 Jo L. Wilson
Jo L. Wilson Ruifeng Zhou1
Ruifeng Zhou1 Andrew Pekosz
Andrew Pekosz- 1W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins University Bloomberg School of Public Health, Baltimore, MD, United States
- 2Deparment of Pediatric Allergy and Immunology, Johns Hopkins Hospital, Baltimore, MD, United States
- 3Adult Emergency Department, Johns Hopkins Hospital, Baltimore, MD, United States
Influenza B Viruses (IBV) have caused an increasing number of cases over the last 15 years. The focus of this study was to assess the role of egg adapted mutants in IBV vaccines on the reactivity of serum from vaccinated or IBV infected individuals. We focused on the 2017-2018 IBV season as this was a significant influenza year with reported low vaccine effectiveness by the CDC. Patient samples were obtained from Johns Hopkins Adult Emergency Room for virus isolation and antigenic characterization. Antigenic characterization was evaluated using neutralizing antibody assays. Viral characterization was carried out using viral genome sequencing and structural modeling, MDCK-SIAT1 growth curves, MDCK Plaque assays and human primary nasal epithelial cell (hNEC) growth curves. In our analysis, we found that in the vaccine strains of both IBV lineages, there was an amino acid change at position 197 (B/Brisbane HA Numbering) that leads to a loss of glycosylation. Our antigenic evaluation shows that there is a significant difference in neutralizing antibody titers between the egg adapted vaccine for the B/Yamagata lineage compared to representative clinical isolates from that season and the cell cultured vaccine. We propose that this loss of a glycosylation site is an important site for propagation in the allantois and that this common site change may play a role in antigenic recognition and therefore immune protection from circulating viruses. Screening egg cultured vaccine viruses for egg adapted mutants, further transitioning vaccine production to mammalian culture models (MDCK) or investigating new models of influenza vaccination may be necessary to improve efficacy of the seasonal influenza vaccine for protection from IBV.
Introduction
Influenza A Virus (IAV) and Influenza B Virus (IBV) co-circulate in the human population causing significant seasonal epidemics. Both viruses can cause upper and lower respiratory disease in humans at a wide range of severity (1). While the overall severity of clinical disease is similar between IAV and IBV (2), IBV does have specific clinical associations including an age predisposition for late childhood, a larger proportion of pediatric deaths and increased risk of the rare acute encephalitis, Reye Syndrome (3–5). IBV overall has been less thoroughly studied compared to IAV, perhaps due to the lack of pandemic potential as IBV is endemic to humans with no true animal reservoir (6). When looking across various surveillance efforts, IBV accounts for about 23% of overall influenza infections over the last two decades with evidence for increasing case numbers in recent years (7).
IBV diverged into two separate genetic lineages – B/Victoria and B/Yamagata – likely in the late 1970s and are defined by differences in their hemagglutinin (HA) gene segment (8). Thorough phylogenetic evaluation of IBV show that these two lineages have taken variant pathways of genetic evolution to maintain circulation (9). B/Victoria viruses show significant structural changes to the HA 160 antigenic loop while B/Yamagata viruses show increased diversity driven by the Neuraminidase (NA) gene (Virk, Jayakumar et al.). Since the SARS-CoV-2 pandemic, the genetic landscape of IBV has changed dramatically. There has been significant antigenic drift in B/Victoria lineage viruses leading to two recent updates of the candidate vaccine virus (CVV) accompanied by a vast reduction in B/Yamagata lineage cases with some suggesting the lineage may be extinct (10).
Hemagglutinin contains two subunits, HA1 and HA2. HA1 contains the receptor binding domain and the dominant antigenic sites whereas HA2 is highly conserved and plays a pivitol role in membrane fusion and viral entry (11). The antigenically dominant sites of IBV HA1 have been defined as the 120-loop, the 150-loop, the 160 loop and the 190 helix (HA1 194-202) (12). These four major structural regions allow for conservation of the global protein structure while being able to maintain frequent amino acid substititions and insertions/deletions to evade the human immune system (13).
Current influenza vaccines induce strain specific immunity with vaccine strain selection based on global surveillance efforts. In the 2017-2018 season, the primary modes of vaccine formulation were embryonated hen’s egg-grown vaccines and Madin-Darby Canine Kidney (MDCK) cell cultured vaccines. Egg grown vaccines made up over 90% of distrubuted doses (14). Recently, there has been an increased focus on shifting vaccine formulation to cell culture vaccines given the known risk of egg-adapted mutations in both IAV and IBV (15–18). Specifically for IBV, egg adaption has been previosly shown to induce loss of the glycosylation site at position 197-199 in the 190 helix which is believed to allow for improved IBV replication in embryonated hen’s eggs through alteration of receptor binding (19–22). An alteration in HA antigenicity has also been documented with the loss of this glycosylation site using ferret antisera in hemagglutin inhibition (HI) assays (17). Cell cultured vaccine also has been shown to have the potential for amino acid changes, primarily in the RNA polymerase, although overall this has been less studied (23).
The primary goal of our study was to investigate IBV vaccines to assess for variation from the CVV in egg and cell cultured vaccines from the 2017-2018 season and potential effects on viral and antigenic characteristics.
Materials and methods
Human sample collection and ethics
The human subjects’ protocol was approved by the Johns Hopkins School of Medicine Institutional Review Board (IRB90001667) and National Institutes of Health Division of Microbiology and Infectious Diseases (protocol 15-0103). Patients were enrolled at the Johns Hopkins Medical Institute (JHMI) Department of Emergency Medicine or on inpatient floors. Symptomatic patients in the emergency department were screened and tested for influenza from triage by clinical providers using a validated clinical decision guideline tool. After written consent was obtained, a nasopharyngeal swab, baseline blood sample, demographics, and vaccination history were collected. Enrolled IBV-positive subjects (presence of IBV was determined by nasal swab RT-PCR) were asked to return for a follow-up visit 3–5 weeks later to collect a convalescent serum sample. For each blood draw, 10 mL of blood was collected in Vacutainer tubes, blood was coagulated at room temperature, and serum was separated and stored at −70°C. Nasopharyngeal swabs were placed in 500 μL infection media (IM) consisting of Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 0.3% BSA, 100U/mL of penicillin and 100ug/mL of streptomycin mixture (Life Technologies) and 2mM Glutamax (Gibco). Samples were frozen at −70°C within 6 hours of collection. These specimen were used to infect human nasal epithelial cells (hNEC) by placing 50-100 uL of nasal swab solution on the apical surface and apical samples were collected on days 3-, 5-, and 7-days post infection. The 3,5- and 7-days post infection samples were used to confirm presence of infectious virus using TCID50 assay. All clinical isolates were subsequently confirmed via sequencing of the HA segment via rtPCR and flow cytometry on virus infected MDCK-Siat cells using HA lineage specific monoclonal antibodies.
Cell lines and primary cells
Madin-Darby Canine Kidney Cells (MDCK)-SIAT1 cells were maintained in complete medium (CM) consisting of Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 100U/mL of penicillin and 100ug/mL of streptomycin mixture (Life Technologies) and 2mM Glutamax (Gibco). Primary Human Nasal Epithelial Cells (Promocell, Heidelberg, Germany) were plated and cultured in serum-free Airway Epithelial Growth Medium (Promocell) with SupplementPack Airway Epithelial Cell Growth Medium without antibiotics. The apical surface of wells was originally coated with 0.03 mg/mL Collagen I, Rat Tail (Gibco). Cells are then plated on 24 well ALI plate with Airway media on the apical and basolateral surface. This media promotes proliferation and inhibits terminal differentiation. The media is changed 24 hours after cell plating and every 48 hours following to maintain cell viability. After 7-10 days, confluence is assessed using visual monitoring by microscopy and objectively measured by Transepithelial Electrical Resistance. When fully confluent (TEER> 400), both apical and basolateral media were removed and ALI differentiation media (Stem Cell Technologies, Pneumacult ALI Basal Medium supplemented with 1X ALI Maintenance Supplement (StemCell Technologies), 0.48 ug/mL Hydrocortisone solution (StemCell Technologies), and 4 ug/mL Heparin sodium salt in PBS (StemCell Technologies) was replaced on the basolateral side only. This change allows full differentiation of human nasal cultures. Media is changed every 48 hours to maintain cell viability. The apical surface of cells is intermittently washed with PBS to remove excess mucus. Full differentiation takes approximately 4 weeks and cells are considered fully differentiated when there is presence of mobile cilia on the cell surface visible with standard microscopy. Cells are used for experiments once considered fully differentiated and remain viable for 4-6 weeks following differentiation. All cells were maintained at 37°C in a humidified incubator supplemented with 5% CO2.
Plaque assay
MDCK cells were grown in complete medium to 100% confluency in 6-well plates. Complete medium was removed, cells were washed twice with PBS containing 100µg/ml calcium and 100µg/ml magnesium (PBS+) and 250µL of inoculum was added. Virus dilution was completed by serially diluting the virus stock 10-fold until 10-6, Cells were incubated at 33°C for 1 hour with rocking every 10 minutes. After 1 hour, the virus inoculum was removed and phenol-red free MEM supplemented with 3% BSA (Sigma), 100U/mL of penicillin and 100ug/mL of streptomycin mixture (Life Technologies), 2mM Glutamax (Gibco) and 5µg/ml N-acetyl trypsin (Sigma), 5mM HEPES buffer and 1% agarose was added. Cells were incubated at 33°C for 3-5 days and then fixed with 4% formaldehyde. After removing the agarose, cells were stained with napthol-blue black. Plaque size was analyzed in Image J.
Fifty percent tissue culture infectious dose (TCID50)
MDCK-SIAT1 cells were seeded in a 96 well plate 2 days before assay and grown to 100% confluence. Cells were washed twice with PBS+ then 180µL of IM was added to each well. Ten-fold serial dilutions of virus from 10-1 to 10-7 was created and then 20µL of the virus dilution was added to the MDCK-SIAT1 cells. Cells were incubated for 6 days at 33°C then fixed with 2% formaldehyde. After fixing, cells were stained with naphthol blue-black, washed and virus titer was calculated using the Reed and Muench method (Reed LJ, 1938).
Lineage determination-RT PCR
We chose a two-step lineage determination process for IBV positive PCR nasopharyngeal swabs. Lineage was also later confirmed using HA sequencing. We used WHO recommended lineage specific primers to create RT-PCR products of specific sizes based on lineage. B/Yamagata viruses generate a 388 bp PCR product and B/Victoria viruses generate 284 bp PCR product that were visualized using an agarose gel. Viral RNA was isolated using Qiagen QIAamp Viral RNA Mini Kit per manufacturer’s protocol. 140ul of nasopharyngeal swab sample was used for each extraction. Concentration of extracted vRNA were measured by NanoDrop and 2ul of vRNA were input into RT-PCR reactions. One-step RT-PCR master mix was prepared with SuperScript™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase per manufacturer’s instruction. All 4 primers were added to the mix at final concentration of 10uM.
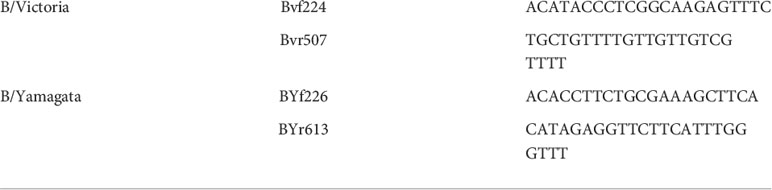
Lineage determination-flow cytometry
MDCK-SIAT-1 cells were plated at about 50% confluency in 6-well tissue culture plates and incubated at 37°C for 24 hours. Cells were washed twice with PBS+ and then infected with 250ul of virus at MOI of 0.2 and incubated at 33°C. At 18 hours post infection, cells were washed twice with PBS and treated with 1X trypsin-EDTA solution (Gibco) for 15 minutes at 37°C. Dislodged cells were collected in a 50mL conical tube and equal volume of complete growth media was added to the tube. The tube was centrifuged at 800g for 4 minutes, followed by 2 washes with 10mL of PBS. One mL of 2% paraformaldehyde was added to each tube and incubated at 4°C for 30 minutes, followed by 2 washes with 10mL of PBS and centrifuged at 900g for 3 minutes. The fixed cells were permeabilized with 1mL of permeabilization buffer (0.2% of Tween-20 in PBS) at 4°C for 15 minutes. Cells were resuspended in FACS buffer (0.3% BSA in PBS) and aliquoted into FACS tubes. 100uL of blocking buffer (1% normal goat serum in FACS buffer) was added to FACS tubes and incubated at room temperature for 30 minutes. Then cells were washed and centrifuged with 2mL of FACS buffer and incubated with the appropriate primary antibody for 45 minutes at room temperature. B/Victoria specific mouse monoclonal antibody BR7B7 (24) was used at 4 ng/ml, and B/Yamagata specific mouse monoclonal antibody WI3E8 was used at 1 ng/ml. Cells were washed twice with 2mL of FACS buffer and incubated with 100uL secondary Goat anti-mouse IgG AF488 at 2ng/ml for 25 minutes at room temperature. Cells were washed with 2mL and resuspended in 500uL of FACS buffer. Samples were run on BD FACSCaliber and histograms were analyzed using FlowJo Software.
HA sequencing and modeling
HA segments from vaccine strains or clinical isolates were sequenced to confirm that no sequence alteration occurred with creation of working stocks using the GISIAD sequence database for sequence comparison when applicable. Viral RNA from each virus isolate was purified using Qiagen vRNA isolation Kit. SuperscriptIII RT-PCR system (ThermoFischer) and used to generate cDNA for sequence analysis. Primers were designed at the 5’ and 3’ noncoding regions of the HA segment with the goal of full sequence coverage.

Additional sequencing primers were designed within the reading frame to ensure full and quality coverage of the entire segment. The cDNA was sent to the Synthesis & Sequencing Facility of the Johns Hopkins University for Sanger sequencing. Geneious (Version 8.0) software was used for sequence analysis. Using FluDB HA Numbering Subtype Conversion, we identified amino acid changes relative to B/Brisbane/60/2008 numbering. We then used PyMol (version 2.4.1) to model AA changes present between clinical strains and vaccine strains and then egg adapted amino acid changes. Hemagglutinin structure of B/Brisbane/60/2008-B/Victoria Lineage (PDB code: 4fqm) was used for modeling as this is the most recent structure of IBV Hemagglutinin published and this structure was used for HA numbering.
Serum microneutralizing antibody assays
Serum samples were first treated (1:3 ratio serum to enzyme) with Receptor Destroying Enzyme (Denka-Seiken) and incubated overnight at 37°C followed by inactivation at 57 °C for 35 minutes. Serum was diluted 2-fold in IM (Dulbecco modified Eagle medium (Sigma), with 10% penicillin/streptomycin (Gibco), 10% L-glutamine (Gibco), 0.5% BSA (Sigma), and 5µg/mL of N-acetyl trypsin (Sigma) at 37°C and 5% CO2) and 100 TCID50 was added for a one-hour incubation at room temperature. Serum Sample/Virus was used to infect a confluent layer of MDCK-SIAT-1 cells. Inoculums were removed after 24 hours, and fresh media (same recipe as above) was added for 96 hours. Plates were fixed and stained as described previously. The Neutralizing Antibody titer was calculated using the highest serum dilution that led to greater than 50% CPE.
Low-MOI infections
Low-multiplicity of infection (MOI) growth curves were performed at an MOI of 0.001 infectious units/cell in MDCK-SIAT1 cells and 0.01 in hNEC cultures. MDCK-SIAT-1 cell infections were performed by incubating inoculum virus solution on the MDCK-SIAT-1 cell monolayer for 2 hours. After the infection, the inoculum was removed and the MDCK-SIAT1 cells were washed three times with PBS+. After washing, IM was added, and the cells were placed at 33°C. At the indicated times post inoculation, IM was removed from the MDCK-SIAT1 cells and frozen at -80°C. Fresh IM was then added. In low-MOI hNEC growth curves, hNECs were acclimated to 33°C or 37°C for 48hrs before infection. The apical surface was washed three times with PBS and the basolateral media was changed at time of infection. hNEC cultures were inoculated at an MOI of 0.01. hNEC cultures were then placed in a 33°C incubator for 2 hours. After incubation, the apical surface of the hNEC culture was washed three times with PBS+. At the indicated times, 100ul of IM without N-acetyl trypsin was added to the apical surface of the hNECs for 10 minutes at 33°C, the IM was harvested and stored at -80°C. Basolateral media was changed every 48 hrs post infection for the duration of the experiment. Infectious virus titers in the apical supernatants were measured with TCID50 assay.
Results
HA sequencing and modeling of vaccine virus and clinical isolates show an egg adapted amino acid change at position 197 across both lineages of IBV in the 2017-2018 season vaccines
The goal of our study was to evaluate for the presence of antigenic changes induced by vaccine production in either egg or cell cultured vaccines. The initial comparison was made through gene segment sequencing of the HA segment of our three comparison viruses per lineage: circulating strain, the egg grown vaccine strain and the cell cultured vaccine. The 2017-2018 season was chosen due to a particularly high incidence of influenza cases that year and additionally clinical reports that suggested that the cell cultured vaccine from that year may have shown better efficacy for IBV protection (25). A limited number of amino acid changes were identified in the circulating virus that were not present in either vaccine virus (Figures 1A, B). The cell cultured vaccine and egg cultured vaccine of both lineages differed at one position, N197, the first amino acid in the 197-199 glycosylation site. The cell cultured vaccine was consistent with the circulating strain at this position (Figures 1A, B). The amino acid change at this position is a loss of a known glycosylation site in the structure (Figure 1C). These findings show a consistent egg adaption-related amino acid change across both lineages of IBV (Figure 1D).
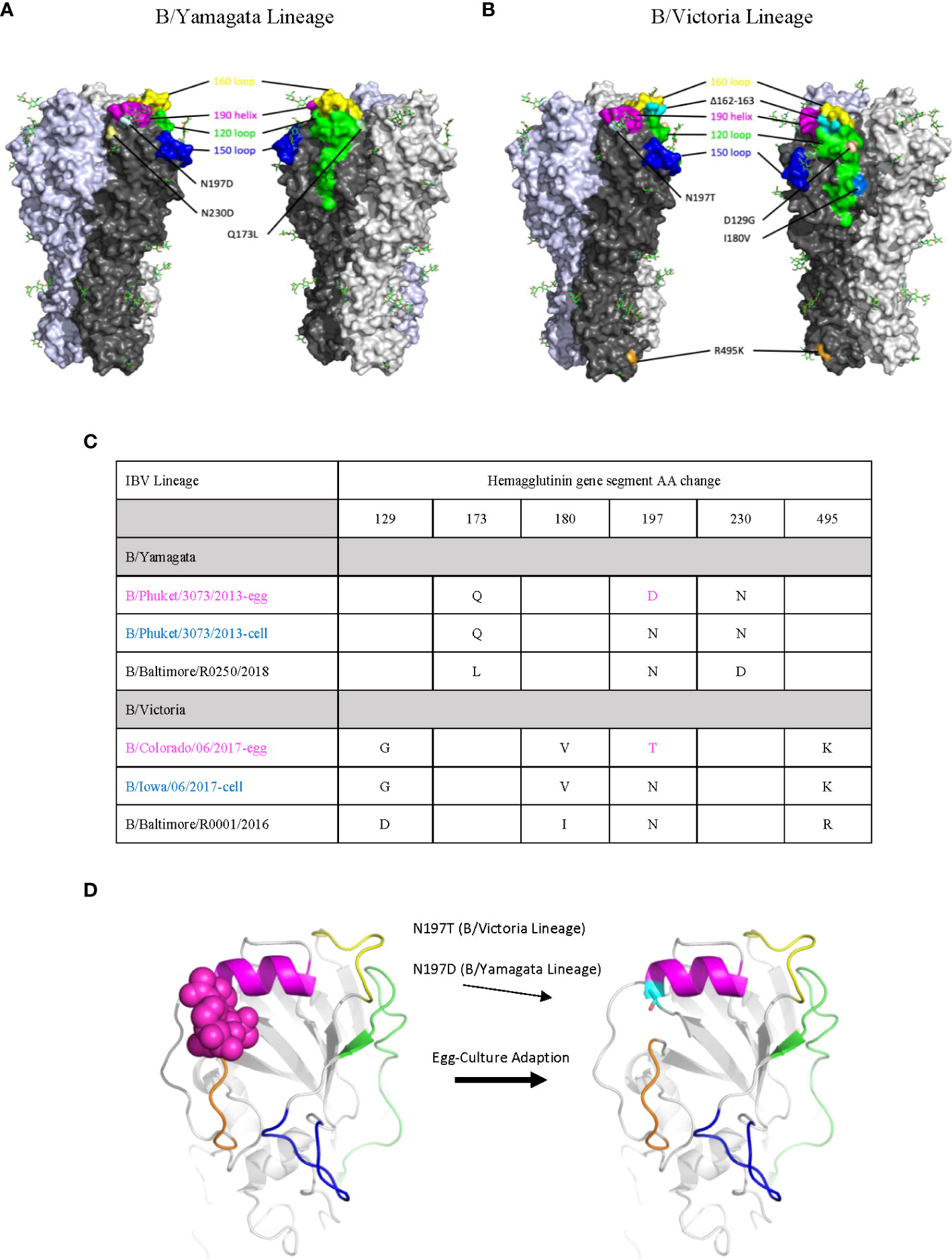
Figure 1 Sequence Analysis of Egg Adapted Vaccine Viruses show loss of Glycosylation Site at Position 197. (A, B) All mutations on the HA surface of egg-cultured vaccine and cell-cultured vaccine strains from the circulating strain in both B/Yamagata (A) and B/Victoria (B) lineages are mapped on the model. A mutation V252M is not shown on the model as the residue is inside the HA trimer structure. The known antigenic sites of IBV are highlighted. The 120 loop is shown in green, the 150 loop is shown in blue, the 160 loop is shown in yellow and the 190 helix is shown in violet. The crystal structure of HA of B/Brisbane/06/2008 was used for modeling and amino acid numbering. (C) Sequence alignment of circulating viruses, egg adapted vaccine strains and cell cultured strains shown. (D) Model of HA detailing egg adapted amino acid change leading to loss of a glycosylation site.
Human serum microneutralization assays (NAb) before and after natural infection with B/Yamagata strains show differential antibody recognition
After confirming that there was a specific egg adapted amino acid change, we hypothesized that this amino acid change in the antigenically active 190 helix might lead to differential post antibody recognition. Although this amino acid change was found across both lineages, study of the B/Yamagata lineage viruses was pursued given the dominance in the season of interest. Individuals with confirmed IBV infection in the 2017-18 Northern Hemisphere influenza season were enrolled in a study that collected serum at the time of IBV infection diagnosis and 28 days later. These paired serum samples were used to determine the antigenic relatedness of egg-adapted vaccine strains, cell culture vaccine strains and circulating IBV strains. In the 2017-2018 season, B/Yamagata was the dominant circulating lineage (26) and all enrolled IBV infections were with B/Yamagata lineage viruses. Neutralizing antibody assays were used to compare neutralization of these viruses with the baseline and convalescent serum. There were statistically significant mean titers comparing baseline and convalescent serum for the clinical isolate and cell cultured vaccine strain, but there was no statistically significant difference between baseline and convalescent serum for the egg cultured vaccine strain (Figure 2A). There was no statistically significant difference in baseline neutralization titer (Figure 2B). Mean neutralization titers between convalescent samples showed a statistically significant difference in the egg cultured vaccine compared to clinical isolate and cell culture vaccine. There was no statistical difference between the cell cultured vaccine and the clinical isolate (Figure 2B). Analyzing the data further, there was a notable difference in titers between previously vaccinated patients during that season in the assessment of egg cultured vaccine. The sample size was too small to evaluate statistical significance between the vaccinated and unvaccinated individuals in these groups.
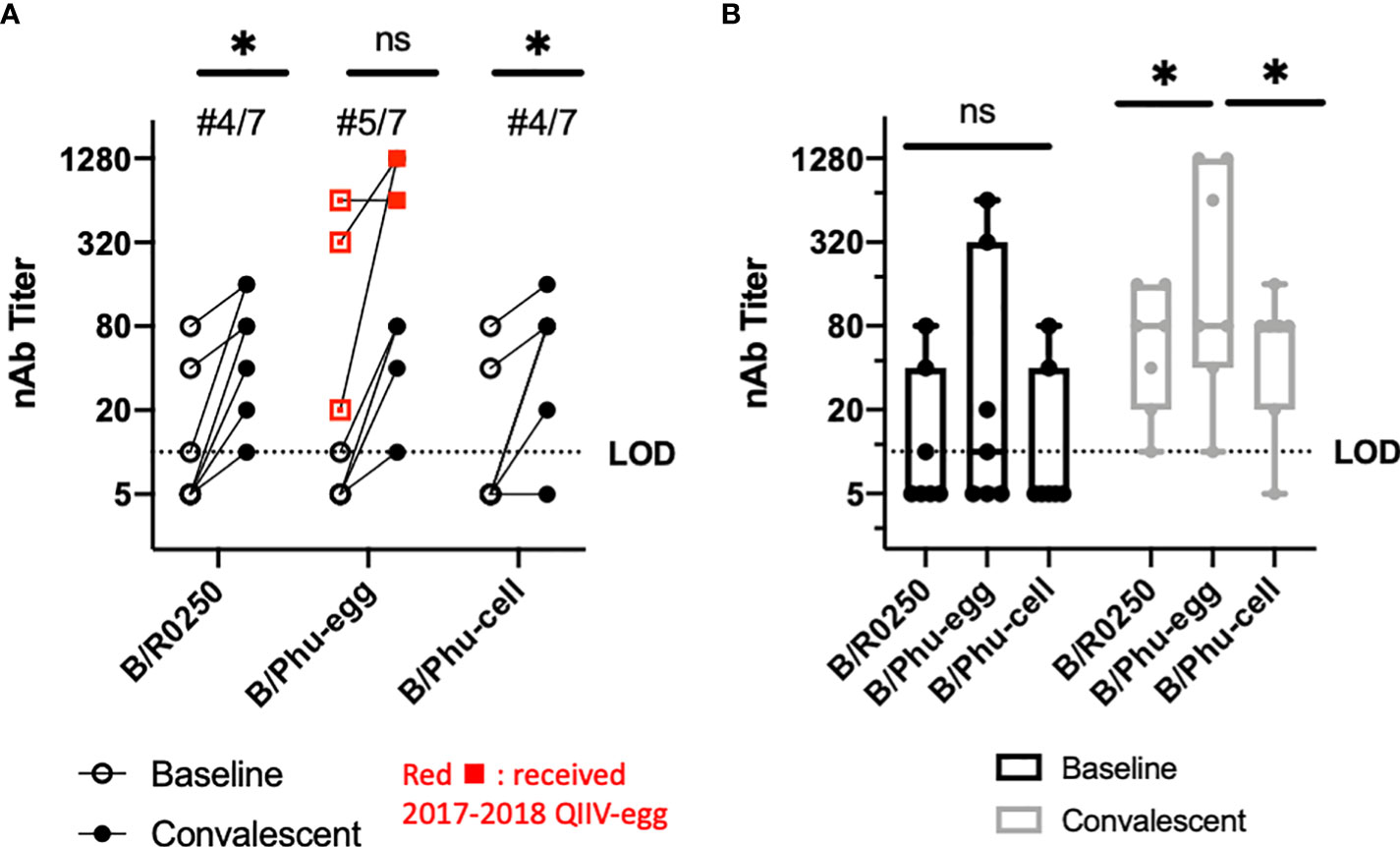
Figure 2 Neutralizing Antibody Assays-IBV infected individuals from 2017-2018 Season. 7 patients with natural IBV infection (B/Yamagata lineage) at Johns Hopkins Hospital had serum samples collected during active infection (baseline) and 4-6 weeks following infection (convalescent). B/Baltimore/R0250/2018 (B/R0250), B/Phuket/3073/2013-egg cultured (B/Phu-egg) and B/Phuket/3073/2013-cell cultured (B/Phu-cell) were used for comparison. (A) Matched values during (baseline) and 4-6 weeks (convalescent) after natural infection with B/Yamagata lineage influenza viruses. Participants shown in red received the egg grown quadrivalent vaccine that season. Paired T-test used to calculate significance (B) Box and Whisker plot comparing median nAb titers during (baseline) and 4-6 weeks after (convalescent) natural infection in our comparison viruses. Two-way Anova with multiple comparisons was used to calculate statistical significance of median values. An * indicates p<0.05 and ns indicates a not significant difference.
Human NAb titers from vaccinated healthcare workers have statistically significant differences in mean titer between the egg grown, cell cultured and clinical isolate from the 2017-2018 season
Given the size of the natural infection cohort, we chose to investigate a larger vaccinated cohort to test our hypothesis. We completed the same serum microneutralization assays on a pre and post vaccination cohort of healthcare workers. These participants were vaccinated with quadrivalent egg cultured IIV and serum microneutralization assay was completed for pre and post vaccination. For each virus, pre and post vaccination serum showed a statistically significant increase in mean titer (Figure 3A). There was a statistically significant difference in mean neutralizing antibody titer between the egg cultured vaccine strain and the other two strains in post-vaccination serum (B/R02050 and B/Phu-cell) (Figure 3B). These data together represent variable antibody recognition of the egg cultured vaccine strain for the 2017-2018 season for B/Yamagata lineage viruses.
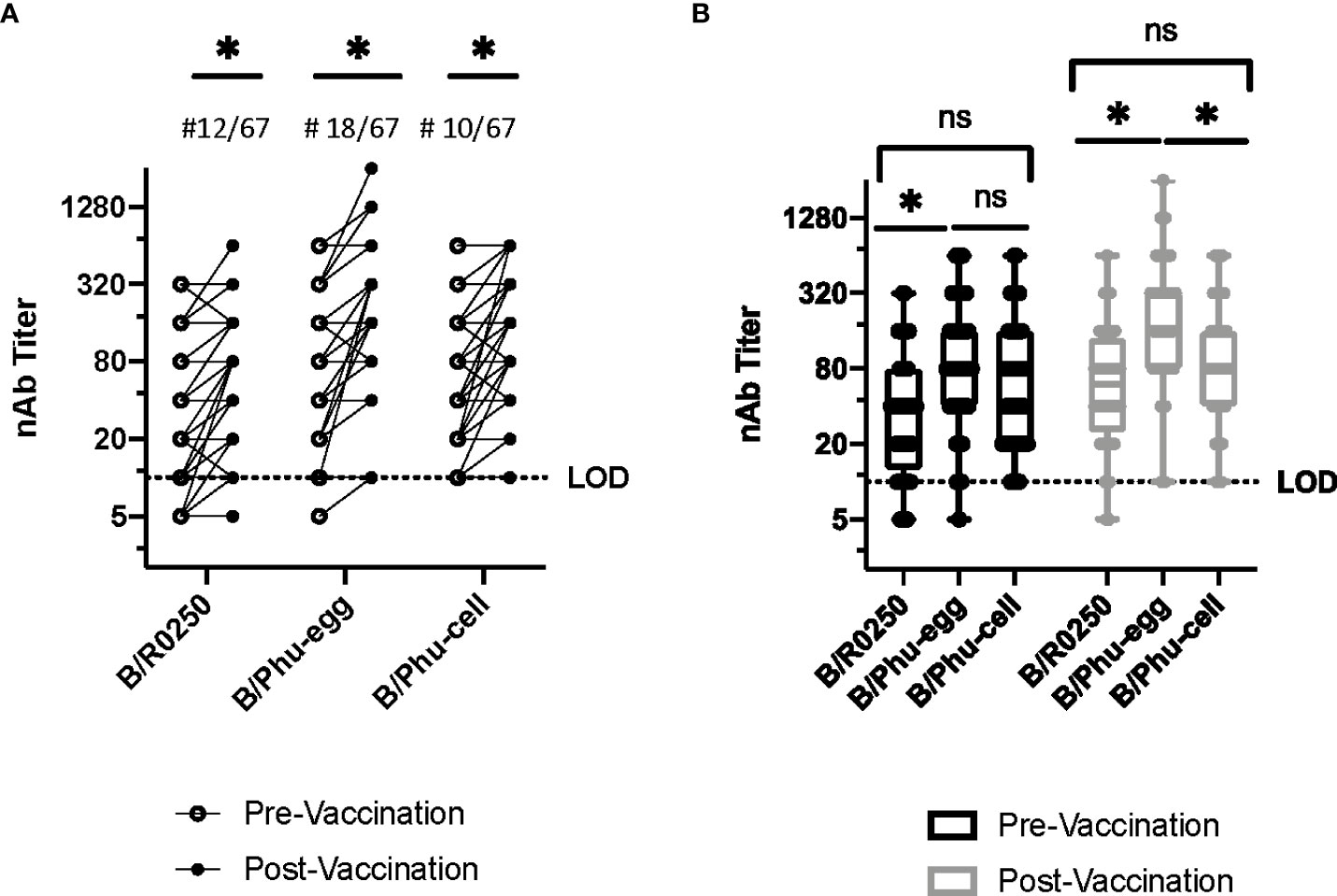
Figure 3 Neutralizing Antibody Assays, Vaccinated Healthcare Workers. A group of 67 healthcare workers had serum samples taken at Johns Hopkins Hospital before vaccination and 4-6 weeks after vaccination in 2019-2020. These serum samples were used to compare the viruses compared above, B/R0250, B/Phu-egg and B/Phu-cell. (A) Comparison of neutralizing antibody titers using the pre-vaccination and post-vaccination serum of patients vaccinated with the quadrivalent IIV. B/Baltimore/R0250/2018 (B/R0250), B/Phuket/3073/2013-egg cultured (B/Phu-egg) and B/Phuket/3073/2013-cell cultured (B/Phu-cell) were used for comparison against patient sera. (A) Matched values pre and post vaccination. Paired T-test used to calculate significance (B) Box and Whisker plot comparing median nAb titers pre and post vaccination. Two-way Anova with multiple comparisons was used to calculate statistical significance of median value. An * indicates p<0.05 and ns indicates a not significant difference.
Plaque assay comparison of clinical strains versus egg grown and cell cultured vaccine show consistently smaller plaque sizes of the cell cultured vaccine strain
Given that amino acid differences between egg-grown vaccine, cell culture grown vaccine and clinical isolates of IBV in the 190 loop could drive differential recognition by neutralizing antibodies, we investigate whether any replication differences could also be attributed to these amino acid mutations. When virus plaque size was determined on MDCK cells, there was a statistically significant difference seen in plaque size in the cell cultured viruses of the Yamagata (Figures 4A, B) lineage viruses when compared to either the egg-grown vaccine or the clinical isolate. Since the cell culture and the clinical isolate both possess the same loss of glycosylation at position 197, we conclude that this mutation is not driving plaque size differences between the three virus strains.
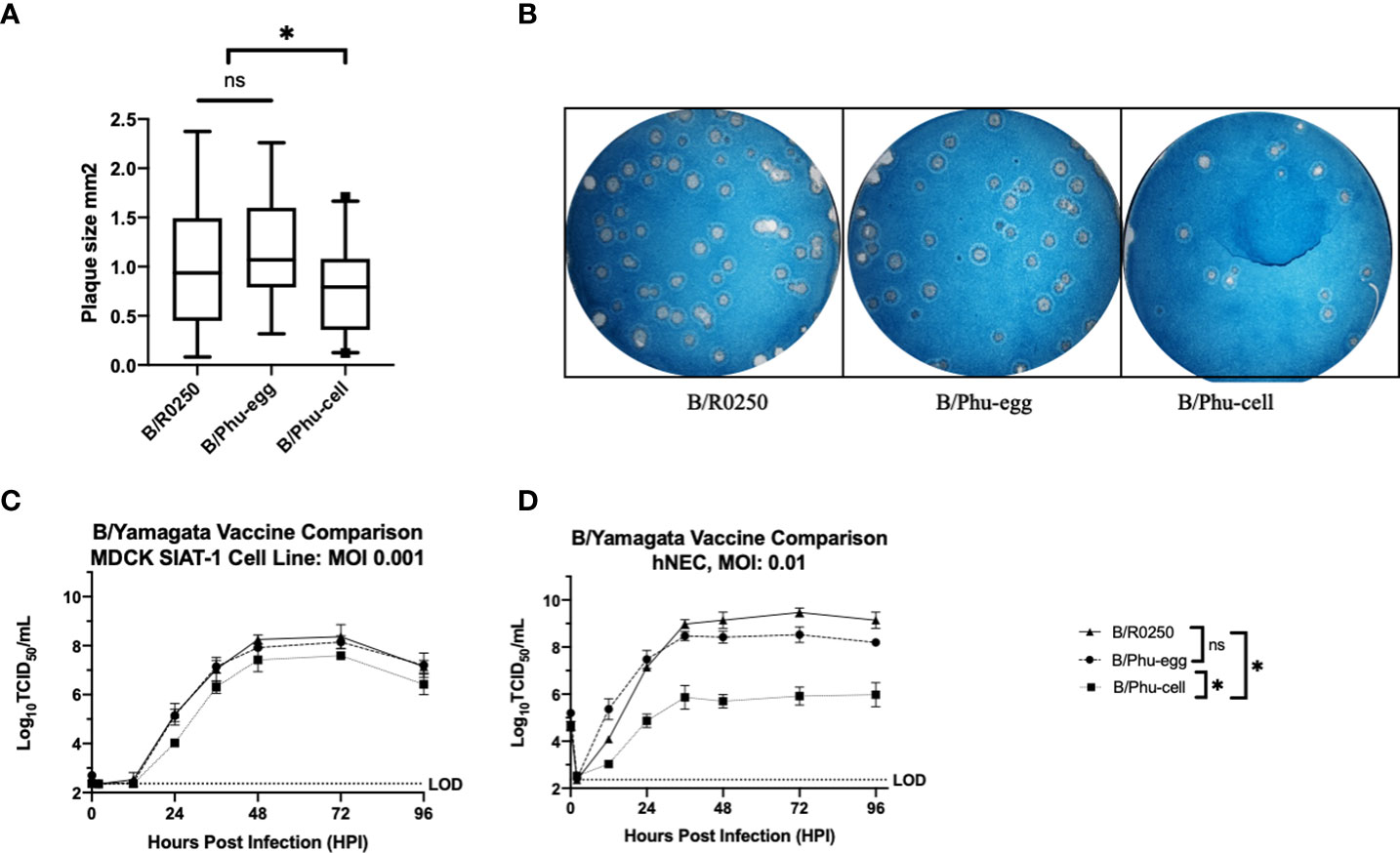
Figure 4 Investigating Viral Phenotype Differences: B/Yamagata (A) Comparison of plaque sizes formed by clinical isolate, egg-adapted vaccine strain and cell-adapted vaccine strain from B/Yamagata, B/R0250, B/Phu-egg and B/Phu-cell respectively. (B) Images of plaques were analyzed with ImageJ. Representative wells shown. Experiments were completed in triplicate. (C) Low MOI Growth Curve comparing B/R0250, B/Phu-egg and B/Phu-cell on MDCK-SIAT-1 Cell line. Growth curves were completed in triplicate. Statistical significance of growth curves calculated on GraphPad Prism using a two-way Anova and Tukey’s multiple comparisons test. (D) Low MOI growth curves on hNEC ALI culture. Growth curves were completed in duplicate. Statistical significance was calculated using the same methods as the MDCK SIAT-1 growth curves. An * indicates p<0.05 and ns indicates a not significant difference.
Low-MOI growth curve analysis show impaired growth kinetics and loss of peak titer in the cell culture vaccine virus of the B/Yamagata lineage
In addition to efficiency of cell to cell spread we assessed infectious virion production over time between these viruses. These growth curves were completed on the MDCK-SIAT1 cell line which overexpresses the enzymes that add alpha 2,6 sialic acid to N-linked glycans, thus making the cell line more susceptible to infection with human influenza virus strains (27). Growth curves were also assessed on human nasal epithelial cells (hNECs) to represent a physiologically relevant cell culture system. There was no significant phenotypic variation with the egg derived vaccine viruses in either the MDCK-SIAT1 or the hNEC growth curves despite the known loss of glycosylation site in the 190 helix. We did see a similar pattern to the plaque assay data in that the cell cultured vaccine virus has lower peak titer and slowed replication compared to the egg cultured virus and the circulating virus (Figures 4C, D). This pattern is seen in both cell culture models, MDCK-SIAT-1 and hNECs. The phenotype of slowed replication appears to be emphasized in the hNEC primary culture model. Again, we hypothesize that the N197D amino acid change seen with egg culture adaption is not having a significant impact on virion production in human nasal cells.
Discussion
The purpose of this study was to investigate IBV vaccines and assess the continued role of egg adapted amino acid changes as has been reported in the literature beginning in the 1980s (19, 20, 22). These questions are necessary to investigate as many of the cited changes throughout history were in antigenically dominant sites and this has had limited investigation in novel IBV vaccines. Our data show that, consistent with historical sites of egg adaption at the 197-199 HA glycoslyation site, novel IBV vaccines B/Phuket/3073/2013 and B/Colorado/06/2017 contain egg adapted amino acid mutations at the same glycosylation site. The historical data as well as the consistent amino acid change across lineages confirms that this site is a weak site for egg cultured amino acid changes.
The site of this amino acid change is in an antigenically dominant site, the 190 helix (12). Given the concerning site for amino acid change from the desired CVV chosen, we investigated the potential effect on antigenic recognition. Neutralizing antibody assays were used to assess antigenic recognition of the clinical isolate, egg adapted vaccine virus and cell cultured virus. Our data show that there is variable antibody recognition in the egg adapted virus using pre and post infection serum. Higher mean anitbody titers in the baseline and convalescent serum from B/Yamagata infected patients was not the expected result as we expected natural infection to have decreased antibody recognition of the egg adapted virus. We went back to assess vaccination history of the natural infection participants. There appears to be two groups that separate by vaccination history. Patients with vaccination that season represent the patients with higher mean titers to the egg adpated virus. We hypothesize that this represents increased protection from vaccine induced antibodies in constrast to antibodies induced from natural infection. This is also consistent with lack of increased mean titer in the comparison viruses of the vaccinated individuals.
This differential antibody recognition was also seen in our vaccination cohort. Together, these data show that there is differential antigenic recognition of the egg adapted vaccine strain, B/Phuket/3073/2013. The clinical isolate and the cell cultured vaccine from that season do not show differential antibody recognition. Given what we found in the antigenically dominant HA sites and the antibody assays, we conclude that the variable antibody detection is secondary to the egg adapted loss of glycosylation at position N197.
We initially hypothesized that the amino acid mutation that was likely effecting antibody binding also might lead to a viral phenotype change. We were surpised to observe that there were no viral phenotype changes observed between the egg adapted vaccine strain and the clinical isolate in plaque assays and growth curves. However, there did exist a decreased cell to cell spread and virion production in the cell cultured vaccine. From this data, we hypothesze that the lack of glycosylation site at the 197-199 position of HA does not have an effect on viral replication kinetics. As has been shown in other studies, this decreased replication efficiency that is seen may lie in cell culture adaption in the RNA Polymerase although this was not specifically pursued in this study.
Given the continued presence of egg adaption in IBV in recent vaccines, potential effect on induced immunity and therefore protection from circulating viruses, we conclude that egg adaption should be screened prior to large production of vaccine virus. Alternatively, as cell cultured vaccine viruses show phenotypic changes but do not seem to have an effect on antigenic recognition, we propose continued transition to the MDCK cell cultured vaccine viruses or entertaining other modes of vaccine formulations for Influenza vaccination.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Johns Hopkins School of Medicine Institutional Review Board (IRB90001667) and National Institutes of Health Division of Microbiology and Infectious Diseases (protocol 15-0103). The patients provided their written informed consent to participate in this study.
Author contributions
The authors confirm contribution to the paper as follows: study conception and design: AP, HL and JW. Data collection was primarily completed by JW and RZ. Clinical specimen provided by RR, KF. Analysis and interpretation of results: JW and AP. Author, draft manuscript preparation: AP and JW. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Johns Hopkins Center for Excellence in Influenza Research and Surveillance (N272201400007C and N7593021C00045). We are also thankful for the salary funding of JW from the following T32 Grants (2T32AI007007-41and 5T32HD044355-18) and a gift from the Richard Eliasberg Family Foundaton.
Acknowledgments
The authors thank the enrollees in our study for making this analysis possible. We also thank the members of the laboratories of Andrew Pekosz, Sabra Klein and Kimberly Davis for comments and discussions on this work. We acknowledge the use of sequences deposited through the efforts of members of the GISAID network.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Paules C, Subbarao K. Influenza. Lancet (2017) 390(10095):697–708. doi: 10.1016/s0140-6736(17)30129-0
2. Irving SA, Patel DC, Kieke BA, Donahue JG, Vandermause MF, Shay DK, et al. Comparison of clinical features and outcomes of medically attended influenza a and influenza b in a defined population over four seasons: 2004-2005 through 2007-2008. Influenza Other Respir Viruses (2012) 6(1):37–43. doi: 10.1111/j.1750-2659.2011.00263.x
3. McCullers JA, Hayden FG. Fatal influenza b infections: time to reexamine influenza research priorities. J Infect Dis (2012) 205(6):870–2. doi: 10.1093/infdis/jir865
4. Tran D, Vaudry W, Moore D, Bettinger JA, Halperin SA, Scheifele DW, et al. Hospitalization for influenza a versus b. Pediatrics (2016) 138(3):e20154643. doi: 10.1542/peds.2015-4643
5. van de Sandt CE, Bodewes R, Rimmelzwaan GF, de Vries RD. Influenza b viruses: not to be discounted. Future Microbiol (2015) 10(9):1447–65. doi: 10.2217/fmb.15.65
6. Osterhaus AD, Rimmelzwaan GF, Martina BE, Bestebroer TM, Fouchier RA. Influenza b virus in seals. Science (2000) 288(5468):1051–3. doi: 10.1126/science.288.5468.1051
7. Caini S, Kusznierz G, Garate VV, Wangchuk S, Thapa B, de Paula Júnior FJ, et al. The epidemiological signature of influenza b virus and its B/Victoria and B/Yamagata lineages in the 21st century. PloS One (2019) 14(9):e0222381. doi: 10.1371/journal.pone.0222381
8. Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. Cocirculation of two distinct evolutionary lineages of influenza type b virus since 1983. Virology (1990) 175(1):59–68. doi: 10.1016/0042-6822(90)90186-u
9. Virk RK, Jayakumar J, Mendenhall IH, Moorthy M, Lam P, Linster M, et al. Divergent evolutionary trajectories of influenza b viruses underlie their contemporaneous epidemic activity. Proc Natl Acad Sci U.S.A. (2020) 117(1):619–28. doi: 10.1073/pnas.1916585116
10. Koutsakos M, Wheatley AK, Laurie K, Kent SJ, Rockman S. Influenza lineage extinction during the COVID-19 pandemic? Nat Rev Microbiol (2021) 19(12):741–2. doi: 10.1038/s41579-021-00642-4
11. Copeland CS, Doms RW, Bolzau EM, Webster RG, Helenius A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol (1986) 103(4):1179–91. doi: 10.1083/jcb.103.4.1179
12. Liu Y, Tan HX, Koutsakos M, Jegaskanda S, Esterbauer R, Tilmanis D, et al. Cross-lineage protection by human antibodies binding the influenza b hemagglutinin. Nat Commun (2019) 10(1):324. doi: 10.1038/s41467-018-08165-y
13. Wang Q, Cheng F, Lu M, Tian X, Ma J. Crystal structure of unliganded influenza b virus hemagglutinin. J Virol (2008) 82(6):3011–20. doi: 10.1128/jvi.02477-07
14. Barr IG, Donis RO, Katz JM, McCauley JW, Odagiri T, Trusheim H, et al. Cell culture-derived influenza vaccines in the severe 2017-2018 epidemic season: a step towards improved influenza vaccine effectiveness. NPJ Vaccines (2018) 3:44. doi: 10.1038/s41541-018-0079-z
15. Powell H, Pekosz A. Neuraminidase antigenic drift of H3N2 clade 3c.2a viruses alters virus replication, enzymatic activity and inhibitory antibody binding. PloS Pathog (2020) 16(6):e1008411. doi: 10.1371/journal.ppat.1008411
16. Rajaram S, Suphaphiphat P, van Boxmeer J, Haag M, Leav B, Iheanacho I, et al. Retrospective assessment of the antigenic similarity of egg-propagated and cell culture-propagated reference influenza viruses as compared with circulating viruses across influenza seasons 2002-2003 to 2017-2018. Int J Environ Res Public Health (2020) 17(15):5423. doi: 10.3390/ijerph17155423
17. Saito T, Nakaya Y, Suzuki T, Ito R, Saito T, Saito H, et al. Antigenic alteration of influenza b virus associated with loss of a glycosylation site due to host-cell adaptation. J Med Virol (2004) 74(2):336–43. doi: 10.1002/jmv.20178
18. Ursin RL, Liu H, Powell HR, Westerbeck JW, Shaw-Saliba K, Sylvia KE, et al. Differential antibody recognition of h3n2 vaccine and seasonal influenza virus strains based on age, vaccine status, and sex in the 2017-2018 season. J Infect Dis (2020) 222(8):1371–82. doi: 10.1093/infdis/jiaa289
19. Lugovtsev VY, Smith DF, Weir JP. Changes of the receptor-binding properties of influenza b virus B/Victoria/504/2000 during adaptation in chicken eggs. Virology (2009) 394(2):218–26. doi: 10.1016/j.virol.2009.08.014
20. Oxford JS, Newman R, Corcoran T, Bootman J, Major D, Yates P, et al. Direct isolation in eggs of influenza a (H1N1) and b viruses with haemagglutinins of different antigenic and amino acid composition. J Gen Virol (1991) 72(Pt 1):185–9. doi: 10.1099/0022-1317-72-1-185
21. Peck H, Laurie KL, Rockman S, Leung V, Lau H, Soppe S, et al. Enhanced isolation of influenza viruses in qualified cells improves the probability of well-matched vaccines. NPJ Vaccines (2021) 6(1):149. doi: 10.1038/s41541-021-00415-3
22. Robertson JS, Naeve CW, Webster RG, Bootman JS, Newman R, Schild GC. Alterations in the hemagglutinin associated with adaptation of influenza b virus to growth in eggs. Virology (1985) 143(1):166–74. doi: 10.1016/0042-6822(85)90105-9
23. Tang CY, Segovia K, McElroy JA, Li T, Guan M, Zhang X, et al. Cell-adapted mutations and antigenic diversity of influenza b viruses in missour -2020 season. Viruses (2021) 13(10):1896. doi: 10.3390/v13101896
24. Verma S, Soto J, Vasudevan A, Schmeisser F, Alvarado-Facundo E, Wang W, et al. Determination of influenza b identity and potency in quadrivalent inactivated influenza vaccines using lineage-specific monoclonal antibodies. PloS One (2017) 12(4):e0175733. doi: 10.1371/journal.pone.0175733
25. Klein NP, Fireman B, Goddard K, Zerbo O, Asher J, Zhou J, et al. Vaccine effectiveness of cell-culture relative to egg-based inactivated influenza vaccine during the 2017-18 influenza season. PloS One (2020) 15(2):e0229279. doi: 10.1371/journal.pone.0229279
26. He G, Yang P, Yan Q, Xiong C. Debate on the compositions of influenza b in northern hemisphere seasonal influenza vaccines. Antimicrob Resist Infect Control (2019) 8:164. doi: 10.1186/s13756-019-0631-2
Keywords: influenza B, vaccines, egg-adaption, cell-culture, human nasal epithelial cells
Citation: Wilson JL, Zhou R, Liu H, Rothman R, Fenstermacher KZ and Pekosz A (2022) Antigenic alteration of 2017-2018 season influenza B vaccine by egg-culture adaption. Front. Virol. 2:933440. doi: 10.3389/fviro.2022.933440
Received: 30 April 2022; Accepted: 27 July 2022;
Published: 12 August 2022.
Edited by:
Aitor Nogales, Centro de Investigación en Sanidad Animal (CISA), SpainReviewed by:
Rodrigo Abreu, LifeArc, United KingdomSteven Rockman, CSL Ltd, Australia
Irina V. Kiseleva, Institute of Experimental Medicine (RAS), Russia
Copyright © 2022 Wilson, Zhou, Liu, Rothman, Fenstermacher and Pekosz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jo L. Wilson, andpbHMxNTRAamh1LmVkdQ==; Andrew Pekosz, YXBla29zejFAamh1LmVkdQ==