 Benni Vargas
Benni Vargas Nicolas Sluis-Cremer
Nicolas Sluis-Cremer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol. , 29 June 2022
Sec. Fundamental Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.917941
This article is part of the Research Topic HIV/SIV basic research update View all 10 articles
The persistence of latent, replication-competent HIV-1 proviruses in resting CD4+ T cells, and other cellular reservoirs, represents a major barrier to a cure. This reservoir is impervious to the immune system and to antiretroviral therapy, but has the potential to produce infectious rebound virus if antiretroviral therapy is interrupted. There are multiple ongoing efforts to identify and/or develop novel therapeutic strategies to eliminate or silence this latent reservoir of HIV-1 infection. One of these strategies is termed “block and lock”. The “block” refers to a therapeutic agent’s capacity to inhibit (or “block”) transcription of HIV-1 proviruses, while the “lock” refers to its capacity to induce permanent silencing of the proviruses, typically via repressive epigenetic modifications. The “block and lock” approach elicits a functional, rather than sterilizing, cure for HIV-1 infection. This review article focuses on therapeutic approaches (i.e., small molecules, nucleic acids and recombinant proteins) that have been identified to block and, in some cases, lock HIV-1 in the latent state. We also touch on critical research that needs to be accomplished to advance this approach into humans.
HIV-1 is a single-stranded positive-sense enveloped RNA retrovirus that causes acquired immunodeficiency syndrome (AIDS), a condition affecting ~ 36 million people worldwide. Antiretroviral therapy (ART) effectively restricts virus replication, and people living with HIV-1 infection on ART lead relatively healthy lives, as virus replication is suppressed to under the limit of detection of clinically used assays (currently, 20 HIV-1 RNA copies/ml of plasma). ART, however, is not curative and low-levels of persistent viremia can be detected by ultra-sensitive assays in the majority of infected individuals (1). Though the source of this viremia is likely to be variable and multi-faceted across each individual, in virologically suppressed individuals it is unclear if there is ongoing viral replication (2) and most replication-competent HIV-1 proviral DNA is found in resting CD4+ T cells (3). Other cell types such as hematopoietic stem cells, macrophages, and monocytes have been shown to possess proviral DNA, though they are not considered a major constituent of the reservoir (4–6). The reservoir of latent HIV-1 that resides in resting CD4+ T cells is thought to constitute the major hurdle to a HIV-1 cure (3) since it is unrecognized by the immune system, unaffected by ART, may produce infectious virus particles, and can contribute to either ongoing viremia during ART or HIV-1 viral rebound if treatment is paused.
Therapeutic approaches to eradicate the reservoir of latent HIV-1 infection in resting CD4+ T cells include (1): transplantation of hematopoietic stem cells engineered to lack the viral co-receptor CCR5 (2); gene editing to cleave the HIV-1 genome; and (3) a “shock and kill” approach that reverses latency thus rendering the infected cells vulnerable to immune clearance mechanisms, including therapeutic or immunomodulatory enhancement strategies (7–10). There are, however, limitations associated with each of these approaches, including scalability (particularly for hematopoietic stem cell treatment), the ability to deliver gene editing vehicles to all cells constituting the reservoir in vivo, and difficulties associated with the viral latency reversal in all, and not just a subset, of infected cells. As an alternative, the “block and lock” strategy involves long-term silencing of HIV-1 proviruses to prevent viral rebound following therapy cessation (11–13). The “block” refers to therapeutic agents, or latency promoting agents (LPAs), that inhibit (or “block”) the transcription of latent HIV-1 proviruses. LPAs can specifically target viral proteins (i.e., Tat or integrase) and, in addition to their “blocking” phenotype, they may also elicit antiviral activity (i.e., inhibit ongoing HIV-1 replication). Alternatively, LPAs could target host proteins and elicit only a “blocking” phenotype. The “lock” refers to the agent’s ability to induce permanent silencing of HIV-1 proviruses, typically via repressive epigenetic modifications. This approach elicits a functional, rather than sterilizing, cure. In this review, we discuss recent reports that encompass the block and lock strategy.
The gene expression of HIV-1 is regulated by viral proteins and host factors. The HIV-1-encoded regulatory protein, trans-activator of transcription (Tat), vastly enhances viral transcription efficiency (14). Tat binds to the trans-activation response (TAR) RNA region that is situated at the 5′ end of HIV-1 transcripts and recruits the RNA polymerase II (Pol) II positive transcription elongation factor p-TEFb (15–17). p-TEFb is a cyclin dependent kinase that consists of cyclin-dependent kinase 9 (CDK9) and cyclin T1. In metabolically active cells, CDK9 and cyclin T1 are secluded in the 7SK ribonucleoprotein (RNP) RNA-protein complex. CDK9 and cyclin T1 are liberated from the 7SK RNP and bind to TAR RNA in complex with Tat and organize the assembly of a super elongation complex (SEC) along with several additional proteins. p-TEFb phosphorylates the 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole sensitivity inducing factor (DSIF) and negative elongation factor (NELF), and importantly the carboxyl terminal domain of the large subunit of RNA Pol II which promotes the transition into the productive synthesis of mRNAs through transcriptional elongation. Of note, CDK9 and cyclin T1 function is decreased in resting CD4+ T cells that harbor HIV-1 proviral DNA. As such therapeutic strategies that further negatively regulate Tat and/or p-TEFb activity would act as potent block and lock agents. HIV-1 transcription is mediated by the 5’-long terminal repeat (LTR) and is often restricted in vivo. Specifically, the binding sites in the LTR for several transcription factors includes nuclear factor kappa B (NF-κB), nuclear factor of activated T cell (NFAT), signal transducer and activator of transcription 5 (STAT5), activator protein 1 (AP-1) and Sp1. In resting CD4+ T cells, many of these viral transcription factors are sequestered, thereby fostering HIV-1 latency (18). Three precisely positioned nucleosomes within the 5′-LTR, namely Nuc-0, Nuc-1, and Nuc-2, also regulate HIV-1 transcription. In particular,the repressive Nuc-1 is positioned immediately downstream of the transcription start site (TSS) and represses viral transcription.
HIV-1 latency is established and regulated through a multitude of host and viral epigenetic and genetic mechanisms. Active viral transcription is mediated by Tat and the multiple activating co-factor complexes, whereas viral latency is promoted by epigenetic regulators that promote nucleosome occupancy at the 5’ HIV-1 LTR promoter region. The class I histone deacetylases (HDACs), HDAC-1, -2 and -3, play a major role in the repressive heterochromatin environment that enforces HIV-1 latency (19–22). Following T cell activation, histone acetylation surrounding Nuc-1 increases significantly, concomitant with removal of the HDACs. Many other histone-modifying enzymes are recruited to the 5’-LTR by host cell transcription factors as well as Tat, of which may also be subject to post-translational modification by various factors. The mammalian SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex, the ATP-dependent BRG1/BRM associated factor (BAF), also contributes to HIV-1 transcriptional repression (23). In latent cells containing proviral DNA, BAF complexes promote nucleosome density immediately downstream of the HIV-1 TSS. The polybromo-associated BAF (PBAF) complex, which is closely related to BAF and possess several of the same subunits, substitutes BAF and induces removal of the repressive Nuc-1, leading to HIV-1 transcription activation and reversing latency. All of the viral proteins and host factors involved in the foundation and maintenance of HIV-1 latency represent viable targets for the development of block and lock cure strategies.
The preservation of cell identity as well as cellular lineage commitment relies on the combination of many factors including signaling pathways, transcription factors, and the genetic/epigenetic machinery. These regulatory factors have many intersections and they modulate the pattern of gene expression. Thus, it is not surprising that these cellular signaling pathways also play a critical role in the conservation of proviral latency and, as described below, several studies have reported that inhibition or activation of key proteins in these pathways can elicit a block and lock effect.
Below, we highlight several small molecules that have been reported to inhibit the reversal of HIV-1 latency and induce a block and lock phenotype. The structures of these small molecules are illustrated in Figure 1. Figure 2 provides a graphical illustration of the molecular mechanisms involved in suppressing viral latency.
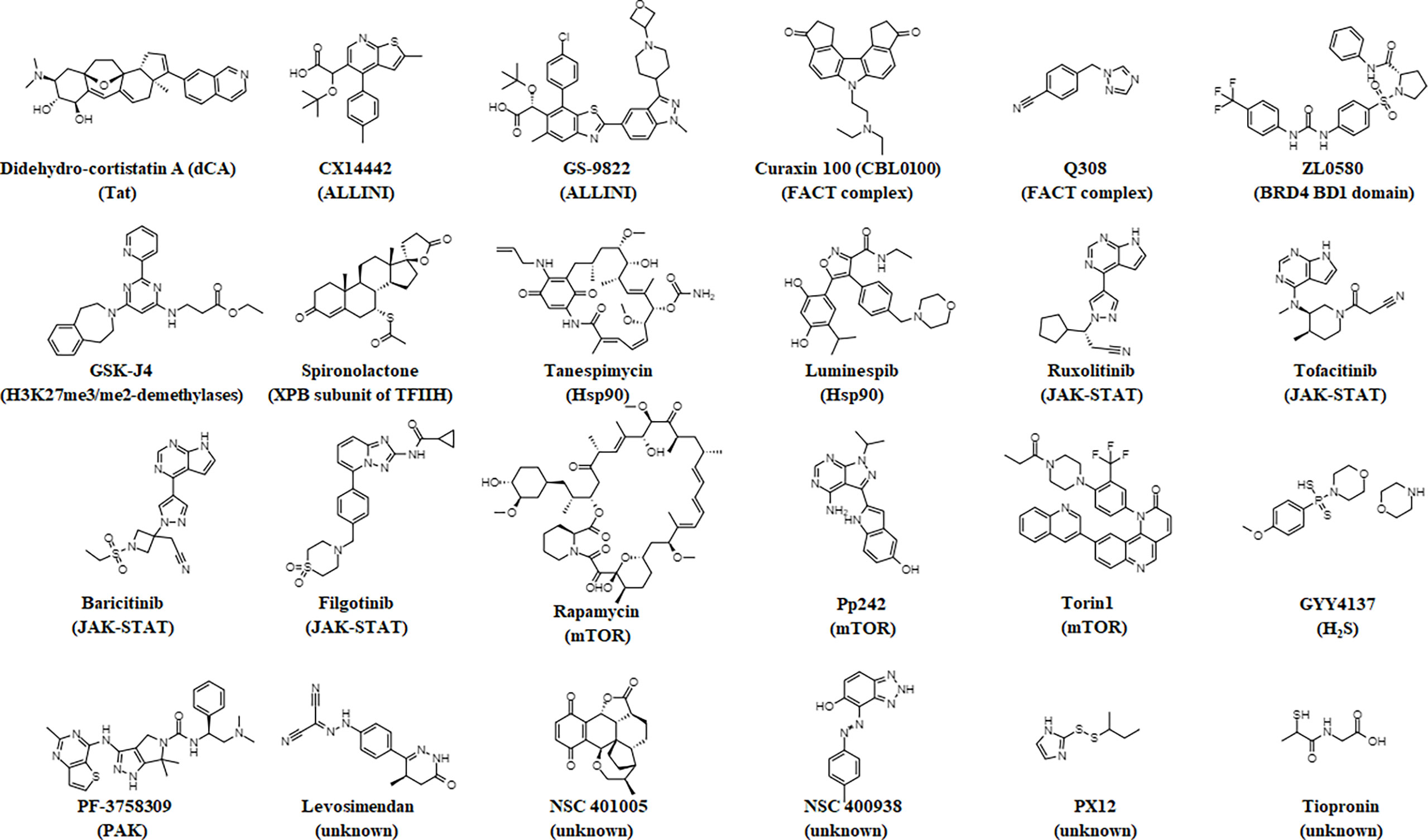
Figure 1 Chemical structures of small molecules that elicit a block and lock phenotype. The molecule’s target is noted in parentheses.
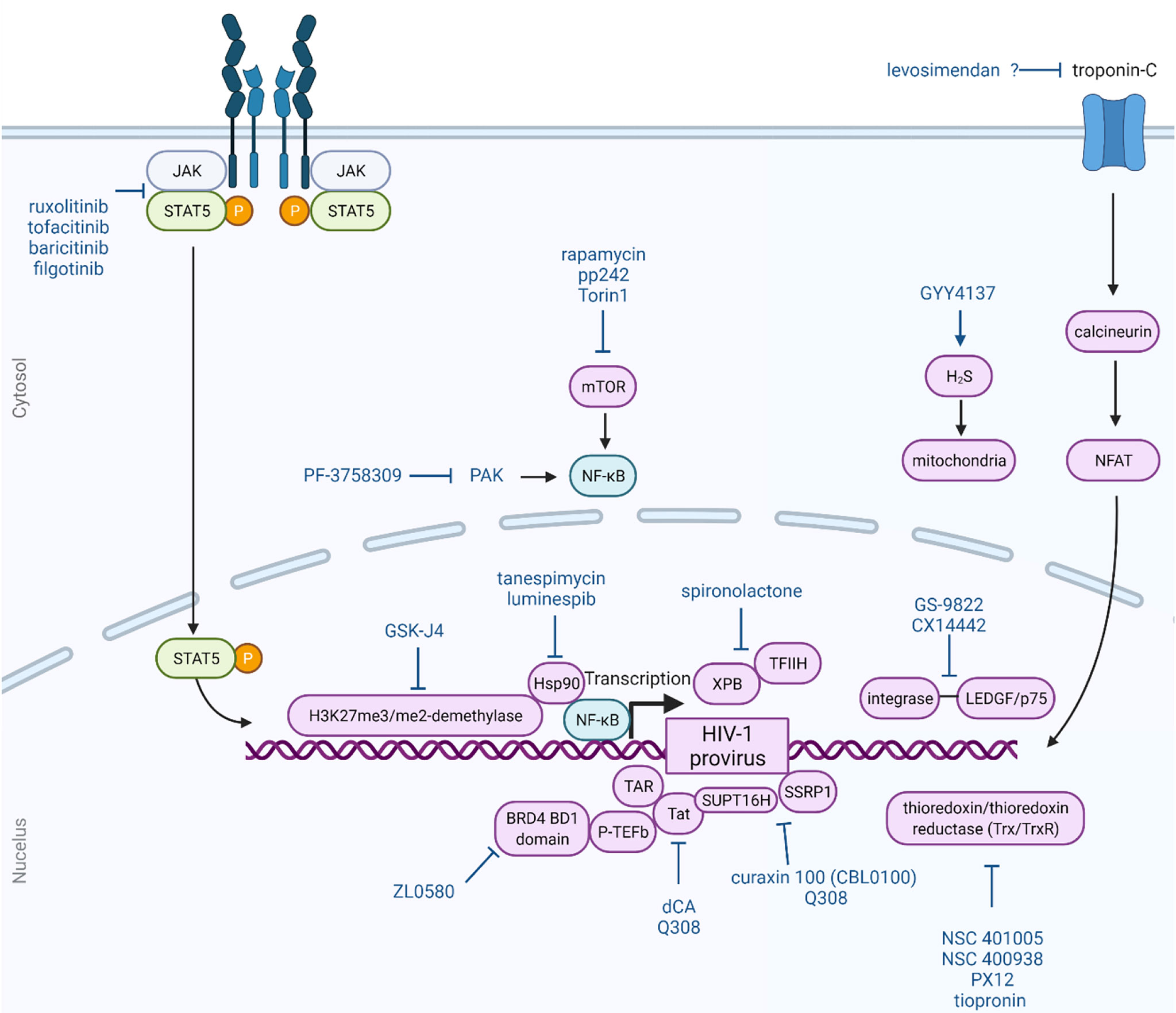
Figure 2 Block and lock approach as a functional cure for HIV-1 infection. The block and lock approach involves long-term durable silencing of viral gene expression. Small molecule can elicit this phenotype by affecting multiple different host and viral proteins and pathways, as illustrated here. This Figure was created using BioRender.com.
dCA is a synthetic analog derived from cortistatin A, which is naturally produced steroidal alkaloid from Corticium simplex. dCA has been shown to bind to the basic domain of Tat and is a potent inhibitor of viral transcription (24). dCA blocks HIV-1 reactivation from latency in primary CD4+ T cells from HIV-1 patients and in several other cellular models of latency (25). Importantly, dCA mediated inhibition of Tat promotes a repressive heterochromatin environment at the HIV-1 promoter, marked by decreased histone H3 lysine 27 (H3K27) acetylation levels at Nuc-1 and limited PBAF recruitment (26). In an in vivo murine model of HIV-1 infection, dCA reduced the viral mRNA burden in mouse tissues, and both reduced and delayed HIV-1 rebound following the cessation of ART (27). HIV-1 resistance to dCA is associated with mutations in the LTR that augment the basal levels of HIV-1 transcription by 10- to 30-fold, and with mutations in the viral accessory proteins Nef and Vpr that increase NF-κB nuclear translocation, which promotes transcription from the HIV-1 promoter (28). Collectively, these studies demonstrate a proof-of-concept that latency promoting agents can be used to suppress ongoing transcriptional events under ART, and can significantly delay and/or reduce viral rebound levels in upon treatment interruption.
HIV-1 preferentially integrates into actively transcribed gene-dense regions. This selectivity is driven by direct interaction between HIV-1 integrase and the lens epithelium–derived growth factor (LEDGF/p75), a host protein that acts as a transcriptional coactivator, binding various proteins to active transcription units through recognition of the trimethylated histone H3 at lysine 36 (H3K36me3) (29). ALLINIs, also referred to as LEDGINs, non-catalytic-site integrase inhibitors (NCINI) or multimeric integrase inhibitors (MINI), bind to HIV-1 integrase at the integrase- LEDGF/p75 interaction site, perturb the viral-host protein interaction, and inhibit HIV-1 integration (30, 31). As expected by an HIV-1 integrase inhibitor, the ALLINI CX14442 was found to reduce the number of integrated proviruses. However, CX14442 also retargeted the site of residual HIV-1 integration away from active chromatin (32). Specifically, proviruses were integrating into less-gene-dense regions with a more repressive epigenetic environment, and were shifted away from H3K36me3, which lowered HIV-1 transcription. Similar results were noted with GS-9822, also an ALLINI, in that residual provirus was both more latent and refractory to reactivation, and were integrated away from genes and gene-dense chromatin, resulting in a more repressive heterochromatin environment (33). Collectively, these studies suggest that if HIV-1-infected individuals are treated early with ART regimens that include an ALLINI, the replication-competent viral reservoir in resting CD4+ T cells will be transcriptionally impaired, thus mimicking a “locked” phenotype.
The FACT proteins suppressor of Ty 16 homolog (SUPT16H) and structure specific recognition protein 1 (SSRP1) represses HIV-1 transcription and induce viral latency (34). Depletion of SUPT16H or SSRP1 protein in cell line models of HIV-1 latency, increases transcriptional initiation and transcriptional elongation and is able to reverse latent HIV-1. Similar observations were reported with a primary CD4+ T cell model of HIV-1 latency. Curaxin 100 (CBL0100) is a small molecule that targets the FACT complex and was found to inhibit both HIV-1 reactivation and replication in in vitro and ex vivo models of virus latency (35). From a mechanistic perspective, CBL0100 targeted HIV-1 transcriptional elongation and decreased the occupancy of FACT and Pol II at the HIV-1 LTR promoter region. Recently, another small molecule, Q308, was also found to inhibit HIV-1 gene transcription mediated by Tat and selectively decreases FACT expression levels (36).
A family of proteins called the bromodomain and extraterminal (BET) proteins are host epigenetic factors marked by 2 N-terminal bromodomains that recognize acetylated histones in chromatin. BRD4, a member of this family, plays a crucial role in HIV-1 transcriptional regulation (37). Specifically, BRD4 competes with Tat for host p-TEFb/CDK9 which results in HIV-1 transcription elongation. The pan-BET inhibitor JQ1, which non-selectively targets both N-terminal bromodomains of all BET proteins, disables the interaction between BRD4 and Tat and reactivates latent HIV-1 infection (38–40). Niu et al. identified ZL0580, a small molecule, that selectively binds to BD1 domain of BRD4. ZL0580 enforced HIV-1 latency in both in vitro and ex vivo cell models of latency by preventing the transactivation of Tat as well as transcription elongation and also induced repressive heterochromatin structure at the HIV-1 LTR promoter (41). Combinatorial treatment of CD4+ T cells derived from aviremic HIV-infected individuals with both ZL0580 and ART demonstrated that ZL0580 promoted suppression of HIV-1and led to a delay in viral rebound after interruption of ART.
H3K27 tri-methylation (H3K27me3) and DNA methylation are correlated with suppression of viral reactivation at the HIV-1 LTR, whereas H3K4 tri-methylation (H3K4me3) is associated with HIV-1 expression (42). Ethyl 3-((6-(4,5-dihydro-1H-benzo[d]azepin-3(2H)-yl)-2-(pyridin-2-yl)pyrimidin-4-yl)amino)propanoate (GSK-J4) is a strong inhibitor of UTX/KDM6A and JMJD3/KDM6B—both of which are the H3K27me3/me2-demethylases. In HIV-1 latently infected cells from patients, GSK-J4 was able to inhibit latent HIV-1 reactivation and promoted methylation of DNAat certain the 5’-LTR region of latent HIV-1 by facilitating engagement of DNA methyltransferase 3 alpha to HIV-1 LTR (43). However, silencing of HIV-1 through epigenetic mechanisms required continuous addition of GSK-J4 and the effect was lost quickly after drug removal. DNA methylation additionally was shortly lost after drug removal, which suggests active and fast demethylation of the DNA in the HIV-1 promoter region. As such, inhibitors of lysine methylases could be used in therapeutic approaches to induce the “block” but not “lock” phenotype.
The transcription factor TFIIH enhances transcription initiation by inducing the DNA strands to open up near the transcription start site and phosphorylating and activating the carboxyl terminal domain of RNA Pol II. The aldosterone, spironolactone, mediates the proteasomal degradation of the XPB subunit of TFIIH as well as represses acute HIV-1 infection in vitro (44). While spironolactone readily inhibits HIV-1 transcription by decreasing Pol II recruitment to the HIV-1 genome, perpetual pretreatment with the drug was not able to maintain sustained epigenetic suppression of HIV-1 upon interruption of the drug, since the virus is reactivated when XPB reemerges. However, spironolactone by itself without ART was found to maintain transcriptional silencing of HIV-1.
Heat shock induces overexpression of Hsp90 and leads to the activation of inducible cellular transcription factors. Specifically, heat shock facilitates HIV-1 transcription through activation of Hsp90 activity, which induces HIV-1-specific essential host cell transcription factors (NF-κB, NFAT, and STAT5), while inhibition of Hsp90 significantly reduces their gene expression (45). Of note, the Hsp90 inhibitors tanespimycin (17-(allylamino)-17-demethoxygeldanamycin) and luminespib (5-[2,4-dihydroxy-5-(propan-2-yl)phenyl]-N-ethyl-4-{4-[(morpholin-4-yl)methyl]phenyl}-1,2-oxazole-3-carboxamide) were found to prevent HIV-1 expression for up to 11 weeks in HIV-1-infected humanized NOD scid IL-2Rγ−/− bone marrow-liver-thymus mice after treatment interruption (46). These data suggest that supplementing ART with Hsp90 inhibitors could possibly prevent rebound viremia from persistent HIV reservoirs.
The replication-competent latent HIV-1 reservoir resides within memory CD4+ T cells — central, transitional, and effector — all of which must be exposed to the γ-C receptor cytokines (IL-2, IL-7 and IL-15) for their long-term persistence (47). STAT-5 phosphorylation (pSTAT5) is induced following the exposure of the respective γ-C receptor cytokines cytokines with their respective receptors which facilitates anti-apoptotic signaling and increased T cell division. Given that there are several binding sites for pSTAT5 within the HIV-1 LTR, IL-2, IL-7 and IL-15 have been shown to enhance viral expression from infected cells. Ruxolitinib and tofacitinib are two FDA-approved Jak inhibitors that were both shown to block reactivation of latent proviruses in a primary CD4+ T cell model of HIV-1 latency (48). pSTAT5 has also been shown to strongly correlate with high levels of integrated viral DNA, and Jak inhibitors can decrease the amount of CD4+ T cells harboring integrated viral DNA (49). Other Jak inhibitors, including baricitinib and filgotinib, also inhibit virus production from latently infected cells, STAT5 phosphorylation and homeostatic proliferation (50, 51). Recently, an AIDS Clinical Trial Group multi-site Phase 2a study (A5336) has evaluated ruxolitinib, which has shown remarkable efficacy and safety in virally suppressed patients living with HIV-1, including a notable reduction in key markers associated with viral persistence (52). Collectively, these studies suggest that Jak inhibitors are a therapeutic strategy to block key events of T cell activation that modulate HIV-1 persistence.
A genome-wide short hairpin (sh) RNA screen identified that the mTOR complex regulates HIV-1 latency (53). Knockdown of the mTOR complex or small molecule inhibition of mTOR activity suppressed latency reversal in several HIV-1 latency models and in patient cells from HIV-infected individuals. This inhibition was due in part to diminished phosphorylation of CDK9. While this study suggests that inhibition of mTOR may have therapeutic potential, other studies have reported that inhibition of mTOR had minimal effects on HIV-1 latency reversal (54, 55).
Reactivation of latent HIV-1 is associated with decreased expression of the H2S producing enzyme cystathionine-γ-lyase and downregulation of endogenous H2S (56). Chemical complementation of cystathionine-γ-lyase activity with a slow-releasing H2S donor, morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate (GYY4137), was found to suppress HIV-1 gene expression by promoting the Keap1-Nrf2 pathway, blocking NF-κB, and engaging the epigenetic silencer, Yin Yang 1 (YY1), to the HIV-1 promoter (56). Combinatorial treatment of GYY4137 and ART prevented viral rebound in latently infected CD4+ T cells from ART-suppressed patients and demonstrated no adverse effect on proviral content or CD4+ T cell subsets, showing that the reduction in viral rebound is due to inhibition of transcription and not due to a reduction of a select group of infected cells.
Our laboratory utilized the 24ST1NLESG cell line model of HIV-1 latency to high-throughput screen a structurally diverse library cell permeable, medically active kinase inhibitors, which targets a comprehensive array of signaling pathways, to identify small molecule inhibitors of HIV-1 latency reversal (55). The screen was carried with or without co-treatment of three latency-reversing agents—prostratin, JQ-1, and panobinostat— each of which having distinct mechanisms of action. We found 12 kinase inhibitors that inhibited HIV-1 latency reversal regardless of the latency reversal agent used in the screen. Of these, PF-3758309, a pan-PAK inhibitor, was identified to be the most potent. The 50% inhibitory concentrations (IC50) in the 24ST1NLESG cells ranged from 0.1 to 1 nM (selectivity indices, >3,300), and for PF-3758309 was also shown to inhibit HIV-1 reversal from latency in CD4+ T cells isolated from HIV-1-infected donors. More recently, we have shown that PAK1 and PAK2 are abundantly expressed in resting CD4+ T cells, and that knockdown of PAK1 and PAK2 in 24ST1NLESG cells inhibits HIV-1 reactivation, mostly likely by decreasing NF-κB expression (unpublished data).
Levosimendan, an FDA-approved drug to currently treat heart failure, was found as an inhibitor of HIV-1 latency reversal via screening of a compound library containing FDA-approved molecules (57). Levosimendan was found to exhibit potent inhibition of HIV-1 latency reversal in several cell lines of latency by affecting the HIV-1 LTR-Tat transcriptional axis, and in primary CD4+ T cells also inhibited both HIV-1 latency reversal and acute replication.
Lu et al. used time-lapse fluorescence microscopy to measure HIV-1 gene-expression dynamics and identify latency promoting agents that would be otherwise disregarded when screening solely for mean gene expression (58). Following a screen of 1,806 compounds they identified NSC 401005, NSC 400938, PX12, and tiopronin, all of which are functionally and structurally similar to inhibitors of thioredoxin reductase, an enzyme whose function is to maintain redox balance in host cells. These molecules reduced reversal of HIV-1 latency in both Jurkat and primary cell models.
The siRNA molecule siPromA targets the multiple NF-κB sites in the HIV-1 LTR promoter to elicit silencing of viral transcription through repressive epigenetic marks (59). si143, which also targets the HIV-1 5’-LTR, similarly induces transcriptional gene silencing (59). Further studies showed that cells expressing PromA, 143, or both, demonstrated significant resistance to viral reactivation that was correlated with significant changes in the levels of H3K27me3, Argonaute RISC Component 1 (AGO1) and HDAC1 in the LTR, which are associated with reduced HIV-1 reactivation (60). LTR362 is an RNA sequence that also targets the multiple NF-κB binding sites in the HIV-1 LTR and has overlap with the siPromA sequence (61). In a humanized mouse model of HIV-1 infection, LTR362 in combination with a HIV-1 glycoprotein 120 (gp120) aptamer and two RNAs targeting the viral mRNAs of Tat and rev inhibited viral reactivation and raised CD4+ T cell counts relative to controls. However, LTR362 did not appear to facilitate epigenetic silencing of the 5′-LTR, with inhibition against HIV-1 infection being related to the post-transcriptional regulation of rev and Tat.
Saayman et al., identified a lncRNA that induced HIV-1 gene silencing in vitro through the recruitment of a chromatin-remodeling complex, involving DNA methyltransferase 3 alpha (DNMT3a), enhancer of zeste homolog 2 (EZH2), and HDAC1 to the HIV-1 LTR promoter (62). Li et al., identified a lncRNA termed NRON that suppressed virus transcription by promoting Tat degradation (63).
Olson et al., sought to silence HIV-1 proviruses by constructing a nuclease-deficient disabled Cas9 (dCas9) linked to the transcriptional repressor domain derived from Kruppel-associated box (KRAB) (64). They found that dCas9-KRAB along with unique guide RNAs (gRNAs) inhibit HIV-1 transcription and expression of latent HIV-1 proviruses, and that this suppression is associated with chromatin changes, including reduced H3 histone acetylation and increased histone H3 lysine 9 trimethylation, epigenetic marks that are correlated with suppression of transcription. Collectively, these studies suggest that genomic approaches could form part of a “toolkit” for block-and-lock strategies.
Yamayoshi et al., developed oligonucleotides that mimic the function of 7SK and showed them to be potent inhibitors of HIV-1 LTR specific transcription (65). Interestingly, the inhibitory effects were shown to be increased by co-transfection with plasmids expressing Tat. Taken together, these data suggest that 7SK mimics may have a potential to be a therapeutic strategy for HIV-1 block and lock strategies.
NullBasic is a 101 amino acid transdominant Tat mutant with an modified basic domain in amino acid positions 49-57, that includes the TAR-binding region, which inhibits viral gene expression via competition with HIV-1 Tat, but also through blocking Rev-mediated transport of HIV-1 mRNA (66). Retroviral vectors that expressed NullBasic were transduced into CD4+ T cells and demonstrated inhibition of HIV-1 transcription and replication via reduced Pol II occupancy at the LTR region and diminished H3K9 acetylation (67). NullBasic retroviral transduction of primary human CD4+ T cells as well as transplantation in a NOD scid gamma mouse model showed viral RNA was undetectable in plasma samples for up to two weeks post-infection and markedly decreased HIV-1 RNA levels in CD4+ T cells derived from tissue (68).
As described above (2. Viral and cellular factors that regulate HIV-1 transcription), Tat binds to the TAR RNA element and facilitates recruitment of the Pol II positive transcription elongation factor p-TEFb. In cells, p-TEFb is held by 7SK snRNP, and Tat can remove p-TEFb from the 7SK snRNP, where it is sequestered and rendered inactive by HEXIM1. Leoz et al., reported that a fusion protein that combined the Tat and HEXIM1 functional domains, strongly reduced HIV-1 gene expression, by competing with the interaction between Tat, TAR and P-TEFb (69). In lymphocyte T cell line models of HIV latency, the fusion protein reduced the spread of infection as well as HIV-1, with minor impacts on cellular metabolism and transcription. This study is a proof-of-concept of innovative approaches to disrupt HIV transcription by competing for RNA-protein interactions by utilizing peptide.
V2O5 nanosheets are similar to natural glutathione peroxidase activity in that they reduce reactive oxygen species correlated with HIV-1 infection (70). V2O5 nanosheets catalyze reactive oxygen species neutralization in HIV-1-infected cells and inhibit HIV-1 reactivation and replication, through modulation of the expression of pathways regulating redox balance, HIV-1 transactivation (e.g., NF-κB), inflammation, and apoptosis. V2O5 nanosheets also block HIV-1 reactivation upon treatment with prostratin of latently infected CD4+ T cells from HIV-infected individuals on ART. V2O5 could be used with other agents to enforce HIV-1 latency and elicit the “block and lock” phenotype.
As described above, numerous diverse approaches have been identified to elicit a “block and lock” phenotype to inhibit reactivation of latent HIV-1 infection in CD4+ T cells. These approaches include small molecules, nucleic acids, and recombinant proteins. Currently, it is unknown whether any single approach will deliver sufficiently long-term silencing of latent proviruses to facilitate a functional HIV-1 cure. As such combination studies using two or more approaches should be rigorously explored in vitro and in appropriate animal models. While some of the therapeutic agents discussed in this application have been tested in humanized mice models of HIV-1 infection, none have progressed to clinical trials, with the exception of the Jak inhibitor ruxolitinib. However, it is not known whether ruxolitinib treatment resulted in block and lock phenotype in the clinical trial, as this measurement was not included as an end point in the study. A key issue for many of the therapeutic agents described in this review relates to target specificity as well as their impact on normal cell function. For example, small molecules that target epigenetic proteins or cell signaling pathways will fundamentally alter the functioning of other cellular processes which, in the long-term, may promote toxicity or adverse events in humans. In this regard, molecules that specifically target viral proteins (e.g. dCA or ALLINIs) may have a therapeutic advantage. To this end, to effectively develop a “block and lock” functional cure strategy the field requires the following advancements; (i) the discovery and development of potent molecules with appropriate safety and pharmacokinetic/pharmacodynamic properties such that they can be used clinically; (ii) the development of delivery systems for these molecules that specifically target latent reservoir cells; and (iii) appropriate clinical trials to assess efficacy and safety in HIV-1-infected individuals.
BV and NS-C co-wrote the review article. All authors contributed to the article and approved the submitted version.
This work was supported by a grant (R21AI57392-01) from the National Institutes of Health in the USA to NS-C.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jacobs JL, Halvas EK, Tosiano MA, Mellors JW. Persistent HIV-1 Viremia on Antiretroviral Therapy: Measurement and Mechanisms. Front Microbiol (2019) 10:2383. doi: 10.3389/fmicb.2019.02383
2. Bale MJ, Kearney MF. Review: HIV-1 Phylogeny During Suppressive Antiretroviral Therapy. Curr Opin HIV AIDS (2019) 14(3):188–93. doi: 10.1097/COH.0000000000000535
3. Siliciano JD, Siliciano RF. In Vivo Dynamics of the Latent Reservoir for HIV-1: New Insights and Implications for Cure. Annu Rev Pathol (2022) 17:271–94. doi: 10.1146/annurev-pathol-050520-112001
4. Kruize Z, Kootstra NA. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front Microbiol (2019) 10:2828. doi: 10.3389/fmicb.2019.02828
5. Mitchell BI, Laws EI, Ndhlovu LC. Impact of Myeloid Reservoirs in HIV Cure Trials. Curr HIV/AIDS Rep (2019) 16(2):129–40. doi: 10.1007/s11904-019-00438-5
6. Sebastian NT, Collins KL. Targeting HIV Latency: Resting Memory T Cells, Hematopoietic Progenitor Cells and Future Directions. Expert Rev Anti Infect Ther (2014) 12(10):1187–201. doi: 10.1586/14787210.2014.956094
7. Lee PH, Keller MD, Hanley PJ, Bollard CM. Virus-Specific T Cell Therapies for HIV: Lessons Learned From Hematopoietic Stem Cell Transplantation. Front Cell Infect Microbiol (2020) 10:298. doi: 10.3389/fcimb.2020.00298
8. Khanal S, Schank M, El Gazzar M, Moorman JP, Yao ZQ. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells. (2021) 10(2):475. doi: 10.3390/cells10020475
9. Karuppusamy KV, Babu P, Thangavel S. The Strategies and Challenges of CCR5 Gene Editing in Hematopoietic Stem and Progenitor Cells for the Treatment of HIV. Stem Cell Rev Rep (2021) 17(5):1607–18. doi: 10.1007/s12015-021-10145-7
10. Xun J, Zhang X, Guo S, Lu H, Chen J. Editing Out HIV: Application of Gene Editing Technology to Achieve Functional Cure. Retrovirol. (2021) 18(1):39. doi: 10.1186/s12977-021-00581-1
11. Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses. (2020) 12(1):84. doi: 10.3390/v12010084
12. Ahlenstiel CL, Symonds G, Kent SJ, Kelleher AD. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front Cell Infect Microbiol (2020) 10:424. doi: 10.3389/fcimb.2020.00424
13. Moranguinho I, Valente ST. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses. (2020) 12(12):1443. doi: 10.3390/v12121443
14. Rice AP. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr Pharm Des (2017) 23(28):4098–102. doi: 10.2174/1381612823666170704130635
15. Asamitsu K, Okamoto T. The Tat/P-TEFb Protein-Protein Interaction Determining Transcriptional Activation of HIV. Curr Pharm Des (2017) 23(28):4091–7. doi: 10.2174/1381612823666170710164148
17. Spector C, Mele AR, Wigdahl B, Nonnemacher MR. Genetic Variation and Function of the HIV-1 Tat Protein. Med Microbiol Immunol (2019) 208(2):131–69. doi: 10.1007/s00430-019-00583-z
18. Mbonye U, Karn J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu Rev Virol (2017) 4(1):261–85. doi: 10.1146/annurev-virology-101416-041646
19. Zaikos TD, Painter MM, Sebastian Kettinger NT, Terry VH, Collins KL. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal While Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J Virol (2018) 92(6):e02110–17. doi: 10.1128/JVI.02110-17
20. Barton KM, Archin NM, Keedy KS, Espeseth AS, Zhang YL, Gale J, et al. Selective HDAC Inhibition for the Disruption of Latent HIV-1 Infection. PloS One (2014) 9(8):e102684. doi: 10.1371/journal.pone.0102684
21. Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A Limited Group of Class I Histone Deacetylases Acts to Repress Human Immunodeficiency Virus Type 1 Expression. J Virol (2009) 3(10):4749–56. doi: 10.1128/JVI.02585-08
22. Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of Histone Deacetylases: Correlation Between Isoform Specificity and Reactivation of HIV Type 1 (HIV-1) From Latently Infected Cells. J Biol Chem (2011) 286(25):22211–8. doi: 10.1074/jbc.M110.180224
23. Mahmoudi T. The BAF Complex and HIV Latency. Transcription. (2012) 3(4):171–6. doi: 10.4161/trns.20541
24. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An Analog of the Natural Steroidal Alkaloid Cortistatin A Potently Suppresses Tat-Dependent HIV Transcription. Cell Host Microbe (2012) 12(1):97–108. doi: 10.1016/j.chom.2012.05.016
25. Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation From Latency. mBio. (2015) 6(4):e00465. doi: 10.1128/mBio.00465-15
26. Li C, Mousseau G, Valente ST. Tat Inhibition by Didehydro-Cortistatin A Promotes Heterochromatin Formation at the HIV-1 Long Terminal Repeat. Epigenet Chromatin (2019) 12(1):23. doi: 10.1186/s13072-019-0267-8
27. Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a "Block-And-Lock" Strategy for HIV-1 Treatment. Cell Rep (2017) 21(3):600–11. doi: 10.1016/j.celrep.2017.09.080
28. Mousseau G, Aneja R, Clementz MA, Mediouni S, Lima NS, Haregot A, et al. Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription. mBio. (2019) 10(4):e01750–18. doi: 10.1128/mBio.01750-18
29. Lesbats P, Engelman AN, Cherepanov P. Retroviral DNA Integration. Chem Rev (2016) 116(20):12730–57. doi: 10.1021/acs.chemrev.6b00125
30. Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, et al. Rational Design of Small-Molecule Inhibitors of the LEDGF/p75-Integrase Interaction and HIV Replication. Nat Chem Biol (2010) 6(6):442–8. doi: 10.1038/nchembio.370
31. Engelman A, Kessl JJ, Kvaratskhelia M. Allosteric Inhibition of HIV-1 Integrase Activity. Curr Opin Chem Biol (2013) 17(3):339–45. doi: 10.1016/j.cbpa.2013.04.010
32. Vranckx LS, Demeulemeester J, Saleh S, Boll A, Vansant G, Schrijvers R, et al. LEDGIN-Mediated Inhibition of Integrase-LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMed. (2016) 8:248–64. doi: 10.1016/j.ebiom.2016.04.039
33. Bruggemans A, Vansant G, Balakrishnan M, Mitchell ML, Cai R, Christ F, et al. GS-9822, a Preclinical LEDGIN Candidate, Displays a Block-and-Lock Phenotype in Cell Culture. Antimicrob Agents Chemother (2021) 65(5):e02328–20. doi: 10.1128/AAC.02328-20
34. Huang H, Santoso N, Power D, Simpson S, Dieringer M, Miao H, et al. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J Biol Chem (2015) 290(45):27297–310. doi: 10.1074/jbc.M115.652339
35. Jean MJ, Hayashi T, Huang H, Brennan J, Simpson S, Purmal A, et al. Curaxin CBL0100 Blocks HIV-1 Replication and Reactivation Through Inhibition of Viral Transcriptional Elongation. Front Microbiol (2017) 8:2007. doi: 10.3389/fmicb.2017.02007
36. Zhou CL, Huang YF, Li YB, Liang TZ, Zheng TY, Chen P, et al. A New Small-Molecule Compound, Q308, Silences Latent HIV-1 Provirus by Suppressing Tat- and FACT-Mediated Transcription. Antimicrob Agents Chemother (2021) 65(12):e0047021. doi: 10.1128/AAC.00470-21
37. Alamer E, Zhong C, Hajnik R, Soong L, Hu H. Modulation of BRD4 in HIV Epigenetic Regulation: Implications for Finding an HIV Cure. Retrovirol. (2021) 18(1):3. doi: 10.1186/s12977-020-00547-9
38. Li Z, Guo J, Wu Y, Zhou Q. The BET Bromodomain Inhibitor JQ1 Activates HIV Latency Through Antagonizing Brd4 Inhibition of Tat-Transactivation. Nucleic Acids Res (2013) 41(1):277–87. doi: 10.1093/nar/gks976
39. Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, et al. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep (2012) 2(4):807–16. doi: 10.1016/j.celrep.2012.09.008
40. Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, et al. BET Bromodomain Inhibition as a Novel Strategy for Reactivation of HIV-1. J Leukoc Biol (2012) 92(6):1147–54. doi: 10.1189/jlb.0312165
41. Niu Q, Liu Z, Alamer E, Fan X, Chen H, Endsley J, et al. Structure-Guided Drug Design Identifies a BRD4-Selective Small Molecule That Suppresses HIV. J Clin Invest (2019) 129(8):3361–73. doi: 10.1172/JCI120633
42. Boehm D, Ott M. Host Methyltransferases and Demethylases: Potential New Epigenetic Targets for HIV Cure Strategies and Beyond. AIDS Res Hum Retroviruses (2017) 33(S1):S8–S22. doi: 10.1089/aid.2017.0180
43. Nguyen K, Dobrowolski C, Shukla M, Cho WK, Luttge B, Karn J. Inhibition of the H3K27 Demethylase UTX Enhances the Epigenetic Silencing of HIV Proviruses and Induces HIV-1 DNA Hypermethylation But Fails to Permanently Block HIV Reactivation. PloS Pathog (2021) 17(10):e1010014. doi: 10.1371/journal.ppat.1010014
44. Mori L, Jenike K, Yeh YJ, Lacombe B, Li C, Getzler A, et al. The XPB Subunit of the TFIIH Complex Plays a Critical Role in HIV-1 Transcription and XPB Inhibition by Spironolactone Prevents HIV-1 Reactivation From Latency. J Virol (2020) 95(4):e01247–20. doi: 10.1128/JVI.01247-20
45. Anderson I, Low JS, Weston S, Weinberger M, Zhyvoloup A, Labokha AA, et al. Heat Shock Protein 90 Controls HIV-1 Reactivation From Latency. Proc Natl Acad Sci USA (2014) 111(15):E1528–37. doi: 10.1073/pnas.1320178111
46. Joshi P, Maidji E, Stoddart CA. Inhibition of Heat Shock Protein 90 Prevents HIV Rebound. J Biol Chem (2016) 291(19):10332–46. doi: 10.1074/jbc.M116.717538
47. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV Reservoir Size and Persistence are Driven by T Cell Survival and Homeostatic Proliferation. Nat Med (2009) 15(8):893–900. doi: 10.1038/nm.1972
48. Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, Schinazi RF. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro. Antimicrob Agents Chemother (2014) 58(4):1977–86. doi: 10.1128/AAC.02496-13
49. Gavegnano C, Brehm JH, Dupuy FP, Talla A, Ribeiro SP, Kulpa DA, et al. Novel Mechanisms to Inhibit HIV Reservoir Seeding Using Jak Inhibitors. PloS Pathog (2017) 13(12):e1006740. doi: 10.1371/journal.ppat.1006740
50. de Armas LR, Gavegnano C, Pallikkuth S, Rinaldi S, Pan L, Battivelli E, et al. The Effect of JAK1/2 Inhibitors on HIV Reservoir Using Primary Lymphoid Cell Model of HIV Latency. Front Immunol (2021) 12:720697. doi: 10.3389/fimmu.2021.720697
51. Yeh YJ, Jenike KM, Calvi RM, Chiarella J, Hoh R, Deeks SG, et al. Filgotinib Suppresses HIV-1-Driven Gene Transcription by Inhibiting HIV-1 Splicing and T Cell Activation. J Clin Invest (2020) 130(9):4969–84. doi: 10.1172/JCI137371
52. Marconi VC, Moser C, Gavegnano C, Deeks SG, Lederman MM, Overton ET, et al. Randomized Trial of Ruxolitinib in Antiretroviral-Treated Adults With Human Immunodeficiency Virus. Clin Infect Dis (2022) 74(1):95–104. doi: 10.1093/cid/ciab212
53. Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe (2016) 20(6):785–97. doi: 10.1016/j.chom.2016.11.001
54. Martin AR, Pollack RA, Capoferri A, Ambinder RF, Durand CM, Siliciano RF. Rapamycin-Mediated mTOR Inhibition Uncouples HIV-1 Latency Reversal From Cytokine-Associated Toxicity. J Clin Invest (2017) 127(2):651–6. doi: 10.1172/JCI89552
55. Vargas B, Giacobbi NS, Sanyal A, Venkatachari NJ, Han F, Gupta P, et al. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob Agents Chemother (2019) 63(2):e01744–18. doi: 10.1128/AAC.01744-18
56. Pal VK, Agrawal R, Rakshit S, Shekar P, Murthy DTN, Vyakarnam A, et al. Hydrogen Sulfide Blocks HIV Rebound by Maintaining Mitochondrial Bioenergetics and Redox Homeostasis. Elife. (2021) 10:e68487. doi: 10.7554/eLife.68487
57. Hayashi T, Jean M, Huang H, Simpson S, Santoso NG, Zhu J. Screening of an FDA-Approved Compound Library Identifies Levosimendan as a Novel Anti-HIV-1 Agent That Inhibits Viral Transcription. Antiviral Res (2017) 146:76–85. doi: 10.1016/j.antiviral.2017.08.013
58. Lu Y, Bohn-Wippert K, Pazerunas PJ, Moy JM, Singh H, Dar RD. Screening for Gene Expression Fluctuations Reveals Latency-Promoting Agents of HIV. Proc Natl Acad Sci USA (2021) 118(11):e2012191118. doi: 10.1073/pnas.2012191118
59. Ahlenstiel C, Mendez C, Lim ST, Marks K, Turville S, Cooper DA, et al. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-Mediated Transcriptional Silencing. Mol Ther Nucleic Acids (2015) 4(10):e261. doi: 10.1038/mtna.2015.31
60. Méndez C, Ledger S, Petoumenos K, Ahlenstiel C, Kelleher AD. RNA-Induced Epigenetic Silencing Inhibits HIV-1 Reactivation From Latency. Retrovirol. (2018) 15(1):67. doi: 10.1186/s12977-018-0451-0
61. Zhou J, Lazar D, Li H, Xia X, Satheesan S, Charlins P, et al. Receptor-Targeted aptamer-siRNA Conjugate-Directed Transcriptional Regulation of HIV-1. Theranostics. (2018) 8(6):1575–90. doi: 10.7150/thno.23085
62. Saayman S, Ackley A, Turner AW, Famiglietti M, Bosque A, Clemson M, et al. An HIV-Encoded Antisense Long Noncoding RNA Epigenetically Regulates Viral Transcription. Mol Ther (2014) 22(6):1164–75. doi: 10.1038/mt.2014.29
63. Li J, Chen C, Ma X, Geng G, Liu B, Zhang Y, et al. Long Noncoding RNA NRON Contributes to HIV-1 Latency by Specifically Inducing Tat Protein Degradation. Nat Commun (2016) 7:11730. doi: 10.1038/ncomms11730
64. Olson A, Basukala B, Lee S, Gagne M, Wong WW, Henderson AJ. Targeted Chromatinization and Repression of HIV-1 Provirus Transcription With Repurposed CRISPR/Cas9. Viruses. (2020) 12(10):1154. doi: 10.3390/v12101154
65. Yamayoshi A, Fukumoto H, Hayashi R, Kishimoto K, Kobori A, Koyanagi Y, et al. Development of 7SK snRNA Mimics That Inhibit HIV Transcription. ChemMedChem. (2021) 16(20):3181–4. doi: 10.1002/cmdc.202100422
66. Apolloni A, Lin MH, Sivakumaran H, Li D, Kershaw MH, Harrich D. A Mutant Tat Protein Provides Strong Protection From HIV-1 Infection in Human CD4+ T Cells. Hum Gene Ther (2013) 24(3):270–82. doi: 10.1089/hum.2012.176
67. Jin H, Li D, Sivakumaran H, Lor M, Rustanti L, Cloonan N, et al. Shutdown of HIV-1 Transcription in T Cells by Nullbasic, a Mutant Tat Protein. mBio. (2016) 7(4):e00518-16. doi: 10.1128/mBio.00518-16
68. Jin H, Sun Y, Li D, Lin MH, Lor M, Rustanti L, et al. Strong In Vivo Inhibition of HIV-1 Replication by Nullbasic, a Tat Mutant. mBio. (2019) 10(4):e01769–19. doi: 10.1128/mBio.01769-19
69. Leoz M, Kukanja P, Luo Z, Huang F, Cary DC, Peterlin BM, et al. HEXIM1-Tat Chimera Inhibits HIV-1 Replication. PloS Pathog (2018) 14(11):e1007402. doi: 10.1371/journal.ppat.1007402
Keywords: HIV-1, latency, functional cure, block and lock, epigenetic
Citation: Vargas B and Sluis-Cremer N (2022) Toward a Functional Cure for HIV-1 Infection: The Block and Lock Therapeutic Approach. Front.Virol. 2:917941. doi: 10.3389/fviro.2022.917941
Received: 11 April 2022; Accepted: 30 May 2022;
Published: 29 June 2022.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Akifumi Takaori-Kondo, Kyoto University, JapanCopyright © 2022 Vargas and Sluis-Cremer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Sluis-Cremer, bnBzMkBwaXR0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.