 Sneha Singh
Sneha Singh Karim Dirani
Karim Dirani Ashok Kumar
Ashok Kumar- 1Department of Ophthalmology, Visual and Anatomical Sciences/Kresge Eye Institute, Wayne State University School of Medicine, Detroit, MI, United States
- 2Department of Biochemistry, Microbiology, and Immunology, Wayne State University School of Medicine, Detroit, MI, United States
Viruses are known to hijack the intracellular organelles, including mitochondria, endoplasmic reticulum, lipid droplets, and cytoskeleton to promote its replication. The host responds to invading viruses by mounting antiviral responses and rearrangement of its organelles. In particular, the mitochondria are one of the target organelles exploited by viruses and their proteins to suppress the host antiviral response. In this review, we have comprehensively summarized the impact of mitochondrial dynamics in modulating antiviral response during emerging and re-emerging RNA virus infections caused by genus Flavivirus (Dengue virus, Zika virus, Hepatitis C virus), and SARS-CoV-2, the causative agent of COVID-19 pandemic. In addition to knowledge gaps in mitochondria-virus interaction studies, we discuss recent advancements in therapeutics regulating the mitochondrial dynamics to combat viral infections.
Introduction
In addition to being a powerhouse, mitochondria play a crucial role in various cellular functions, including cell-cycle control, cell development, and apoptosis (1–3). Mitochondria take a central stage in cellular metabolism since the tricarboxylic acid cycle (TCA), fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), calcium buffering, and heme synthesis occur within the mitochondria (4). Due to the overarching role in maintaining cellular homeostasis and innate immune response, the tight regulation of mitochondrial function is crucial during cellular stress stimuli and pathogen invasion.
During viral infection, pattern recognition receptors (PRRs) trigger the production of interferons (IFNs). However, mitochondrial antiviral-signaling protein (MAVS) acts as a central hub for signal transduction initiated by RIG-I-like receptors, involved in the recognition of viral RNA (Figure 1).
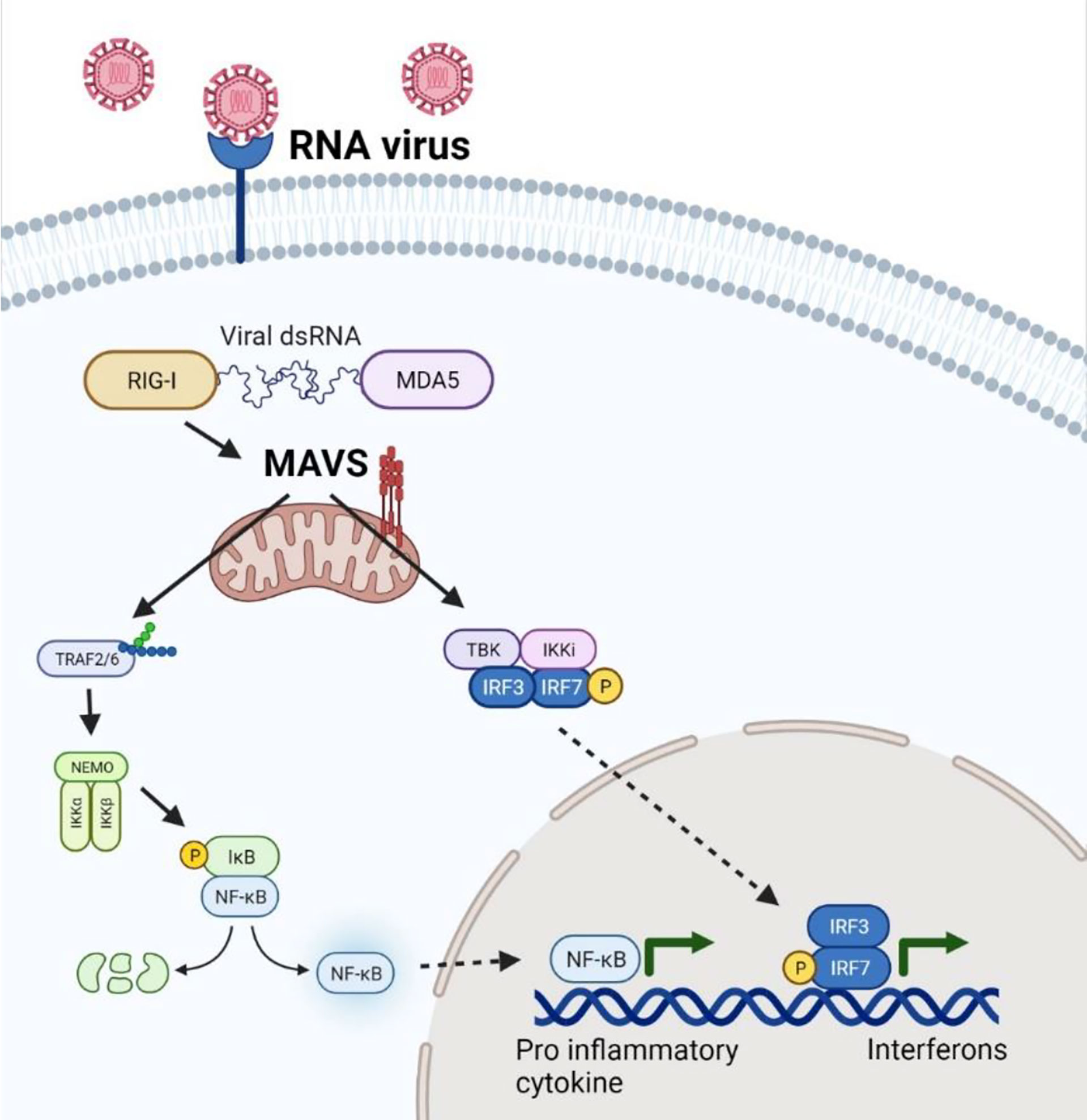
Figure 1 MAVS-mediated Antiviral and Inflammatory response during RNA virus infection. MAVS acts as a central hub to recognize the RIG-I mediated sensing of viral RNA in the host cell leading to a trigger of NFkB and IRFs activation leading to transcription of genes regulating inflammatory and antiviral response during viral infection.
Amongst viral pathogens, RNA viruses are the leading cause of human infections and are responsible for major epidemics and pandemics, including the ongoing COVID-19. In particular, the RNA viruses can rapidly mutate resulting in the evolution of new variants which can escape from the host immune surveillance (5). Several studies show that viral infection alters mitochondrial dynamics, a synchronized process to combat extracellular threats and maintain cellular homeostasis (6–9).
Mitochondrial dynamics encompass the process of mitochondrial elongation (through fusion) and mitochondrial division (through fission). In addition, the damaged mitochondria are removed by mitochondria-selective autophagy, a process called mitophagy. Together, the synchronization of these processes maintains the health of the cell and mitochondria-regulated host metabolism. In the following section, we briefly discuss these three processes.
a) Mitochondrial Fusion
Mitochondrial fusion is the process of integration of individual mitochondria into a single organelle. The process involves the fusion of a) outer mitochondrial membrane (OMM) mediated by Mitofusin 1 (MFN1) and Mitofusin 2 (MFN2); b) inner mitochondrial membrane (IMM) mediated by Optic atrophy protein 1 (OPA1) (10–14). MFN1 and MFN2 are normally expressed in the same cell and can initiate mitochondrial fusion independently, suggesting their redundant role (15).
The fusion of the mitochondria stimulates RLR-mediated-MAVS signaling along with the interaction of MAVS and STING at mitochondria-associated membranes (MAMs) with the endoplasmic reticulum (16). Koshiba et al. revealed that mitochondrial fusion and mitochondrial membrane potential regulated by MFN1 and MFN2, respectively, are essential for MAVS-mediated signaling (17, 18). Moreover, the deletion of MFN1 or MFN2, reduced viral-induced IFNs and pro-inflammatory cytokines, thereby increasing the viral replication. Suppression of mitochondrial fusion is usually favored by the virus for its proliferation and evasion of the antiviral innate immune signaling as evidenced in SARS-CoV, SARS-CoV-2, Influenza, and HIV infection studies (19–21). Mitochondrial elongation, therefore, exacerbates viral-infection induced-RIG-I-dependent antiviral innate immunity and aids in reducing viral replication (Figure 2).
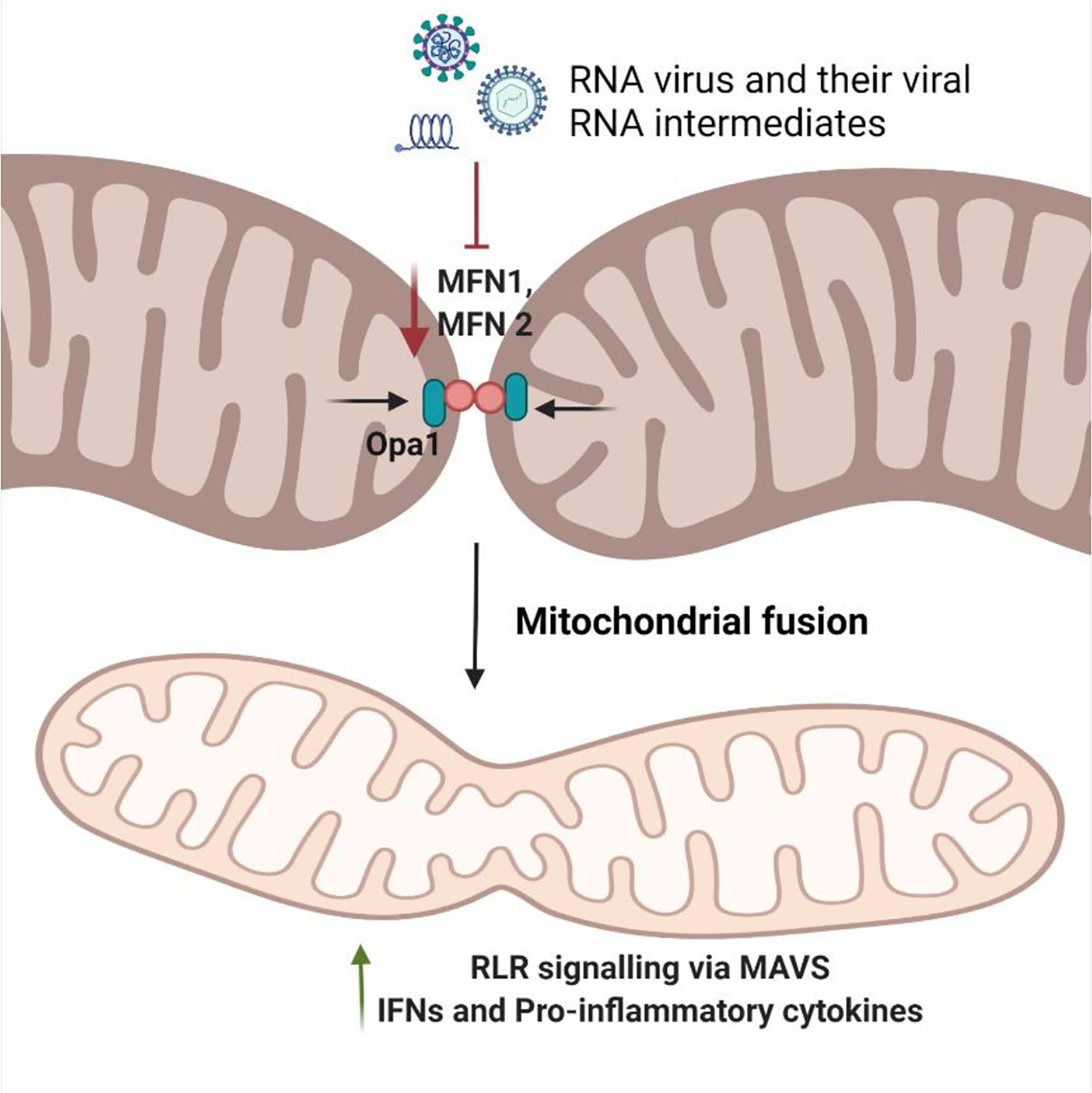
Figure 2 Modulation of mitochondrial fusion by RNA viruses. Mitochondrial fusion is mediated by OPA1 and mitofusins (MFNs). Their synchronous effect allows functional tubules to rejoin the mitochondrial network at large. During viral infection, host cells respond by promoting mitochondrial fusion to enhances the innate antiviral immune response via increased MAVS-mediated pathway. The RNA virus and the viral RNA intermediates try to decrease the levels of MFN1/MFN2 to inhibit the process of mitochondrial fusion, thereby reducing the RLR antiviral response to enhance their replication.
b) Mitochondrial Fission
The splitting up of mitochondria into smaller organelles is known as mitochondrial fission. It initiates with the recruitment of dynamin-related protein (Drp1) to mitochondria upon its post-translational modification (phosphorylation, nitrosylation, and sumoylation) (22, 23). Drp1 is primarily cytosolic but migrates to the outer mitochondrial membrane (OMM) to initiate mitochondrial fission by binding to Fis1. Mitochondrial fission cascade can also be triggered independent of Drp1, by endoplasmic reticulum (ER) tubules and actin filaments (24), wherein close contacts between ER and mitochondria-associated membranes (MAMs) interact with the fission apparatus (25, 26). The mitochondrial fission is complemented by mitophagy for the exclusion of the damaged portion of the organelle (24, 27) (Figure 3).
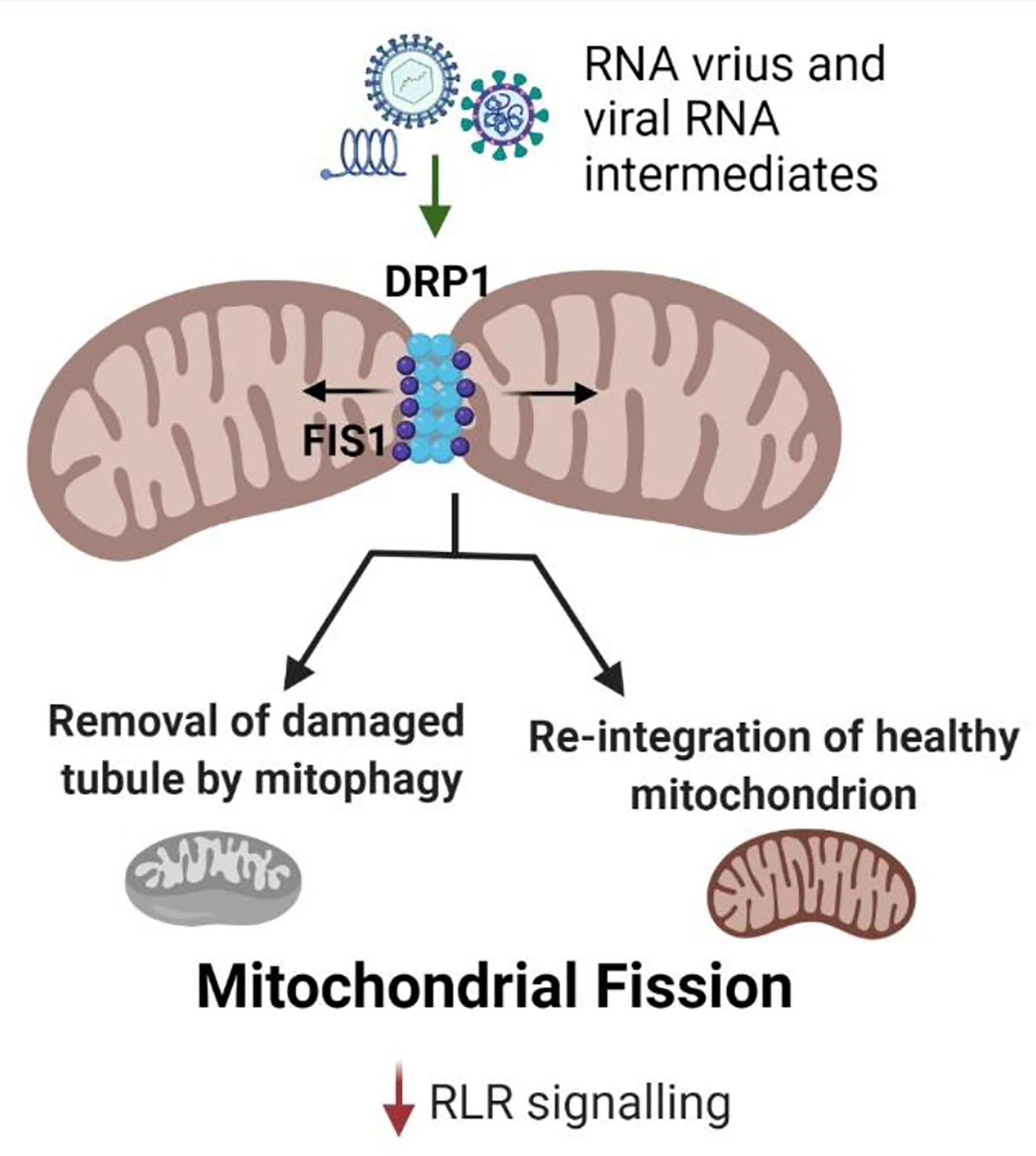
Figure 3 Mitochondrial fission during RNA virus infection. Drp1 is the key protein involved in mitochondrial fission which allows the removal of the damaged tubule from the mitochondrial network. The damaged tubule undergoes mitophagy mediated recycling. Mitochondrial fission reduces MAVS signaling resulting in diminished antiviral response. RNA viruses and their viral RNA intermediates activate Drp1 to facilitate mitochondrial fission to reduce the RLR antiviral signaling and thereby, enhance their replication.
Smaller mitochondrial size diminishes the RLR (RIG-I-like receptor) signaling (9). Thus, depletion of Drp1 prevents mitochondrial fission and boosts antiviral response (28, 29). Furthermore, the association of ER and mitochondria leads to the stimulation of cytosolic RNA sensors- RIG-I and MDA5 in a MAVS-dependent manner during mitochondrial fission (7). Viral dsRNA intermediates promote mitochondrial fission leading to a decrease in RLR signaling activated by the viral RNA exposure to the host immune system, thereby enabling the virus replication (28). Likewise, during bacterial infection, mitochondrial fragmentation is promoted, reducing the host immune response to enable their intracellular survival (21).
c) Mitophagy
Mitophagy represents the selective autophagy of faulty or damaged mitochondria to preserve homeostasis of mitochondrial dynamics at large. Mitochondrial fission upon stress, infection, or pathological diseases leads to the trigger of mitophagy as a final cellular rescue response (16, 30, 31).
Mitophagy is facilitated by two independent pathways with differences in their requirement on ubiquitin (Ub), namely the PTEN-induced kinase 1 (PINK1)/Parkin pathway and receptor-mediated pathway (32, 33). PINK1/Parkin pathway is an Ub-dependent pathway mediated by two key proteins: a) PINK1, a mitochondrial serine/threonine kinase, and b) an E3 ligase termed Parkin, a signal amplifier in response to PINK1 activation (34). In regular mitochondrial functioning, cytosolic PINK1 tagged with a mitochondrial target sequence (MTS) translocates to the IMM by specific outer and inner membrane-associated translocases, TOM, and TIM, respectively. PINK1 is degraded through proteolysis in a process comprising the elimination of MTS by mitochondrial processing protease (MPP) and cleavage by presenilin-associated rhomboid-like protease (PARL) (27). However, the loss of membrane potential (ΔΨm) in damaged mitochondria decreases the activity of TOM and TIM leading to the stabilization of PINK1 on the OMM (30, 35–38). PINK1 and Parkin work synchronously to facilitate Ub-tagging of damaged mitochondrial membranes. Consequently, the dysfunctional mitochondria are engulfed by a phagophore leading to the formation of a mitophagosome that ultimately transports it to a lysosome. Mitophagy can also occur in a receptor-mediated pathway that includes the receptors on OMM and IMM, including BNIP3, NIX, FUNDC1, PHB-2, and others (Figure 4).
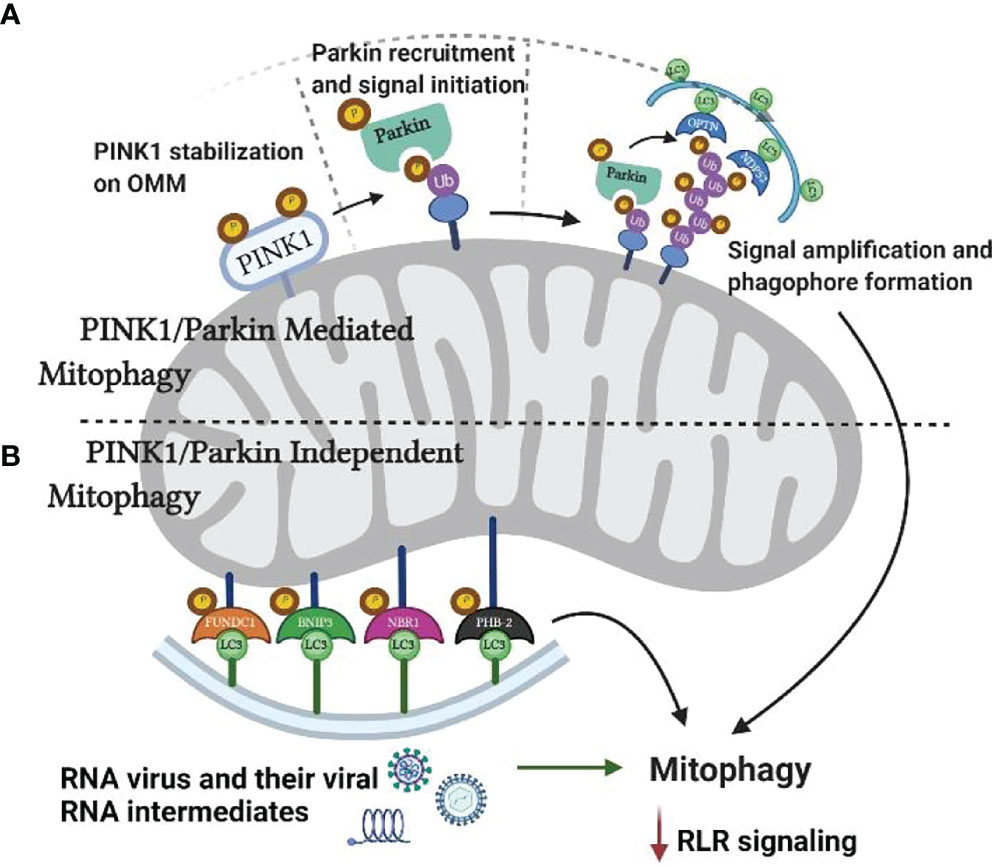
Figure 4 Mitophagy and RNA virus infection. Mitochondria can undergo mitophagy either by PINK1/Parkin dependent or independent pathways. As increased rate of mitophagy reduces RLR mediated antiviral response, RNA viruses and their viral RNA intermediates increase mitophagy by targeting PINK1 and other key proteins.
Some viruses (HBV, HCV, NDV, measles) shift the mitochondrial dynamics towards fission and mitophagy to favor viral replication and reduce overall mitochondrial mass to lessen the host antiviral response (39–42). However, the functional involvement of mitophagy in the antiviral innate immune response is still in infancy but holds significant promise in identifying a potential antiviral therapeutic target.
Impact of Emerging and Re-Emerging RNA Viruses on Mitochondrial Dynamics
In the following section, we will summarize and discuss the impact of mitochondrial dynamics on antiviral response, and viral replication during infection caused by Dengue virus, Zika virus, Hepatitis C virus, and SARS-CoV-2 (Figure 5 and Table 1).
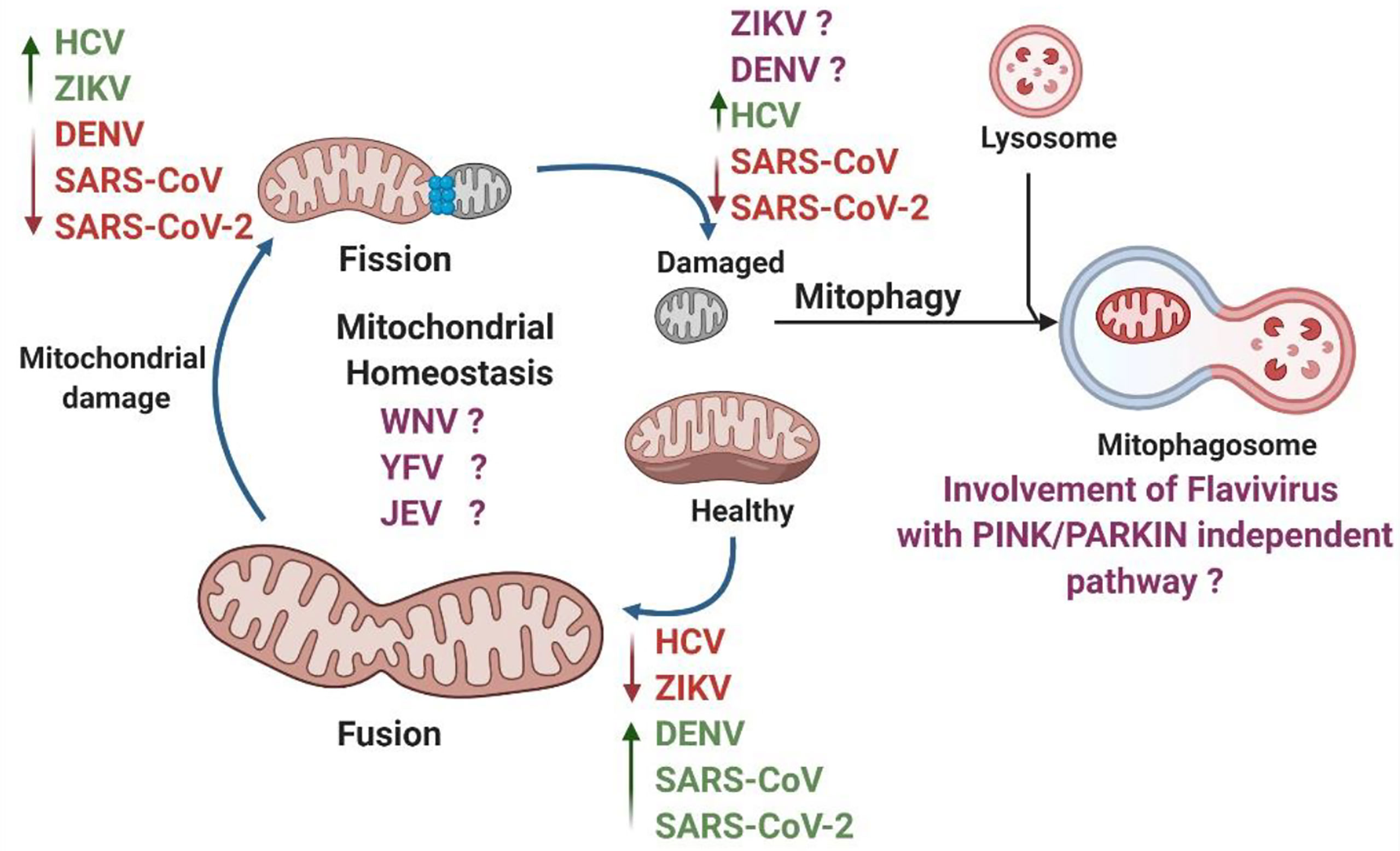
Figure 5 Summary of RNA viruses impacting various processes of mitochondrial dynamics. Under static conditions, the mitochondria exist in a tubular network. However, physiological stress and viral infection can cause damage to the mitochondria. The damaged mitochondria undergo fission and recycled via mitophagy. The remaining functional tubules rejoin the mitochondrial network through the fusion process. Viruses can hijack these processes to aid in their replication. Individual RNA viruses are shown to increase (in green) or decrease (in red) mitochondrial dynamics. There are many flavivirus with unknown effects on mitochondria (in purple).
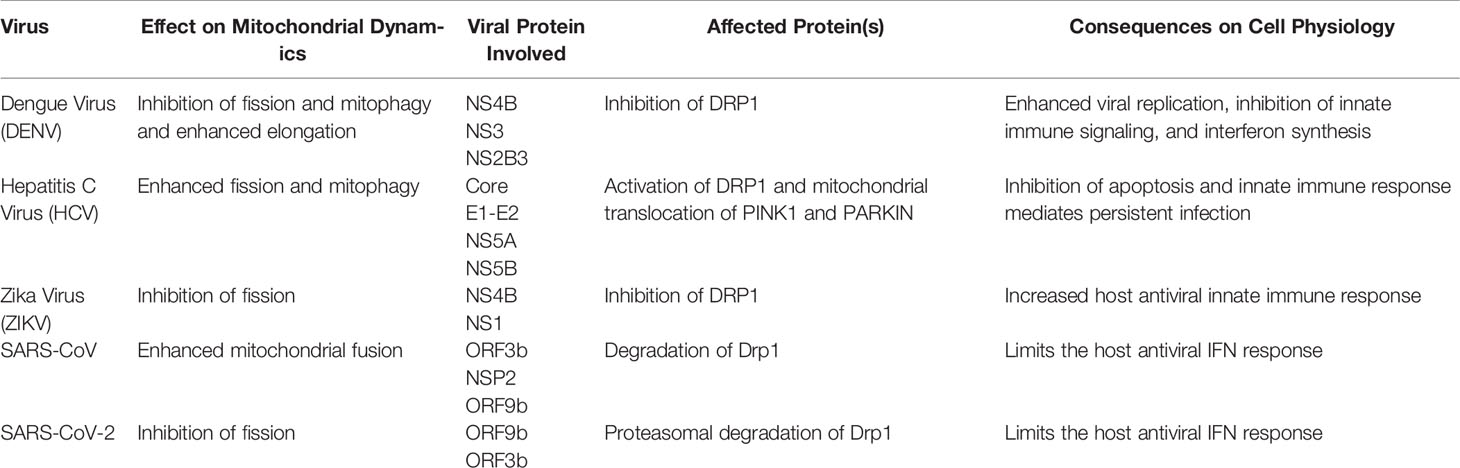
Table 1 Involvement of Flaviviruses and the role of viral proteins in causing an alteration in mitochondrial dynamics.
Dengue Virus
Dengue virus (DENV) is an arthropod-borne RNA virus comprised of a positive single-stranded RNA. DENV belongs to the genus Flavivirus and is responsible for epidemics in tropical and sub-tropical regions around the globe with an estimated 100 million symptomatic cases per year (43). The viral genome codes for three structural proteins – Capsid (C), Envelope (E), and Pre-membrane (PrM) and seven nonstructural proteins NS1, 2A, 2B, 3, 4A, 4B, and 5, aiding in viral replication.
DENV’s effect on mitochondrial dynamics has been associated with an increase in antiviral immune evasion (19, 20, 44, 45) with contradicting studies demonstrating their effect on mitochondrial morphology. Yu et al. showed that DENV NS2B3 protein partially cleaves MFN1 and MFN2, attenuating the interferon responses resulting in increased viral replication and cell death (18–20). However, recent studies demonstrate that DENV NS4B and NS3 proteins enhance mitochondrial fusion along with a reduction in mitochondrial fission via degradation of the total- and p616-Drp1. The induction of mitochondrial fusion degrades the integrity of MAMs, and the sites of ER-mitochondria interaction, alleviating RIG-I dependent activation of IFN response, thereby promoting DENV replication. Inversely, these findings were verified by knocking down Mfn2, which led to mitochondrial fragmentation and increased production of IFN-λ1, and impaired DENV replication (19). Moreover, it was reported that the induction of mitochondrial fission and subsequent fragmentation with the use of a potent mitochondria uncoupling reagent, Carbonyl cyanide cholorophenylhydrazone (CCCP) or via overexpression of activated Drp1 led to a reduction in viral replication, suggesting that mitochondrial elongation is beneficial in DENV replication (20). Furthermore, even in mosquito cells, DENV infection increased the mitochondrial fusion by increasing the MFNs and no alteration of Drp1 levels (46).
Zika Virus
Like DENV, Zika virus (ZIKV) also belongs to the family of Flaviviridae and shares similar characteristics. Although identified in 1947, most ZIKV studies were performed during an outbreak in Brazil in 2016 as it was linked to causing congenital malformations, including ocular complications (47–52). Currently, there are no vaccines or specific antiviral drugs available to treat ZIKV diseases.
ZIKV NS2B3 and NS3 proteins were shown to downregulate the expression of MAVS, IFN (specifically IFNβ), and ISGs (53). ZIKV NS3 protein prevents the transport of RIG-I and MDA5 to the mitochondria by binding to the 14-3-3 binding motif of MAVS, thereby inhibiting the RLR signaling pathway (54). However, there are contradicting reports on the effect of mitochondrial dynamics on antiviral immunity against ZIKV. ZIKV infection caused mitochondrial elongation, which was enhanced by the knockdown of Drp1 (19, 55). However, in human retinal pigment epithelial cells, ZIKV infection increased mitochondrial fission (56). On the other hand, a recent study showed that ZIKV infection in astrocytes leads to ROS imbalance, mitochondrial functional defects, and DNA breakage leading to neurological disorders without any effect on its morphology (57). ZIKV NS1 protein triggered abnormal mitochondrial fragmentation and a decrease in MFN2 levels contributed to ZIKV-induced cell death in neuronal cells (58).
Hepatitis C Virus
Hepatitis C virus (HCV) is an important human pathogen belonging to the family Flaviviridae with the single stranded-RNA genome. Approximately 71 million people are clinically infected with HCV, resulting in nearly 400,000 deaths annually due to liver cirrhosis and hepatocellular carcinoma (HCC) (59). The HCV genome encodes ten proteins – four structural proteins C, E1, E2, and p7 along with six non-structural proteins, NS2, 3, 4A, 4B, 5A, and 5B (60). The transmission of HCV is primarily via intravenous drug use, blood transfusions, and unsterilized medical pieces of equipment. There are multiple candidates for HCV prophylactic vaccines, however, none are available for use (61). There is a highly effective antiviral drug, Daclatasvir with a curing rate of 95% available to date (62).
HCV NS3/4A protease cleaves the MAVS protein and inhibits the formation of the MAVS signalosome, leading to diminished immune response and IFN production (63). HCV infection promotes mitochondrial fission and mitophagy to prevent the spread of virus-induced mitochondrial damage. This allows the maintenance of an adequate cellular environment for viral dissemination and the prevention of apoptosis. HCV NS5A triggers mitochondrial fragmentation, loss of mitochondrial membrane potential, and Parkin translocation to the mitochondria, leading to mitophagy (64). Furthermore, NS5A protein inhibits the activity of electron transport chain (ETC) enzyme complex I leading to increased mitochondrial calcium uptake, mitochondrial permeability, and ROS production (41, 64). HCV stimulates the synthesis of the Ub-dependent proteins - PINK1 and Parkin and triggers their translocation to the IMM and subsequent mitophagy. The induced mitophagy can enhance HCV-regulated inhibition of oxidative phosphorylation (40). The HCV core (C) protein inhibits mitophagy by sequestering Parkin (65). The underlying mechanism of how HCV and its core protein mediate these effects remains to be characterized. HBV/HCV alters mitochondrial dynamics to enhance mitochondrial fission and mitophagy and keep a check on the mitochondrial injury, thereby contributing to persistent HCV infection (41). HBV/HCV-induced mitophagy leads to attenuation of IFN signaling through which the increased PARKIN-MAVS interaction cripples the innate immunity (39–41). Interestingly, Kim et al. have shown a promising protective role of Ginsenoside Rg3 (G-Rg3) treatment against HCV-induced mitophagy, which follows mitochondrial fission (66).
The role of mitophagy in regulating flavivirus infection has not been studied in-depth and the functional involvement of various flavivirus proteins in inducing mitophagy would aid in a better understanding of the mechanisms at large.
SARS-CoV-2
SARS-CoV-2, the causative agent of the COVID-19 pandemic is a positive-single-stranded RNA virus belonging to the betacoronaviridae family shared with SARS-CoV and MERS-CoV (67). The virus can transmit via respiratory droplets formed during sneezing, talking, or coughing along with the contact of our mucosal surface with contaminated objects and ocular surface (68–70). The SARS-CoV-2 genome encodes for four structural proteins Spike (S), Envelope (E), Nucleocapsid (N), and Membrane (M) protein along with 16 non-structural proteins (NSP 1-16), and eight auxiliary proteins (ORF 3a, 3b, 6, 7a, 7b, 8, 9b, and 14) (71, 72).
SARS-CoV-2 infection is associated with altered mitochondrial dynamics resulting in oxidative stress, pro-inflammatory cytokine production, and cell death. The morphology of mitochondria in SARS-CoV-2 infected cells is significantly displaced and arranged around the dsRNA regions in the cytoplasm. The intra-cristal space, as well as the matrix, is expanded leading to thinner mitochondria (73). The virulence factors ORF9b and dsRNA of SARS-CoV as well as SARS-CoV-2 localize in the mitochondria and targets the MAVS signalosome, degrading the TRAF3 and TRAF6 signaling molecules, thereby hampering the antiviral response (74, 75). SARS-CoV ORF9b trigger degradation of Drp1 leading to mitochondrial fusion limiting the host cell IFN response against the virus (21). Similarly, SARS-CoV-2 triggers inhibition of mitochondrial fission to facilitate its replication. Protein-protein interaction studies have indicated that SARS-CoV-2 ORF9b interacts with TOMM70, a mitochondrial import receptor that plays a critical role in modulating interferon response (71, 76). ORF9b localizes to mitochondria and causes mitochondrial elongation by triggering ubiquitination and proteasomal degradation of Drp1, thereby inhibiting fission. ORF9b also targets the MAVS signalosome by usurping PCBP2 and AIP4 to trigger the degradation of MAVS, TRAF3, and TRAF6, thereby limiting the host antiviral interferon response (74, 77). In addition, SARS-CoV-2 NSP13 and 9C protein may also be involved in altering the innate immune response by regulation of MAVS signal transduction (71). The SARS-CoV NSP2 protein interacts with PHB, PHB2, and STOML2 while SARS-CoV-2 ORF3b interacts with STOML2 to regulate mitochondrial homeostasis, mitophagy, and mitochondrial fusion and finally alter the innate immune response of the host (77–79). For SARS-CoV-2, the ORF9b protein induces autophagy by interaction with Prohibitins (PHBs). One of the SARS-CoV-2 viral proteins ORF3a includes a 20nt base sequence, which could target the host USP30 transcript, a mitochondrial deubiquitinase involved in mitochondrial homeostasis, and mitophagy (71, 80).
The complexity of SARS-CoV-2 infection has increased due to evasion from vaccine acquired immunity and the evolution of numerous viral variants sweeping the world. The study of various mutants on host immunity and the organelles should be given importance and kept on track for therapeutic intervention.
Conclusions
Mitochondria are a network of dynamic organelles with recurrent cycles of fission and fusion. These processes help in intermixing, content distribution, maintenance of energy homeostasis, and mitochondrial functional capacity. Mitochondrial fission and fusion constitute a major part of mitochondrial dynamics while mitochondrial quality control is regulated by mitophagy (81). Amongst the flaviviruses, the alteration of mitochondrial dynamics has been studied in HCV, DENV, and ZIKV while it remains unknown for other emerging flaviviruses such as WNV, JEV, and YFV (Figure 5).
A few of the viruses (HCV, Influenza A) and their viral proteins induce the cleavage of MAVS from mitochondria, thereby reducing their ability to induce interferon response (82–84). The viruses (HCV, ASFV, HIV-1) also alter the intracellular distribution of mitochondria either by concentrating the mitochondria near the viral factories to meet the energy demand during viral replication or by cordoning off the mitochondria within the cytoplasm to prevent the release of the mediators of apoptosis (8). These cellular functions are performed to provide energy for viral replication and release of progeny virion. However, mechanisms regulating mitochondrial dynamics during flavivirus, and SARS-CoV-2 infection have not been studied to date.
Interestingly, intracellular calcium concentration also regulates mitochondrial dynamics since the calcium-dependent phosphatase calcineurin dephosphorylates Drp1, facilitating the recruitment of Drp1 to the mitochondria and the consequent mitochondrial fission (85). The involvement of calcium channels and the variation in the concentration of calcium ions has not been studied in the process of mitochondrial fission with flaviviruses and SARS-CoV-2, an interesting area to be explored in detail.
Since mitochondria are the source of energy and play an important role in antiviral immunity, the damage to mitochondrial DNA may help in evading the mitochondrial antiviral immune response (86). Indeed, several viruses (HSV-1, HCV, EBV, HIV) degrade host mitochondrial DNA (mtDNA) to augment their genome replication (86–91). Also, studies have reported the enrichment of SARS-CoV, and SARS-CoV-2 viral RNA in mitochondria and nucleolus, implicating their role in regulating the viral life cycle, ranging from virion assembly to disruption of host-mitochondrial function (92, 93). Interestingly, the viral ORFs can release mtDNA in the cytoplasm and activate the inflammasome pathway, thereby suppressing innate and adaptive immunity (94). However, there is a lack of studies on the effect of flaviviruses on mtDNA, which would shed light on its role in mitochondrial dynamics and antiviral immunity.
Given the importance of mitochondrial dynamics in various cellular processes, pharmacological modulators of mitochondrial dynamics have been employed in combination with direct-acting antivirals (DAAs) to combat viral infection. Several therapies (Tenofovir, Zalcitabine, and Didanosine) against human immunodeficiency virus (HIV) infection exert antiviral activity by modulating mitochondrial function (95, 96). Among RNA viruses, mitochondrial hyper-fusion drug Mito-C and 8-O-(E-p-methoxycinnamoyl) harpagide (MCH), have been reported to possess antiviral activity against influenza virus by influencing mitochondrial dynamics (97, 98). However, further studies are needed to investigate their effects against other RNA viruses including flaviviruses and SARS-CoV-2. A notable DAA, Sofosbuvir can competitively block the HCV NS5B polymerase and effectively inhibit HCV-RNA synthesis (99). While Sofosbuvir has proven efficacious, it has also been shown to destabilize mitochondrial membrane potential and further induce mitochondrial fission. However, a novel ginsenoside (G-Rg3) has been shown to inhibit HCV-induced abnormal mitochondrial fission and stabilize mitochondrial membrane potential, further potentiating the therapeutic effect of Sofosbuvir (66). As previously stated, it has been shown that SARS-CoV-2 alters mitochondrial dynamics to diminish the host immune response. Perhaps the addition of a pharmacological agent such as G-Rg3 to aid in the stabilization of mitochondrial dynamics may prove beneficial in the acute treatment of SARS-CoV-2 and other RNA viruses and warrants further investigation.
While these recent findings have allowed us to identify potential therapeutic targets, further studies are needed to decipher how these viruses alter mitochondria. Moreover, elucidation of the role of emerging flavivirus structural and nonstructural protein involvement in regulating mitochondrial dynamics could provide therapeutic advances with the potential to reduce the viral disease burden on the human population. These studies aid in developing therapeutic approaches in the absence of a vaccine candidate against several RNA viruses and their emerging variants of concerns.
Future directions
a. Studies related to mitochondrial dynamics in RNA viruses of public health concern including JEV, WNV, and YFV.
b. Interplay of intracellular calcium in regulating mitochondrial dynamics during RNA viral infections.
c. The interaction of RNA virus proteins with mtDNA in regulating mitochondrial dynamics and antiviral response.
d. Therapeutic strategies to block viral replication in host cells by regulating mitochondrial dynamics.
e. Effect of chronic or long-term viral infection (e.g., Long COVID) on mitochondria.
Author Contributions
AK conceived the idea, and SS and KD wrote the first draft. AK and SS revised the manuscript to final form. All authors read and approved the manuscript.
Funding
Research in our laboratory is supported in parts by National Institute of Health (NIH) Grants (R21AI135583, R01EY026964, and R01 EY027381 to A.K.), NIH Core Grant P30EY004068 (to Linda D. Hazlett), and an unrestricted grant from Research to Prevent Blindness Inc. (to Kresge Eye Institute, Wayne State University).
Author Disclaimer
The authors have made every effort to ensure that appropriate and justified references have been added to the best of their knowledge. The authors would like to apologize to the groups whose references would have been overlooked while writing the review article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank other members of the lab for their helpful discussions.
References
1. Pinkoski MJ, Waterhouse NJ, Green DR. Mitochondria, Apoptosis and Autoimmunity. Curr Dir Autoimmun (2006) 9:55–73. doi: 10.1159/000090772
2. Horbay R, Bilyy R. Mitochondrial Dynamics During Cell Cycling. Apoptosis (2016) 21:1327–35. doi: 10.1007/s10495-016-1295-5
3. Nilsson MI, Tarnopolsky MA. Mitochondria and Aging-The Role of Exercise as a Countermeasure. Biol (Basel) (2019) 8(2):40. doi: 10.3390/biology8020040
4. Mishra P, Chan DC. Metabolic Regulation of Mitochondrial Dynamics. J Cell Biol (2016) 212:379–87. doi: 10.1083/jcb.201511036
5. Sanjuan R, Domingo-Calap P. Mechanisms of Viral Mutation. Cell Mol Life Sci (2016) 73:4433–48. doi: 10.1007/s00018-016-2299-6
6. Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral Control of Mitochondrial Apoptosis. PloS Pathog (2008) 4:e1000018. doi: 10.1371/journal.ppat.1000018
7. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial-Associated Endoplasmic Reticulum Membranes (MAM) Form Innate Immune Synapses and are Targeted by Hepatitis C Virus. Proc Natl Acad Sci United States America (2011) 108:14590–5. doi: 10.1073/pnas.1110133108
8. Anand SK, Tikoo SK. Viruses as Modulators of Mitochondrial Functions. Adv Virol (2013) 2013:738794. doi: 10.1155/2013/738794
9. Pourcelot M, Arnoult D. Mitochondrial Dynamics and the Innate Antiviral Immune Response. FEBS J (2014) 281:3791–802. doi: 10.1111/febs.12940
10. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, Encoding a Dynamin-Related GTPase, is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat Genet (2000) 26:211–5. doi: 10.1038/79944
11. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, is Mutated in Dominant Optic Atrophy. Nat Genet (2000) 26:207–10. doi: 10.1038/79936
12. Wong ED, Wagner JA, Gorsich SW, Mccaffery JM, Shaw JM, Nunnari J. The Dynamin-Related GTPase, Mgm1p, is an Intermembrane Space Protein Required for Maintenance of Fusion Competent Mitochondria. J Cell Biol (2000) 151:341–52. doi: 10.1083/jcb.151.2.341
13. Santel A, Fuller MT. Control of Mitochondrial Morphology by a Human Mitofusin. J Cell Sci (2001) 114:867–74. doi: 10.1242/jcs.114.5.867
14. Chan DC. Dissecting Mitochondrial Fusion. Dev Cell (2006) 11:592–4. doi: 10.1016/j.devcel.2006.10.009
15. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and are Essential for Embryonic Development. J Cell Biol (2003) 160:189–200. doi: 10.1083/jcb.200211046
16. Mohanty A, Tiwari-Pandey R, Pandey NR. Mitochondria: The Indispensable Players in Innate Immunity and Guardians of the Inflammatory Response. J Cell Commun Signal (2019) 13:303–18. doi: 10.1007/s12079-019-00507-9
17. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial Membrane Potential is Required for MAVS-Mediated Antiviral Signaling. Sci Signal (2011) 4:ra7. doi: 10.1126/scisignal.2001147
18. Yu CY, Liang JJ, Li JK, Lee YL, Chang BL, Su CI, et al. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PloS Pathog (2015) 11:e1005350. doi: 10.1371/journal.ppat.1005350
19. Chatel-Chaix L, Cortese M, Romero-Brey I, Bender S, Neufeldt CJ, Fischl W, et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe (2016) 20:342–56. doi: 10.1016/j.chom.2016.07.008
20. Barbier V, Lang D, Valois S, Rothman AL, Medin CL. Dengue Virus Induces Mitochondrial Elongation Through Impairment of Drp1-Triggered Mitochondrial Fission. Virology (2017) 500:149–60. doi: 10.1016/j.virol.2016.10.022
21. Tiku V, Tan MW, Dikic I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol (2020) 30:263–75. doi: 10.1016/j.tcb.2020.01.006
22. Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, et al. S-Nitrosylation of Dynamin-Related Protein 1 Mediates Mutant Huntingtin-Induced Mitochondrial Fragmentation and Neuronal Injury in Huntington's Disease. Antioxid Redox Signal (2013) 19:1173–84. doi: 10.1089/ars.2012.4928
23. Melatti C, Pieperhoff M, Lemgruber L, Pohl E, Sheiner L, Meissner M. A Unique Dynamin-Related Protein is Essential for Mitochondrial Fission in Toxoplasma Gondii. PloS Pathog (2019) 15:e1007512. doi: 10.1371/journal.ppat.1007512
24. Van Der Bliek AM, Shen Q, Kawajiri S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb Perspect Biol (2013) 5(6). doi: 10.1101/cshperspect.a011072
25. Friedman JR, Lackner LL, West M, Dibenedetto JR, Nunnari J, Voeltz GK. ER Tubules Mark Sites of Mitochondrial Division. Science (2011) 334:358–62. doi: 10.1126/science.1207385
26. Korobova F, Ramabhadran V, Higgs HN. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science (2013) 339:464–7. doi: 10.1126/science.1228360
27. Jin SM, Youle RJ. PINK1- and Parkin-Mediated Mitophagy at a Glance. J Cell Sci (2012) 125:795–9. doi: 10.1242/jcs.093849
28. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial Dynamics Regulate the RIG-I-Like Receptor Antiviral Pathway. EMBO Rep (2010) 11:133–8. doi: 10.1038/embor.2009.258
29. Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, et al. Virus-Infection or 5'ppp-RNA Activates Antiviral Signal Through Redistribution of IPS-1 Mediated by MFN1. PloS Pathog (2010) 6:e1001012. doi: 10.1371/journal.ppat.1001012
30. Durcan TM, Fon EA. The Three 'P's of Mitophagy: PARKIN, PINK1, and Post-Translational Modifications. Genes Dev (2015) 29:989–99. doi: 10.1101/gad.262758.115
31. Gkikas I, Palikaras K, Tavernarakis N. The Role of Mitophagy in Innate Immunity. Front Immunol (2018) 9:1283. doi: 10.3389/fimmu.2018.01283
32. Khaminets A, Behl C, Dikic I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol (2016) 26:6–16. doi: 10.1016/j.tcb.2015.08.010
33. Georgakopoulos ND, Wells G, Campanella M. The Pharmacological Regulation of Cellular Mitophagy. Nat Chem Biol (2017) 13:136–46. doi: 10.1038/nchembio.2287
34. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature (2015) 524:309–14. doi: 10.1038/nature14893
35. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PloS Biol (2010) 8:e1000298. doi: 10.1371/journal.pbio.1000298
36. Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The Mitochondrial Intramembrane Protease PARL Cleaves Human Pink1 to Regulate Pink1 Trafficking. J Neurochem (2011) 117:856–67. doi: 10.1111/j.1471-4159.2011.07253.x
37. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J Cell Biol (2014) 205:143–53. doi: 10.1083/jcb.201402104
38. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin is Activated by PINK1-Dependent Phosphorylation of Ubiquitin at Ser65. Biochem J (2014) 460:127–39. doi: 10.1042/BJ20140334
39. Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PloS Pathog (2013) 9:e1003722. doi: 10.1371/journal.ppat.1003722
40. Kim SJ, Syed GH, Siddiqui A. Hepatitis C Virus Induces the Mitochondrial Translocation of Parkin and Subsequent Mitophagy. PloS Pathog (2013) 9:e1003285. doi: 10.1371/journal.ppat.1003285
41. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, et al. Hepatitis C Virus Triggers Mitochondrial Fission and Attenuates Apoptosis to Promote Viral Persistence. Proc Natl Acad Sci USA (2014) 111:6413–8. doi: 10.1073/pnas.1321114111
42. Meng G, Xia M, Wang D, Chen A, Wang Y, Wang H, et al. Mitophagy Promotes Replication of Oncolytic Newcastle Disease Virus by Blocking Intrinsic Apoptosis in Lung Cancer Cells. Oncotarget (2014) 5:6365–74. doi: 10.18632/oncotarget.2219
43. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The Global Distribution and Burden of Dengue. Nature (2013) 496:504–7. doi: 10.1038/nature12060
44. Mclean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-Induced Autophagy Protects Cells Against Death and Enhances Virus Replication. J Biol Chem (2011) 286:22147–59. doi: 10.1074/jbc.M110.192500
45. Datan E, Roy SG, Germain G, Zali N, Mclean JE, Golshan G, et al. Dengue-Induced Autophagy, Virus Replication and Protection From Cell Death Require ER Stress (PERK) Pathway Activation. Cell Death Dis (2016) 7:e2127. doi: 10.1038/cddis.2015.409
46. Santana-Roman ME, Maycotte P, Uribe-Carvajal S, Uribe-Alvarez C, Alvarado-Medina N, Khan M, et al. Monitoring Mitochondrial Function in Aedes Albopictus C6/36 Cell Line During Dengue Virus Infection. Insects (2021) 12:934. doi: 10.3390/insects12100934
47. Abbasi AU. Zika Virus Infection; Vertical Transmission and Foetal Congenital Anomalies. J Ayub Med Coll Abbottabad (2016) 28:1–2.
48. Panchaud A, Stojanov M, Ammerdorffer A, Vouga M, Baud D. Emerging Role of Zika Virus in Adverse Fetal and Neonatal Outcomes. Clin Microbiol Rev (2016) 29:659–94. doi: 10.1128/CMR.00014-16
49. Singh PK, Guest JM, Kanwar M, Boss J, Gao N, Juzych MS, et al. Zika Virus Infects Cells Lining the Blood-Retinal Barrier and Causes Chorioretinal Atrophy in Mouse Eyes. JCI Insight (2017) 2:e92340. doi: 10.1172/jci.insight.92340
50. Singh PK, Khatri I, Jha A, Pretto CD, Spindler KR, Arumugaswami V, et al. Determination of System Level Alterations in Host Transcriptome Due to Zika Virus (ZIKV) Infection in Retinal Pigment Epithelium. Sci Rep (2018) 8:11209. doi: 10.1038/s41598-018-29329-2
51. Singh S, Kumar A. Ocular Manifestations of Emerging Flaviviruses and the Blood-Retinal Barrier. Viruses (2018) 10 (10):530. doi: 10.3390/v10100530
52. Singh PK, Kasetti RB, Zode GS, Goyal A, Juzych MS, Kumar A. Zika Virus Infects Trabecular Meshwork and Causes Trabeculitis and Glaucomatous Pathology in Mouse Eyes. mSphere (2019) 4(3):e00173–19. doi: 10.1128/mSphere.00173-19
53. Li W, Li N, Dai S, Hou G, Guo K, Chen X, et al. Zika Virus Circumvents Host Innate Immunity by Targeting the Adaptor Proteins MAVS and MITA. FASEB J (2019) 33:9929–44. doi: 10.1096/fj.201900260R
54. Riedl W, Acharya D, Lee JH, Liu G, Serman T, Chiang C, et al. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe (2019) 26:493–503.e496. doi: 10.1016/j.chom.2019.09.012
55. Seong RK, Lee JK, Cho GJ, Kumar M, Shin OS. mRNA and miRNA Profiling of Zika Virus-Infected Human Umbilical Cord Mesenchymal Stem Cells Identifies miR-142-5p as an Antiviral Factor. Emerg Microbes Infect (2020) 9:2061–75. doi: 10.1080/22221751.2020.1821581
56. Russo CA, Torti MF, Marquez AB, Sepulveda CS, Alaimo A, Garcia CC. Antiviral Bioactivity of Resveratrol Against Zika Virus Infection in Human Retinal Pigment Epithelial Cells. Mol Biol Rep (2021) 48:5379–92. doi: 10.1007/s11033-021-06490-y
57. Ledur PF, Karmirian K, Pedrosa C, Souza LRQ, Assis-De-Lemos G, Martins TM, et al. Zika Virus Infection Leads to Mitochondrial Failure, Oxidative Stress and DNA Damage in Human iPSC-Derived Astrocytes. Sci Rep (2020) 10:1218. doi: 10.1038/s41598-020-57914-x
58. Yang S, Gorshkov K, Lee EM, Xu M, Cheng YS, Sun N, et al. Zika Virus-Induced Neuronal Apoptosis via Increased Mitochondrial Fragmentation. Front Microbiol (2020) 11:598203. doi: 10.3389/fmicb.2020.598203
59. Chu JYK, Ou JJ. Autophagy in HCV Replication and Protein Trafficking. Int J Mol Sci (2021) 22(3):1089. doi: 10.3390/ijms22031089
60. Lindenbach BD, Rice CM. The Ins and Outs of Hepatitis C Virus Entry and Assembly. Nat Rev Microbiol (2013) 11:688–700. doi: 10.1038/nrmicro3098
61. Hartlage AS, Kapoor A. Hepatitis C Virus Vaccine Research: Time to Put Up or Shut Up. Viruses (2021) 13(8):1596. doi: 10.3390/v13081596
62. Gitto S, Gamal N, Andreone P. NS5A Inhibitors for the Treatment of Hepatitis C Infection. J Viral Hepat (2017) 24:180–6. doi: 10.1111/jvh.12657
63. Horner SM, Gale M Jr. Regulation of Hepatic Innate Immunity by Hepatitis C Virus. Nat Med (2013) 19:879–88. doi: 10.1038/nm.3253
64. Jassey A, Liu CH, Changou CA, Richardson CD, Hsu HY, Lin LT. Hepatitis C Virus Non-Structural Protein 5a (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells (2019) 8(4):290. doi: 10.3390/cells8040290
65. Hara Y, Yanatori I, Ikeda M, Kiyokage E, Nishina S, Tomiyama Y, et al. Hepatitis C Virus Core Protein Suppresses Mitophagy by Interacting With Parkin in the Context of Mitochondrial Depolarization. Am J Pathol (2014) 184:3026–39. doi: 10.1016/j.ajpath.2014.07.024
66. Kim SJ, Jang JY, Kim EJ, Cho EK, Ahn DG, Kim C, et al. Ginsenoside Rg3 Restores Hepatitis C Virus-Induced Aberrant Mitochondrial Dynamics and Inhibits Virus Propagation. Hepatology (2017) 66:758–71. doi: 10.1002/hep.29177
67. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus From Patients With Pneumonia in China 2019. N Engl J Med (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
68. Sawant OB, Singh S, Wright RE 3rd, Jones KM, Titus MS, Dennis E, et al. Prevalence of SARS-CoV-2 in Human Post-Mortem Ocular Tissues. Ocul Surf (2020) 19:322–9. doi: 10.1101/2020.10.05.20201574
69. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z, et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N Engl J Med (2020) 382:1177–9. doi: 10.1056/NEJMc2001737
70. Singh S, Garcia G, Shah R, Kramerov AA, Wright RE 3rd, Spektor TM, et al. SARS-CoV-2 and its Beta Variant of Concern Infect Human Conjunctival Epithelial Cells and Induce Differential Antiviral Innate Immune Response. Ocul Surf (2021) 23:184–94. doi: 10.1016/j.jtos.2021.09.007
71. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O'meara MJ, et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature (2020) 583(7816):459–468. doi: 10.1038/s41586-020-2286-9
72. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome Composition and Divergence of the Novel Coronavirus, (2019-Ncov) Originating in China. Cell Host Microbe (2020) 27:325–8. doi: 10.1016/j.chom.2020.02.001
73. Cortese M, Lee JY, Cerikan B, Neufeldt CJ, Oorschot VMJ, Kohrer S, et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe (2020) 28:853–66.e855. doi: 10.1016/j.chom.2020.11.003
74. Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J Immunol (2014) 193:3080–9. doi: 10.4049/jimmunol.1303196
75. Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang C, et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol (2021) 12:780768. doi: 10.3389/fmicb.2021.780768
76. Liu XY, Wei B, Shi HX, Shan YF, Wang C. Tom70 Mediates Activation of Interferon Regulatory Factor 3 on Mitochondria. Cell Res (2010) 20:994–1011. doi: 10.1038/cr.2010.103
77. Shenoy S. Coronavirus (Covid-19) Sepsis: Revisiting Mitochondrial Dysfunction in Pathogenesis, Aging, Inflammation, and Mortality. Inflamm Res (2020) 69:1077–85. doi: 10.1007/s00011-020-01389-z
78. Cornillez-Ty CT, Liao L, Yates JR 3rd, Kuhn P, Buchmeier MJ. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 2 Interacts With a Host Protein Complex Involved in Mitochondrial Biogenesis and Intracellular Signaling. J Virol (2009) 83:10314–8. doi: 10.1128/JVI.00842-09
79. Perrin-Cocon L, Diaz O, Jacquemin C, Barthel V, Ogire E, Ramiere C, et al. The Current Landscape of Coronavirus-Host Protein-Protein Interactions. J Transl Med (2020) 18:319. doi: 10.1186/s12967-020-02480-z
80. Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS-CoV-2 Hijacking of Host Mitochondria in COVID-19 Pathogenesis. Am J Physiol Cell Physiol (2020) 319:C258–67. doi: 10.1152/ajpcell.00224.2020
81. Westermann B. Mitochondrial Fusion and Fission in Cell Life and Death. Nat Rev Mol Cell Biol (2010) 11:872–84. doi: 10.1038/nrm3013
82. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an Adaptor Protein in the RIG-I Antiviral Pathway and is Targeted by Hepatitis C Virus. Nature (2005) 437:1167–72. doi: 10.1038/nature04193
83. Chen Z, Benureau Y, Rijnbrand R, Yi J, Wang T, Warter L, et al. GB Virus B Disrupts RIG-I Signaling by NS3/4A-Mediated Cleavage of the Adaptor Protein MAVS. J Virol (2007) 81:964–76. doi: 10.1128/JVI.02076-06
84. Ohman T, Rintahaka J, Kalkkinen N, Matikainen S, Nyman TA. Actin and RIG-I/MAVS Signaling Components Translocate to Mitochondria Upon Influenza A Virus Infection of Human Primary Macrophages. J Immunol (2009) 182:5682–92. doi: 10.4049/jimmunol.0803093
85. Cereghetti GM, Stangherlin A, Martins De Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc Natl Acad Sci U.S.A. (2008) 105:15803–8. doi: 10.1073/pnas.0808249105
86. Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. Herpes Simplex Virus Eliminates Host Mitochondrial DNA. EMBO Rep (2007) 8:188–93. doi: 10.1038/sj.embor.7400878
87. De Mendoza C, Sanchez-Conde M, Timmermans E, Buitelaar M, De Baar MP, Gonzalez-Lahoz J, et al. Mitochondrial DNA Depletion in HIV-Infected Patients is More Pronounced With Chronic Hepatitis C and Enhanced Following Treatment With Pegylated Interferon Plus Ribavirin. Antivir Ther (2005) 10:557–61. doi: 10.1177/135965350501000410
88. Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C Virus Triggers Mitochondrial Permeability Transition With Production of Reactive Oxygen Species, Leading to DNA Damage and STAT3 Activation. J Virol (2006) 80:7199–207. doi: 10.1128/JVI.00321-06
89. Wiedmer A, Wang P, Zhou J, Rennekamp AJ, Tiranti V, Zeviani M, et al. Epstein-Barr Virus Immediate-Early Protein Zta Co-Opts Mitochondrial Single-Stranded DNA Binding Protein to Promote Viral and Inhibit Mitochondrial DNA Replication. J Virol (2008) 82:4647–55. doi: 10.1128/JVI.02198-07
90. Corcoran JA, Saffran HA, Duguay BA, Smiley JR. Herpes Simplex Virus UL12.5 Targets Mitochondria Through a Mitochondrial Localization Sequence Proximal to the N Terminus. J Virol (2009) 83:2601–10. doi: 10.1128/JVI.02087-08
91. Ming-Ju H, Yih-Shou H, Tzy-Yen C, Hui-Ling C. Hepatitis C Virus E2 Protein Induce Reactive Oxygen Species (ROS)-Related Fibrogenesis in the HSC-T6 Hepatic Stellate Cell Line. J Cell Biochem (2011) 112:233–43. doi: 10.1002/jcb.22926
92. Somasundaran M, Zapp ML, Beattie LK, Pang L, Byron KS, Bassell GJ, et al. Localization of HIV RNA in Mitochondria of Infected Cells: Potential Role in Cytopathogenicity. J Cell Biol (1994) 126:1353–60. doi: 10.1083/jcb.126.6.1353
93. Becker JT, Sherer NM. Subcellular Localization of HIV-1 Gag-Pol mRNAs Regulates Sites of Virion Assembly. J Virol (2017) 91(6):e02315–16. doi: 10.1128/JVI.02315-16
94. Prasun P. COVID-19: A Mitochondrial Perspective. DNA Cell Biol (2021) 40:713–9. doi: 10.1089/dna.2020.6453
95. Birkus G, Hitchcock MJ, Cihlar T. Assessment of Mitochondrial Toxicity in Human Cells Treated With Tenofovir: Comparison With Other Nucleoside Reverse Transcriptase Inhibitors. Antimicrob Agents Chemother (2002) 46:716–23. doi: 10.1128/AAC.46.3.716-723.2002
96. Stoker ML, Newport E, Hulit JC, West AP, Morten KJ. Impact of Pharmacological Agents on Mitochondrial Function: A Growing Opportunity? Biochem Soc Trans (2019) 47:1757–72. doi: 10.1042/BST20190280
97. Kwon EB, Yang HJ, Kim YS, Li W, Choi JG. 8-O-(E-P-Methoxycinnamoyl)Harpagide Inhibits Influenza A Virus Infection by Suppressing Intracellular Calcium. Molecules (2021) 26(4):1029. doi: 10.3390/molecules26041029
98. Pila-Castellanos I, Molino D, Mckellar J, Lines L, Da Graca J, Tauziet M, et al. Mitochondrial Morphodynamics Alteration Induced by Influenza Virus Infection as a New Antiviral Strategy. PloS Pathog (2021) 17:e1009340. doi: 10.1371/journal.ppat.1009340
Keywords: flavivirus, zika virus, dengue virus, hepatitis C virus, SARS-CoV-2, COVID-19, mitochondria, mitophagy
Citation: Singh S, Dirani K and Kumar A (2022) Intricacy of Mitochondrial Dynamics and Antiviral Response During RNA Virus Infection. Front.Virol. 2:918806. doi: 10.3389/fviro.2022.918806
Received: 12 April 2022; Accepted: 17 May 2022;
Published: 16 June 2022.
Edited by:
Amelia K Pinto, Saint Louis University, United StatesReviewed by:
Juan C De La Torre, The Scripps Research Institute, United StatesJoão Mamede, Rush University, United States
Copyright © 2022 Singh, Dirani and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashok Kumar, YWt1bWFAbWVkLndheW5lLmVkdQ==