 Sayaka Sukegawa
Sayaka Sukegawa Hiroaki Takeuchi
Hiroaki Takeuchi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol. , 25 July 2022
Sec. Fundamental Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.934892
This article is part of the Research Topic HIV/SIV basic research update View all 10 articles
HIV-1 targets the monocyte/macrophage lineage and CD4+ T cells for its replication. The efficiency of infection, replication, and cell-to-cell spread differs between these cell types. These differences are caused by various factors such as viral tropism, viral proteins, host factors, and cell proliferation. However, the precise mechanisms of how macrophages influence HIV-1 infection have not been fully elucidated. Macrophages are long-lived cells susceptible to infection predominantly with R5-tropic strains of HIV-1. Although co-receptor use switches from CCR5 to CXCR4 in up to 50% of patients during AIDS progression, R5-tropic strains remain predominant in the remaining patients. Compared to HIV-1-infected T cells, infected macrophages are less susceptible to HIV-induced cytopathic effects and survive for more than a few weeks. Efforts to cure HIV-1 may be thwarted by the existence of reservoir cells that cannot be targeted by ART. Resting CD4+ T lymphocytes are thought to be the primary reservoir cells, but recent studies demonstrated that monocyte/macrophage lineage cells may also act as viral reservoirs. This review will focus on the impact of monocytes/macrophages during HIV-1 replication, the establishment of the reservoirs, and recent approaches toward HIV-1 eradication by specifically targeting monocyte/macrophage lineage cells.
Macrophages have a crucial role in capturing foreign entities by endocytosing or phagocytosing pathogens as the first line of defense. They contribute to the adaptive immune response by presenting antigens to T cells, and to innate immunity by regulating inflammation via cytokine secretion (1). Macrophages express several receptors on their plasma membranes, including pattern-recognition receptors (PRRs), which contribute significantly to recognizing pathogens for phagocytosis. Phagocytosed proteins are processed and presented in histocompatibility complex class II (MHC class II) to activate CD4+ T lymphocytes, while exogenous proteins such as viral proteins are presented on histocompatibility complex class I (MHC class I) to activate CD8+ T lymphocytes. In addition, the phagocytosed proteins can be presented on MHC class I, which is called cross-presentation. Macrophages and dendritic cells (DCs) play a critical role as antigen-presenting cells (APCs). Once pathogens are phagocytosed, these cells degrade and ingest exogenous proteins, then cross-present antigens on MHC class I to activate CD8+ cytotoxic T lymphocytes (2). PRRs function as sensors that rapidly initiate innate immune responses after binding with pathogen-associated molecular patterns (PAMPs) (3). Interferons (IFNs), secreted after the recognition of PAMPs by PRRs, have the potential to inhibit HIV-1 infection. IFNs also induce the expressions of several interferon-stimulated genes (ISGs), including SAMHD1, APOBEC3, Mx2 → MX2, tetherin, SERINC, and Siglec-1, which are well-known host factors that restrict HIV-1 replication (4). The functions of these host factors in HIV-1 replication are described in detail in the following section.
Despite their role in the clearance of pathogens, monocytes/macrophages as well as CD4+ T cells are targeted by HIV-1 for its replication. HIV-1 entry into the host cell begins with the attachment of viral envelope (Env) to CD4+ T-cell surface receptors, resulting in the exposure of the V3 loop of Env-gp120 following conformational changes induced by attachment. Sequentially, chemokine receptors CXCR4 or CCR5 are bound by Env-gp120, allowing insertion of viral Env-gp41 into the cell membrane and entrance of the viral capsid core into the host cell (5, 6). Macrophages are long-lived cells expressing CD4 as the primary receptor and chemokine receptors CXCR4 and CCR5 as co-receptors imbuing susceptibility to infection with HIV-1. The R5- and dual-tropic HIV-1 strains preferentially infect macrophages and memory-type CD4+ T lymphocytes by utilizing the CCR5 co-receptor, whereas most other CD4+ T lymphocytes are infected with X4-tropic viruses utilizing CXCR4. Co-receptor usage is thus one of the factors determining virus susceptibility, with several other factors such as differences in host cell proliferation and the existence of viral protein or host factors being significantly associated with disease progression after HIV-1 infection (7, 8). Generally, R5-tropic viruses are predominant at the early stages of infection and are associated with cell-to-cell transmission in individuals, whereas X4-tropic viruses usually emerge later (9). At this time, co-receptor usage switches from CCR5 to CXCR4, according to disease progression from acute to chronic infection in up to 50% of AIDS patients (9). However, R5-tropic virus predominance is sustained in the remaining patients. It is still unclear which conditions or factors are associated with facilitating this switch in tropism, but the series of HIV-1 infections with X4- or R5/X4-dual tropic HIV-1 are strongly associated with progression to AIDS by accelerating the reduction of CD4+ T cells (9).
Several factors are involved in the regulation of HIV replication, and a sufficient supply of nucleotides (dNTPs) is essential for efficient reverse transcription and the subsequent production of proviral DNA. Because the concentration of cellular dNTPs is strictly regulated by host cell cycle status, the characteristics of reverse transcription in non-dividing myeloid cells and activated CD4+ T cells are different. Cell cycle status greatly affects the level of ribonucleotide reductase subunit R2, which determines the amount of dNTPs available for reverse transcription (RT) (10). In addition, sterile alpha motif domain and HD domain-containing protein 1 (SAMHD1) was reported to be a host factor restricting HIV-1 and Vpx-deficient HIV-2 replication at the RT stage by depleting intracellular dNTPs in non-dividing cells such as DCs, macrophages, and resting CD4+ T cells (11–13) (Figure 1). It was reported that dNTP levels are approximately 130- to 250-fold lower in macrophages than in activated CD4+ T cells (14). HIV-2 Vpx counteracts the function of SAMHD1 by its degradation in a proteasome-dependent manner. In HIV-1, cyclin L2 promotes the proteasomal degradation of SAMHD1 (15). Thus far, the important roles of several other host factors including APOBEC3F, APOBEC3G, tetherin, and MX2 have also been implicated in HIV-1 replication in monocytes/macrophages (Figure 1). The apolipoprotein B editing complex (APOBEC) is a family of cellular cytidine deaminases. The APOBEC3 family is incorporated into nascent virions, which induce G-to-A hypermutation in the target cell. The viral cDNA produced by this mutation leads to HIV-1 genome changes, resulting in the inhibition of HIV-1 infection (16–18). HIV-1 Vif counteracts the function of this through proteasomal degradation. Macrophages express APOBEC3G, APOBEC3F, and APOBEC3DE, which are all upregulated by IFN-α stimulation. Similar to other APOBEC family members, APOBEC3A (A3A) is also upregulated by stimulation with IFN-α secreted during innate immune responses (18, 19). Interestingly, A3A is highly expressed in monocytes, whereas its expression is weak in fully differentiated macrophages (19). As the result of the silencing of A3A, HIV-1 infected monocytes increase virus replication. This differential expression level of A3A is tightly connected to the susceptibility to HIV-1 infection (16, 19). Myxovirus-resistance protein 2 (MX2, also known as MXB) is another well-known host viral restriction factor expressed in monocytes/macrophages and CD4+ T cells. MX2 protein limits HIV-1 replication by restricting nuclear import and repressing proviral DNA integration into host chromatin (20–23) (Figure 1). This protein is a member of the RNA polymerase II-associated factor 1 (PAF1) family, identified in HeLa-CD4 cells by siRNA screening to identify restriction host factors in HIV-1 replication. A previous study reported that PAF1 was expressed in monocytes/macrophages in addition to T lymphocytes, and that the PAF1 complex (PAF1c) had a role in the inhibition of HIV replication by repressing RT and proviral DNA integration (24). There are two possible mechanisms of PAF1c restriction: first, according to their observations, a large quantity of PAF1 complex is localized in the nucleus. The interaction of PAF1 and RNA polymerase II is thought to be involved in mRNA transcription, elongation, and stability (25); second, the interaction with SKI8 (WDR61), which is part of the SKI complex, is associated with mRNA decay (25). Furthermore, the interactions of PAF1/SKI8 with DNA and RNA in the cytoplasm allow it to function as a PRR. Tetherin (BST-2, also known as CD317) is highly expressed on the surface of macrophages, in contrast to its absence or low-level expression by CD4+ T cells. Tetherin plays a crucial role in retaining progeny virions in infected cells and preventing their release (Figures 1 and 2A). However, Vpu, an accessory protein of HIV-1, antagonizes this function by downregulating tetherin by interfering with the trafficking of newly synthesized and recycling of tetherin proteins from the plasma membrane (26–29). In 2015, the serine incorporator (SERINC) family, especially SERINC3 and SERINC5 members, was newly identified as a host factor antagonized by Nef protein, which inhibited infection by progeny viruses (Figure 2C). The mechanism involves the incorporation of these proteins into newly synthesized virus particles, resulting in interference with their fusion to secondary target cells (30, 31). Furthermore, Zutz et al. revealed that endogenous SERINC5 is highly expressed in differentiated monocyte-derived macrophages at levels sufficient to exert antiviral effects in these primary cells but not in monocytes themselves (32). To date, it is clear that Env glycoprotein has a crucial role in the function of SERINC3/5, but its precise mechanisms of action remain incompletely understood (33–36). Neuropilin-1 (NRP-1), a transmembrane protein, was newly identified in 2021 by Wang et al., and shown to inhibit progeny virus infection (Figure 2C) (37). This protein is highly expressed on the cell surface of macrophages and DCs but not on stimulated or resting CD4+ T cells. Virion-incorporated NRP-1 inhibits their attachment to secondary target cells and consequently contributes to the suppression of infectivity, affecting HIV-1 transmission in a myeloid lineage cell-specific manner.
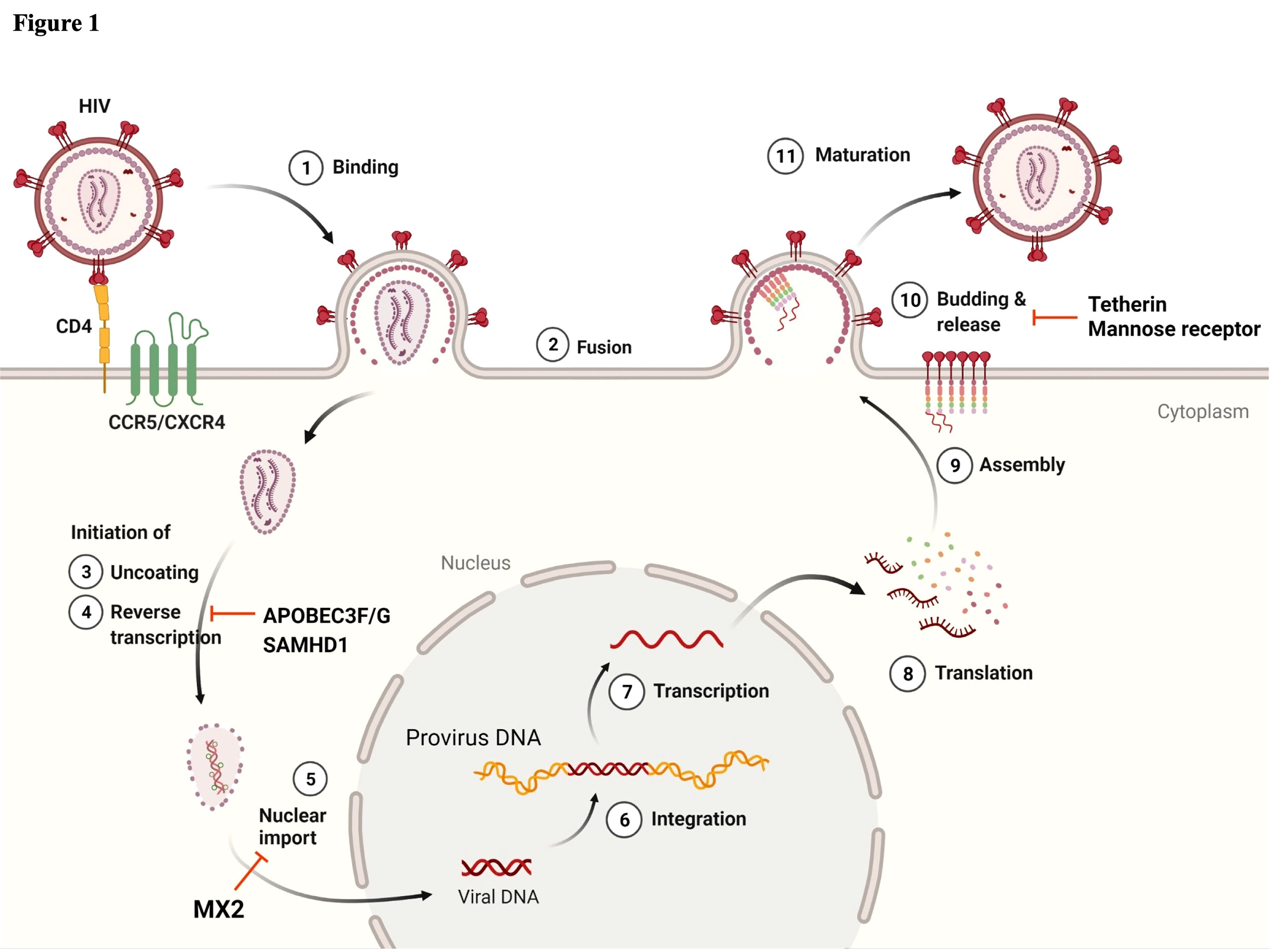
Figure 1 Schematic of the HIV-1 life cycle and host factors regulating HIV-1 replication in myeloid lineage cells. ① Viral infection begins by interactions between the viral envelope (Env) and the host cell receptor, CD4, and subsequently with the co-receptors CXCR4 or CCR5. ② This binding enables the fusion of virus to the host cell membrane and the release of viral cores into the cytoplasm. Once the viral core is released, ③ the uncoating and ④ reverse transcription (RT) of viral RNA are initiated in the cytoplasm. Virion-incorporated APOBEC3F/G and target cell SAMHD1 play a role in the inhibition of RT. ⑤ Viral content is transported to the nuclear pore, and uncoating and RT may then be completed. MX2 inhibits nuclear import. Following the completion of RT, ⑥ the viral genome is integrated into the host chromatin DNA. ⑦ Proviral DNA is transcribed and ⑧ translated into the Gag polyprotein. ⑨ Gag polyproteins are assembled at the plasma membrane and ⑩ initiate the viral budding and release of immature virions. Tetherin and mannose receptor (hMRC1) contribute to the inhibition of viral release. ⑪ For viral release from the cell membrane, viral protease cleaves Gag polyproteins, and mature virions are entirely released from the cell membrane. Image created with BioRender.com.
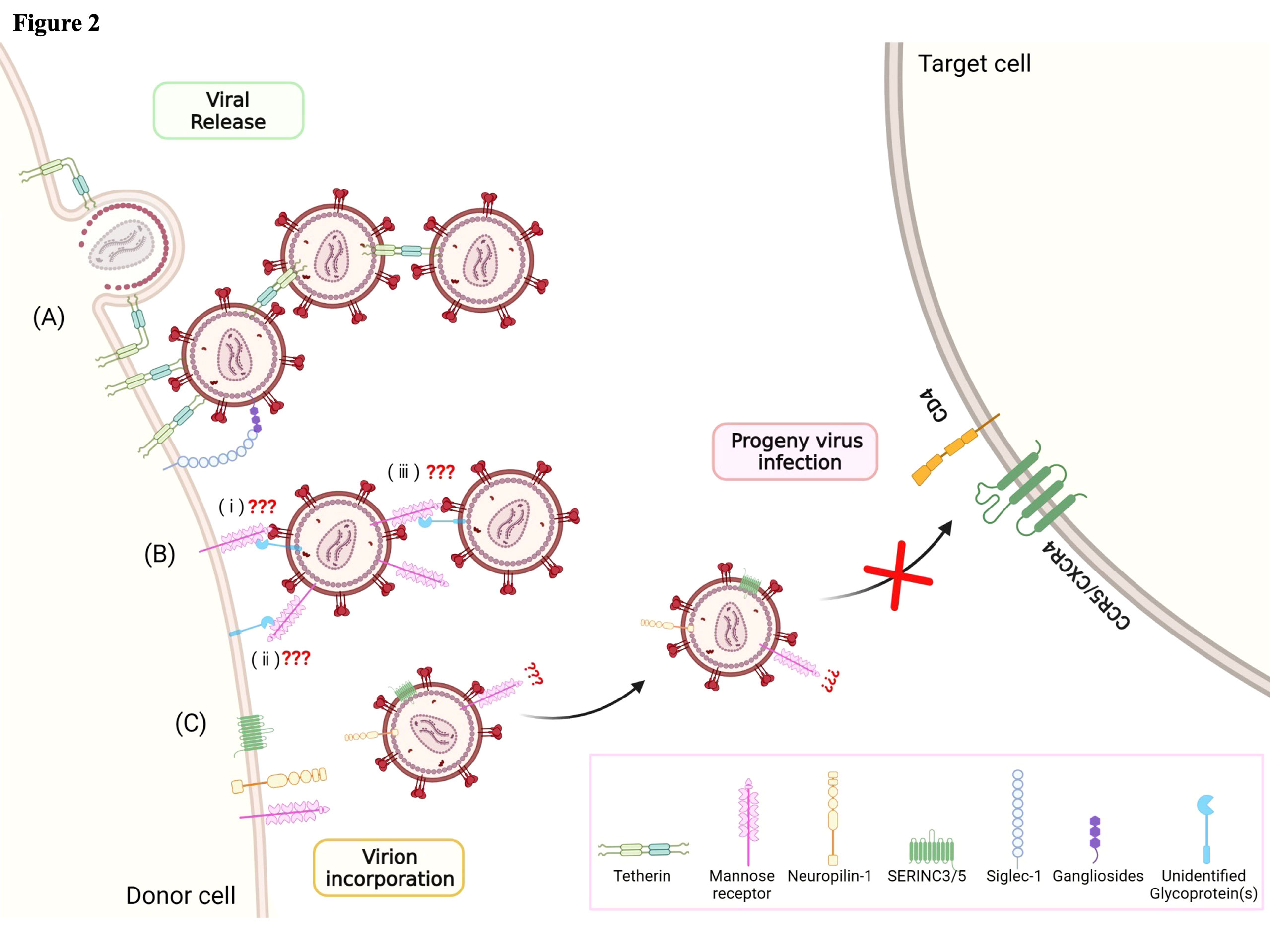
Figure 2 Host factors regulating the late phase of the HIV-1 life cycle, from viral release to progeny virus infection. Various cellular host factors including tetherin, mannose receptor (hMRC1), neuropilin-1 (NRP-1), SERINC3/5, and Siglec-1 are highly expressed on the surface of the plasma membrane. (A) Tetherin and Siglec-1 play a role in capturing viral particles at the budding step. (B) hMRC1 also inhibits viral particle release. It is hypothesized that (i) hMRC1 expressed on the cell surface may interact with Env glycoprotein and/or unidentified host glycoprotein(s) associated with viral particles. (ii) Unidentified host glycoprotein(s) expressed on the cell surface may interact with hMRC1 associated with viral particles. (iii) Viral particle-associated hMRC1 may interact with Env glycoprotein and/or unidentified host glycoprotein(s). However, the precise mechanisms are still unclear. (C) Virion-incorporated SERINC3/5 and NRP-1 contribute to the inhibition of progeny virus infection through interference with the binding step. Image created with BioRender.com.
In addition to the host factors described above, several viral accessory proteins, including Vif, Vpr, Vpu, Vpx, and Nef, also contribute to the regulation of HIV-1 replication in macrophages. Although the functions of most of these viral accessary proteins are well characterized, the precise role of Vpr is still poorly understood. Vpr has multiple biological functions, including the nuclear import of viral preintegration complex (PIC), repression of HIV-1 transcription, G2 cell cycle arrest, enhancement of the expression and processing of Env glycoprotein in macrophages, and induction of host protein degradation by recruitment of the E3 ubiquitin ligase complex (1, 38). Vpr is packaged into nascent virions (39) through interactions with the P6 domain of the viral Gag precursor (40). Vpr primarily localizes to the nucleus/nuclear envelope in PBMCs and macrophages (41, 42), and an observation by Desai et al. indicated that a substantial amount of Vpr incorporated into virus particles was released from the PIC and accumulated in the nucleus, suggesting a role for Vpr in the early stages of HIV-1 infection (43). However, the effect of virion-incorporated Vpr in the first round of infection of macrophages has not been revealed (44, 45). Vpr has a critical role in HIV-1 replication in monocytic cell lines including THP-1 or primary macrophages, but not in CD4+ T cells (i.e., Vpr-defective HIV-1 is severely restricted in macrophages) (46). Indeed, Mashiba et al. recently reported that Vpr promoted HIV-1 infection in non-dividing cells such as macrophages (44, 45). This evidence suggests the existence of host factors suppressing HIV-1 replication, which is counteracted by Vpr. Numerous host factors neutralized by Vpr have been identified including HTLF, CCDC137, MCM10, TET2, and LAPTM5 (44, 47–50). Despite the functional analyses of these Vpr-associated host factors, the exact role of Vpr in HIV-1 replication, especially in macrophages, is unclear and further study is required.
As described by Law et al., in addition to cell-free transmission, HIV-1 spreads via cell-to-cell contact through virological synapses (VS) in vivo (41). These are formed when the viral envelopes on the plasma membrane of HIV-1-infected cells bind to uninfected CD4+ T cells (51). Modes of cell-to-cell transmission are classified according to the cellular structures involved, i.e., filopodial bridges, membrane nanotubes (also known as TNT), and virological synapses (Figure 3A). The key feature of this mode of spread is that, in general, the effectiveness of HIV-1 infection is >10-fold greater than for cell-free infection of CD4+ T cells (52–56). Similar to the cell-to-cell transmission between CD4+ T cells, infected macrophages/monocyte-derived dendritic cells (MDDCs) recruit CD4, CCR5, LFA-1, ICAM-1, Gag proteins, and envelope glycoproteins, at the site of cell–cell contact, described as VS formation. More recently, it was reported that cell-to-cell contact between infected macrophages/MDDCs and CD4+ T cells was stabilized by interactions between gp120-CD4 and LFA-1-ICAM-1 and might be very effective for HIV-1 transmission (Figure 3C) (57). Previous studies suggested that DC-SIGN, a C-type lectin expressed on immature DCs, has a role in capturing HIV-1 particles through interactions with envelope glycoproteins and the subsequent transfer of infectious particles to other cells (Figure 3B) (58). Another member of the C-type lectin family, sialic acid-binding Ig-like lectin 1 (Siglec-1, also known as CD169), expressed on the surface of DCs and macrophages, also captures infectious particles via sialyl lactose-containing gangliosides exposed on the envelope, and thus also contributes to cell-to-cell transmission (Figure 2A) (59). Furthermore, Dupont et al. recently reported that TNT was highly positive for Siglec-1 and was required for TNT-mediated HIV-1 transmission in macrophages, as shown by silencing Siglec-1 (Figure 3A) (60). As described above, infected macrophages also significantly contribute to the cell-to-cell transmission through the formation of VS between infected macrophages and uninfected CD4+ T cells, which leads to the transfer of a high multiplicity of HIV-1 to the latter (61). Several groups also reported that this mode of infection is less sensitive to certain classes of antiretroviral therapies (ARTs) and reverse transcriptase (RT) inhibitors (62–64), and is more resistant to neutralizing antibodies against specific Env epitopes (65, 66).
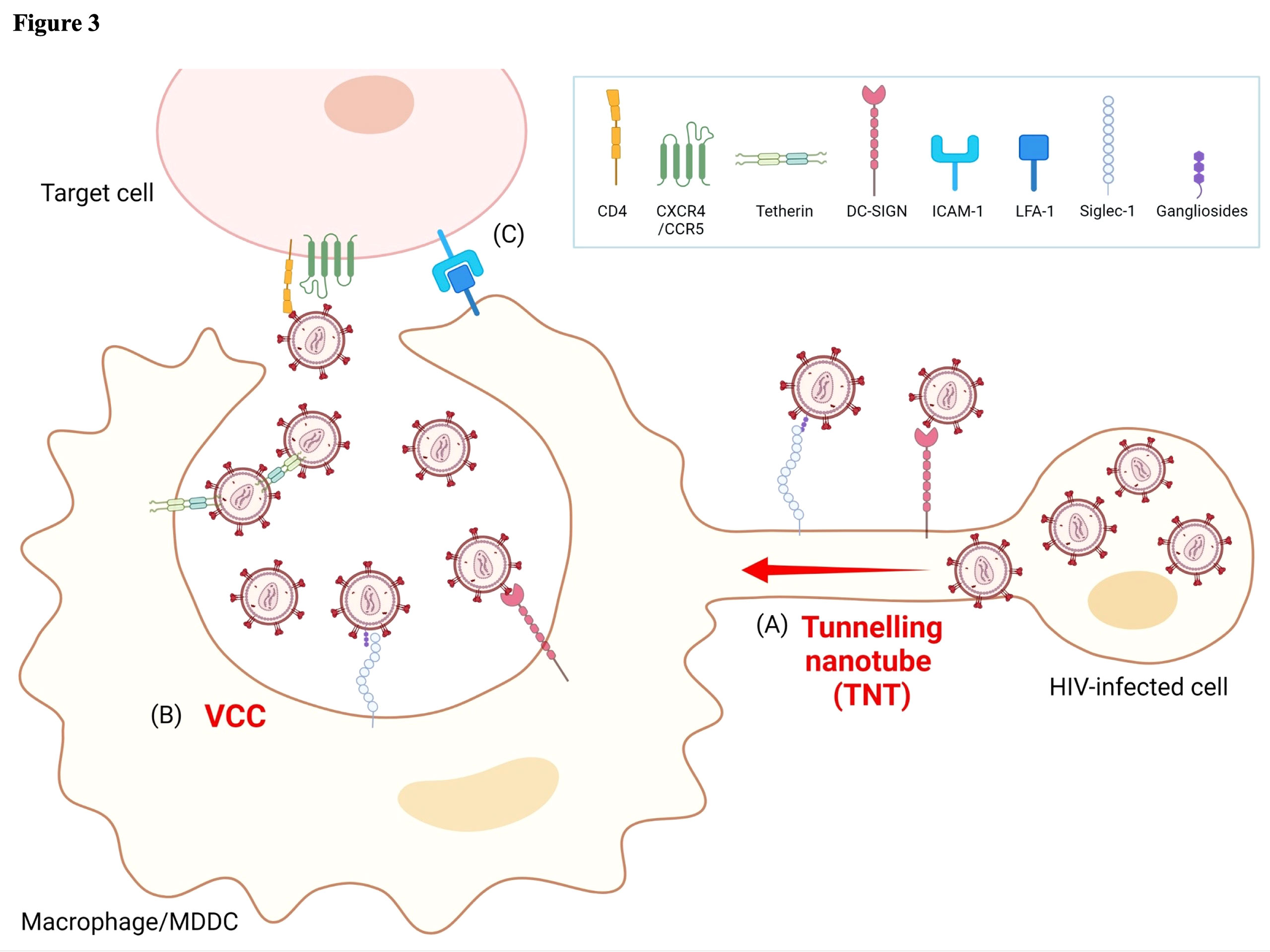
Figure 3 Models of HIV-1 cell-to-cell transmission in myeloid lineage cells. (A) HIV-1-infected cells form tunneling nanotubes (TNT) to transport infectious viral particles to other target cells by utilizing attachment receptors such as Siglec-1 or DC-SIGN. (B) Infected macrophages form unique virus-containing particles (VCC) and retain infectious viral particles through the association of tetherin, Siglec-1, or DC-SIGN with Env glycoprotein. When encountering other target cells, VCC containing either free virions or host factor(s)-associated virions may release infectious virus. (C) The binding of adhesion molecules LFA-1 and ICAM-1 has a well-characterized role in stabilizing virological synapse (VS) formation and contributes to cell-to-cell HIV-1 transmission. Image created with BioRender.com.
In contrast to the mechanisms of viral assembly in infected CD4+ T cells, viral particles accumulate on the plasma membrane and in intracellular tetraspanin-enriched virus-containing compartments (VCCs) in productively infected macrophages (Figure 3B) (67–70). VCCs rapidly change transmission mode to VS in a cytoskeleton-dependent manner. The structural features of VCC are similar to those of late endosomes or multivesicular bodies (MVB), but are distinguished from them in terms of acidity. Thus, VCCs are non-acidic and non-degrading compartments (71–73). As described in Section 2, tetherin is a critical host factor regulating HIV-1 release that is highly expressed on the surface of macrophages (Figures 1 and 2A), but not or only at a low level on CD4+ T cells (26, 27). Thus, tetherin effectively restricts HIV-1 release from the cell surface of macrophages rather than from CD4+ T cells, and this function is antagonized by the viral protein Vpu (27). In addition to the inhibition of cell-free viruses, tetherin has a role in forming VCC and potentially contributes to restricting cell-to-cell transmission (Figure 3B). Tetherin retains HIV-1 virions at the plasma membrane, which are subsequently internalized into VCC (74–76). Recently, Hammonds et al. reported that lectins such as Siglec-1, expressed on DCs and macrophage cell surfaces, play a crucial role in forming VCC (Figure 3B) (72). That study demonstrated that VCC formation is not necessary for macrophage infection but is necessary for cell-to-cell viral transmission from infected macrophages to CD4+ T cells (72). HIV-1 virions captured and internalized in VCCs through Siglec-1 are potentially protected from innate immune responses. Thus, captured HIV-1 infectious virions can transfer to target CD4+ T cells through VS, a mechanism independent of normal virus replication (77, 78). Human mannose receptor C-type 1 (hMRC1) also significantly contributes to the spread of infection through cell-to-cell transmission. Its function in HIV-1 infection is described in detail in the next section.
The human mannose receptor C-type1 (hMRC1), designated CD260, is a 175-kDa type I transmembrane glycoprotein belonging to the C-type lectin family. It is expressed on most tissue macrophage surfaces, DCs, selected lymphatic cells, liver endothelial cells, and vaginal epithelial cells (79). This protein contains three types of extracellular domains, namely, CR (cysteine-rich domain), FNII (fibronectin type II repeat), and eight tandem CRD (C-type carbohydrate recognition domains) (79). The CRD domains have a crucial role in capturing foreign bodies, including HIV-1. Recently, it was shown to be involved in capturing pathogens including bacteria and fungi, as well as viruses like HIV-1 (79). In HIV-1 infection, hMRC1 binds to viral Env through interactions mediated by glycosylated mannose residues on Env (80–82). Intriguingly, unlike typical virus entry, this uptake of HIV-1 mediated by hMRC1 does not lead to productive infection (82, 83), but the phagocytosed pathogen contributes to antigen presentation (84, 85). However, Nguyen and Hildreth provided evidence that hMRC1 on the macrophage surface facilitates virus transmission to CD4+ T cells (81). Several factors regulate this function of hMRC1, including viral proteins such as Vpr, Tat, and Nef, as well as unidentified host factors, but the precise mechanisms remain unclear. Sukegawa et al. demonstrated that hMRC1 inhibits virus release from HIV-1-infected macrophages in a BST-2-independent manner (86) (Figure 2B). Interestingly, the amount of endogenously expressed hMRC1 was significantly downregulated after virus infection. It is assumed that the virus counteracts the function of hMRC1-mediated inhibition of virion release by removing hMRC1 from the cell surface. This reduction of hMRC1 has been reported in several other papers, both in vivo and in vitro. Koziel et al. reported a modest reduction of hMRC mRNA in HIV-1-positive patients (87). Other investigators have reported the relevance of viral proteins such as Nef, Tat, and Vpr. Vigerust et al. reported that Nef induced the surface repression of hMRC1 without affecting steady-state levels but interfered with the recycling of mannose receptors to the cell surface (88). In addition, HIV-1 Tat was reported to inhibit transcription from the rat mannose receptor promoter (89). However, recently, Lubow et al. reported that the contribution of Tat to the downregulation of mannose receptors was insignificant (53). Furthermore, they implicated the involvement of Nef and Vpr in the downregulation of hMRC1 and indicated that cooperation between Vpr and Nef induced a synergistic reduction of hMRC1 (53). Thus, Vpr downregulates the transcription of hMRC1, and Nef removes hMRC1 from the cell surface through lysosomal degradation, causing a further reduction of hMRC1. It was also demonstrated that the downregulation of Env expression by hMRC1 was counteracted by Vpr through rescue from lysosomal degradation, which increased the formation of VS and facilitated cell-to-cell transmission between macrophages and CD4+ T cells (53, 56). This evidence demonstrates the significant impact of hMRC1 during HIV-1 replication in macrophages and on the normal function of host immune defense by capturing pathogens.
As noted above, CD4+T lymphocytes are considered the primary HIV-1 reservoir cells. The contribution of monocytes/macrophages remains controversial (7). However, there is evidence showing the contribution of tissue-resident macrophages in establishing HIV-1 reservoirs. The earliest study was reported by Gartner et al. in 1986 (90). Due to the difficulty of obtaining primary tissue-resident macrophages, non-human primate models have been widely used to investigate the association of infectious virus dynamics with disease progression in vivo (90–92). In 2001, Igarashi et al. confirmed the contribution of macrophages in HIV-1 reservoirs utilizing SHIV in a macaque model (93). That report observed the persistence of SHIV infection in tissue macrophages under the depletion of CD4+ T cells according to disease progression in the macaque. Furthermore, the administration of potent RT inhibitors effectively blocked HIV-1 circulating in CD4+ T cells but not in macrophages, demonstrating that tissue macrophages can sustain viral replication independently.
Reservoirs in macrophages start to become established a few days after the initial HIV-1 infection and are sustained during the asymptomatic stage of disease in these long-lived cells (7). In contrast to the loss of CD4+ T cells during the progression to AIDS, infected macrophages are less susceptible to HIV-induced cytopathic effects (7) and are more resistant to CD8+ cytotoxic T lymphocyte (CTL)-mediated killing (94). Hence, they may survive for more than a few weeks (1) and can therefore persistently infect cells as they are also poorly targeted by ART (i.e., they are less susceptible to some antiretroviral drugs). In fact, recent in vivo studies revealed the contribution of monocytes/macrophages as reservoirs. Honeycutt et al. developed humanized myeloid-only mouse models (MoM) unable to support human lymphocyte development (95). Using these models, they reported the detection of rebound viremia in three of nine mice 7 weeks after the discontinuation of ART (96). This report thus provided novel evidence that monocytes/macrophages can be a source of rebound viremia following ART cessation. Furthermore, Ganor et al. demonstrated that integrated HIV-1 DNA, HIV-1 RNA, and p24 were predominantly detected in urethral macrophages rather than CD4+ T lymphocytes in HIV-1-infected patients on ART (97).
The central nervous system (CNS, microglia) is also considered another tissue-resident macrophage reservoir (98, 99). Within a few days after primary infection, HIV-1 entered the CNS through the blood–brain barrier (BBB) via infected monocytes rather than CD4+ T cells (100, 101). In the CNS, HIV-1 persists predominantly in tissue-resident macrophages, such as microglia and perivascular macrophages differentiated from monocytes. More recently, Joseph et al. reported the isolation of R5-tropic virus from the cerebrospinal fluid (CSF) of ART-treated patients, suggesting that HIV-1 is continuously replicating in CNS reservoirs (102, 103). As with other reservoir cells, HIV-1-infected microglia are also long-lived, and unlike other hematopoietic reservoirs, the turnover of microglia is very slow, with only 28% of these cells renewed annually (104). Thus, HIV-1-infected microglia potentially sustain lifetime reservoirs in infected individuals. As described above, monocytes/macrophages, as well as CD4+ T cells, contribute to the persistence of infection and represent reservoirs in vivo. Importantly, some antiretroviral drugs such as protease inhibitors, saquinavir, and ritonavir exhibited a 2- to 10-fold lower activity in infected macrophages relative to chronically infected lymphocytes (105, 106). Moreover, Gavegnano et al. showed that the intracellular concentrations of nucleoside analog active metabolites in macrophages were 5- to 140-fold lower than in lymphocytes, contributing to significantly weaker antiviral activity (107). These features may also contribute to the persistence of HIV-1-infected macrophages and result in the establishment of HIV-1 reservoirs and the evolution of drug-resistant viruses.
Viral replication in HIV-infected individuals is controlled by combination antiretroviral therapy (cART), which mainly targets viral enzymes. However, latently infected reservoirs and drug-resistant viruses are still a significant barrier to curing HIV-1. To this end, several strategies such as gene therapy using CRISPR, vaccines to boost anti-HIV immune responses, and immunotherapy with broadly neutralizing antibodies have been investigated. In addition to these strategies, the “Shock and Kill” approach to reducing the size of reservoirs is a current challenge, whereby latency-reversing agents (LRAs) reactivate latently infected provirus (the shock) and induce cell death mediated by cytopathic effects, apoptosis, or CTL response (the kill), combined with ART treatment to prevent the occurrence of a new infection. This strategy is still under development (108), and some small-molecule chemical compounds, including histone deacetylase inhibitors (HDACi) (e.g., SAHA/vorinostat) (109), bromodomain and extraterminal domain inhibitors (BETi) (e.g., JQ1) (110), and protein kinase C (PKC) agonists [e.g., Prostratin, PEP-005 (ingenole-3-angelate) and bryostatin-1] (111), are being intensively investigated as candidate LRAs. Several groups have reported that using an LRA, active via a single mechanism, might not lead to effective viral reactivation in reservoir cells, and combinations of drug candidates with different mechanisms of action will be needed to enhance proviral activation. Currently, combining the BRD4 inhibitor (JQ1) and PKC agonist(s) (e.g., Prostratin, PEP-005) might be the most effective means of reactivating the HIV-1 provirus (7, 108). Despite the potent effects of LRA or a combination of LRAs on ex vivo or in vitro analyses, multiple clinical trials have failed to reduce latent reservoir size sufficiently, and none have shown superior therapeutic effects compared with current cART (112). A reason for this may be that different responses to LRAs by several types of latent reservoirs (7, 113) are influenced by the size and chromosomal location of the integrated provirus (114) in HIV-1 reservoirs. Therefore, the development of new LRAs or combination therapies of several LRAs to induce efficient provirus reactivation remains an unmet need.
HT and SS contributed to the conception and wrote all sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by: grant 21fk0410023s0303, 22fk0410052s0101 for Research Program on HIV/AIDS from Japan Agency for Medical Research and Development to HT, Takeda Science Foundation to SS. The founder had no role in decision to publish or preparation of the manuscript
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lubow J, Collins KL. Vpr is a VIP: HIV vpr and infected macrophages promote viral pathogenesis. Viruses (2020) 12(8):809. doi: 10.3390/v12080809
2. Muntjewerff EM, Meesters LD, van den Bogaart G. Antigen cross-presentation by macrophages. Front Immunol (2020) 11:1276. doi: 10.3389/fimmu.2020.01276
3. Yin X, Langer S, Zhang Z, Herbert KM, Yoh S, König R, et al. Sensor sensibility-HIV-1 and the innate immune response. Cells (2020) 9(1):254. doi: 10.3390/cells9010254
4. Rojas M, Luz-Crawford P, Soto-Rifo R, Reyes-Cerpa S, Toro-Ascuy D. The landscape of IFN/ISG signaling in HIV-1-Infected macrophages and its possible role in the HIV-1 latency. Cells (2021) 10(9):2378. doi: 10.3390/cells10092378
5. Chen B. Molecular mechanism of HIV-1 entry. Trends Microbiol (2019) 27:878–91. doi: 10.1016/j.tim.2019.06.002
6. Moeser M, Nielsen JR, Joseph SB. Macrophage tropism in pathogenic HIV-1 and SIV infections. Viruses (2020) 12(10):1077. doi: 10.3390/v12101077
7. Hendricks CM, Cordeiro T, Gomes AP, Stevenson M. The interplay of HIV-1 and macrophages in viral persistence. Front Microbiol (2021) 12:646447. doi: 10.3389/fmicb.2021.646447
8. Borrajo A, Ranazzi A, Pollicita M, Bellocchi MC, Salpini R, Mauro MV, et al. Different patterns of HIV-1 replication in MACROPHAGES is led by Co-receptor usage. Medicina (Kaunas) (2019) 55(6):297. doi: 10.3390/medicina55060297
9. He S, Wu Y. Relationships between HIV-mediated chemokine coreceptor signaling, cofilin hyperactivation, viral tropism switch and HIV-mediated CD4 depletion. Curr HIV Res (2019) 17:388–96. doi: 10.2174/1570162X17666191106112018
10. Kennedy EM, Amie SM, Bambara RA, Kim B. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J Biol Chem (2012) 287:14280–8. doi: 10.1074/jbc.M112.348482
11. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by vpx. Nature (2011) 474:654–7. doi: 10.1038/nature10117
12. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol (2012) 13:223–8. doi: 10.1038/ni.2236
13. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature (2011) 474:658–61. doi: 10.1038/nature10195
14. Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem (2004) 279:51545–53. doi: 10.1074/jbc.M408573200
15. Kyei GB, Cheng X, Ramani R, Ratner L. Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe (2015) 17:98–106. doi: 10.1016/j.chom.2014.11.009
16. Berger G, Durand S, Fargier G, Nguyen XN, Cordeil S, Bouaziz S, et al. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PloS Pathog (2011) 7:e1002221. doi: 10.1371/journal.ppat.1002221
17. Ikeda T, Molan AM, Jarvis MC, Carpenter MA, Salamango DJ, Brown WL, et al. HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J Gen Virol (2019) 100:1140–52. doi: 10.1099/jgv.0.001276
18. Peng G, Greenwell-Wild T, Nares S, Jin W, Lei KJ, Rangel ZG, et al. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood (2007) 110:393–400. doi: 10.1182/blood-2006-10-051763
19. Stavrou S, Ross SR. APOBEC3 proteins in viral immunity. J Immunol (2015) 195:4565–70. doi: 10.4049/jimmunol.1501504
20. Buffone C, Kutzner J, Opp S, Martinez-Lopez A, Selyutina A, Coggings SA, et al. The ability of SAMHD1 to block HIV-1 but not SIV requires expression of MxB. Virology (2019) 531:260–8. doi: 10.1016/j.virol.2019.03.018
21. Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature (2013) 502:559–62. doi: 10.1038/nature12542
22. Wang X, Wang H, Liu MQ, Li JL, Zhou RH, Zhou Y, et al. IFN-λ inhibits drug-resistant HIV infection of macrophages. Front Immunol (2017) 8:210. doi: 10.3389/fimmu.2017.00210
23. Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature (2013) 502:563–6. doi: 10.1038/nature12653
24. Liu L, Oliveira NM, Cheney KM, Pade C, Dreja H, Bergin AM, et al. A whole genome screen for HIV restriction factors. Retrovirology (2011) 8:94. doi: 10.1186/1742-4690-8-94
25. Zhu B, Mandal SS, Pham AD, Zheng Y, Erdjument-Bromage H, Batra SK, et al. The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev (2005) 19:1668–73. doi: 10.1101/gad.1292105
26. Schindler M, Rajan D, Banning C, Wimmer P, Koppensteiner H, Iwanski A, et al. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology (2010) 7:1. doi: 10.1186/1742-4690-7-1
27. Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U.S.A. (2009) 106:2868–73. doi: 10.1073/pnas.0813223106
28. Schmidt S, Fritz JV, Bitzegeio J, Fackler OT, Keppler OT. HIV-1 vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. mBio (2011) 2:e00036–00011. doi: 10.1128/mBio.00036-11
29. Dubé M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA. HIV-1 vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic (2011) 12:1714–29. doi: 10.1111/j.1600-0854.2011.01277.x
30. Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, et al. HIV-1 nef promotes infection by excluding SERINC5 from virion incorporation. Nature (2015) 526:212–7. doi: 10.1038/nature15399
31. Usami Y, Wu Y, Göttlinger HG. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by nef. Nature (2015) 526:218–23. doi: 10.1038/nature15400
32. Zutz A, Schölz C, Schneider S, Pierini V, Münchhoff M, Sutter K, et al. SERINC5 is an unconventional HIV restriction factor that is upregulated during myeloid cell differentiation. J Innate Immun (2020) 12:399–409. doi: 10.1159/000504888
33. Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem (2017) 292:6014–26. doi: 10.1074/jbc.M117.777714
34. Beitari S, Ding S, Pan Q, Finzi A, Liang C. Effect of HIV-1 env on SERINC5 antagonism. J Virol (2017) 91(4):e02214–16. doi: 10.1128/JVI.02214-16
35. Chen YC, Sood C, Marin M, Aaron J, Gratton E, Salaita K, et al. Super-resolution fluorescence imaging reveals that serine incorporator protein 5 inhibits human immunodeficiency virus fusion by disrupting envelope glycoprotein clusters. ACS Nano (2020) 14:10929–43. doi: 10.1021/acsnano.0c02699
36. Zhang X, Shi J, Qiu X, Chai Q, Frabutt DA, Schwartz RC, et al. CD4 expression and env conformation are critical for HIV-1 restriction by SERINC5. J Virol (2019) 93. doi: 10.1128/JVI.00544-19
37. Wang S, Zhao L, Zhang X, Zhang J, Shang H, Liang G. Neuropilin-1, a myeloid cell-specific protein, is an inhibitor of HIV-1 infectivity. Proc Natl Acad Sci U.S.A. (2022) 119(2):e2114884119. doi: 10.1073/pnas.2114884119
38. Fabryova H, Strebel K. Vpr and its cellular interaction partners: R we there yet? Cells (2019) 8(11):1310. doi: 10.3390/cells8111310
39. Cohen EA, Dehni G, Sodroski JG, Haseltine WA. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J Virol (1990) 64:3097–9. doi: 10.1128/jvi.64.6.3097-3099.1990
40. Bachand F, Yao XJ, Hrimech M, Rougeau N, Cohen EA. Incorporation of vpr into human immunodeficiency virus type 1 requires a direct interaction with the p6 domain of the p55 gag precursor. J Biol Chem (1999) 274:9083–91. doi: 10.1074/jbc.274.13.9083
41. Lu YL, Spearman P, Ratner L. Human immunodeficiency virus type 1 viral protein r localization in infected cells and virions. J Virol (1993) 67:6542–50. doi: 10.1128/jvi.67.11.6542-6550.1993
42. Jacquot G, Le Rouzic E, David A, Mazzolini J, Bouchet J, Bouaziz S, et al. Localization of HIV-1 vpr to the nuclear envelope: impact on vpr functions and virus replication in macrophages. Retrovirology (2007) 4:84. doi: 10.1186/1742-4690-4-84
43. Desai TM, Marin M, Sood C, Shi J, Nawaz F, Aiken C, et al. Fluorescent protein-tagged vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology (2015) 12:88. doi: 10.1186/s12977-015-0215-z
44. Wang Q, Su L. Vpr enhances HIV-1 env processing and virion infectivity in macrophages by modulating TET2-dependent IFITM3 expression. mBio (2019) 10(4):e01344–19. doi: 10.1128/mBio.01344-19
45. Mashiba M, Collins DR, Terry VH, Collins KL. Vpr overcomes macrophage-specific restriction of HIV-1 env expression and virion production. Cell Host Microbe (2014) 16:722–35. doi: 10.1016/j.chom.2014.10.014
46. Balliet JW, Kolson DL, Eiger G, Kim FM, McGann KA, Srinivasan A, et al. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: Mutational analysis of a primary HIV-1 isolate. Virology (1994) 200:623–31. doi: 10.1006/viro.1994.1225
47. Lahouassa H, Blondot ML, Chauveau L, Chougui G, Morel M, Leduc M, et al. HIV-1 vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc Natl Acad Sci U.S.A. (2016) 113:5311–6. doi: 10.1073/pnas.1600485113
48. Zhang F, Bieniasz PD. HIV-1 vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. Elife (2020) 9:e55806. doi: 10.7554/eLife.55806
49. Romani B, Shaykh Baygloo N, Aghasadeghi MR, Allahbakhshi E. HIV-1 vpr protein enhances proteasomal degradation of MCM10 DNA replication factor through the Cul4-DDB1[VprBP] E3 ubiquitin ligase to induce G2/M cell cycle arrest. J Biol Chem (2015) 290:17380–9. doi: 10.1074/jbc.M115.641522
50. Zhao L, Wang S, Xu M, He Y, Zhang X, Xiong Y, et al. Vpr counteracts the restriction of LAPTM5 to promote HIV-1 infection in macrophages. Nat Commun (2021) 12:3691. doi: 10.1038/s41467-021-24087-8
51. Piguet V, Sattentau Q. Dangerous liaisons at the virological synapse. J Clin Invest (2004) 114:605–10. doi: 10.1172/JCI22812
52. Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol (2008) 6:815–26. doi: 10.1038/nrmicro1972
53. Lubow J, Virgilio MC, Merlino M, Collins DR, Mashiba M, Peterson BG, et al. Mannose receptor is an HIV restriction factor counteracted by vpr in macrophages. Elife (2020) 9:e51035. doi: 10.7554/eLife.51035
54. Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol (2011) 85:7169–76. doi: 10.1128/JVI.00231-11
55. Martin N, Welsch S, Jolly C, Briggs JA, Vaux D, Sattentau QJ. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J Virol (2010) 84:3516–27. doi: 10.1128/JVI.02651-09
56. Collins DR, Lubow J, Lukic Z, Mashiba M, Collins KL. Vpr promotes macrophage-dependent HIV-1 infection of CD4+ T lymphocytes. PloS Pathog (2015) 11:e1005054. doi: 10.1371/journal.ppat.1005054
57. Lopez P, Koh WH, Hnatiuk R, Murooka TT. HIV Infection stabilizes macrophage-T cell interactions to promote cell-cell HIV spread. J Virol (2019) 93(18):e00805–19. doi: 10.1128/JVI.00805-19
58. Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell (2000) 100:587–97. doi: 10.1016/S0092-8674(00)80694-7
59. Izquierdo-Useros N, Lorizate M, Puertas MC, Rodriguez-Plata MT, Zangger N, Erikson E, et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PloS Biol (2012) 10:e1001448. doi: 10.1371/journal.pbio.1001448
60. Dupont M, Souriant S, Balboa L, Vu Manh TP, Pingris K, Rousset S, et al. Tuberculosis-associated IFN-I induces siglec-1 on tunneling nanotubes and favors HIV-1 spread in macrophages. Elife (2020) 9:e52535. doi: 10.7554/eLife.52535
61. Groot F, Welsch S, Sattentau QJ. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood (2008) 111:4660–3. doi: 10.1182/blood-2007-12-130070
62. Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature (2011) 477:95–8. doi: 10.1038/nature10347
63. Duncan CJ, Russell RA, Sattentau QJ. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. Aids (2013) 27:2201–6. doi: 10.1097/QAD.0b013e3283632ec4
64. Agosto LM, Zhong P, Munro J, Mothes W. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PloS Pathog (2014) 10:e1003982. doi: 10.1371/journal.ppat.1003982
65. Schiffner T, Sattentau QJ, Duncan CJ. Cell-to-cell spread of HIV-1 and evasion of neutralizing antibodies. Vaccine (2013) 31:5789–97. doi: 10.1016/j.vaccine.2013.10.020
66. Dufloo J, Bruel T, Schwartz O. HIV-1 cell-to-cell transmission and broadly neutralizing antibodies. Retrovirology (2018) 15:51. doi: 10.1186/s12977-018-0434-1
67. Carter CA, Ehrlich LS. Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol (2008) 62:425–43. doi: 10.1146/annurev.micro.62.081307.162758
68. Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol (2007) 177:329–41. doi: 10.1083/jcb.200609050
69. Nkwe DO, Pelchen-Matthews A, Burden JJ, Collinson LM, Marsh M. The intracellular plasma membrane-connected compartment in the assembly of HIV-1 in human macrophages. BMC Biol (2016) 14:50. doi: 10.1186/s12915-016-0272-3
70. Mlcochova P, Pelchen-Matthews A, Marsh M. Organization and regulation of intracellular plasma membrane-connected HIV-1 assembly compartments in macrophages. BMC Biol (2013) 11:89. doi: 10.1186/1741-7007-11-89
71. Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P. HIV-1 buds and accumulates in "nonacidic" endosomes of macrophages. Cell Host Microbe (2007) 2:85–95. doi: 10.1016/j.chom.2007.06.011
72. Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, et al. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PloS Pathog (2017) 13:e1006181. doi: 10.1371/journal.ppat.1006181
73. Tan J, Sattentau QJ. The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends Microbiol (2013) 21:405–12. doi: 10.1016/j.tim.2013.05.001
74. Giese S, Marsh M. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PloS Pathog (2014) 10:e1004189. doi: 10.1371/journal.ppat.1004189
75. Chu H, Wang JJ, Qi M, Yoon JJ, Chen X, Wen X, et al. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe (2012) 12:360–72. doi: 10.1016/j.chom.2012.07.011
76. Blanchet FP, Stalder R, Czubala M, Lehmann M, Rio L, Mangeat B, et al. TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology (2013) 10:6. doi: 10.1186/1742-4690-10-6
77. Dupont M, Sattentau QJ. Macrophage cell-cell interactions promoting HIV-1 infection. Viruses (2020) 12(5):492. doi: 10.3390/v12050492
78. Martinez-Picado J, McLaren PJ, Telenti A, Izquierdo-Useros N. Retroviruses as myeloid cell riders: What natural human siglec-1 "Knockouts" tell us about pathogenesis. Front Immunol (2017) 8:1593. doi: 10.3389/fimmu.2017.01593
79. Azad AK, Rajaram MV, Schlesinger LS. Exploitation of the macrophage mannose receptor (CD206) in infectious disease diagnostics and therapeutics. J Cytol Mol Biol (2014) 1(1):1000003. doi: 10.13188/2325-4653.1000003
80. Fanibunda SE, Modi DN, Gokral JS, Bandivdekar AH. HIV gp120 binds to mannose receptor on vaginal epithelial cells and induces production of matrix metalloproteinases. PloS One (2011) 6:e28014. doi: 10.1371/journal.pone.0028014
81. Nguyen DG, Hildreth JE. Involvement of macrophage mannose receptor in the binding and transmission of HIV by macrophages. Eur J Immunol (2003) 33:483–93. doi: 10.1002/immu.200310024
82. Trujillo JR, Rogers R, Molina RM, Dangond F, McLane MF, Essex M, et al. Noninfectious entry of HIV-1 into peripheral and brain macrophages mediated by the mannose receptor. Proc Natl Acad Sci U.S.A. (2007) 104:5097–102. doi: 10.1073/pnas.0611263104
83. Pontow SE, Kery V, Stahl PD. Mannose receptor. Int Rev Cytol (1992) 137b:221–44. doi: 10.1073/pnas.0611263104
84. Geijtenbeek TB, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol (2016) 16:433–48. doi: 10.1038/nri.2016.55
85. Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol (2005) 26:104–10. doi: 10.1016/j.it.2004.12.001
86. Sukegawa S, Miyagi E, Bouamr F, Farkašová H, Strebel K. Mannose receptor 1 restricts HIV particle release from infected macrophages. Cell Rep (2018) 22:786–95. doi: 10.1016/j.celrep.2017.12.085
87. Koziel H, Eichbaum Q, Kruskal BA, Pinkston P, Rogers RA, Armstrong MY, et al. Reduced binding and phagocytosis of pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J Clin Invest (1998) 102:1332–44. doi: 10.1172/JCI560
88. Vigerust DJ, Egan BS, Shepherd VL. HIV-1 nef mediates post-translational down-regulation and redistribution of the mannose receptor. J Leukoc Biol (2005) 77:522–34. doi: 10.1189/jlb.0804454
89. Caldwell RL, Egan BS, Shepherd VL. HIV-1 tat represses transcription from the mannose receptor promoter. J Immunol (2000) 165:7035–41. doi: 10.4049/jimmunol.165.12.7035
90. Gartner S, Markovits P, Markovitz DM, Betts RF, Popovic M. Virus isolation from and identification of HTLV-III/LAV-producing cells in brain tissue from a patient with AIDS. Jama (1986) 256:2365–71. doi: 10.1001/jama.1986.03380170081023
91. Micci L, Alvarez X, Iriele RI, Ortiz AM, Ryan ES, McGary CS, et al. CD4 depletion in SIV-infected macaques results in macrophage and microglia infection with rapid turnover of infected cells. PloS Pathog (2014) 10:e1004467. doi: 10.1371/journal.ppat.1004467
92. Cai Y, Sugimoto C, Liu DX, Midkiff CC, Alvarez X, Lackner AA, et al. Increased monocyte turnover is associated with interstitial macrophage accumulation and pulmonary tissue damage in SIV-infected rhesus macaques. J Leukoc Biol (2015) 97:1147–53. doi: 10.1189/jlb.4A0914-441R
93. Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc Natl Acad Sci U.S.A. (2001) 98:658–63. doi: 10.1073/pnas.98.2.658
94. Clayton KL, Collins DR, Lengieza J, Ghebremichael M, Dotiwala F, Lieberman J, et al. Resistance of HIV-infected macrophages to CD8(+) T lymphocyte-mediated killing drives activation of the immune system. Nat Immunol (2018) 19:475–86. doi: 10.1038/s41590-018-0085-3
95. Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest (2016) 126:1353–66. doi: 10.1172/JCI84456
96. Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, et al. HIV Persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med (2017) 23:638–43. doi: 10.1038/nm.4319
97. Ganor Y, Real F, Sennepin A, Dutertre CA, Prevedel L, Xu L, et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol (2019) 4:633–44. doi: 10.1038/s41564-018-0335-z
98. Williams DW, Anastos K, Morgello S, Berman JW. JAM-a and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+ monocytes in HIV-infected individuals. J Leukoc Biol (2015) 97:401–12. doi: 10.1189/jlb.5A0714-347R
99. Kruize Z, Kootstra NA. The role of macrophages in HIV-1 persistence and pathogenesis. Front Microbiol (2019) 10:2828. doi: 10.3389/fmicb.2019.02828
100. León-Rivera R, Veenstra M, Donoso M, Tell E, Eugenin EA, Morgello S, et al. Central nervous system (CNS) viral seeding by mature monocytes and potential therapies to reduce CNS viral reservoirs in the cART era. mBio (2021) 12(2):e03633–20. doi: 10.1128/mBio.03633-20
101. Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology (1992) 42:1736–9. doi: 10.1212/WNL.42.9.1736
102. Akiyama H, Gummuluru S. HIV-1 persistence and chronic induction of innate immune responses in macrophages. Viruses (2020) 12(7):711. doi: 10.3390/v12070711
103. Joseph SB, Kincer LP, Bowman NM, Evans C, Vinikoor MJ, Lippincott CK, et al. Human immunodeficiency virus type 1 RNA detected in the central nervous system (CNS) after years of suppressive antiretroviral therapy can originate from a replicating CNS reservoir or clonally expanded cells. Clin Infect Dis (2019) 69:1345–52. doi: 10.1093/cid/ciy1066
104. Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, et al. The lifespan and turnover of microglia in the human brain. Cell Rep (2017) 20:779–84. doi: 10.1016/j.celrep.2017.07.004
105. Wong ME, Jaworowski A, Hearps AC. The HIV reservoir in monocytes and macrophages. Front Immunol (2019) 10:1435. doi: 10.3389/fimmu.2019.01435
106. Perno CF, Newcomb FM, Davis DA, Aquaro S, Humphrey RW, Caliò R, et al. Relative potency of protease inhibitors in monocytes/macrophages acutely and chronically infected with human immunodeficiency virus. J Infect Dis (1998) 178:413–22. doi: 10.1086/515642
107. Gavegnano C, Detorio MA, Bassit L, Hurwitz SJ, North TW, Schinazi RF. Cellular pharmacology and potency of HIV-1 nucleoside analogs in primary human macrophages. Antimicrob Agents Chemother (2013) 57:1262–9. doi: 10.1128/AAC.02012-12
108. Maina EK, Adan AA, Mureithi H, Muriuki J, Lwembe RM. A review of current strategies towards the elimination of latent HIV-1 and subsequent HIV-1 cure. Curr HIV Res (2021) 19:14–26. doi: 10.2174/1570162X18999200819172009
109. Matalon S, Rasmussen TA, Dinarello CA. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol Med (2011) 17:466–72. doi: 10.2119/molmed.2011.00076
110. Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a tat-independent mechanism. Cell Cycle (2013) 12:452–62. doi: 10.4161/cc.23309
111. Sánchez-Duffhues G, Vo MQ, Pérez M, Calzado MA, Moreno S, Appendino G, et al. Activation of latent HIV-1 expression by protein kinase c agonists. a novel therapeutic approach to eradicate HIV-1 reservoirs. Curr Drug Targets (2011) 12:348–56. doi: 10.2174/138945011794815266
112. Rodari A, Darcis G, Van Lint CM. The current status of latency reversing agents for HIV-1 remission. Annu Rev Virol (2021) 8:491–514. doi: 10.1146/annurev-virology-091919-103029
113. Grau-Expósito J, Luque-Ballesteros L, Navarro J, Curran A, Burgos J, Ribera E, et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PloS Pathog (2019) 15:e1007991. doi: 10.1371/journal.ppat.1007991
Keywords: monocyte, macrophage, HIV-1, immunity, CD4+ T cells, viral tropism, HIV-1 reservoir
Citation: Sukegawa S and Takeuchi H (2022) Toward the unveiling of HIV-1 dynamics: Involvement of monocytes/macrophages in HIV-1 infection. Front. Virol. 2:934892. doi: 10.3389/fviro.2022.934892
Received: 03 May 2022; Accepted: 29 June 2022;
Published: 25 July 2022.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Yasuko Tsunetsugu Yokota, Tokyo University of Technology, JapanCopyright © 2022 Sukegawa and Takeuchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroaki Takeuchi, aHRha2UubW9sdkB0bWQuYWMuanA=; Sayaka Sukegawa, c2F5YWthLnN1a2VnYXdhLm1vbHZAdG1kLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.