silencing efficiency of different siRNAs targeting the same gene can vary significantly and result in incomplete gene knockdown that complicates the validation of a host factor. Similarly, siRNA delivery can trigger the innate immune response due to the recognition of RNA via innate immune sensing (described later) and lead to a proinflammatory or antiviral state phenotype (15). To overcome RNAi screening limitations, validation of candidate gene sets should include at least two independent reagents in at least two separate assays (14) besides to the characterization of false positives by the use of non-targeting RNAi controls to evaluate nonspecific off side effects (9, 16). Despite its limitations, genome-wide RNAi screens have successfully provided valuable data such as druggable targets for emerging viruses like the ubiquitin ligase CBLL1 involved in West Nile Virus (WNV) internalization (17) or the de novo pyrimidine synthesis pathway as HDF for genome replication and transcription of Ebola virus (EBOV) (18).
2.1.2 Haploid Cell Screening
Gene silencing for the assessment of potential viral-host factors has to deal with difficulty to generate efficient bi-allelic mutants in diploid cells (6). In contrast, the development of haploid screening takes advantage of the presence of a single allele for each gene in haploid cells to induce efficient gene inactivation, silenced by lentiviruses/retrovirus or transposon-mediated insertional mutagenesis, to generate null mutants for most nonessential genes. Haploid genetic screens have been a useful tool for identifying many HDF. As representative examples, haploid screening identified the cytidine monophosphate N-acetylneuraminicacid synthase (CMAS) and the solute carrier family 35-member A2 (SCL35A2) as receptors for the influenza-hemagglutinin binding. Indeed, gene silencing of SCL35A2 and CMAS turned mutant cells almost complete resistant to influenza infection (6). Similarly, haploid screening found that mutations associated to the endo/lysosomal cholesterol transporter protein Niemann- Pick 1 (NPC1) rendered Hap1 mutant cells resistant to EBOV and Marburg virus infection (19). More recently, insertional mutagenesis in latent HIV-1 pseudohaploid KBM7 cell line (Hap-Lat) allowed the identification of up to 69 novel candidate gene associated to the maintaining of the HIV latency, leading to the identification of several druggable host-factors (e.g. ADK, NF1 and GRIK5) as well as the postulation of the GRIK5 inhibitor topiramate as a viable new latency reversal agent (20).
Although these significant achievements, haploid screens are limited to haploid or near-haploid cell type availability (chronic myeloid cell line KBM7 and HAP-1 for humans) which may limit the biological relevance of the identified candidate in the context of a natural viral infections (13).
2.1.3 CRISPR Screening Technologies
The prokaryotic Clustered regularly interspaced short palindromic repeats/CRISPR - CRISPR associated proteins (CRISPR/Cas) constitute an adaptive immune system that protects bacteria against bacteriophages and plasmids (21). CRISPR/Cas9 allows to edit host cell genome in almost any cell type and it has encouraged many potential applications in biological research in vitro and in vivo, including the virology field, where it is being used for the characterization of HDF through loss- or gain- of function studies.
2.1.3.1. CRISPR-Cas9Knock-Out Screening
The mechanism of action of CRISPR/Cas9 from Streptococcus pyogenesis is extensively described elsewhere (11, 22, 23). Briefly, the identification of HDF through CRISPRKO is based in the use of available or customized synthetic guide RNA (sgRNA) libraries which upon recognition of the target site, induce double strand breaks (DSB), followed by non-homologous end joining (NHEJ) repairing, frameshift mutations and disruption of the gene function (24, 25). Current libraries include distinct number of target genes, from selected candidates to genome-wide approaches; distinct number of sgRNA targeting per gene; different targeted positions within the gene (ORF, promoter, etc.) and also may different in the delivery system (plasmid transfection, pseudovirus infection, etc). Usually, once CRISPRKO is performed, KO cells undergo a functional readout through virus infection followed by a selection process due to virus-induced cell death. The surviving cells are then collected for KO characterization analysis (11, 13, 26). Of note, potential silencing of genes essential for cell survival and growth hampers the study of their role as HDF and arose as one of the limitations of the CRISPR-KO screening studies, besides to the large-scale readout requirements. As example, CRISPR screenings have identified divergent HDF among Flaviviridae family: endoplasmic reticulum (ER) associated multiprotein complexes are critical for DENV, ZIKV and WNV while HCV is dependent on RNA-binding proteins and enzymes involved in metabolism (27). More recently, CRISPR screening has identified GATA6 gene as an HDF involved in the regulation of ACE2 expression required for SARS-CoV-2, suggesting GATA6 as a potential anti-SARS-CoV-2 therapeutic strategy (Ma ‘ayan 28).
CRISPR technologies are under constant development, broadening the potential applications in the virus-host interactions field. Recently, optimization of CRISPR system using Cas12 instead of Cas9, allowed the development of an alternative PAM, which targets T’-rich gene sequences (29). Moreover, gene editing is expanding to other applications as for example the direct silencing of mRNA using the class VI CRISPR/Cas13 systems which hybridizes and cleaves directly the target RNA without the requirement for a PAM sequence (30), as an alternative to siRNA (13).
2.1.3.2 CRISPR Agonist Screenings
Recently developed modifications on the catalytic domains of the Cas9 have resulted in an inactive endonuclease form or “dead” Cas9 (dCas9) but with preserved capacity to interact with sgRNA and bind to targeted DNA sites. Combining sgRNA complementary to proximal sites of the gene promoters and transcription activators (e.g. VP64, p65 or MS2) fused to the dCas9 allow the CRISPR agonist system to recruit transcription factors and initiate gene expression as an alternative to traditional DNA transfection approaches. Several CRISPR agonist systems are commercially available (CRISPR VP64-p65-Rta, CRISPR-Synergistic Activation Mediator, CRISPR-SunTag) which rely on their transcription activators fused to the Cas9 or their capacity to recruit cellular transcription factors as described in (25, 31).
2.2 Genomic-Based Approaches
As a result of our continued interactions with pathogens, our genomes have been shaped through processes of co-evolution, with pathogen-imposed selection pressures leading to selection signatures in ancient and modern human genomes. Thus, understanding how human genetics influence infectious disease susceptibility offers the opportunity for new insights into potential drug targets, and therapies.
As genetic technology has developed, a raft of genetic loci influencing susceptibility to infectious diseases have been discovered through genome-wide association studies (GWAS), including for HIV infection, TB, hepatitis and malaria (32). However, it has become clear that, although human genetics play a role in disease susceptibility, in contrast to other syndromes, GWAS of case–control design may not be the most powerful method to tease out complex host–pathogen relationships. The in-depth understanding of single-gene disorders facilitated by next-generation sequencing technologies has enabled us to better understand the functional mechanisms of host defense to infection through experiments of nature resulting in loss of function (LOF) or, in increasingly recognized instances, gain of function (GOF) of discrete genes. More recently, the development and availability of multi-omics experimental techniques promise to deliver similar mechanistic understanding of the functional importance of genome-level variation at genomic loci implicated in disease pathogenesis. In addition, insights from study of inborn errors of immunity and multi-omics profiling together with developments in analytical methods are further advancing our knowledge of this important area.
2.2.1 Genome-Wide Association Studies
GWAS is useful unbiased methodology for the identification of novel HDF based on the evaluation of hundreds of thousands of genetic variants across the genomes of many individuals. Generally, GWAS aim to associate characteristic allelic differences (genotypes) to specific phenotypes by considering sequence variations in the human genome, as single-nucleotide polymorphisms (SNPs) (33). Applied to virology, deep analysis of genetic heterogeneity between individuals is combined with the clinical data from cohorts and longitudinal data grouped according to their susceptibility to a specific infection and clinical outcome (34). In that sense, GWAS analysis establishing the HIV viral load and disease progression as set points, found significant SNPs respectively associated to the HLA class I region (HLA-B*57, HLA-C), zinc ribbon domain-containing protein 1 (ZNRD1) and ring finger protein 39 (RNF39) as key cellular components for HIV control (34–36). Similarly, SNPs related to interferon gene IFNL3 (formerly interleukin-28B) or IFNL4, are strongly associated with the response to PEG-IFN/RBV treatment against HCV (37, 38). Nowadays, several initiatives are mapping of the genetic architecture associated to the Covid-19, as for example the COVID-19 Host Genetics Initiative (39) or The Severe Covid-19 GWAS Group (40). The latter has already identified genetic variants associated to genes SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6 and XCR1 in Covid-19 patients with respiratory failure. Similarly, GWAS has been useful to identify inborn deficiencies in TLR3/TLR7-dependent type I IFN response in patients suffering Covid-19 pneumonia (41). Further studies will elucidate the functional role and application of present and forthcoming GWAS data for the treatment of SARS-CoV-2/Covid-19.
However, GWAS has methodological limitations besides to the careful inclusion criteria and quality controls requirements. First, population stratification, defined by the undetected presence of systematically genetic diversity in the samples of study that infer inaccurate associations, requires the use of specific analysis to correct stratification. Methods such as principal component analysis or linear mixed models that consider both common and rare variants are advisable (33). Secondly, polygenic statistical models should be considered in cases where thousands of genetic variants might impact in underlying biological process. Finally, identified candidate genes will require additional functional validation to establish unambiguous causality, by combining GWAS data with functional genomics technologies, as siRNA, CRISPR or alternative technologies (42).
2.2.2 Genome-Wide Gene Expression Screening: RNA-Sequencing
The development of Next Generation sequencing has enabled the high throughput assessment of changes in the abundance of mRNA transcripts in a cell population as a result of the perturbation induced by a specific virus. The analysis of differential gene expression through RNA sequencing (RNA-seq) screening requires the use of computational tools to identify the candidate genes modulated by the presence of the virus. Often RNA sequencing screening results in a huge number of genes that will require the use of computational tools to, initially, establish the criteria to define the statistical differentially gene modulation (up- or down- regulation) and integrate/assign gene expression patterns or clusters between the experimental conditions (virus, cell type, time-point, etc.). Next, the use of data bases allows the assignment of the function of genes for which no role has not been previously reported as well as the gene networks modulated due to the infection. The use of transcriptome analysis has provided valuable data to identify host factors involved in DNA replication and chromatin remodeling in HIV infected macrophages (reviewed in (43), the implication of Zaire EBOV proteins VP35 and VP24 in the inhibition of IRF3 and STAT1/STAT2 signaling pathways respectively (44, 45) or, more recently, leaded to the characterization of the host response patterns upon SARS-CoV-2 infection and the upregulation of the protease TMPRSS2 as host factor involved in the replication of SARS-CoV-2 (46).
2.2.3 Single-Cell RNA-Sequencing
Recent advances in sequencing technologies combined to mathematical modeling and computer science have revolutionized the application of “omics” in the virus – host interaction field. Single-cell RNA sequencing (scRNA-Seq) has evolved to overcome the limitations of the population studies such as the heterogeneity in cell cycle or activation that might mask differential expression patterns (12). The scRNA-Seq analysis is based on the cell component of the host-virus interaction and provides an integrative view of the virus–host landscape which can comprise the identification of susceptible cell types to viral infection, characterization of novel immune subtypes involved in viral process, changes in gene expression (e.g. proinflammatory response) and mapping of cellular interacting networks activated or repressed due to virus infection (47).
High-throughput scRNA-Seq workflow is feasible as a result of the use of automated processes for biased selection of cell populations according to cell surface markers, size and/or shape by fluorescence activated cell sorting (FACS) or Magnetic activated cell sorting (MACS), coupled to commercially available optimized RNA processing protocols which handle with the low amount of RNA material for the RNA extraction, reverse transcription, library preparation and sequencing (8, 12). Inclusion of quality controls in scRNA-Seq approaches are compulsory to ensure data reliability and noise reduction prior to the data analysis. Nowadays, available computational packages that include tools such as Principal Component Analyses (PCA) or t-distributed Stochastic Neighbor Embedding (t-SNE) allow a user-friendly intuitive visualization of data clustering previous to further detailed/deep analysis.
2.3 Proteomics
Proteomics approaches allow the unbiased mapping of protein-protein interactions and signaling perturbations due to infection, and ultimately enable structure-function studies. The understanding of these virus–host protein interactions is crucial for the development of treatments and preventive measures against infectious diseases. Over the past decade, proteomic approaches have become prime contributors to the discovery and understanding of virus–host interactions that represent anti‐ and pro‐pathogenic cellular responses (extensively reviewed (10).
2.3.1 Yeast Two-Hybrid
The yeast two hybrid system (Y2H) is a valuable method for the determination of viral-host binary protein interactions. The principle of the method is based in the reconstitution of a split protein from fragments to restore its function (48). The proteins to be evaluated (bait and prey) are fused to fragments of a split protein, as for example the Gal4 transcriptor activator, where its DNA binding domain and Gal4 activation domain are fused respectively to bait and prey protein and then co-expressed in budding yeast cells. If protein interaction happens, Gal4 function is restored and detected by a reporter gene. This method allows large scale and automatization pipeline for high-throughput screening (48, 49). Y2H screening approaches can be performed testing a complex mixture of preys available through a cDNA library or, alternatively array-based screening for predefined individual bait vs prey pair assessment. In that sense, Y2H has successfully identified HDFs interacting with the HIV-1 integrase, such as LEDGF/p75, Transportin-SR2 (TNPO3) and von Hippel-Lindau binding protein 1 (VBP1) during the integration process (50). Similarly, Y2H identified the interaction between Vpx (SIV/HIV-2) or Vpr (HIV-1) with Cyclin L2 as HDF regulating viral replication in quiescent cells and macrophages through a DCAF1 and SAMHD1 dependent mechanisms (51).
Nevertheless, Y2H has some limitations: Since the system is set up to co-express prey and bait proteins in yeast, it may result in misfolding or lack post-translational modifications, especially those related to membrane compartments, impairing interaction detection and resulting in false negative events. Similarly, the co-expression of proteins in a non-natural environment may also result in false positives. Although the percentage of false positive events is similar to other screening methodologies post-filtering analyses are required for a reliable data of biologically relevant virus-host protein interactions (52).
2.3.2 Affinity Purification-Mass Spectrometry – Based Proteomics
Affinity purification-mass spectrometry (AP-MS), a low-biased protein interaction-based approach, provides an integrative view of the virus-host interaction network. Briefly, AP-MS rely in the of use engineered affinity purification tags fused to either the N- or C-terminus of the target viral protein to efficiently purify the protein(s) of interest. The tagged protein together with its interacting proteins are affinity purified, trypsinized into peptide fragments, separated by liquid chromatography and analyzed by mass spectrometer (MS). Peptide intensity spectra will allow the identification of the proteins present in the mixture (53, 54). To overcome technical difficulties in affinity purification, commonly used C-terminal green fluorescent protein- or FLAG- tag among others tags, have been progressively replaced for tandem affinity purification (TAP) tag systems. TAP is based in two contiguous epitope tags are fused to the targeted protein followed by two-stage purification. TAP greatly reduces the nonspecific binding contamination, improving downstream MS analysis (55).
Optimal AP-MS experimental design can also provide spatial and temporal information of the virus-host interactions. For example, chemical cross-linking stabilizes transient virus-host macromolecular complexes (commonly associated to signaling cascades regulation or enzyme-substrate binding) or the selective enrichment of subcellular organelles or compartments, render a dynamic virus-host interaction into a detectable process by AP-MS (7, 54). Indeed, AP-MS also has been shown to be informative about viral RNA-host protein interaction, changes in virus-host protein abundance and post-translational modifications as a result of a viral infection (7, 26, 54).
The high sensitivity of the AP-MF generates a huge number of candidates which will require data processing prior to functional characterization. First, it is recommended to filter a true interaction from a contaminant by computational algorithms, such as significance analysis of interaction (SAINT), and/or by checking in protein interaction databases (E.j. CRAPome, VirusMINT, others). Next, computational tools (e.j. Search Tool for Retrieval of interacting gens/proteins database;STRING) are useful to visualize predictive functional associations, pathways and biological processes modified by viral infection prior to functional validation of the candidates.
2.3.3 Advanced Imaging for Virus-Host Interaction Studies
Advanced imaging methodologies, including improved high-resolution light microscopy, confocal microscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM) or cryo-EM, among others, take advantage of new optical imaging tools to perform spatio-temporal assessment of the viral replication cycle, for a better understanding of viral structures and interactions within the cell between host proteins and viral counterparts. These techniques have been extensively reviewed elsewhere (56–61).
Among them, it is worth to mention Imaging Flow cytometry (IFC), a technique which merges flow cytometry and fluorescence microscopy, enabling a high-throughput analysis of cell morphology besides to a multi-channel fluorescence imaging of single cell (see (57) for review). Applied to virology, ICF is useful to study subtle virus-host interactions, such as viral entry interactions, tracking intracellular distribution of viral proteins or nucleic acids, with a high resolution. ICF has been useful to characterize the HCMV immediate-early 1 and 2 (IE1/2) viral antigen as a countermeasure against antiviral restriction factor SAMHD1 in infected macrophages (62).
Another technique that bridges structure to function is the single-molecule Förster resonance energy transfer imaging (smFRET) (reviewed in (56). Briefly, FRET refers to a non-radioactive energy transfer between a donor and acceptor fluorophores, site-specifically labeling the molecules of interest, excited by a laser and fluorescence recorded by total internal fluorescence microscope (TIRF) or confocal microscopy. As example, smFRET has been used to study conformational changes of the HIV-1 surface envelope protein (Env) upon engaging with cellular CD4 receptor and coreceptors CCR5/CXCR4, providing useful information for the development of vaccines seeking the induction of broadly neutralizing antibodies (bnAbs) or antibody therapies with bnAbs (56, 63, 64).
Advanced imaging technologies may provide a huge amount of relevant information of viral dynamics but also will require consistent data analysis to prevent imaging artifacts in conjunction with conventional functional and biochemical assays for an integrative validation of the results (56).
3 Innate Immune Modulation as a Putative Host Directed Antiviral Therapy
Innate immune cells, including macrophages, dendritic cells, neutrophils, innate lymphoid cells among other cell types, constitute the front line of the defense against pathogen infections. These cells express germline-encoded pattern recognition receptors (PRRs) that recognize evolutionary conserved microbial structures, named pathogen-associated molecular patterns (PAMPs), as well as endogenous molecules released from damaged cells, known as damage associated molecular patterns (DAMPs) (65, 66). The knowledge of novel PPRs and pathways is under constant development, due to review scope limitations, the PRR families discussed here are the Toll-like receptors (TLRs), retinoic acid-inducible gene I-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and the cytosolic dsDNA sensors (CDS). Nonetheless, other PRRs such as the C-type lectin receptors (CLRs) and absent in melanoma 2 (AIM2)-like receptors among other PRRs, might be also interesting as alternative for host-based antiviral approaches and therefore, reference (67) is recommended.
The ligands and signaling mechanisms differ in the different PRRs families. However, they do share common features: upon a PAMPs or DAMPs recognition, signaling cascades will generally lead to the production of type I interferon (IFN-I), the induction of a huge amount of interferon stimulated genes (ISGs) and the release of proinflammatory cytokines and chemokines, which in an autocrine and paracrine manner, will promote an antiviral state, the pathogen clearance as well as the priming of the adaptive immunity (66, 68). Interestingly, the PRRs sensing pathways have been shown to be interconnected. First, the activation of the signaling cascades upon ligand binding of the different sort of PPRs often converge in the activation of the same downstream molecular intermediates. For example, main PRR signaling routes often converge at the MAVS, IKK complex or IKK-related proteins among other signal transduction elements or transcription factors (NF-kB, IRF3, IRF7) to promote gene transcription. Secondly, although PRRs do have some degree of pathogen specialization, in some cases can sense a broader sort of ligands than expected, being involved in the sensing of distinct pathogens, from DNA to RNA viruses or even bacteria. Altogether, overlapping sensing and signaling represents opportunity for the development of targeted pan-antiviral therapies.
3.1 IFN-Based Therapies
The antiviral activity of interferon (IFN) was first reported in 1954 (69) and 1957 (70–72), and described as a host factor responsible of virus inactivation. Since then, significant advances have shed light on the intricate signaling pathways conforming innate immune response, which led to the development of distinct types of therapeutic approaches, from targeting IFN itself to more specific interventions targeting key innate immune proteins and pathways.
The interferon family is divided in three classes: type-I IFN, the largest IFN family in humans, which comprises IFNα and IFNβ among others (73); type-II IFN or IFNγ (74) and type-III IFN or IFNλs, which were characterized in 2003 (75, 76). Whereas type-I and –III are mainly produced by infected cells, IFNγ is produced in T and NK cells upon antigenic and/or mitogenic stimuli (74) and therefore seems to be part of the adaptive immune response, in contrast to the innate immune nature of the other two classes (77, 78).
Due to the role of IFN in inhibiting viral infection and/or spreading, some viruses have evolved in order to overcome or avoid its effects (reviewed in (79) and (80)). In general, viral immune evasion strategies can be classified in (i) avoid detection by corresponding intracellular sensors, (ii) blockade of intracellular signaling pathways leading to IFN production and (iii) masking PAMPs through chemical modification. The identification and description of the host cellular proteins involved in these strategies might represent also interesting targets for therapeutic intervention.
Examples of viral immune evasion strategies include, the host factor TRIM25 interaction with non-structural protein 1 (NS1) of influenza A virus, leading to inhibition of RIG-I signaling pathway and IFN induction (67, 81). Other examples include the Sendai virus, in which viral respirovirus C protein binds to IFNAR2 and therefore inhibits JAK1/TYK2 activation (82). On the other hand, albeit all known flavivirus encode the NS5 protein to overcome JAK-STAT signaling and IFN-associated response the underlying mechanism of mediated by NS5 protein differs significantly among the Flavivirus family (see (83) for review). In that sense, DENV NS5 triggers proteasomal degradation of STAT2 through the binding to the host ubiquitin protein ligase E3 component N-recognin 4 protein (UBR4). Similarly, ZIKV NS5 triggers proteasomal degradation of STAT2 in a mechanism that remains unclear (84). Alternatively, NS5 of other Flavivirus, such as the WNV, interacts with the host protein prolidase, an enzyme participating in the metabolism of collagen, to impair IFNAR1 maturation and cell surface expression, leading to an upstream blocking of the JAK-STAT pathway. Although targeting at the NS5 has been proposed as direct-acting antiviral intervention (18, 85), its multiple host counterparts still remain to explored as HDTs for the management of flavivirus infections.
Similarly, filovirus such as EBOV, inhibits IFN-response at different levels (86). First, EBOV VP35 circumvent innate sensing by RLRs due to its capacity to bind double-stranded RNA (dsRNA), blocking RIG-I and MDA-5 recognition. Indeed, VP35 has been shown to prevent PACT-induced RIG-I ATPase activity and RIG-I activation (87, 88). Moreover, as alternative host-based intervention to be explored, VP35 also impairs TBK-1/IKKϵ interaction with IRF3, blocking IRF3 dimerization, phosphorylation and nuclear translocation (86). Other EBOV proteins also participate in the IFN-evasion mechanisms as for example VP24 which has been described to arrest nuclear transportation of tyrosine phosphorylated STAT1 through the interaction with cellular protein NPI-1 (86).
Recently, in the context of SARS-Cov-2 infection, several SARS-CoV-2 accessory proteins ORF3b, ORF6, ORF7a and ORF8 have been described to act as IFN-I antagonists to impair host immune response as reviewed in (89). Among these viral proteins, ORF6 localizes at the nuclear pore complex (NPC) and interacts with host proteins Rae1 and Nup98, forming a complex that inhibits cytoplasm-nucleus trafficking of transcription factors STAT1 and STAT2, thus blocking their nuclear translocation and suppressing IFN associated response (90, 91).
In summary, the existence of viral-encoded immune evasion strategies to bypass the IFN response underlines the importance of the IFN pathway and supports the idea of developing therapeutic strategies through the targeted modulation of involved host factors. In that sense, the modulation of IFN response, and specifically the JAK-STAT pathway, as potential intervention target for HDT are extensively reviewed in (92). As of now, there are 12 IFN-based treatments, belonging to 8 distinct IFN types (Table 1) approved by the FDA for the treatment of diseases with viral or malignant origin, including hepatitis B or C, genital warts and other infections.
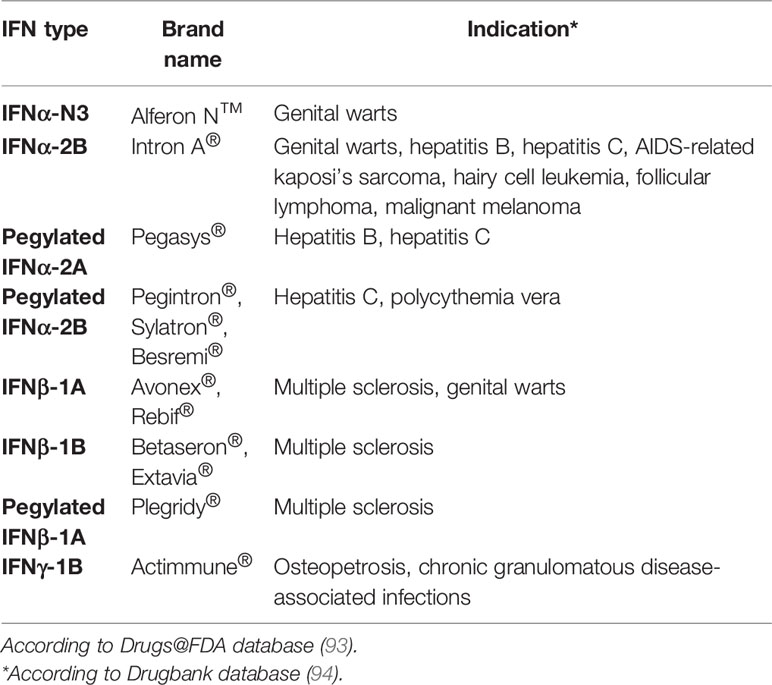
Table 1 IFN-based treatments approved by the Food and Drug Administration (FDA).
Hepatitis B (HBV) and C (HCV) viruses can cause chronic infection, characterized by elevated levels of alanine aminotransferase (ALT) and viral antigens. Treatment with IFNα monotherapy showed a reduction in those levels in clinical trials with chronic hepatitis B (95) or C (96) infection, even after therapy interruption, proving it to be a successful treatment. However, due to the short half-life of IFNα, frequent injection administration was required. To ameliorate that inconvenience, pegylated forms of IFNα in which polyethylene glycol (PEG) is covalently attached to the IFN molecule were formulated, achieving a higher half-life and sustained blood levels (97). Genital warts, also called condylomata acuminate, are a common sexually transmitted disease (STD) caused by human papillomavirus (HPV) in where a papillomatous proliferation of the skin and mucosa takes place. Clinical trials using IFN as treatment showed effectivity in reducing the wart area thanks to its antiviral and antiproliferative properties (98, 99). AIDS-related Kaposi’s sarcoma is a type of cancer that can be caused by the infection with human herpesvirus 8 (HHV-8) in the context of HIV/AIDS-related immune suppression. Due to the viral and proliferative nature of the disease, IFN was used in clinical trials with patients with AIDS-related Kaposi’s sarcoma, showing disease improvement (100, 101). In chronic granulomatous disease (CGD) phagocytes are able to digest but not kill certain microorganisms, causing a susceptibility towards infections that may lead to death. In this context, treatment with IFNγ in clinical trials showed a reduction of the frequency of CGD-related infections (102).
Despite having a well-described pathway against infections and being approved as treatment for certain viral diseases, IFN-based therapies are far from being considered the ideal pan-antiviral. In fact, its side effects prompted a shift in the standard therapy against HCV towards the recently developed oral direct-acting antivirals (DAAs) targeting viral proteins (103). Moreover, there have also been reports on IFN resistance by some viruses. DENV would be an example as DENV-infected cells display a resistance towards the antiviral effect of IFN (104, 105) even though cell pre-treatment with type-I IFN regulates infection in vitro (106). Other examples of IFN resistance include HIV-1: recently, it has been reported that HIV-1 isolates from patients display IFN resistance at different stages of the infection course with the highest degree being observed after in the viral rebound after treatment interruption (107). Furthermore, characterization of HIV-1 from donor and recipient pairs revealed that transmitted viruses showed higher type-I IFN resistance (108). Taking all this data together, it becomes evident that IFN-based therapies may not be the best treatment for certain viral infections and that there is a need to either produce novel, more potent and selective IFN-based therapies or to shift completely the focus towards other targets in the pursue of effective antiviral treatments.
3.2 PRR-Based Therapies
3.2.1 Toll-Like Receptors Agonists
TLRs exposed in the cellular membrane (TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10) generally (but not exclusively) bind to microbial elements from bacteria, mycoplasma, fungi and viruses respectively. TLRs located in intracellular endosomal compartments recognize nucleic acids characteristic of virus and bacteria such as dsRNA (TLR3), ssRNA (TLR7/8) and unmethylated DNA with CpG motifs (TLR9) (109). TLR structure and signaling transduction are widely described elsewhere (68, 110–112).
To date few TLR agonists (TLRas) have been approved by the FDA (or equivalent regulatory agencies) for their use in humans (mostly in immunotherapy against cancer). This includes TLR2/4a bacillus Calmette-Guérin (BCG, an attenuated strain of Mycobacterium bovis), Picibanil (OK432 from Streptococcus pyogenes) and monophosphoryl lipid A (derivative of Salmonella minnesota LPS) and the Imidazoquinoline derivative Imiquimod as TLR7a (113). In the regard of viral infections, many TLRas are currently under study (summarized in Table 2), mainly focusing the engagement of intracellular TLR3, TLR7/8 and TLR9. The polyriboisosinic:polyribocytidylic acid (poly(I:C)) is a TLR3a that mimics dsRNA and is highly capable of inducing IFN production through TLR3 engagement. However, poly(I:C) triggers unspecific activation of both TLR3 and RIG-I and/or MDA5 pathways, raising toxicity concerns due to exacerbated secretion of proinflammatory cytokines. Improved poly(I:C) derivatives, named poly(IC12U) and poly(ICLC), have been designed to reduce the level of toxicity by increasing the specificity of the ligand for TLR3. Poly(ICLC) has a chemical modification (poly-l-lysine in carboxymethylcellulose) that improves its resistance to nucleolytic hydrolysis, resulting in extended and boosted agonist TLR3 activity (112). Interestingly, poly(ICLC) has been described to induce innate immunity genes related to the NFkB pathway, the inflammasome and the complement cascade. Similarly, poly(IC12U), which exclusively engages TLR3, has been shown to increase immunization efficacy when used as adjuvant in vaccines against H5N1 and HSV-2 (129). Other alternative synthetic TLR3a, such as ARNAX, incorporates an GpC phosphorothioate oligodeoxynucleotide that together with a short length of the RNA, results in binding to TLR3 without MDA5 activation (130). ARNAX has been proved to induce potent cDCs stimulation and to promote mucosal IgA production as adjuvant for influenza vaccination in the mice model (131).

Table 2 TLR agonists under study against viral infections.
The modulation of TLR7/8 pathway is another interesting mechanism to be considered. Imiquimod (brand name Aldara™) is FDA-approved TLR7/8a (mainly a TLR7a) that is currently in use for the treatment of HPV- and HSV-induced genital and perianal warts (111). In vitro antiviral activity of Imiquimod against HIV-1 has also been reported in pretreated human macrophages in a mechanism involving the downregulation of HIV entry coreceptors CCR5 and CD4 (123). Imiquimod derivative resiquimod (R848) has been proposed for its use in combination to nucleoside analogues for the treatment of noroviruses (124) as well as adjuvant in HIV-1 vaccines using HIV abortive RNAs seeking priming of DCs (125). Similar data has been reported for 3M-052, a recently developed synthetic TLR7/8a, used as adjuvant of Env-based vaccine that elicits robust and durable protection versus HIV in the non-human primate (Rhesus macaque) model of infection (126).
Regarding TLR9, the synthetic CpG oligodeoxynucleotide (CpG-ODN) 1018 mimics natural ligand of TLR9 and is currently used as adjuvant for the HBV vaccine Heplisav-B, (132). CpG 1018 potential as adjuvant in vaccines against COVID-19 has been also proposed in mice models (127). As an improvement of CpG-ODNs, development of MGN1703, a TLR9a based on a “dumbbell-shape” structure made of covalently closed DNA has improved tolerability and safety profile of MGN1703 compared to previous CpG-ODNs. Preclinical data published by (128) indicate that MGN1703 treatment of PBMCs from HIV+ individuals results in higher natural killer (NK) activation and NK function, restricting HIV+ spread in autologous CD4+ T cells. Indeed, authors postulate a dual role of TLR9 activation by MGN1703 as antiviral and latency reversing agent of HIV.
In addition to the modulation of intracellular TLRs, agonists binding to cell surface TLRs are gaining attention as therapeutic approaches against viral infections. For example, Pam3CKS4, agonist of TLR1/2, is able to activate NFkB pathway in hepatocytes challenged with hepatitis Delta virus (HDV) or coinfected with HDV/HBV, inhibiting total HDV genome, antigenome RNA and viral protein levels in an agonist dose-dependent manner with reduced toxicity. Therefore, Pam3CKS4 postulates as a potential therapy for the management of chronically HDV/HBV infections (114). More recently, the role of the TLR2 and TLR4 signaling pathways is under debate in the context of the current SARS-Cov-2 pandemics. Recent experimental evidences pointed that SARS-CoV-2 proteins belonging to envelope (133) and/or spike (134) trigger the expression of proinflammatory cytokines through the TLR2 signaling pathway. Indeed, topically administration at the respiratory tract of the TLR2/6a INNA-051 (a pegylated synthetic analogue of the diacylated lipopeptide, S-[2,3-bis(palmitoyloxy)propyl] cysteine (Pam2Cys), reduces viral RNA level in nose and throat in the ferret model of infection (115). Regarding TLR4, the S1 subunit of the spike protein of SARS-Cov-2 has been predicted to bind to TLR4 in sillico studies. Authors suggest that interaction with TLR4 would enhance ACE2 expression to facilitate viral entry (135, 136). Although all these studies remain to be confirmed, the role and modulation of TLR2 and TLR4 pathways in the context of SARS-Cov-2 should be explored as potential therapeutic intervention (68, 111, 134).
3.2.2 Intracellular RNA Sensors
Retinoic acid-inducible gene I-like receptors (RLRs) family is composed of retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5) and LGP2. RLR are cytoplasmic sensors expressed in nearly all cell types that upon virus-derived RNA binding, interact with the mitochondrial antiviral signaling protein or MAVS (also known as IPS-1) to induce an antiviral environment (see (137, 138) for review). RLRs can be activated by a broad spectrum of viruses including ssRNA(+) Flavivirirus (WNV; HVC; Zika virus) or Coronavirus (SARS coronavirus); ssRNA(-) Filovirus (EBOV, Marburg virus); ssRNA (RT) retrovirus (HIV) but also viral RNA derived from dsDNA viruses such as Herpervirus (HSV-1, Epstein–Barr) (138) Hence, there is an increasing interest to explore RIG-I activation as a therapeutic approach. To date, three classes of RIG-I agonists have been described according to their chemical structures: Nucleotide-based, RNA-based and small molecular compounds (139). A member belonging to the first family, the dinucleotide-derived small molecule SB9200 (Inarigir soproxil) has been described to elicit dose-dependent activation of RIG-I (and also NOD2) that triggers induction of IFNα/β and ISG in liver. Initial studies of SB9200 showed antiviral activity against rodent woodchuck hepatitis virus (WHV) in in vivo challenged woodchuck sequentially treated with SB9200 and then with Entecavir (ETV). SB9200 pretreatment resulted in improved viral clearance (140). Next, Jones et al., (141) associated RIG-I activation by SB9200 to HCV inhibition in both HCV replicon system as in patient-derived virus models of infection. Hence, SB9200 was active against several HCV genotypes, including the NS5A resistant associated genotype (141). Indeed, the phase 1 clinical trial NCT01803308 showed an association between the decline in viral RNA and the levels of SB9200 detection in plasma on naïve adult with chronic hepatitis C. Concomitant with reported activity versus other RNA viruses including norovirus, respiratory syncytial virus (RSV) and influenza and HBV, RIG-I activation becomes a relevant mechanism to be considered as a pan-antiviral therapeutic approach (139, 141).
The RNA-based RIG-I ligands include 5′-triphosphorylated and diphosphorylated short dsRNAs. These ligands have been described to induce immune activation in several in vitro models of infection (142), including the inhibition of VSV, Dengue (DENV) and vaccinia virus due to 5’-ppp-RNA treatment in human lung epithelial A549 cells, impairment of HIV in ex vivo lymphocyte CD4+ T cells, inhibition of HCV in Huh 7.5 or the promotion of protective antiviral response that prevents DENV and CHIKV infection in primary myeloid cells (143). In that direction, the edition of the structure, length and sequence of RNA-based RIG-I is providing novel agent with potentiated antiviral activity and specificity for RIG-I such as the M8 consisting in a 99-nucleotide, uridine-rich hairpin 5’ppp-RNA that provides stronger antiviral activity against influenza, DENV and CHIKV compared to poly(I:C) in both in vitro and in vivo models (144). The pan antiviral potential of triggering RLRs has been reported by the use of synthetic RNAs mimicking structural domains of non-coding regions (NCRs) of the foot-and mouth disease virus (FMDV), which induce IFN protective response against FMDV, WNV and Rift Valley Fever virus (RVFV) in in vitro and mice models respectively (145–147). Recently, another improved RNA-based RIG-I agonist named stem-loop RNA 14 (SLR14) has shown to provide prophylactic and post-infection resistance in K18-ACE2 mice infected with SARS-Cov-2. Intravenously administration of SLR14 resulted in the sustained reduction of viral replication in the lung and trachea at 3-8 dpi. Combination of SLR14 with anti-IFNAR antibody administration completely abolished SARS-CoV-2 replication. Efficacy of SLR14 was conserved against several antibody evading variants of concern, including B.1.1.7, B.1.351, P.1, B.1.526, and B.1.617.2 (148).
Regarding small molecular compounds, pretreatment with isoflavone compounds Kin100 and Kin101 showed anti-HCV and anti-Influenza activity in Huh7 and MRC5 cellular models respectively (149). Similarly, hydroxiquinole KN1400 and derivative agents trigger MAVS-IRF3 dependent induction of IFN-β response, providing prophylactic and therapeutic protection against WNV, DENV and HCV as well as EBOV virus in THP cells (150, 151). Indeed, activation of the RIG-I pathway at the MAVS-IRF3 level is an interesting option to circumvent evasion mechanisms to the RIG-I sensing like EBOV or influenza among others as reviewed in (67, 152). Further studies, considering each particular viral infection and cell type, will be required to explore the potential of RLRs agonists as new pan antiviral compounds.
3.2.3 Cytosolic dsDNA Sensors
The cyclic GMP-AMP synthase (cGAS) – stimulator of interferon genes (STING) pathway (cGAS-STING) and the IFN-γ-inducible protein 16 (IFI16) are cytoplasmic sensors of dsDNA from DNA viruses (e.g: HBV, HSV-1) and retrovirus (e.g: HIV-1) as a result from reverse transcription of their RNA genome (67, 153, 154).
Cyclic dinucleotides (CDNs), natural ligands of cGAS-STING, have been studied to activate cGAS-STING pathway. However, their susceptibility to hydrolysis by phosphodiesterases and poor tissue diffusion profile, discouraged their application in the clinic. Conversely, dimeric amidobenzimidazole (diABZI), a new class of synthetic small molecule that behaves as STING agonist, exerts broad in vitro antiviral activity against human parainfluenza 3 (PIV) and rhinovirus 16 (RHV), two causative agents of respiratory infections in humans which lack of approved antiviral treatments or vaccines. Antiviral activity of diABZI involves distinct mechanisms associated to IFN response and autophagy (155). Recently, diABZI has shown broad antiviral activity against human Cov-OC43 and SARS-Cov-2 in in vitro challenged A549 and A549-ACE2 cell lines respectively (156). These results have been confirmed so far by at least two articles. First, diABZI showed potent anti-SARS-Cov-2 activity in both human primary bronchial epithelial cells and in vivo challenged mice by the transient activation of IFN-I signaling. Indeed, diABZI was active against South African (B1.351) variant of concern indicating potential broad activity to other SARS-Cov-2 variants as well as to other coronaviruses (157). Additionally, diABZI also inhibited SARS-Cov-2 in lung epithelial cells and in K18-ACE2 transgenic mice through a short activation of STING and a time-limited cytokine production, involving lymphocyte and myeloid cells activation to resolve viral infection (158).
Since many cGAS-STING agonists are currently under study in clinical trials as immunooncotherapy and inflammatory agents and are showing promising safety and immune activation profiles (159, 160), it is likely that further research will elucidate the application of new cGAS-STING agonist candidates for the management of viral infections
3.2.4 Alternative Intervention Targets: NOD-Like Receptors
NLRs recognize a broad array of PAMPs motifs including bacterial cell wall component γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) by NOD1, muramyl dipeptide (MDP) by NOD2 or poly(I:C) through NLRP3 (161–164). NLRs also recognize DAMPs signals associated to ER stress as a result of viral infections. This non-canonical pathway leads to the interaction with MAVS and IRF-3 dependent induction of IFN-α/β for viral clearance (165). Moreover, some NLRs (e.g NLRP3) form a specialized multiprotein complex, named inflammasome, that triggers inflammation and, in some cases, cell death as a response to danger signals associated to viral infections (164, 166).
Among NLR immunomodulators, MDP (NOD2 agonist) has been widely studied to confer protection against HIV, HSV, vaccinia virus or influenza (161). However, its pyrogenic and arthritogenic undesired side effects in humans, led to the development of MDP derivatives (L18-MDP, MDP-LysL18, murabutide and others) that have been proposed as adjuvants as reviewed in (167). Similarly, the use of leukotriene B4, an endogenous lipid mediator of inflammation, has been found to activate the NOD2-dependent pathway interacting with MAVS-IRF3, eliciting an IFN-β protective response to influenza A virus (IAV) (168).
Interestingly, modulation of NLRs play a central role limiting the timing and amplitude of the immune response. As example, complications in influenza A virus (IAV) infection are often associated to the prolonged presence of Th17 cells and neutrophils at the sites of infection that result in exacerbated inflammation and tissue damage. In mice challenged with IAV, treatment with MDP (NOD2 agonist) promotes higher levels of CXC12 and CCL5 chemokines to recruit Treg to the lung and concomitant with TGFβ secretion. This anti-inflammatory environment limits the presence of Th17 cells and infiltrated neutrophils to the site of infection, preserving tissue integrity that facilitates infection resolution (169). The regulatory functions of NLRs are not restricted to NOD2. Other NLR belonging to the NLRC family, such as NLRX1 or NLRC3, negatively regulate RIG-I-MAVS interplay or STING signaling respectively as reviewed in (164). Therefore, NLRs dual role is likely to be explored to elicit innate response but also as limiting agents of overwhelming inflammation and tissue damage (153).
3.3 Signaling Pathways
The molecular basis of antiviral innate immune response and pathogen sensing consist in a complex network with multiple layered and interconnected regulatory mechanisms. Deciphering the molecular basis of this virus-host interplay offers the possibility to explore the modulation of the immune signaling pathways as potential HDTs.
In that sense, the modulation of the NF-κB pathway and its regulation through the IKK complex and the IKK related kinases TBK1 and IKKϵ, has been postulated as HDT. The pathway is implicated in many biological processes, including cell growth, metabolism, apoptosis, cell cycle and cell migration and invasion (170). Interestingly, IKK – TBK1/IKKϵ interplay is a confluent downstream regulator of multiple pathogen sensing pathways as described in Figure 2 (see (170, 171) for review). As examples of the potential of IKK modulation as HDT, siRNA of IkBα has been shown to induce HIV reactivation in latently infected cells (172, 173). Similarly, TBK-1/IKKϵ inhibitors have been reported to inhibit HIV replication in human primary macrophages (174) as well as a broad antiviral activity against diverse sort of virus including HSV-1 and HSV2 (175), HCV (176), RVFV (177) or Venezuelan equine encephalitis virus infection (VEEV) (178).
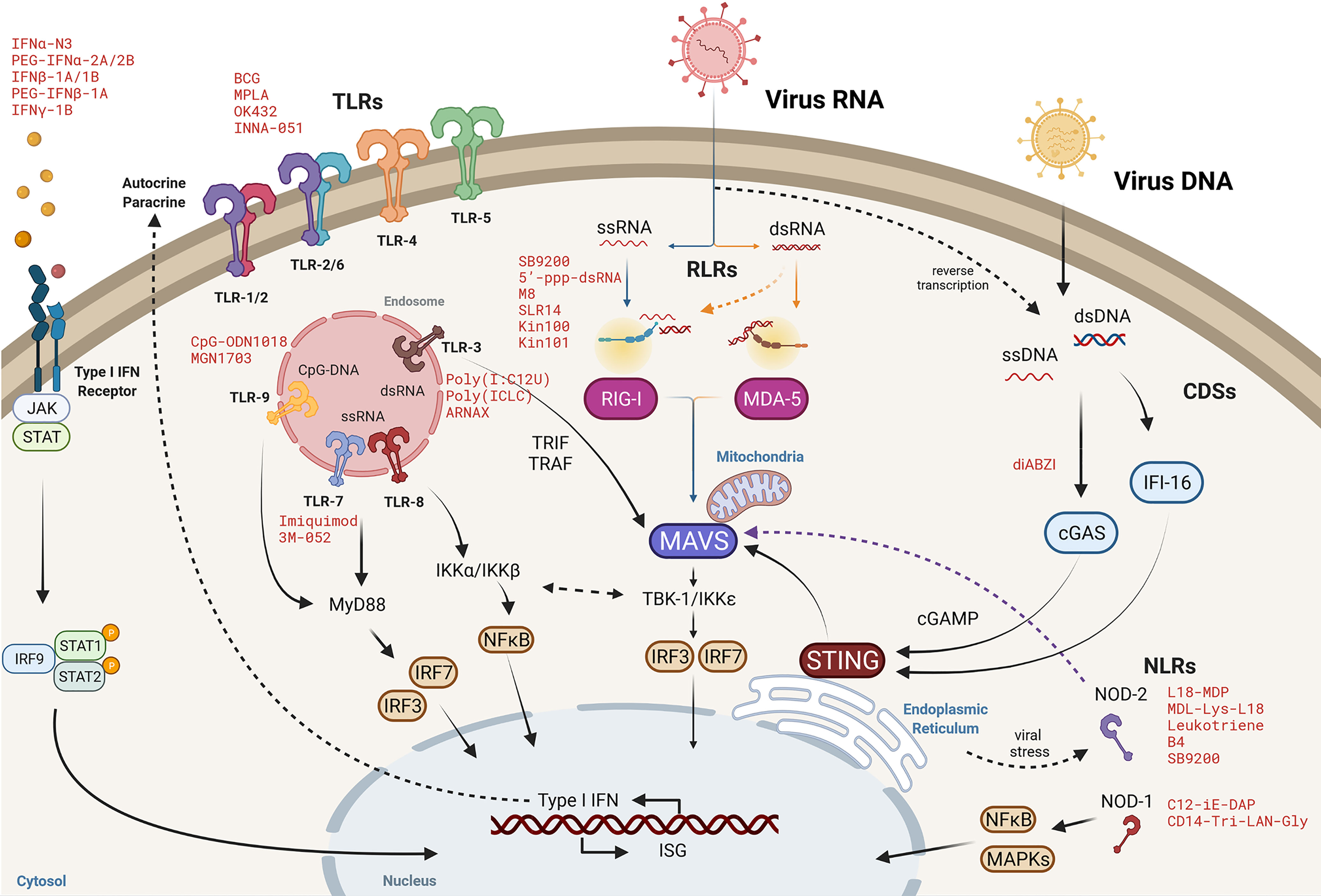
Figure 2 Cytosolic viral RNA recognition by pattern recognition receptors. Molecular pathways of Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), cytosolic DNA sensors (CDSs) and NOD-like receptors (NLRs) converge at different downstream signaling elements, such as at mitochondrial antiviral signaling protein (MAVS) or the TBK1/IKKϵ axis to activate nuclear factor-κB (NF-κB) and interferon regulatory factor (IRF-3/IRF-7) and then, orchestrate a robust innate immune response by the expression of type I IFN and interferon stimulated genes (ISG) responses to clear viral infection. Representative agonists mimicking viral PAMPs or DAMPs are denoted in red as potential intervention approaches to promote an antiviral cellular environment.
Another key signaling pathway in the innate immune response is that of the Janus activated kinase (JAK) – signal transducer and activator of transcription (STAT), which is activated in response to IFN and leads to the expression of the aforementioned ISGs (179, 180). Due to the myriad of different processes regulated by ISGs, distinct effects have been reported for modulators of the JAK-STAT pathway. Those that may qualify as putative HDT include the modulation of the pathway by the JAK inhibitors (JAKi) ruxolitinib and tofacitinib as a mean to inhibit HIV-1 replication and latency reactivation (181, 182) or the JAKi baricitinib, which was authorized for emergency use in COVID-19 patients in order to block the cytokine overproduction, but has also been reported to inhibit viral entry (183) [see (92) for review].
4 Concluding Remarks and Future Perspective
Antiviral drug resistance is an increasing long-lasting unsolved concern as prolonged drug exposure led to the selection of resistant strains and limited treatment efficacy. Host directed therapies have been proposed as an alternative to virus-centered therapeutics to overcome the appearance of drug resistance and represent also an interesting approach for the development of pan-antivirals. However, identification of druggable host targets, with limited undesired effects upon pharmacological targeting is not a straight-forward question and nowadays, few molecules have been introduced in the clinical practice. One of the main caveats of HDT is the correct identification and validation of host cell targets within the myriad of processes and proteins that interact with viral replication cycles, which can only be solved with an in-depth characterization of virus-host interactions. Further research elucidating the complex innate immunity regulation, interconnections of the different signaling routes and cell responses for each specific viral infection, is required to explore the role of innate immunity among putative HDT. Moreover, the specificity of the course of each viral infection, drug tolerability, pharmacokinetics and therapeutic window must be included in these forthcoming studies to validate the use of HDTs as new antivirals.
Huge strides have been made in understanding the innate immune response following infection with different viruses. However, not all the signaling events are defined fully and further details of the proteins required and how they are regulated are likely to be revealed. It is also clear that the outcome of a virus–host innate system interaction is complex and depends upon the particular virus as well as the host species. Also, it is now appreciated that immune responses for at least some viral infections requires the contribution of many PRRs. How all these signals are integrated in natural infection is unknown. Because innate immune signaling can mediate harmful immune responses following viral infection, further studies should reveal drug targets of innate immune signaling pathways that may be modulated in order to improve outcome to different viral infections. Conversely, given the many promising innate immune agonists that have been reported to have anti-viral activities, there is reason to be optimistic that such strategies may be employed as treatments for viral infection.
Author Contributions
Writing and original draft preparation, RB, EG-V and EB. Writing—review and editing, RB, EG-V and EB. Supervision, RB and EB. Funding acquisition, RB and EB All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported in part by grants from the Instituto de Salud Carlos III, Fondo de Investigación Sanitaria (FIS) PI19/00194 co-financed by FEDER. EG-V is a research fellow from PERIS (SLT017/20/000090). EB and RB are research fellows from ISCIII-FIS (CPII19/00012; CP19/00011).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Health TLP. COVID-19 Pandemic: What’s Next for Public Health? Lancet Public Health (2022) 7:e391. doi: 10.1016/S2468-2667(22)00095-0
2. Andrei G. Vaccines and Antivirals: Grand Challenges and Great Opportunities. Front Virol (2021) 0:666548. doi: 10.3389/FVIRO.2021.666548
3. de Clercq E, Li G. Approved Antiviral Drugs Over the Past 50 Years. Clin Microbiol Rev (2016) 29:695–747. doi: 10.1128/CMR.00102-15
4. Tompa DR, Immanuel A, Srikanth S, Kadhirvel S. Trends and Strategies to Combat Viral Infections: A Review on FDA Approved Antiviral Drugs. Int J Biol Macromol (2021) 172:524–41. doi: 10.1016/J.IJBIOMAC.2021.01.076
5. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-Directed Therapies for Bacterial and Viral Infections. Nat Rev Drug Discovery (2018) 17:35–56. doi: 10.1038/NRD.2017.162
6. Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, et al. Haploid Genetic Screens in Human Cells Identify Host Factors Used by Pathogens. Sci (1979) (2009) 326:1231–5. doi: 10.1126/SCIENCE.1178955/SUPPL_FILE/CARETTE.SOM.PDF
7. Greco TM, Diner BA, Cristea IM. The Impact of Mass Spectrometry-Based Proteomics on Fundamental Discoveries in Virology. Annu Rev Virol (2014) 1(1):581–604. doi: 10.1146/annurev-virology-031413-085527
8. Ciuffi A, Rato S, Telenti A. Single-Cell Genomics for Virology. Viruses (2016) 8(5):123. doi: 10.3390/v8050123
9. Perreira JM, Meraner P, Brass AL. “Functional Genomic Strategies for Elucidating Human-Virus Interactions: Will CRISPR Knockout RNAi and Haploid Cells?,” in. Adv Virus Res (2016) 94:1–51. doi: 10.1016/bs.aivir.2015.11.001
10. Jean Beltran PM, Federspiel JD, Sheng X, Cristea IM. Proteomics and Integrative Omic Approaches for Understanding Host-Pathogen Interactions and Infectious Diseases. Mol Syst Biol (2017) 13:922. doi: 10.15252/MSB.20167062
11. Puschnik AS, Majzoub K, Ooi YS, Carette JE. A CRISPR Toolbox to Study Virus-Host Interactions. Nat Rev Microbiol (2017) 15(6):351–64. doi: 10.1038/nrmicro.2017.29
12. Cristinelli S, Ciuffi A. The Use of Single-Cell RNA-Seq to Understand Virus–Host Interactions. Curr Opin Virol (2018) 29:39–50. doi: 10.1016/j.coviro.2018.03.001
13. Kirby EN, Shue B, Thomas PQ, Beard MR. CRISPR Tackles Emerging Viral Pathogens. Viruses (2021) 13(11):2157. doi: 10.3390/v13112157
14. Ramage H, Cherry S. Virus-Host Interactions: From Unbiased Genetic Screens to Function. Annu Rev Virol (2015) 2(1):497–524. doi: 10.1146/annurev-virology-100114-055238
15. Meng Z, Lu M. RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Front Immunol (2017) 8:331. doi: 10.3389/fimmu.2017.00331
16. Badia R, Ballana E, Esté JA, Riveira-Muñoz E. Antiviral Treatment Strategies Based on Gene Silencing and Genome Editing. Curr Opin Virol (2017) 24:46–54. doi: 10.1016/j.coviro.2017.04.001
17. Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, et al. RNA Interference Screen for Human Genes Associated With West Nile Virus Infection. Nature (2008) 455:242. doi: 10.1038/NATURE07207
18. Martin S, Chiramel AI, Schmidt ML, Chen YC, Whitt N, Watt A, et al. A Genome-Wide siRNA Screen Identifies a Druggable Host Pathway Essential for the Ebola Virus Life Cycle. Genome Med (2018) 10:1–14. doi: 10.1186/S13073-018-0570-1/FIGURES/6
19. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature (2011) 477:340–3. doi: 10.1038/NATURE10348
20. Röling M, Sisakht MM, Ne E, Moulos P, Crespo R, Stoszko M, et al. A Two-Color Haploid Genetic Screen Identifies Novel Host Factors Involved in HIV-1 Latency. mBio (2021) 12(6):e0298021. doi: 10.1128/MBIO.02980-21
21. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Sci (1979) (2012) 337(6096):816–21. doi: 10.1126/science.1225829
22. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome Engineering Using the CRISPR-Cas9 System. Nat Protoc (2013) 8:2281–308. doi: 10.1038/nprot.2013.143
23. Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophy (2017) 46:505–29. doi: 10.1146/ANNUREV-BIOPHYS-062215-010822
24. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Sci (1979) (2014) 343(6166):84–87. doi: 10.1126/science.1247005
25. Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. Genome-Scale CRISPR-Cas9 Knockout and Transcriptional Activation Screening. Nat Protoc (2017) 12(4):828–863. doi: 10.1038/nprot.2017.016
26. Schenider WM, Hoffmann H-H. Flavivirus–host Interactions: An Expanding Network of Proviral and Antiviral Factors. Curr Opin Virol (2022) 52:71–7. doi: 10.1016/j.coviro.2021.11.007
27. Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, et al. Genetic Dissection of Flaviviridae Host Factors Through Genome-Scale CRISPR Screens. Nature (2016) 535:159–63. doi: 10.1038/nature18631
28. Israeli M’, Finkel Y, Yahalom-Ronen Y, Paran N, Chitlaru T, Israeli O, et al. Genome-Wide CRISPR Screens Identify GATA6 as a Proviral Host Factor for SARS-CoV-2 via Modulation of ACE2. Nat Commun (2022) 13:1–16. doi: 10.1038/s41467-022-29896-z
29. Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell (2015) 163(3):759–71. doi: 10.1016/j.cell.2015.09.038
30. Wang F, Wang L, Zou X, Duan S, Li Z, Deng Z, et al. Advances in CRISPR-Cas Systems for RNA Targeting, Tracking and Editing. Biotechnol Adv (2019) 37:708–29. doi: 10.1016/J.BIOTECHADV.2019.03.016
31. Papikian A, Liu W, Gallego-Bartolomé J, Jacobsen SE. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat Commun (2019) 10(1):729. doi: 10.1038/s41467-019-08736-7
32. Chapman SJ, Hill AVS. Human Genetic Susceptibility to Infectious Disease. Nat Rev Genet (2012) 13:175–88. doi: 10.1038/NRG3114
33. Uffelmann E, Huang QQ, Munung NS, de Vries J, Okada Y, Martin AR, et al. Genome-Wide Association Studies. Nat Rev Methods Primers (2021) 1)1:1–21. doi: 10.1038/s43586-021-00056-9
34. van Manen D, van ’t Wout AB, Schuitemaker H. Genome-Wide Association Studies on HIV Susceptibility, Pathogenesis and Pharmacogenomics. Retrovirology (2012) 9:1–8. doi: 10.1186/1742-4690-9-70/TABLES/1
35. Fellay J, Ge D, Shianna Kv, Colombo S, Ledergerber B, Cirulli ET, et al. Common Genetic Variation and the Control of HIV-1 in Humans. PloS Genet (2009) 5:1000791. doi: 10.1371/JOURNAL.PGEN.1000791
36. McLaren PJ, Fellay J. HIV-1 and Human Genetic Variation. Nat Rev Genet (2021) 22:645–57. doi: 10.1038/S41576-021-00378-0
37. Nahon P, Cobat A. Human Genetics of HCV Infection Phenotypes in the Era of Direct-Acting Antivirals. Hum Genet (2020) 139:855–63. doi: 10.1007/S00439-020-02136-4
38. Gao Y, Nepal N, Jin SZ. Toll-Like Receptors and Hepatitis C Virus Infection. Hepatobiliary Pancreat Dis Int (2021) 20:521–9. doi: 10.1016/J.HBPD.2021.07.011
39. Niemi MEK, Karjalainen J, Liao RG, Neale BM, Daly M, Ganna A, et al. Mapping the Human Genetic Architecture of COVID-19. Nature (2021) 600:472–7. doi: 10.1038/s41586-021-03767-x
40. Genomewide Association Study of Severe Covid-19 With Respiratory Failure. Genomewide Association Study of Severe Covid-19 with Respiratory Failure New Engl J Med (2020) 383:1522–34. doi: 10.1056/NEJMoa2020283
41. Zhang Q, Bastard P, Karbuz A, Gervais A, Tayoun AA, Aiuti A, et al. Human Genetic and Immunological Determinants of Critical COVID-19 Pneumonia. Nature (2022) 603:587–98. doi: 10.1038/s41586-022-04447-0
42. Johnson R, Burch KS, Hou K, Paciuc M, Pasaniuc B, Sankararaman S. Estimation of Regional Polygenicity From GWAS Provides Insights Into the Genetic Architecture of Complex Traits. PLoS Comput Biol (2021) 17:e1009483. doi: 10.1371/JOURNAL.PCBI.1009483
43. Deshiere A, Joly-Beauparlant C, Breton Y, Ouellet M, Raymond F, Lodge R, et al. Global Mapping of the Macrophage-HIV-1 Transcriptome Reveals That Productive Infection Induces Remodeling of Host Cell DNA and Chromatin. Sci Rep 2017 7:1 (2017) 7:1–13. doi: 10.1038/s41598-017-05566-9
44. Hartman AL, Dover JE, Towner JS, Nichol ST. Reverse Genetic Generation of Recombinant Zaire Ebola Viruses Containing Disrupted IRF-3 Inhibitory Domains Results in Attenuated Virus Growth In Vitro and Higher Levels of IRF-3 Activation Without Inhibiting Viral Transcription or Replication. J Virol (2006) 80:6430. doi: 10.1128/JVI.00044-06
45. Hartman AL, Ling L, Nichol ST, Hibberd ML. Whole-Genome Expression Profiling Reveals That Inhibition of Host Innate Immune Response Pathways by Ebola Virus Can Be Reversed by a Single Amino Acid Change in the VP35 Protein. J Virol (2008) 82:5348 doi: 10.1128/JVI.00215-08
46. Sun J, Ye F, Wu A, Yang R, Pan M, Sheng J, et al. Comparative Transcriptome Analysis Reveals the Intensive Early Stage Responses of Host Cells to SARS-CoV-2 Infection. Front Microbiol (2020) 11:593857/BIBTEX. doi: 10.3389/FMICB.2020.593857/BIBTEX
47. Luo G, Gao Q, Zhang S, Yan B. Probing Infectious Disease by Single-Cell RNA Sequencing: Progresses and Perspectives. Comput Struct Biotechnol J (2020) 18:2962–71. doi: 10.1016/j.csbj.2020.10.016
48. Mehla J, Caufield JH, Uetz P. Making the Right Choice: Critical Parameters of the Y2H Systems. Methods Mol Biol (2018) 1794:17–28. doi: 10.1007/978-1-4939-7871-7_2
49. Striebinger H, Koegl M, Bailer SM. A High-Throughput Yeast Two-Hybrid Protocol to Determine Virus-Host Protein Interactions. Virus-Host Interact (2013) 1064:1. doi: 10.1007/978-1-62703-601-6_1
50. Rain JC, Cribier A, Gérard A, Emiliani S, Benarous R. Yeast Two-Hybrid Detection of Integrase-Host Factor Interactions. Methods (2009) 47:291–7. doi: 10.1016/J.YMETH.2009.02.002
51. Kyei GB, Cheng X, Ramani R, Ratner L. Cyclin L2 Is a Critical HIV Dependency Factor in Macrophages That Controls SAMHD1 Abundance. Cell Host Microbe (2015) 17:98. doi: 10.1016/J.CHOM.2014.11.009
52. Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, et al. An Experimentally Derived Confidence Score for Binary Protein-Protein Interactions. Nat Methods (2009) 6:91–7. doi: 10.1038/NMETH.1281
53. Tucker G, Loh PR, Berger B. A Sampling Framework for Incorporating Quantitative Mass Spectrometry Data in Protein Interaction Analysis. BMC Bioinf (2013) 14:299. doi: 10.1186/1471-2105-14-299
54. Luo Y, Muesing MA. Mass Spectrometry-Based Proteomic Approaches for Discovery of HIV-Host Interactions. Future Virol (2014) 9(11):979–92. doi: 10.2217/FVL.14.86
55. Adelmant G, Garg BK, Tavares M, Card JD, Marto JA. Tandem Affinity Purification and Mass Spectrometry (TAP-MS) for the Analysis of Protein Complexes. Curr Protoc Protein Sci (2019) 96:e84. doi: 10.1002/CPPS.84
56. Lu M, Risco C. Single-Molecule FRET Imaging of Virus Spike–Host Interactions. Viruses (2021) 13:332. doi: 10.3390/V13020332
57. McClelland RD, Culp TN, Marchant DJ. Imaging Flow Cytometry and Confocal Immunofluorescence Microscopy of Virus-Host Cell Interactions. Front Cell Infect Microbiol (2021) 11:749039/BIBTEX. doi: 10.3389/FCIMB.2021.749039/BIBTEX
58. Risco C. Application of Advanced Imaging to the Study of Virus-Host Interactions. Viruses (2021) 13(10):1958. doi: 10.3390/V13101958
59. Saffarian S. Application of Advanced Light Microscopy to the Study of HIV and Its Interactions With the Host. Viruses (2021) 13(2):223. doi: 10.3390/V13020223
60. Wolff G, Bárcena M. Multiscale Electron Microscopy for the Study of Viral Replication Organelles. Viruses (2021) 13(2):197. doi: 10.3390/V13020197
61. Shepherd DC, Dalvi S, Ghosal D. From Cells to Atoms: Cryo-EM as an Essential Tool to Investigate Pathogen Biology, Host-Pathogen Interaction, and Drug Discovery. Mol Microbiol (2022) 117:610–7. doi: 10.1111/MMI.14820
62. Businger R, Deutschmann J, Gruska I, Milbradt J, Wiebusch L, Gramberg T, et al. Human Cytomegalovirus Overcomes SAMHD1 Restriction in Macrophages via Pul97. Nat Microbiol (2019) 4:2260–72. doi: 10.1038/S41564-019-0557-8
63. Lu M, Ma X, Castillo-Menendez LR, Gorman J, Alsahafi N, Ermel U, et al. Associating HIV-1 Envelope Glycoprotein Structures With States on the Virus Observed by smFRET. Nature (2019) 568:415–9. doi: 10.1038/s41586-019-1101-y
64. Cale EM, Driscoll JI, Lee M, Gorman J, Zhou T, Lu M, et al. Antigenic Analysis of the HIV-1 Envelope Trimer Implies Small Differences Between Structural States 1 and 2. J Biol Chem (2022) 298:101819. doi: 10.1016/j.jbc.2022.101819
65. Ni G, Ma Z, Damania B. cGAS and STING: At the Intersection of DNA and RNA Virus-Sensing Networks. PLoS Pathogens (2018) 14(8):e1007148. doi: 10.1371/journal.ppat.1007148
66. Cai C, Tang YD, Xu G, Zheng C. The Crosstalk Between Viral RNA- and DNA-Sensing Mechanisms. Cell Mol Life Sci (2021) 78(23):7427–34. doi: 10.1007/s00018-021-04001-7
67. Chan YK, Gack MU. Viral Evasion of Intracellular DNA and RNA Sensing. Nat Rev Microbiol (2016) 14:360. doi: 10.1038/NRMICRO.2016.45
68. Diamond MS, Kanneganti T-D. Innate Immunity: The First Line of Defense Against SARS-CoV-2. Nat Immunol (2022) 23:165–76. doi: 10.1038/S41590-021-01091-0
69. Nagano Y, Kojima Y, Sawai Y. Immunité Et Interférence Dans La Vaccine; Inhibition De L’infection Dermique Par Le Virus Inactivé [Immunity and Interference in Vaccinia; Inhibition of Skin Infection by Inactivated Virus]. Comptes Rendus Des Seances Soc Biol Ses Filiales (1954) 148:750–2.
70. Isaacs A, Lindenmann J. Virus Interference. I. The Interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi: 10.1098/RSPB.1957.0048
71. Isaacs A, Lindenmann J, Valentine RC. Virus Interference. II. Some Properties of Interferon. Proc R Soc London Ser B - Biol Sci (1957) 147:268–73. doi: 10.1098/rspb.1957.0049
72. Lindenmann J, Burke DC, Isaacs A. Studies on the Production, Mode of Action and Properties of Interferon. Br J Exp Pathol (1957) 38:551.
73. Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their Actions. Annu Rev Biochem (2003) 56:727–77. doi: 10.1146/ANNUREV.BI.56.070187.003455
74. Farrar MA, Schreiber RD. The Molecular Cell Biology of Interferon-Gamma and Its Receptor. Annu Rev Immunol (2003) 11:571–611. doi: 10.1146/ANNUREV.IY.11.040193.003035
75. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-λs Mediate Antiviral Protection Through a Distinct Class II Cytokine Receptor Complex. Nat Immunol (2002) 4(1):69–77. doi: 10.1038/ni875
76. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and Their Class II Cytokine Receptor IL-28r. Nat Immunol 2002 4:1 (2002) 4:63–8. doi: 10.1038/ni873
77. Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol (2018) 10(11):a028423. doi: 10.1101/CSHPERSPECT.A028423
78. Walter MR. The Role of Structure in the Biology of Interferon Signaling. Front Immunol (2020) 11:606489/BIBTEX. doi: 10.3389/FIMMU.2020.606489/BIBTEX
79. García-Sastre A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe (2017) 22:176–84. doi: 10.1016/j.chom.2017.07.012
80. Rojas JM, Alejo A, Martín V, Sevilla N. Viral Pathogen-Induced Mechanisms to Antagonize Mammalian Interferon (IFN) Signaling Pathway. Cell Mol Life Sci (2020) 4(78):1423–44. doi: 10.1007/S00018-020-03671-Z
81. Lamotte LA, Tafforeau L. How Influenza A Virus NS1 Deals With the Ubiquitin System to Evade Innate Immunity. Viruses (2021) 13:2309. doi: 10.3390/V13112309
82. Kitagawa Y, Yamaguchi M, Kohno M, Sakai M, Itoh M, Gotoh B. Respirovirus C Protein Inhibits Activation of Type I Interferon Receptor-Associated Kinases to Block JAK-STAT Signaling. FEBS Lett (2020) 594:864–77. doi: 10.1002/1873-3468.13670
83. Best SM. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J Virol (2017) 91(3):e01970-16. doi: 10.1128/JVI.01970-16
84. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe (2016) 19:882–90. doi: 10.1016/J.CHOM.2016.05.009
85. Obi JO, Gutiérrez-Barbosa H, Chua JV, Deredge DJ. Current Trends and Limitations in Dengue Antiviral Research. Trop Med Infect Dis (2021) 6(4):180. doi: 10.3390/TROPICALMED6040180
86. Jain S, Martynova E, Rizvanov A, Khaiboullina S, Baranwal M. Structural and Functional Aspects of Ebola Virus Proteins. Pathogens (2021) 10(10):1330. doi: 10.3390/PATHOGENS10101330
87. Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, et al. Mutual Antagonism Between the Ebola Virus VP35 Protein and the RIG-I Activator PACT Determines Infection Outcome. Cell Host Microbe (2013) 14:74–84. doi: 10.1016/J.CHOM.2013.06.010/ATTACHMENT/0EE285E0-EC8A-4EE9-B356-7A2E1537F00C/MMC1.PDF
88. Woolsey C, Menicucci AR, Cross RW, Luthra P, Agans KN, Borisevich V, et al. A VP35 Mutant Ebola Virus Lacks Virulence But Can Elicit Protective Immunity to Wild-Type Virus Challenge. Cell Rep (2019) 28:3032–3046.e6. doi: 10.1016/J.CELREP.2019.08.047/ATTACHMENT/2AB40FEE-8113-4152-86F3-C8B237B2CBD0/MMC1.PDF
89. Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front Immunol (2021) 12, 2698. doi: 10.3389/FIMMU.2021.708264/BIBTEX
90. Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS-CoV-2 Orf6 Hijacks Nup98 to Block STAT Nuclear Import and Antagonize Interferon Signaling. Proc Natl Acad Sci USA (2020) 117:28344–54. doi: 10.1073/PNAS.2016650117
91. Miyamoto Y, Itoh Y, Suzuki T, Tanaka T, Sakai Y, Koido M, et al. SARS-CoV-2 ORF6 Disrupts Nucleocytoplasmic Trafficking to Advance Viral Replication. Commun Biol (2022) 5:483. doi: 10.1038/S42003-022-03427-4
92. Ezeonwumelu IJ, Garcia-Vidal E, Ballana E. JAK-STAT Pathway: A Novel Target to Tackle Viral Infections. Viruses (2021) 13(12):2379. doi: 10.3390/v13122379
93. Drugs@FDA: FDA-Approved Drugs. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (Accessed March 7, 2022).
94. DrugBank Online | Database for Drug and Drug Target Info. Available at: https://go.drugbank.com/ (Accessed March 7, 2022).
95. Wong DKH, Cheung AM, O’Rourke K, Naylor CD, Detsky AS, Heathcote J. Effect of Alpha-Interferon Treatment in Patients With Hepatitis B E Antigen-Positive Chronic Hepatitis B. A Meta-Analysis. Ann Intern Med (1993) 119:312–23. doi: 10.7326/0003-4819-119-4-199308150-00011
96. Hoofnagle JH, Mullen KD, Jones DB, Rustgi V, Di Bisceglie A, Peters M, et al. Treatment of Chronic non-A,non-B Hepatitis With Recombinant Human Alpha Interferon. A Preliminary Report. N Engl J Med (1986) 315:1575–8. doi: 10.1056/NEJM198612183152503
97. Chevaliez S, Pawlotsky JM. Interferon-Based Therapy of Hepatitis C. Adv Drug Deliv Rev (2007) 59:1222–41. doi: 10.1016/J.ADDR.2007.07.002
98. Gall SA, Hughes CE, Trofatter K. Interferon for the Therapy of Condyloma Acuminatum. Am J Obstet Gynecol (1985) 153:157–63. doi: 10.1016/0002-9378(85)90103-6
99. Eron LJ, Judson F, Tucker S, Prawer S, Mills J, Murphy K, et al. Interferon Therapy for Condylomata Acuminata. N Engl J Med (1986) 315:1059–64. doi: 10.1056/NEJM198610233151704
100. Volberding PA, Mitsuyasu RT, Golando JP, Spiegel RJ. Treatment of Kaposi’s Sarcoma With Interferon Alfa-2b (Intron® a). Cancer (1987) 59:620–5. doi: 10.1002/1097-0142(19870201)59:3+<620::AID-CNCR2820591309>3.0.CO;2-5
101. Fischl MA, Finkelstein DM, He W, Powderly WG, Triozzi PL, Steigbigel RT. A Phase II Study of Recombinant Human Interferon-α2a and Zidovudine in Patients With AIDS-Related Kaposi’s Sarcoma. J Acquir Immune Defic Syndr Hum Retrovirol (1996) 11:379–84. doi: 10.1097/00042560-199604010-00008
102. Group*, T. I. C. G. D. C. S. A Controlled Trial of Interferon Gamma to Prevent Infection in Chronic Granulomatous Disease. New Engl J Med (1991) 324:509–16. doi: 10.1056/nejm199102213240801
103. Solbach P, Wedemeyer H. The New Era of Interferon-Free Treatment of Chronic Hepatitis C. Viszeralmedizin. (2015) 31(4):290–6. doi: 10.1159/000433594.
104. Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, et al. Dengue Virus Type 2 Antagonizes IFN-Alpha but Not IFN-Gamma Antiviral Effect via Down-Regulating Tyk2-STAT Signaling in the Human Dendritic Cell. J Immunol. (2005) 174(12):8163–72. doi: 10.4049/jimmunol.174.12.8163
105. Ye H, Duan X, Yao M, Kang L, Li Y, Li S, et al. USP18 Mediates Interferon Resistance of Dengue Virus Infection. Front Microbiol. (2021) 12:682380. doi: 10.3389/fmicb.2021.682380.
106. Diamond MS, Roberts TG, Edgil D, Lu B, Ernst J, Harris E. Modulation of Dengue Virus Infection in Human Cells by Alpha, Beta, and Gamma Interferons. J Virol. (2000) 74(11):4957–66. doi: 10.1128/jvi.74.11.4957-4966.2000.
107. Gondim MVP, Sherrill-Mix S, Bibollet-Ruche F, Russell RM, Trimboli S, Smith AG, et al. Heightened Resistance to Host Type 1 Interferons Characterizes HIV-1 at Transmission and After Antiretroviral Therapy Interruption. Sci Transl Med. (2021) 13(576):eabd8179. doi: 10.1126/scitranslmed.abd8179
108. Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, et al. Resistance to Type 1 Interferons Is a Major Determinant of HIV-1 Transmission Fitness. Proc Natl Acad Sci USA (2017) 114(4):E590–E599. doi: 10.1073/pnas.1620144114
109. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
110. Martinsen JT, Gunst JD, Højen JF, Tolstrup M, Søgaard OS. The Use of Toll-Like Receptor Agonists in HIV-1 Cure Strategies. Front Immunol (2020) 11:1112. doi: 10.3389/FIMMU.2020.01112
111. Farooq M, Batool M, Kim MS, Choi S. Toll-Like Receptors as a Therapeutic Target in the Era of Immunotherapies. Front Cell Dev Biol (2021) 9:756315. doi: 10.3389/FCELL.2021.756315
112. Komal A, Noreen M, El-Kott AF. TLR3 Agonists: RGC100, ARNAX, and Poly-IC: A Comparative Review. Immunol Res (2021) 69:312–22. doi: 10.1007/S12026-021-09203-6
113. Vacchelli E, Galluzzi L, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial Watch: FDA-Approved Toll-Like Receptor Agonists for Cancer Therapy. OncoImmunology (2012) 1:894–907. doi: 10.4161/ONCI.20931
114. Michelet M, Alfaiate D, Chardès B, Pons C, Faure-Dupuy S, Engleitner T, et al. Inducers of the NF-κb Pathways Impair Hepatitis Delta Virus Replication and Strongly Decrease Progeny Infectivity In Vitro. JHEP Rep (2021) 4(3):100415. doi: 10.1016/J.JHEPR.2021.100415
115. Proud PC, Tsitoura D, Watson RJ, Chua BY, Aram MJ, Bewley KR, et al. Prophylactic Intranasal Administration of a TLR2/6 Agonist Reduces Upper Respiratory Tract Viral Shedding in a SARS-CoV-2 Challenge Ferret Model. EBioMedicine (2021) 63:103153. doi: 10.1016/J.EBIOM.2020.103153
116. Ichinohe T, Kawaguchi A, Tamura Si, Takahashi H, Sawa H, Ninomiya A, et al. Intranasal Immunization With H5N1 Vaccine Plus Poly I:Poly C12U, a Toll-Like Receptor Agonist, Protects Mice Against Homologous and Heterologous Virus Challenge. Microbes Infect (2007) 9:1333–40. doi: 10.1016/J.MICINF.2007.06.007
117. Overton ET, Goepfert PA, Cunningham P, Carter WA, Horvath J, Young D, et al. Intranasal Seasonal Influenza Vaccine and a TLR-3 Agonist, Rintatolimod, Induced Cross-Reactive IgA Antibody Formation Against Avian H5N1 and H7N9 Influenza HA in Humans. Vaccine (2014) 32:5490–5. doi: 10.1016/J.VACCINE.2014.07.078
118. Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity (2019) 50:907–23. doi: 10.1016/J.IMMUNI.2019.03.025
119. Takeda Y, Takaki H, Fukui-Miyazaki A, Yoshida S, Matsumoto M, Seya T. Vaccine Adjuvant ARNAX Promotes Mucosal IgA Production in Influenza HA Vaccination. Biochem Biophys Res Commun (2018) 506:1019–25. doi: 10.1016/J.BBRC.2018.10.166
120. Edwards L, Ferenczy A, Eron L, Baker D, Owens ML, Fox TL, et al. Self-Administered Topical 5% Imiquimod Cream for External Anogenital Warts. Arch Dermatol (1998) 134:25–30. doi: 10.1001/ARCHDERM.134.1.25
121. Gilbert J, Drehs MM, Weinberg JM. Topical Imiquimod for Acyclovir-Unresponsive Herpes Simplex Virus 2 Infection. Arch Dermatol (2001) 137:1015–7.
122. Martinez V, Molina JM, Scieux C, Ribaud P, Morfin F. Topical Imiquimod for Recurrent Acyclovir-Resistant HSV Infection. Am J Med (2006) 119:e9–e11. doi: 10.1016/J.AMJMED.2005.06.037
123. Meng FZ, Liu JB, Wang X, Wang P, Hu WH, Hou W, et al. Tlr7 Activation of Macrophages by Imiquimod Inhibits Hiv Infection Through Modulation of Viral Entry Cellular Factors. Biol (Basel) (2021) 10(7):661. doi: 10.3390/BIOLOGY10070661/S1
124. Tuipulotu DE, Netzler NE, Lun JH, Mackenzie JM, White PA. TLR7 Agonists Display Potent Antiviral Effects Against Norovirus Infection via Innate Stimulation. Antimicrob Agents Chemother (2018) 62(5):e02417-17. doi: 10.1128/AAC.02417-17
125. Stunnenberg M, van Hamme JL, Zijlstra-Willems EM, Gringhuis SI, Geijtenbeek TBH. Crosstalk Between R848 and Abortive HIV-1 RNA-Induced Signaling Enhances Antiviral Immunity. J Leukoc Biol (2022). doi: 10.1002/JLB.4A0721-365R. Epub ahead of print.
126. Kasturi SP, Ur Rasheed MA, Havenar-Daughton C, Pham M, Legere T, Sher ZJ, et al. 3M-052, a Synthetic TLR-7/8 Agonist, Induces Durable HIV-1 Envelope-Specific Plasma Cells and Humoral Immunity in Nonhuman Primates. Sci Immunol (2020) 5(48):eabb1025. doi: 10.1126/SCIIMMUNOL.ABB1025
127. Kuo TY, Lin MY, Coffman RL, Campbell JD, Traquina P, Lin YJ, et al. Development of CpG-Adjuvanted Stable Prefusion SARS-CoV-2 Spike Antigen as a Subunit Vaccine Against COVID-19. Sci Rep (2020) 10:1–10. doi: 10.1038/s41598-020-77077-z
128. Offersen R, Nissen SK, Rasmussen TA, Østergaard L, Denton PW, Søgaard OS, et al. A Novel Toll-Like Receptor 9 Agonist, MGN1703, Enhances HIV-1 Transcription and NK Cell-Mediated Inhibition of HIV-1-Infected Autologous CD4+ T Cells. J Virol (2016) 90:4441. doi: 10.1128/JVI.00222-16
129. Owen AM, Fults JB, Patil NK, Hernandez A, Bohannon JK. TLR Agonists as Mediators of Trained Immunity: Mechanistic Insight and Immunotherapeutic Potential to Combat Infection. Front Immunol (2021) 11:622614. doi: 10.3389/FIMMU.2020.622614
130. Matsumoto M, Seya T. TLR3: Interferon Induction by Double-Stranded RNA Including Poly(I:C). Adv Drug Deliv Rev (2008) 60:805–12. doi: 10.1016/J.ADDR.2007.11.005
131. Ong GH, Lian BSX, Kawasaki T, Kawai T. Exploration of Pattern Recognition Receptor Agonists as Candidate Adjuvants. Front Cell Infect Microbiol (2021) 11:745016. doi: 10.3389/fcimb.2021.745016
132. A Two-Dose Hepatitis B Vaccine for Adults (Heplisav-B). JAMA (2018) 319:822–3. doi: 10.1001/JAMA.2018.1097
133. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 Senses the SARS-CoV-2 Envelope Protein to Produce Inflammatory Cytokines. Nat Immunol 2021 22:7 (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
134. Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 Spike Protein Induces Inflammation via TLR2-Dependent Activation of the NF-κb Pathway. Elife (2021) 10:68563. doi: 10.7554/ELIFE.68563
135. Aboudounya MM, Heads RJ. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflammation (2021) 2021:8874339. doi: 10.1155/2021/8874339
136. Pantazi I, Al-Qahtani AA, Alhamlan FS, Alothaid H, Matou-Nasri S, Sourvinos G, et al. SARS-CoV-2/ACE2 Interaction Suppresses IRAK-M Expression and Promotes Pro-Inflammatory Cytokine Production in Macrophages. Front Immunol (2021) 12:683800. doi: 10.3389/FIMMU.2021.683800
137. Bruns AM, Horvath CM. LGP2 Synergy With MDA5 in RLR-Mediated RNA Recognition and Antiviral Signaling. Cytokine (2015) 74(2):198–206. doi: 10.1016/j.cyto.2015.02.010
138. Rehwinkel J, Gack MU. RIG-I-Like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20(9):537–51. doi: 10.1038/s41577-020-0288-3
139. Yong HY, Luo D. RIG-I-Like Receptors as Novel Targets for Pan-Antivirals and Vaccine Adjuvants Against Emerging and Re-Emerging Viral Infections. Front Immunol (2018) 9:1379/BIBTEX. doi: 10.3389/FIMMU.2018.01379/BIBTEX
140. Suresh M, Korolowicz KE, Balarezo M, Iyer RP, Padmanabhan S, Cleary D, et al. Antiviral Efficacy and Host Immune Response Induction During Sequential Treatment With SB 9200 Followed by Entecavir in Woodchucks. PLoS One (2017) 12(1):e0169631. doi: 10.1371/JOURNAL.PONE.0169631
141. Jones M, Cunningham ME, Wing P, DeSilva S, Challa R, Sheri A, et al. SB 9200, a Novel Agonist of Innate Immunity, Shows Potent Antiviral Activity Against Resistant HCV Variants. J Med Virol (2017) 89:1620–8. doi: 10.1002/JMV.24809
142. Goulet ML, Olagnier D, Xu Z, Paz S, Belgnaoui SM, Lafferty EI, et al. Systems Analysis of a RIG-I Agonist Inducing Broad Spectrum Inhibition of Virus Infectivity. PLoS Pathog (2013) 9:e1003298. doi: 10.1371/JOURNAL.PPAT.1003298
143. Olagnier D, Scholte FEM, Chiang C, Albulescu IC, Nichols C, He Z, et al. Inhibition of Dengue and Chikungunya Virus Infections by RIG-I-Mediated Type I Interferon-Independent Stimulation of the Innate Antiviral Response. J Virol (2014) 88:4180–94. doi: 10.1128/JVI.03114-13
144. Chiang C, Beljanski V, Yin K, Olagnier D, ben Yebdri F, Steel C, et al. Sequence-Specific Modifications Enhance the Broad-Spectrum Antiviral Response Activated by RIG-I Agonists. J Virol (2015) 89:8011–25. doi: 10.1128/JVI.00845-15
145. Rodríguez-Pulido M, Martín-Acebes MA, Escribano-Romero E, Blázquez AB, Sobrino F, Borrego B, et al. Protection Against West Nile Virus Infection in Mice After Inoculation With Type I Interferon-Inducing RNA Transcripts. PLoS One (2012) 7(11):e49494. doi: 10.1371/JOURNAL.PONE.0049494
146. Lorenzo G, Rodríguez-Pulido M, López-Gil E, Sobrino F, Borrego B, Sáiz M, et al. Protection Against Rift Valley Fever Virus Infection in Mice Upon Administration of Interferon-Inducing RNA Transcripts From the FMDV Genome. Antiviral Res (2014) 109:64–7. doi: 10.1016/J.ANTIVIRAL.2014.06.010
147. Borrego B, Rodríguez-Pulido M, Revilla C, Álvarez B, Sobrino F, Domínguez J, et al. Synthetic RNAs Mimicking Structural Domains in the Foot-And-Mouth Disease Virus Genome Elicit a Broad Innate Immune Response in Porcine Cells Triggered by RIG-I and TLR Activation. Viruses (2015) 7:3954–73. doi: 10.3390/V7072807
148. Mao T, Israelow B, Lucas C, Vogels CBF, Gomez-Calvo ML, Fedorova O, et al. A Stem-Loop RNA RIG-I Agonist Protects Against Acute and Chronic SARS-CoV-2 Infection in Mice. J Exp Med (2022) 219(1):e20211818. doi: 10.1084/JEM.20211818
149. Bedard KM, Wang ML, Proll SC, Loo YM, Katze MG, Gale M Jr, et al. Isoflavone Agonists of IRF-3 Dependent Signaling Have Antiviral Activity Against RNA Viruses. J Virol. (2012) 86(13):7334–44. doi: 10.1128/JVI.06867-11
150. Pattabhi S, Wilkins CR, Dong R, Knoll ML, Posakony J, Kaiser S, et al. Targeting Innate Immunity for Antiviral Therapy Through Small Molecule Agonists of the RLR Pathway. J Virol (2015) 90:2372–87. doi: 10.1128/JVI.02202-15
151. Green RR, Wilkins C, Pattabhi S, Dong R, Loo Y, Gale M. Transcriptional Analysis of Antiviral Small Molecule Therapeutics as Agonists of the RLR Pathway. Genom Data (2016) 7:290–2. doi: 10.1016/J.GDATA.2016.01.020
152. Dutta S, Das N, Mukherjee P. Picking Up a Fight: Fine Tuning Mitochondrial Innate Immune Defenses Against RNA Viruses. Front Microbiol (2020) 11:1990/BIBTEX. doi: 10.3389/FMICB.2020.01990/BIBTEX
153. Jiang M, Jiang M, Chen P, Chen P, Wang L, Li W, et al. cGAS-STING, an Important Pathway in Cancer Immunotherapy. J Hematol Oncol (2020) 13(1):81. doi: 10.1186/S13045-020-00916-Z
154. Gan Y, Li X, Han S, Liang Q, Ma X, Rong P, et al. The cGAS/STING Pathway: A Novel Target for Cancer Therapy. Front Immunol (2022) 12:795401. doi: 10.3389/FIMMU.2021.795401
155. Zhu Q, Hu H, Liu H, Shen H, Yan Z, Gao L. A Synthetic STING Agonist Inhibits the Replication of Human Parainfluenza Virus 3 and Rhinovirus 16 Through Distinct Mechanisms. Antiviral Res (2020) 183:104933. doi: 10.1016/J.ANTIVIRAL.2020.104933
156. Liu W, Reyes HM, Yang JF, Li Y, Stewart KM, Basil MC, et al. Activation of STING Signaling Pathway Effectively Blocks Human Coronavirus Infection. J Virol (2021) 95(12):e00490-21. doi: 10.1128/jvi.00490-21
157. Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological Activation of STING Blocks SARS-CoV-2 Infection. Sci Immunol (2021) 6(59):eabi9007. doi: 10.1126/sciimmunol.abi9007
158. Humphries F, Shmuel-Galia L, Jiang Z, Wilson R, Landis P, Ng SL, et al. A Diamidobenzimidazole STING Agonist Protects Against SARS-CoV-2 Infection. Sci Immunol (2021) 6(59):eabi9002. doi: 10.1126/sciimmunol.abi9002
159. le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial Watch: STING Agonists in Cancer Therapy. Oncoimmunology (2020) 9(1):1777624. doi: 10.1080/2162402X.2020.1777624
160. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/S41577-021-00524-Z
161. Geddes K, Magalhães JG, Girardin s. E. Unleashing the Therapeutic Potential of NOD-Like Receptors. Nat Rev Drug Discovery (2009) 8(6)465–79. doi: 10.1038/nrd2783
162. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of Innate Immune Antiviral Responses by Nod2. Nat Immunol (2009) 10:1073–80. doi: 10.1038/NI.1782
163. Lupfer C, Thomas PG, Kanneganti T-D. Nucleotide Oligomerization and Binding Domain 2-Dependent Dendritic Cell Activation is Necessary for Innate Immunity and Optimal CD8+ T Cell Responses to Influenza A Virus Infection. J Virol (2014) 88:8946–55. doi: 10.1128/JVI.01110-14
164. Kienes I, Weidl T, Mirza N, Chamaillard M, Kufer TA. Role of NLRs in the Regulation of Type I Interferon Signaling, Host Defense and Tolerance to Inflammation. Int J Mol Sci (2021) 22:1–28. doi: 10.3390/IJMS22031301
165. Wiese KM, Coates BM, Ridge KM. The Role of Nucleotide-Binding Oligomerization Domain-Like Receptors in Pulmonary Infection. Am J Respir Cell Mol Biol (2017) 57:151–61. doi: 10.1165/RCMB.2016-0375TR
166. Zhao C, Zhao W. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front Immunol (2020) 11:211/BIBTEX. doi: 10.3389/FIMMU.2020.00211/BIBTEX
167. Maisonneuve C, Bertholet S, Philpott DJ, de Gregorio E. Unleashing the Potential of NOD- and Toll-Like Agonists as Vaccine Adjuvants. Proc Natl Acad Sci USA (2014) 111(34):12294–9. doi: 10.1073/pnas.1400478111
168. le Bel M, Gosselin J. Leukotriene B4 Enhances NOD2-Dependent Innate Response Against Influenza Virus Infection. PLoS One (2015) 10(10):e0139856. doi: 10.1371/JOURNAL.PONE.0139856
169. Egarnes B, Gosselin J. Contribution of Regulatory T Cells in Nucleotide-Binding Oligomerization Domain 2 Response to Influenza Virus Infection. Front Immunol (2018) 9:132/FULL. doi: 10.3389/FIMMU.2018.00132/FULL
170. Clark K, Peggie M, Plater L, Sorcek RJ, Young ERR, Madwed JB, et al. Novel Cross-Talk Within the IKK Family Controls Innate Immunity. Biochem J (2011) 434(1):93–104. doi: 10.1042/BJ20101701
171. Antonia RJ, Hagan RS, Baldwin AS. Expanding the View of IKK: New Substrates and New Biology. Trends Cell Biol (2021) 31:166–78. doi: 10.1016/j.tcb.2020.12.003
172. Fernandez G, Zaikos TD, Khan SZ, Jacobi AM, Behlke MA, Zeichner SL. Targeting I B Proteins for HIV Latency Activation: The Role of Individual I B and NF- B Proteins. J Virol (2013) 87:3966–78. doi: 10.1128/jvi.03251-12
173. Khan SZ, Gasperino S, Zeichner SL. Nuclear Transit and HIV LTR Binding of NF-κb Subunits Held by Iκb Proteins: Implications for HIV-1 Activation. Viruses (2019) 11:1–14. doi: 10.3390/v11121162
174. Pujantell M, Riveira-Munoz E, Badia R, Castellvi M, Garcia-Vidal E, Sirera G, et al. RNA Editing by ADAR1 Regulates Innate and Antiviral Immune Functions in Primary Macrophages. Sci Rep (2017) 7(1):13339. doi: 10.1038/s41598-017-13580-0
175. Su AR, Qiu M, Li YL, Xu WT, Song SW, Wang XH, et al. BX-795 Inhibits HSV-1 and HSV-2 Replication by Blocking the JNK/p38 Pathways Without Interfering With PDK1 Activity in Host Cells. Acta Pharmacol Sin (2017) 38(3):402–14. doi: 10.1038/aps.2016.160
176. Li Q, Pène V, Krishnamurthy S, Cha H, Liang TJ. Hepatitis C Virus Infection Activates an Innate Pathway Involving IKK-α in Lipogenesis and Viral Assembly. Nat Med (2013) 19(6):722–9. doi: 10.1038/nm.3190
177. Narayanan A, Kehn-Hall K, Senina S, Lundberg L, van Duyne R, Guendel I, et al. Curcumin Inhibits Rift Valley Fever Virus Replication in Human Cells. J Biol Chem (2012) 287(40):33198–214. doi: 10.1074/jbc.M112.356535
178. Amaya M, Voss K, Sampey G, Senina S, de la Fuente C, Mueller C, et al. The Role of Ikkβ in Venezuelan Equine Encephalitis Virus Infection. PLoS One (2014) 9(2):e86745. doi: 10.1371/journal.pone.0086745
179. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
180. Schoggins JW. Interferon-Stimulated Genes: What Do They All Do? Annu Rev Virol. (2019) 6(1):567–584. doi: 10.1146/annurev-virology-092818-015756
181. Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, Schinazi RF. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation in Vitro. Antimicrob Agents Chemother. (2014) 58(4):1977–86. doi: 10.1128/AAC.02496-13.
182. Gavegnano C, Brehm JH, Dupuy FP, Talla A, Ribeiro SP, Kulpa DA, et al. Novel Mechanisms to Inhibit HIV Reservoir Seeding Using Jak Inhibitors. PLoS Pathog. (2017) 13(12):e1006740. doi: 10.1371/journal.ppat.1006740
Keywords: antiviral drug-resistance, host factors, host-directed therapies, innate immunity, emerging viruses
Citation: Badia R, Garcia-Vidal E and Ballana E (2022) Viral-Host Dependency Factors as Therapeutic Targets to Overcome Antiviral Drug-Resistance: A Focus on Innate Immune Modulation. Front.Virol. 2:935933. doi: 10.3389/fviro.2022.935933
Received: 04 May 2022; Accepted: 06 June 2022;
Published: 05 July 2022.
Edited by:
Mayte Coiras, Instituto de Salud Carlos III (ISCIII), SpainReviewed by:
Margarita Sáiz, Spanish National Research Council (CSIC), SpainMarco Sgarbanti, National Institute of Health (ISS), Italy
Copyright © 2022 Badia, Garcia-Vidal and Ballana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Badia, cmJhZGlhQGlyc2ljYWl4YS5lcw==; Ester Ballana, ZWJhbGxhbmFAaXJzaWNhaXhhLmVz