 Alvin Crespo-Bellido
Alvin Crespo-Bellido Siobain Duffy
Siobain Duffy
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 17 June 2022
Sec. Viral Diversification and Evolution
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.899608
This article is part of the Research Topic Insights in Viral Diversification and Evolution: 2021 View all 5 articles
The rapidly expanding phylum Cressdnaviricota contains circular, Rep-encoding single-stranded (CRESS) DNA viruses that are organized within seven established families, but many CRESS DNA virus sequences are not taxonomically defined. We hypothesized that genes in CRESS DNA virus ambisense genomes exhibit strand-specific signatures due to a cytosine to thymine transition bias that can help determine the orientation of the genome: which strand is packaged and is in the “virion sense”. To identify broad strand-specific patterns across genera, we performed compositional analyses of codon usage across the two major opposite sense open reading frames of 712 reference viruses. Additionally, we developed a statistical test to identify relative codon overrepresentation between ambisense sequence pairs for each classified virus exemplar and an additional 137 unclassified CRESS DNA viruses. Codons clustered by the identity of their third-position nucleotide, displaying both strand- and genus-specific patterns across Cressdnaviricota. Roughly 70% of virion-sense sequences have a relative overrepresentation of thymine-ending codons while ~80% of anti-sense sequences display a relative overrepresentation of adenine-ending codons (corresponding to a relative overrepresentation of thymine in these genes as packaged). Thirteen of the 137 unclassified viruses show strong evidence of having the rarer circovirus-like genome orientation, and likely represent novel genera or families within Cressdnaviricota. Given the strong strand-specific patterns of relative codon overrepresentation, the results suggest that the relative codon overrepresentation test can serve as a tool to help corroborate the genome organization of unclassified CRESS DNA viruses.
The rise of metagenomics has revolutionized our understanding of the expansive distribution of viruses (1). With innovative high-throughput sequencing methods, many novel groups of uncultivated viruses have been discovered during the past decades. Circular, Rep-encoding single-stranded DNA (CRESS DNA) viruses are among the viral groups with a drastic increase in discovery rate (2). Once thought to be relatively rare within the virosphere, the application of the phi29 polymerase for rolling circle amplification of circular DNA templates in metagenomic studies have revealed the remarkable breadth and ubiquity of CRESS DNA viruses (3–5). To keep up with the increasing rate of data collection, there has been a paradigm-shift from traditional virus taxonomy involving biological features like host range and virion morphology to sequence-guided classification (6, 7).
CRESS DNA viruses are characterized by their small, circular genomes, which can be monopartite (comprised of one segment) or multipartite (comprised of many segments) (2). Seven families (i.e., Bacilladnaviridae, Circoviridae, Geminiviridae, Genomoviridae, Nanoviridae, Smacoviridae and Redondoviridae) and six groups of unclassified CRESS DNA viruses that infect or are associated with eukaryotes were recently classified in phylum Cressdnaviricota based on phylogenetic analysis of the homologous HUH endonuclease that functions as a replication initiation protein (Rep) (8). Cressdnaviricota includes the smallest known capsid-encoding, eukaryotic-infecting pathogens in the virosphere, with hosts as diverse as diatoms, fungi, plants, and animals (2). However, many are known from sequence alone and do not have any definitive host, as is the case for viruses in Smacoviridae, Redondoviridae and unclassified CRESS DNA viruses. While viruses in some genera may have up to 10 open reading frames (ORFs), their genomes minimally encode for two proteins: the homologous Rep and a non-orthologous coat protein (CP) (2). CRESS DNA viruses’ elevated substitution rates (9–12) and a mechanistic predisposition for recombination (13–16) have facilitated their rapid evolution and emergence. Excepting the nanoviruses which encode one gene per genomic segment, viruses in the established families of this diverse group have ambisense genomes, meaning they encode proteins in both directions with respect to a single origin of replication. Specifically, Rep and CP are always transcribed in opposite directions, with CP typically encoded in the template (or virion) sense strand (transcribed from the complement of the packaged ssDNA genome) and Rep encoded in anti-sense in the virion genome (capable of direct transcription from the virion sense strand). Since the orientation of these genes can be an important criterion in assigning a novel species to a genus, as is the case for the members of Circoviridae (17), identifying strand-specific traits for CRESS DNA viruses might facilitate genome characterization.
For this study, we wanted to determine whether there are patterns of unequal usage of synonymous codons, or codon usage bias (CUB), across Cressdnaviricota and whether those patterns could help classify unclassified CRESS DNA viruses with ambisense genomes. CUB is the joint consequence of mutation, selection, and genetic drift (18–21), and, thus, revealing broad patterns in codon preference can give significant insights into processes shaping viral genomes. Factors implicated in shaping viral CUB include selection to match the host codon usage to optimize the use of the host tRNA pool (i.e., translational selection) (22, 23), selection against sequences activating innate host immunity such as CpG dinucleotides (24, 25), and mutational biases (26, 27). Previous studies in ssDNA bacteriophages show a preference for thymine-ending codons that is consistent with a cytosine-to-thymine (c→t) transition bias and that differs from the codon preferences of their hosts (28, 29). Cardinale etal. (30) further showed in begomoviruses that thymine-ending codons (nnt) are preferred in the virion-sense CP and adenine-ending codons (nna) are preferred in the anti-sense Rep, suggesting that a uniform mutational bias (not translational selection) is the main driver of CUB in viruses of this genus. Given the fact that unpaired DNA is highly vulnerable to spontaneous oxidative cytosine deamination, which can lead to increased c→t transitions during replication (31), we hypothesized that the c→t mutational bias will be present across Cressdnaviricota. If this mutational pressure proves to be significant for many CRESS DNA viruses, we believe that CUB could be exploited to aid taxonomic classification by imprinting ambisense gene pairs with strand-specific biases. We expect an overrepresentation for nnt codons in virion-sense and, in complement, nna codons in anti-sense. Additionally, we expect these biases to reflect a reduction in nnc codons in the virion-sense and a complementary reduction of nng codons in the anti-sense. Alternatively, if this mutational bias is not a dominant feature, then CUB of uncultured CRESS DNA viruses may match host CUB and help determine the ecology of these uncharacterized viruses.
Here we describe the Rep and CP codon usage patterns for circoviruses and cycloviruses based on their differences in genome organization, followed by the patterns exhibited by viruses of other CRESS DNA virus genera. Lastly, we extend the analyses to unclassified CRESS DNA viruses and report relative codon overrepresentation patterns. Results broadly reveal that codon usage is both genus- and strand-specific in Cressdnaviricota, indicating that additional factors besides a c→t mutational bias influence codon preference. We nonetheless observed that there is a general trend of relative codon overrepresentation of nnt codons in the virion-sense and of nna codons in ORFs encoded in anti-sense on the virion single-stranded genome of genera in Cressdnaviricota. When applied to unclassified CRESS DNA viral genomes, relative codon overrepresentation tests corroborate that 13 distinct unclassified CRESS DNA viruses encode for Rep in the virion-sense and CP in the anti-sense, a genome organization that is presently unique to circoviruses. This suggests that the circovirus-like genomic orientation is more common than our current taxonomy suggests.
Coding sequences of all ambisense CRESS DNA viruses from genera with at least 20 species exemplars in the ICTV MSL36 were downloaded from the NCBI RefSeq database. The CRESS DNA virus genera analyzed here are Begomovirus, Circovirus, Cyclovirus, Gemycircularvirus, Mastrevirus and Porprismacovirus. Only complete genomes with annotated Rep and CP were selected for analysis; only mastreviruses that had annotations of the spliced version of Rep were included (sample sizes in Table 1). For each genus, Rep and CP sequences were further parsed into separate FASTA files and analyzed separately (except in the principal component analysis detailed in 2.5). For comparison, the coding sequences of the ambisense DNA-B segment of bipartite begomoviruses (i.e., the virion-sense nuclear shuttle protein, NSP, and the anti-sense movement protein, MP) were also analyzed. Gene annotations in the opposite sense of the defined genome orientation for each genus (see Figure 1) were verified for potential incorrect classification by BLASTing the ORFs. Thirty-one misannotated sequences of classified members of Cressdnaviricota were found to be in reverse complement in GenBank; accessions and their GenBank orientation are detailed in Supplemental File 1.
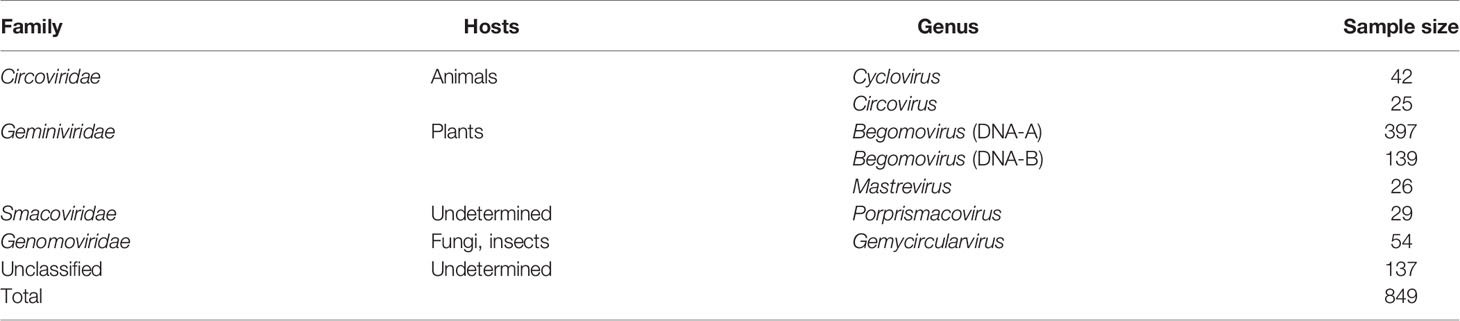
Table 1 Sample sizes by genera of CRESS DNA virus RefSeq sequences used in this study.
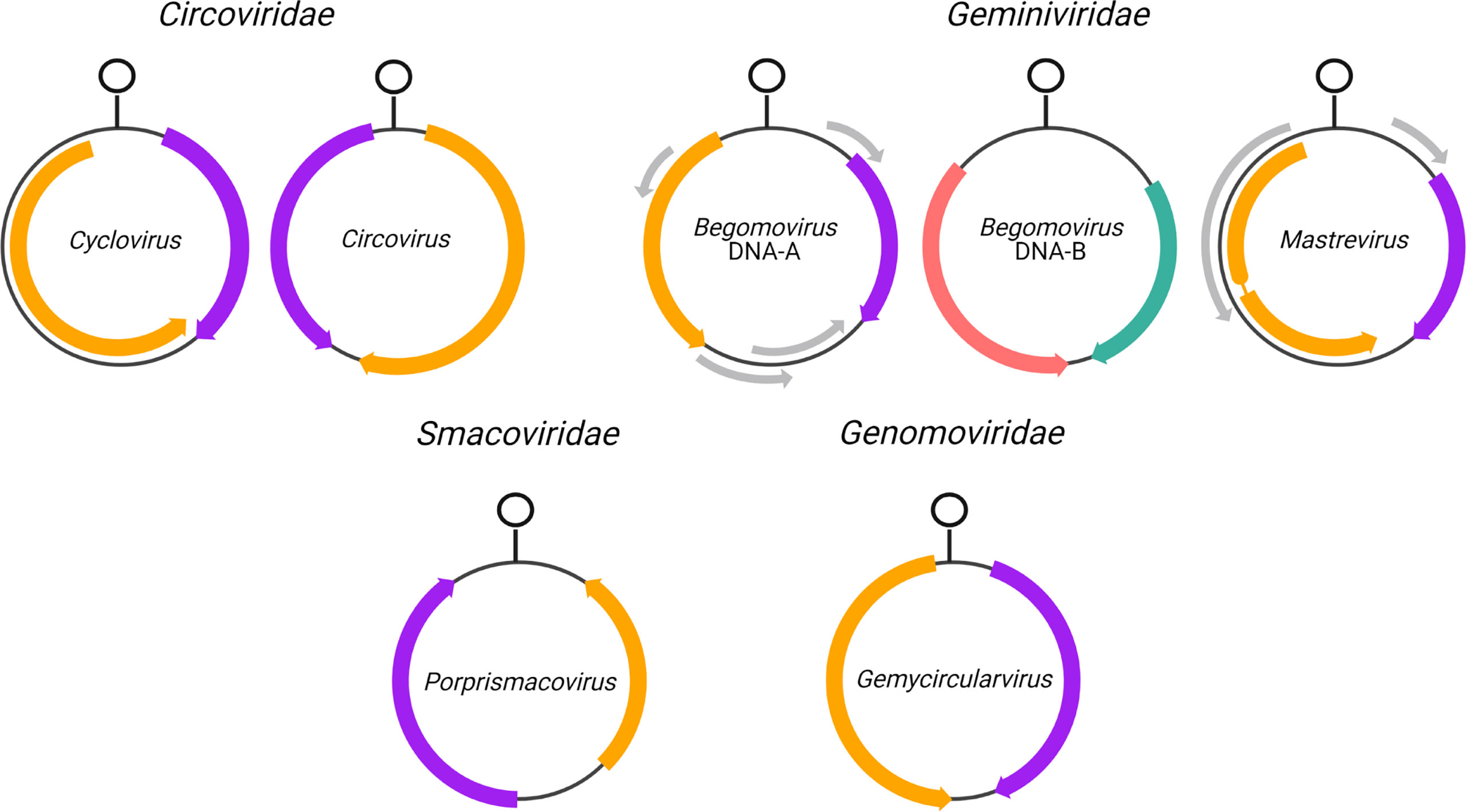
Figure 1 Genome organization of CRESS-DNA virus genera analyzed in this study. Rep and CP open reading frames (ORFs) are shown in orange and purple, respectively, while MP is shown in pink and NSP is shown in keppel green forbegomovirus DNA-B segments. Arrows indicate the direction of transcription relative to the stem loop structure where rolling-circle replication is inititiated. Grey arrows indicate additional ORFs not analyzed in this study. The thin line interrupting Rep in Mastrevirus indicates an intron.
The complete genomes of unclassified ambisense CRESS DNA viruses with Rep and CP annotations were downloaded via the NCBI Taxonomy Browser (https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi) in July 2021. Redundant sequences were removed from the data set using a 95% sequence identity cutoff with CD-Hit (32) in the CD HIT Suite (33). The correct orientation of the ORFs for each virus was verified based on the stem loop harboring the putative origin of replication predicted by the StemLoop-Finder tool (34). Viruses in which the software could not predict a stem loop were removed from the data set. If predicted motifs did not match the canonical stem loop sequences commonly found in CRESS DNA viruses (2), genomes were manually examined for sequences that did match and were used to correctly orient the genomes. Annotated Rep and CP sequences were then extracted and split into virion-sense and anti-sense groups for analyses.
The proportions of nnt, nna, nng and nnc codons (where n=any nucleotide, t=thymine, a= adenine, g=guanine and c=cytosine) were calculated for all Rep and CP (in the case of begomovirus DNA-B, MP and NSP) sequences using the Biostrings package in R version 4.0.3 (35). Mann-Whitney U tests with a Bonferroni-corrected significance cutoff of 0.05 were used to identify statistically significant differences between codon frequencies.
Relative synonymous codon usage (RSCU) values and effective number of codons (Nc) were calculated using DAMBE6 (36). The RSCU value for a codon is the ratio between that codon’s observed and expected frequencies, assuming equal usage of all synonymous codons for each amino acid (37). Following convention, codons with RSCU values <0.6 were considered underrepresented, whereas codons with RSCU values >1.6 were considered overrepresented. RSCU heatmaps were visualized using the ComplexHeatmap R package (38).
Nc is a gene-specific CUB index that reveals the degree to which the entire genetic code is used (39). The theoretical values of Nc are at their minimum when exactly one codon is used in each synonymous codon family and at their maximum when all synonymous codons are used equally. In the DAMBE6 implementation of the Nc calculation, which breaks 6-fold compound codon families into 2-fold and 4-fold codon families (40), values range from 23 (when synonymous codon usage is maximally biased) to 61 (when all codons are used uniformly).
To assess potential constraints on CUB imposed by amino acid composition, grand averages of hydropathy (GRAVY) and aromaticity (AROMA) scores were calculated with CodonW version 1.4.2 (41). A GRAVY score is calculated as the average of the sum of the hydropathic indices of each amino acid in a sequence (42). Values range from -2 to 2, with negative values indicating hydrophilicity while positive values indicate hydrophobicity. The AROMA scores reflect the aromaticity of a protein, defined as the relative frequency of aromatic amino acids in a sequence (43). Higher AROMA values indicate increased aromaticity.
Principal component analysis (PCA) was performed on the RSCU values using the stats package in R. A PCA is a dimensionality reduction statistical method that transforms a large set of variables into linear combinations (known as principal components) that account for as much of the variance as possible. In this case, the RSCU values of 59 codons (excluding Met and Trp, which do not have synonymous codons, and STOP codons) were reduced into two principal components and plotted against each other using the factoextra package in R.
A correlation analysis was performed to identify the relationships between frequencies of codons ending in a given nucleotide (nnt, nna, nng, nnc), amino acid composition (i.e., GRAVY and AROMA scores) and CUB indices (i.e., Nc and the RSCU PC1 and PC2). Pearson’s correlation coefficients were calculated using the Hmisc package and matrices were visualized using the corrplot package, both in R.
Hypergeometric tests were performed to assess whether nnt, nna, nng and nnc codons of each sequence were statistically overrepresented relative to the major opposite sense sequence of the same genomic segment. The hypergeometric test uses the hypergeometric distribution (i.e., the probability distribution of k success states in n draws without replacement) to calculate the statistical significance of having drawn a specific k successes (out of n draws) from a given population. We used this test to identify which sub-populations are over- or under-represented in a sample. Using nnt codons as an example, the test determines the probability of randomly drawing the observed number of nnt codons for ORF 1 or ORF 2 in the global population of codons (defined as the sum of all codons in ORF 1 and ORF 2). Our hypergeometric tests were designed to ignore Met and Trp codons in the calculation. The tests were carried out using the phyper function in the R stats package. The R script is available on Github at https://github.com/acrespo-virevol/Cressdnaviricota-codon-usage.
A pairwise nucleotide identity matrix was calculated for complete Rep sequences using SDT v1.2 (44) with default settings to assess evolutionary relationships between samples.
A total of 849 ambisense viruses, which include 6 recognized CRESS DNA virus genera across Cressdnaviricota and a subset of unclassified CRESS DNA virus sequences, were analysed in this study (Table 1). The coding region organization is mostly conserved across established genera, with all viruses (except those in Circovirus) harboring the putative origin of replication on the CP-encoding strand (Figure 1). Among unclassified CRESS DNA viruses, genomes have organizations with either CP or Rep in the virion-sense, as detailed below. We first present results for members of the Circoviridae genera, Cyclovirus and Circovirus, given that one of the main distinguishing features between them is the Rep and CP orientation of their ambisense genomes. We follow that with the analyses of several other CRESS DNA virus genera and, lastly, CUB analyses for unclassified CRESS DNA virus sequences.
Circoviridae is comprised of viruses from two established genera: Cyclovirus and Circovirus. Cycloviruses have been identified only via sequencing and are believed to infect invertebrates based on the presence of endogenous virus elements within arthropod genomes (45). Some circoviruses, on the other hand, are well-characterized and include economically important pathogens of livestock such as porcine circovirus (17, 46). These genera only encode the two major proteins, Rep and CP, and the main distinguishing feature between them is that they have opposite gene orientations (Figure 1). We focused our analyses on these genera as a proof of concept; if the mutational bias affects both ORFs uniformly, they will have similar strand-specific biases regardless of what gene is encoded.
We first calculated the distribution of codons in the Rep and CP sequences of circo- and cycloviruses(Figure 2; Supplemental Table 1). For cyclovirus CP sequences (which are in the virion-sense), the nnt codon distribution exhibited the highest median value (41.0%) while nnc had the lowest (13.2%). Conversely, nna was highest (32.6%) for cyclovirus Reps while nng was the lowest (15.8%). The distributions in both comparisons were significantly different (Mann-Whitney U; p-value <0.05). The differences in CP nna and nng and the complementary Rep nnt and nnc counts were not significantly different (Supplemental Table 2), indicating they are more evenly distributed. These results suggest an enrichment of thymine accompanied by a depletion of cytosine (which in anti-sense are equivalent to adenine and guanine, respectively), an observation that is consistent with strong c→t mutational bias on the virion sense of the genome.
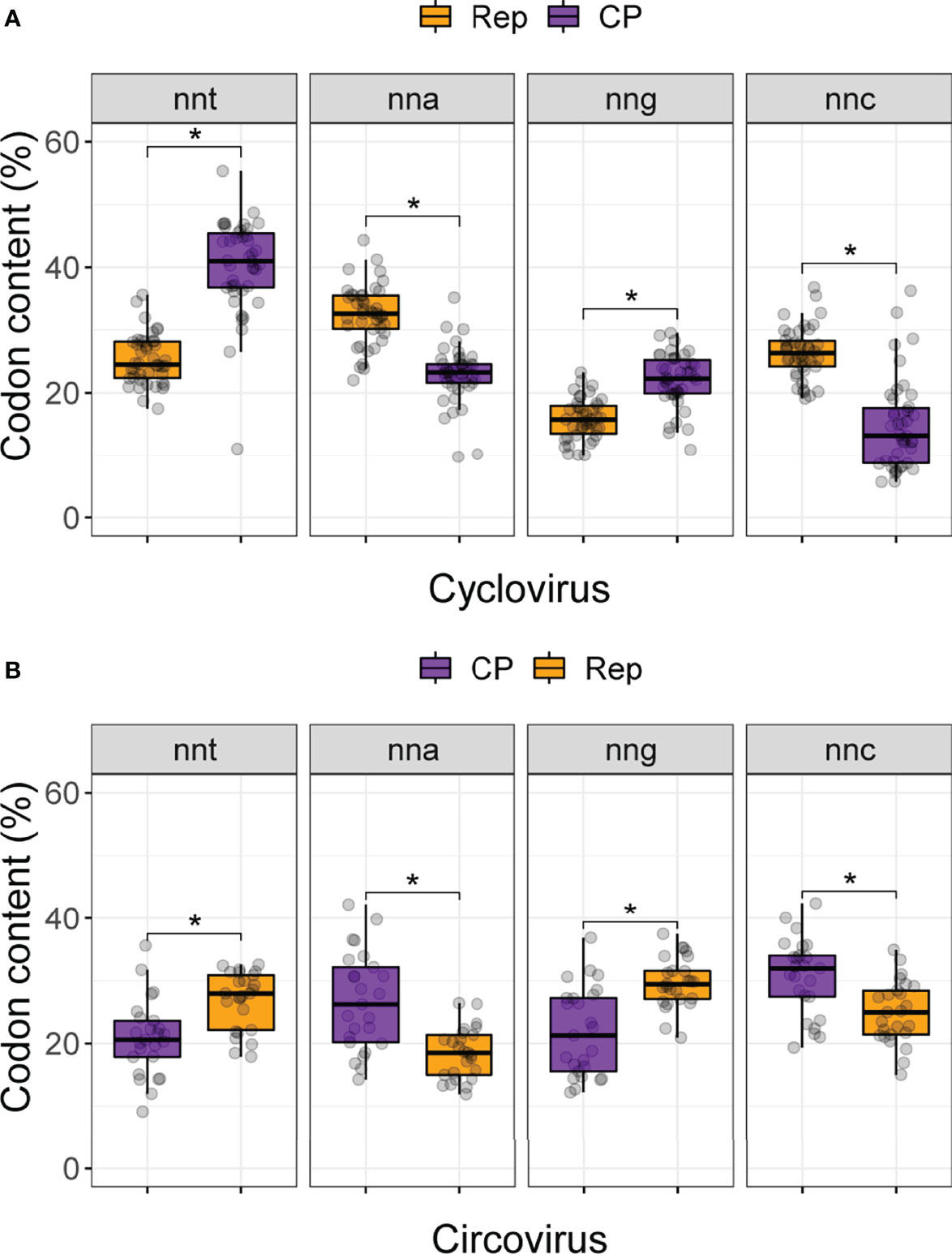
Figure 2 Codon content for (A) cyclovirus and (B) circovirus RefSeq Rep and CP sequences. Codons are grouped by nucleotide identity at the third position (i.e., nnt, nna, nng and nnc, where n, any nucleotide; t, thymine; a, adenine; g, guanine and c, cytosine). Asterisks denote significant differences (p < 0.05) between the Rep and CP distributions based on Bonferroni-corrected Mann Whitney U tests.
Interestingly, for circoviruses, it was nng that had the highest median value (29.4%), followed by nnt (28.0%), nnc (25%) and nna (18.5%) for the virion-sense Rep (Figure 2B). Only nna content was significantly lower than the rest (Supplemental Table 2). The anti-sense CP had complementary results with a median high for nnc (32.0%), followed by nna (26.2%), nng (21.3%) and, lastly, nnt (20.6%). No difference was observed between CP nnt and nng, yet they were both significantly lower than nnc content. These results suggest that circovirus coding sequences do not broadly conform to the expectation that nnt will be the most highly enriched codons, and generally have increased guanine content and are depleted of adenine at synonymous sites.
RSCU was used as a codon-based CUB index to identify under- and overrepresented codons in the Rep and CP sequences of members of Circoviridae (Figure 3). For cyclovirus CP sequences, nnt codons were generally overrepresented (RSCU >1.6) while nnc codons were underrepresented (RSCU <0.6) (Figure 3A; Supplemental File 2). In turn, cyclovirus Rep sequences showed nna and nng codons were typically over- and underrepresented, respectively. One sequence, corresponding to Duck associated cyclovirus 1 (DuACyV1; accession NC_034977), displayed a noticeably different RSCU profile (Figure 3A). Examination of the DuACyV1 CP RSCU values shows that only nna and nnc codons are overrepresented while a significant portion of nnt codons (10/16) are underrepresented (Supplemental Table 3). In the DuACyV1 Rep, most of both over- and underrepresented codons were a-ending.
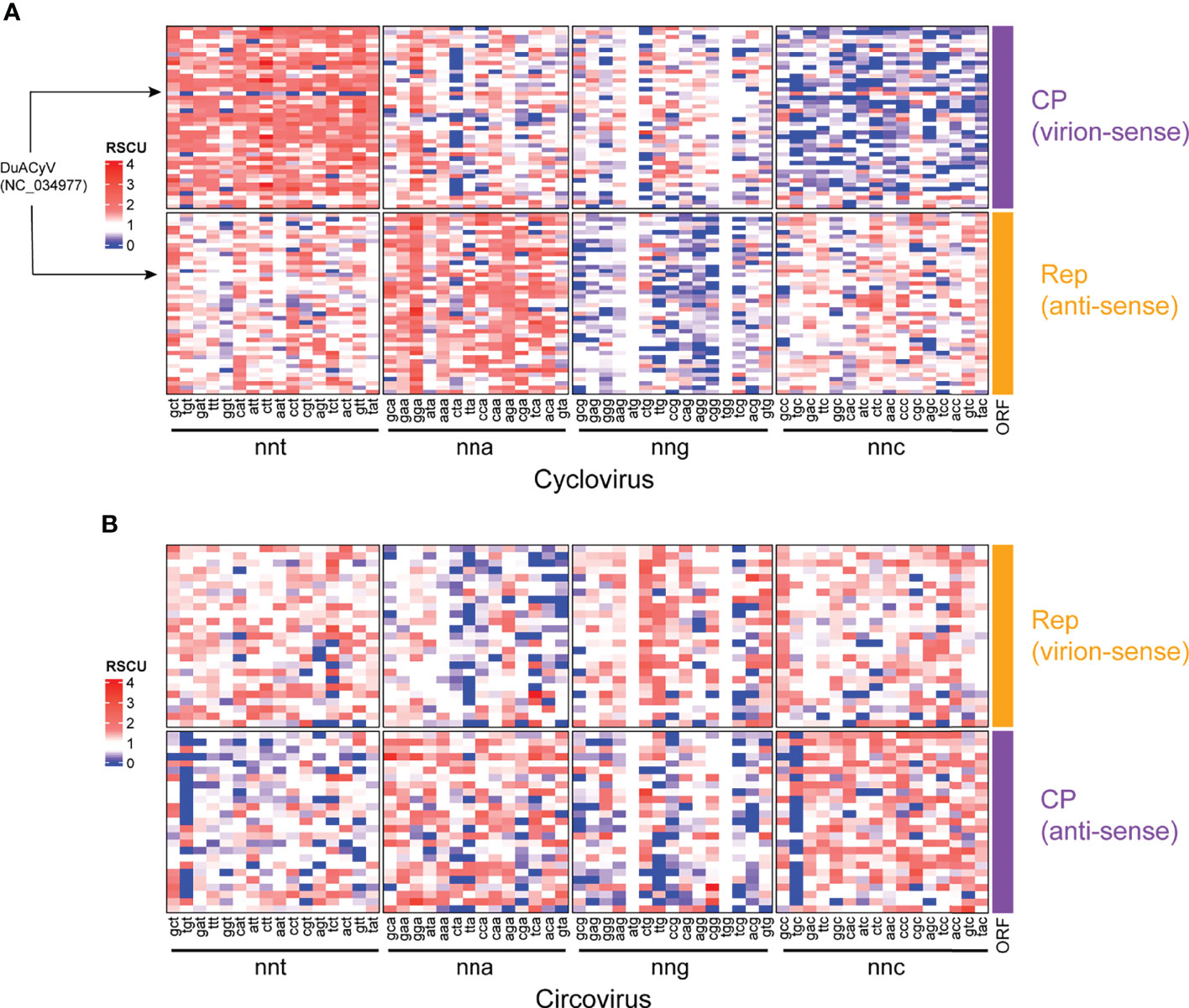
Figure 3 RSCU heatmaps for (A) cyclovirus and (B) circovirus RefSeq Rep and CP sequences. Each row represents a coding sequence, either Rep or CP, as indicated. Columns represent codons, which are grouped by nucleotides at the third position (i.e., nnt, nna, nng and nnc, where n, any nucleotide; t, thymine; a, adenine; g, guanine and c, cytosine). Underrepresented codons (RSCU < 0.6) are shown in blue, middling codons (RSCU=0.61-1.59) are in white and overrepresented codons (RSCU >1.6) are in shades of red.
The RSCU profiles of circoviruses were less conserved across both ORFs, with no broad patterns in preferred relative codon usage based on codons grouped by the identity of their third nucleotide (Figure 3B; Supplemental File 2). For circovirus Rep sequences, nna codons seemed generally more underrepresented than in any other group while there was a general tendency of underrepresentation of nnt and nng codons across the CP sequences.
We conducted a PCA to understand the variance in RSCU profiles of viruses in Circoviridae (Figure 4; Supplemental File 3). In both genera, we find that PC1 generally separates sequences into Rep and CP clusters (Figures 4A, B). PC1 largely separates sequences according to nnt/nng and nna/nnc RSCU in both genera, as reflected by the contributions of individual codons to PC1 (Figures 4C, D). This separation potentially reflects the c→t mutational bias, given that nnt codons have negative PC1 scores while nnc codons have positive PC1 scores, and the inverse relationship is observed for the complementary nna and nng codons. For circoviruses, PC2 seems to be separating sequences by hosts, as most of the bird-associated virus sequences have negative PC2 scores while a majority of the mammalian and fish circovirus sequences have positive PC2 scores (Supplemental File 3). In contrast, no apparent clustering by host source was observed for cycloviruses, which is consistent with phylogenetic analyses that show cyclovirus sequences do not cluster according to host of isolation and their associations might not be indicative of definitive host range (17).
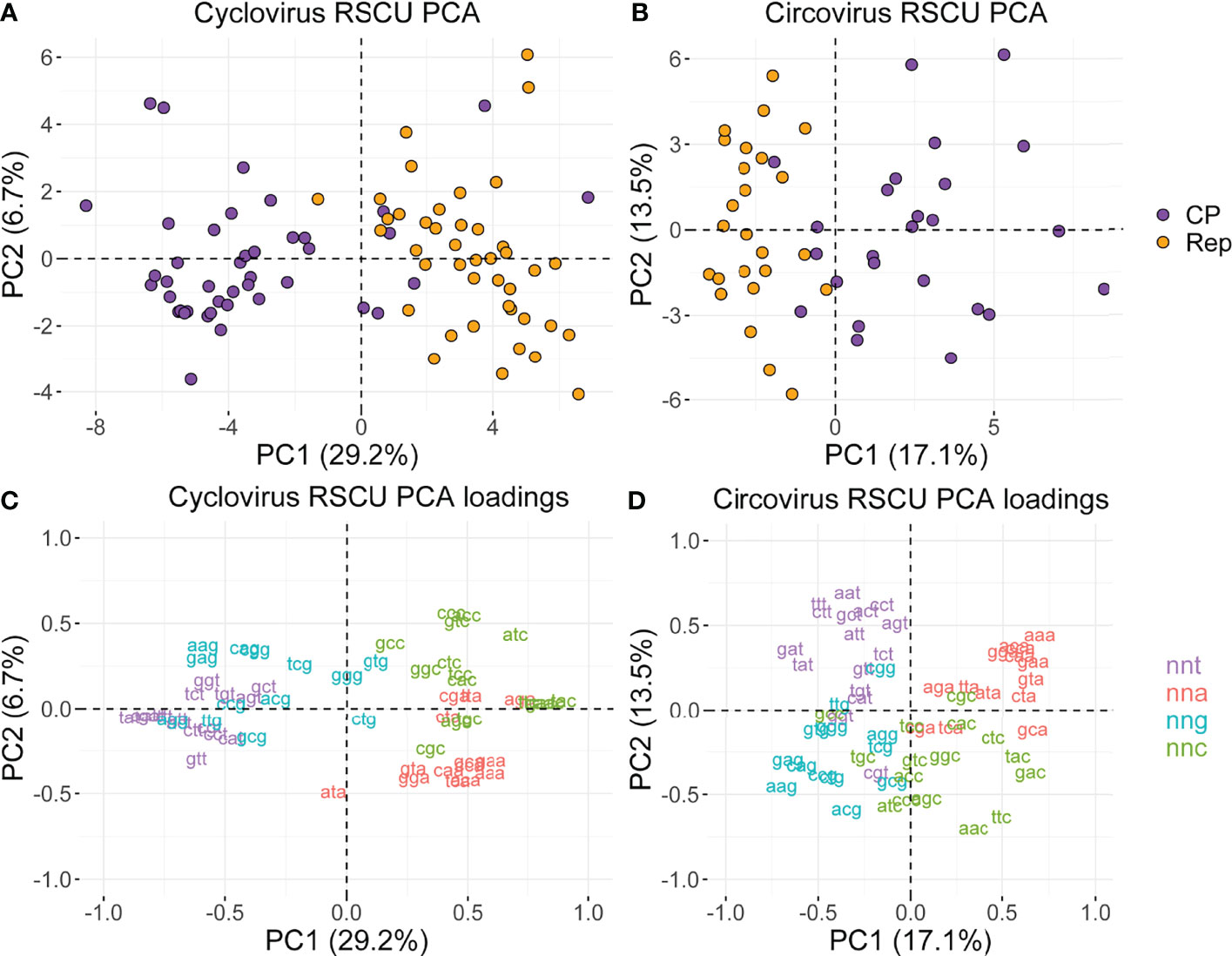
Figure 4 (A, B) PCA plots based on RSCU values of the cyclovirus and circovirus RefSeq Rep and CP sequences, and (C, D) codon contributions to PC1 and PC2. (A-D) The percentages next to PC1 and PC2 indicate the proportion of the variance in the data explained by that principal component. (C, D) Coordinates correspond to the contribution of the labeled codons to the principal components.
We performed correlation analyses between nucleotide composition, amino acid composition and codon usage indices of coding sequences for viruses in Circoviridae (Figure 5). Even though the represented species within each genus may have disparate host ranges and host influence on codon usage might weaken the relationships between compared variables, we were able to identify base composition constraints as strong determinants of codon usage variation.
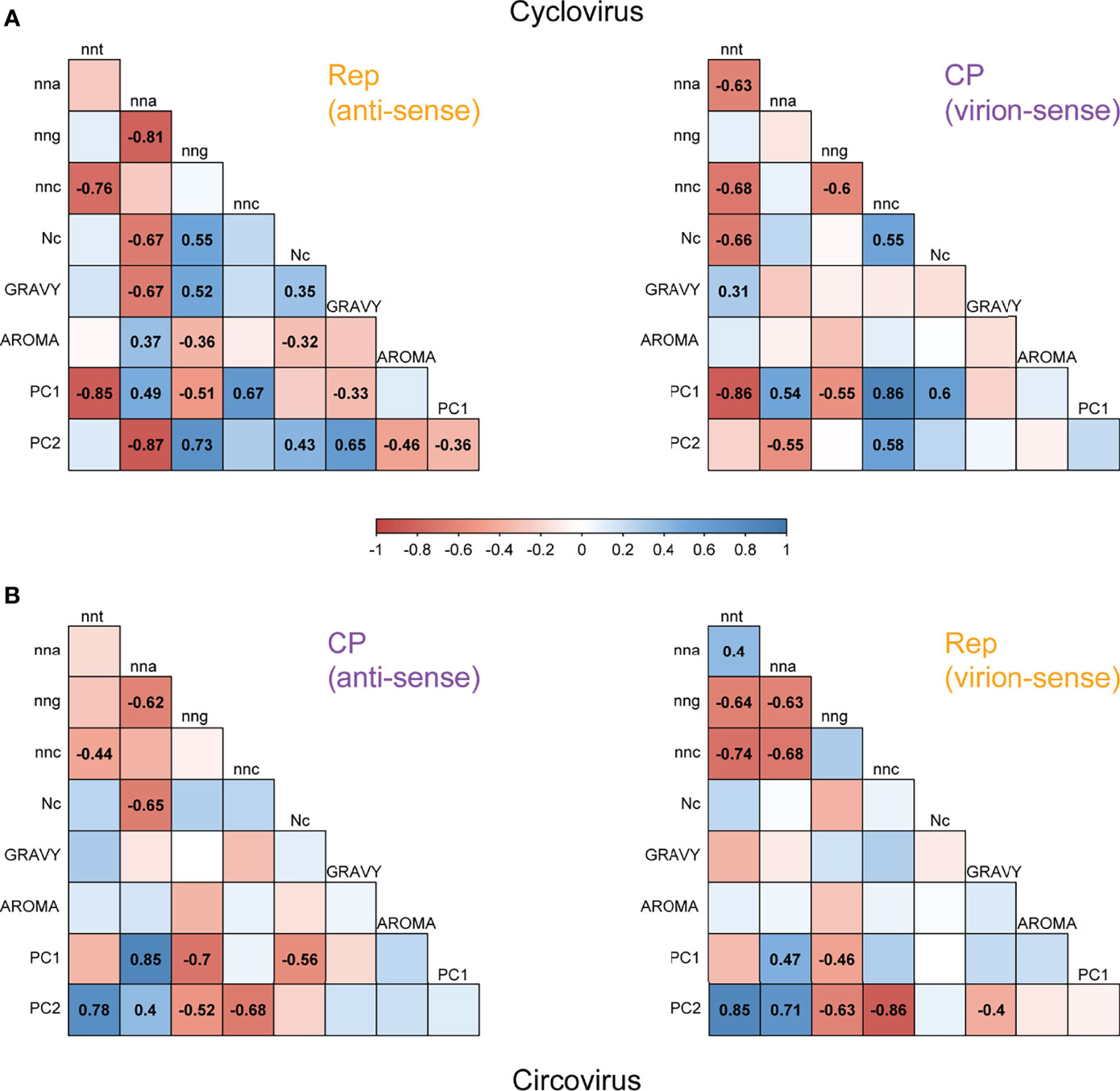
Figure 5 Correlation matrices of codon proportions, amino acid composition and CUB measures for (A) cyclovirus and (B) circovirus RefSeq Rep and CP sequences. Colored boxes represent the scale of Pearson correlation coefficients, with shades of red representing negative correlations and shades of blue representing positive correlations. Only the coefficients for statistically significant correlations (p < 0.05) are shown. The shown scale applies to both panels.
Between cyclovirus CP codon contents, the strongest correlation was observed between nnc and nnt (r= -0.68), which is in accordance with a c→t mutational bias (Figure 5A). However, similar correlations were observed between nnt-nna (r= -0.63), and nnc-nng (r= -0.60), suggesting other mutational constraints may be influencing codon usage for some species. The strongest relationship between codon proportions in cyclovirus Rep sequences corresponds to nng and nna (r= -0.81), which is a complementary pattern to the correlation in CP. Yet, nnc-nnt also had a strong negative correlation (r= -0.76). Nc was negatively correlated with nnt in CP (r= -0.66) and with nna in Rep (r= -0.67), reinforcing that constraints on codon usage correlate with the proportions of nnt codons in virion sense and nna codons in anti-sense. GRAVY scores had a significant negative correlation with Rep nna (r= -0.67), indicating a selective constraint associated with nna codon usage. The RSCU PC1 had strong and inverse correlations with CP nnc (r= 0.86) and nnt (r= -0.86) as well as Rep nnc (r= 0.67) and nnt (r= -0.85), which shows that PC1 is mostly influenced by nnc and nnt codon content. The strongest relationships with PC2 were observed with Rep nna (r= -0.87) and nng (r= 0.73), implying that separation along PC2 is mainly due to nna and nng content of Rep sequences.
The strand-specific relationships observed for circoviruses (Figure 5B) resemble those of cycloviruses in terms of codon content. The strongest correlation for the virion-sense Rep sequences is the negative correlation between nnc and nnt (r= -0.74) while the strongest correlation for the anti-sense CP sequences is between nng and nna (r= -0.62). These trends are consistent with the virion- and anti-sense patterns of cycloviruses, suggesting that the c→t bias on the virion sense of the genome influences synonymous codon usage in both ORFs for viruses in Circoviridae. However, the circovirus Rep sequences also showed significant negative correlations between nnc-nna (r= -0.68), nng-nnt (r= -0.64) and nng-nna (r= -0.63), suggesting other biases may be present. Regarding codon composition, Nc is only significantly correlated with the Rep nna content (r= -0.65). No strong significant correlations are found between amino acid composition indices and any other variables. In contrast to cycloviruses, PC1 is mainly correlated with CP nna (r= 0.85) and nng (r= -0.7), implying PC1 mostly explains the variance in nna and nng content in the CP sequences. PC2 has strong positive correlations with nnt and negative correlations with nnc in both Rep and CP, indicating that variance in PC2 largely corresponds to the nnt and nnc content across both ORFs. In addition, strong correlations were observed between PC2 and Rep nna (r= 0.71) and nng (r= -0.63), which means that PC2 further explains variance in the circovirus Rep sequences based on nna and nng content.
Hypergeometric tests were used to evaluate the relative overrepresentation of codons in the Rep and CP sequences of individual Circoviridae RefSeqs (Figure 6; Supplemental File 4). The expectation is that nnt codons will consistently be overrepresented in virion-sense sequences while nna codons will be overrepresented in the anti-sense sequences due to the assumed influence of c→t mutation bias in shaping codon usage.
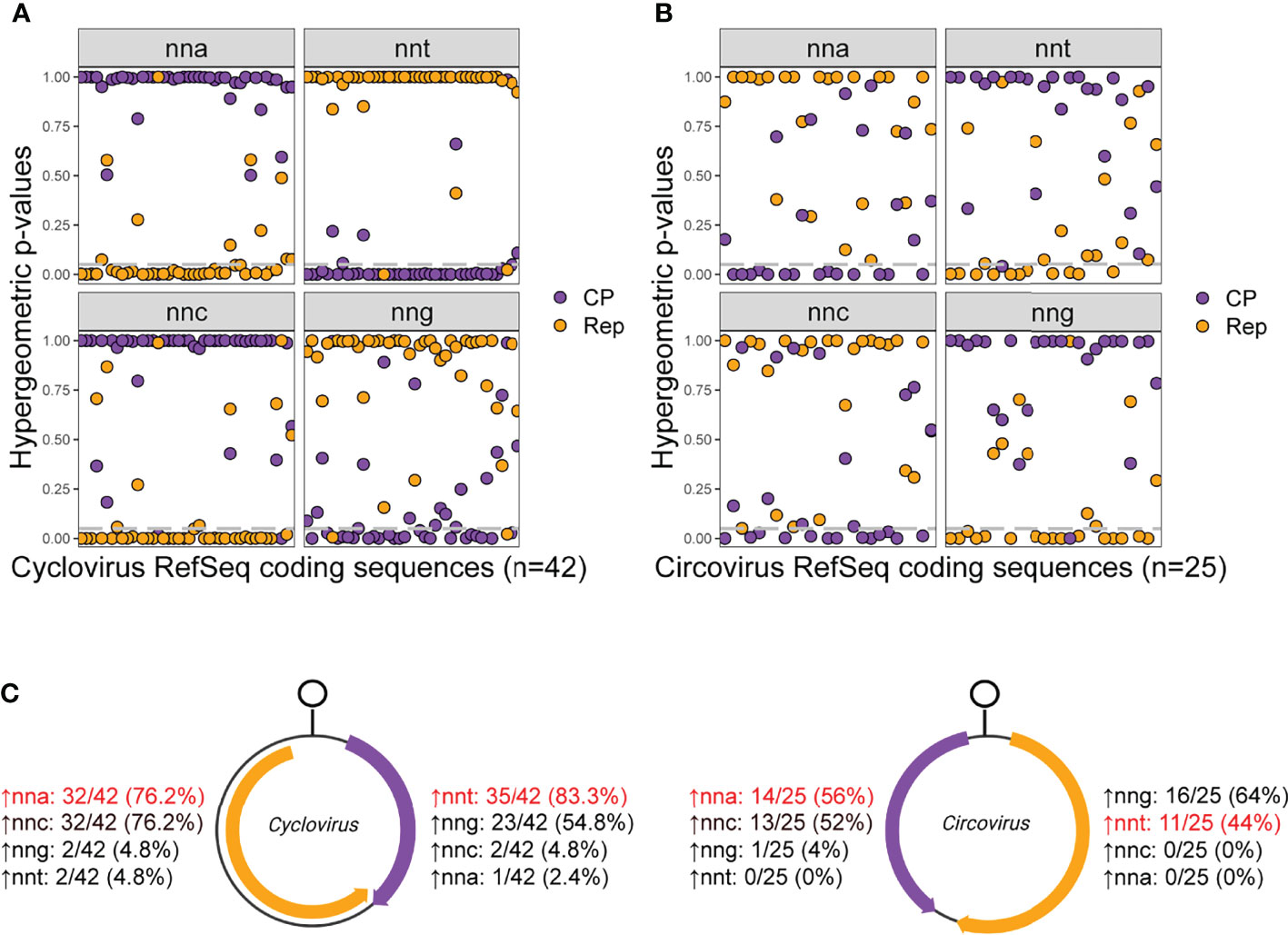
Figure 6 Hypergeometric test p-values for relative codon overrepresentation of (A) cyclovirus and (B) circovirus RefSeq coding sequences and (C) the number of Rep and CP sequences with relative overrepresentation of nnt, nna, nng and nnc codons. (A, B) Grey dashed lines indicate a hypergeometric p-value cutoff of 0.05. (C) Percentages indicate the number of sequences in the data set that show relative overrepresentation of the corresponding codon. The codons in red are the ones expected to be most overrepresented in each strand if c→t mutational bias is the predominant process shaping codon usage.
Most of the cycloviruses showed a relative overrepresentation of nnt codons in their virion-sense CP (83.3% of sequences) and of nna codons in their anti-sense Rep (76.2%) (Figures 6A, C). There were three coding sequences that showed contrary results: two Rep sequences with a relative overrepresentation of nnt codons, corresponding to Spider associated cyclovirus 1 (SpACyV1; accession NC_040324) and DuACyV1 (accession NC_034977; the DuACyV1 CP also showed an overrepresentation of nna codons). Many viruses also showed relative nng overrepresentation in the virion-sense and nnc overrepresentation in the anti-sense. However, this is likely a by-product of c→t mutational bias acting on the virion-sense strand of the genome, as nng is not enriched in the virion-sense, but nng (which reflects cytosine) is depopulated in the anti-sense (illustrated in Figure 2A). A similar scenario is observed for nnc codons, where nnc codons are not enriched in the anti-sense but are depleted in the virion-sense. Therefore, relative overrepresentation of nng in the virion-sense and nnc in the anti-sense is due to a depletion of their complementary base in the opposite sense.
A majority (56%) of circovirus anti-sense sequences showed nna overrepresentation (Figures 6B, C). In contrast to cycloviruses, nnt codons were not overrepresented in most virion-sense sequences (44%) but nng codons were (64%). These results are consistent with nng being the most frequent codon, on average, in the virion-sense (Figure 2B). Only the Starling circovirus (StCV; accession NC_008033) anti-sense CP sequence showed nnt codon overrepresentation, and no virion-sense sequences have relative nna codon overrepresentation.
There are varying relative codon overrepresentation trends observed in ambisense genes for begomoviruses, mastreviruses, porprsimacoviruses and gemycircularviruses (Figure 7; Supplemental File 4). All viruses for these families possess genomes with a cyclovirus-like orientation (with Rep in anti-sense). These trends and relevant sequence composition analyses related to synonymous codon usage were examined.
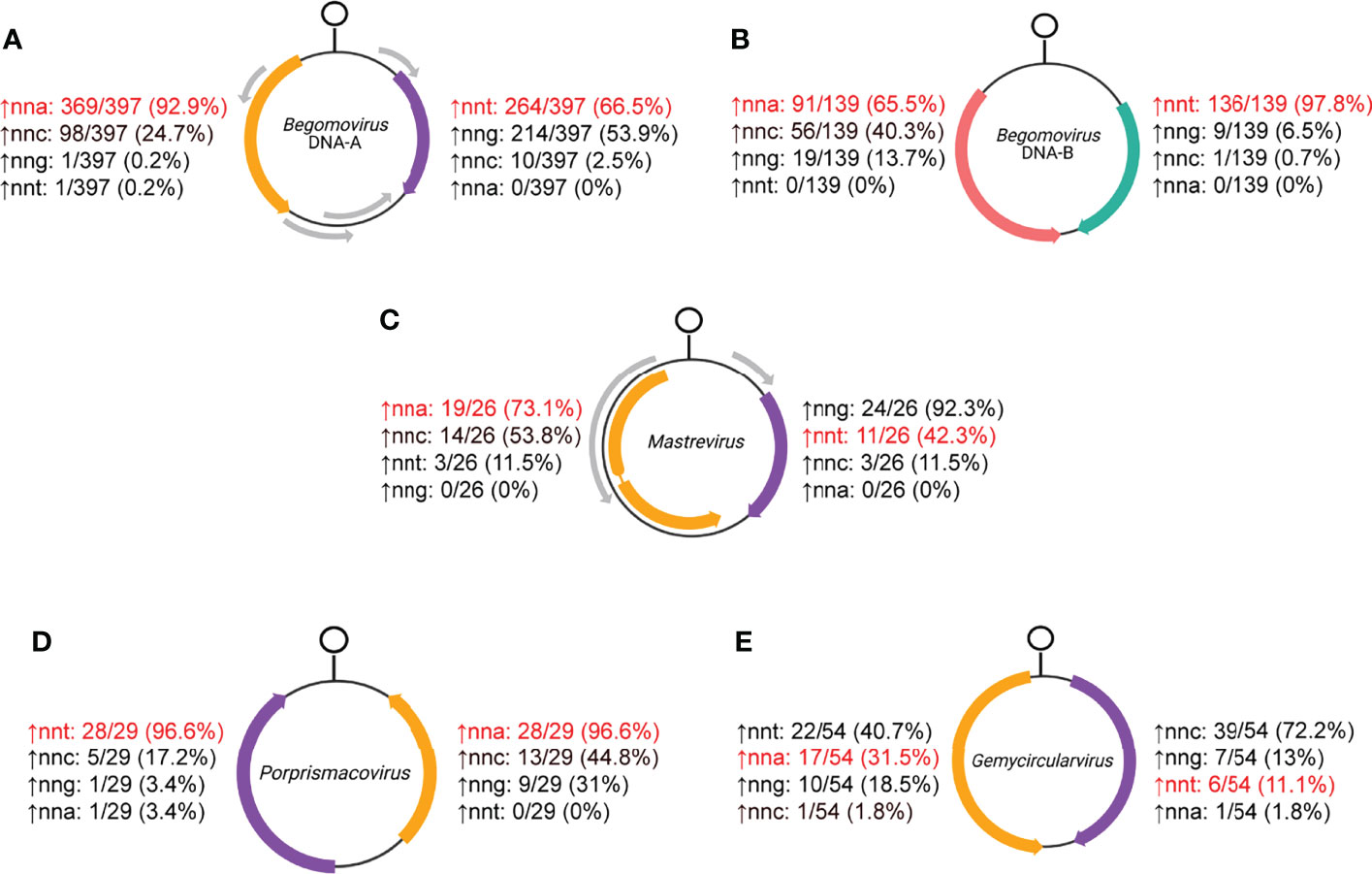
Figure 7 Hypergeometric test results of relative overrepresentation of nnt, nna, nng and nnc codons for (A) begomovirus DNA-A, (B) begomovirus DNA-B, (C) mastrevirus, (D) posprismacovirus and (E) gemycircularvirus RefSeq coding sequences. Rep and CP open reading frames (ORFs) are shown in orange and purple, respectively, while MP is shown in pink and NSP is shown in keppel green for begomovirus DNA-B segments. Percentages indicate the number of sequences in the data set that show relative overrepresentation of the corresponding codon. The codons in red are the ones expected to be most overrepresented in each strand if c→t mutational bias is the predominant process shaping codon usage.
Begomoviruses (family Geminiviridae) are whitefly-transmitted CRESS DNA viruses that infect dicotyledonous plants (or dicots) and severely constrain crop production in tropical and subtropical regions around the world (47). Their genomes can be monopartite or bipartite, and in bipartite genomes their segments are denominated DNA-A and DNA-B. As monopartite and bipartite DNA-A begomovirus segments are homologous, we will refer to them jointly as DNA-A throughout this manuscript. On the virion-sense strand, DNA-A has a CP gene, and often a partially overlapping gene that encodes the precoat protein. The anti-sense strand of DNA-A encodes the Rep and additional proteins involved in transcription, replication, and RNA-silencing suppression. The DNA-B segment encodes two proteins involved in systemic movement: a nuclear shuttle protein (NSP) in the virion-sense and a movement protein (MP) in the anti-sense. We extended our analyses to exemplars of both segments to compare their codon usage patterns.
Consistent with a c→t mutational bias, the hypergeometric tests show that DNA-A segments usually have a relative nnt overrepresentation in the virion-sense and almost always have nna overrepresentation in the anti-sense (Figure 7A). Codon composition analyses reveal an enrichment of nnt, low usage of nnc and a depletion of nna codons in the virion-sense while there is relatively even usage of all bases except for a decrease in nng in the anti-sense for DNA-A segments (Figure S1A). While the c→t bias may explain a significant fraction of the overrepresentation patterns, the higher number of anti-sense sequences displaying nna overrepresentation relative to the number of virion-sense sequences with nnt overrepresentation is largely due to the observed virion-sense adenine depletion. DNA- B segments also typically displayed overrepresentation of nnt in the virion-sense and nna codons in the anti-sense, but it was the virion-sense sequences that nearly always showed the expected (nnt) relative overrepresentation (Figure 7B). Compared to DNA-A, DNA-B segments had distinct codon composition patterns: increased nnt and decreased nnc usage in the virion-sense NSP (in accordance with a c→t bias), and similar (although statistically different) usage of all codon types aside from an increase in nna for the anti-sense MP (Figure S1B). Overall, codon usage in the virion-sense does not complement the usage in the anti-sense for either segment. The contrasting results between segments and between ambisense genes suggest that codon usage is evolving in a segment- and strand-specific manner in begomoviruses.
The RSCU analyses are consistent with the codon composition analyses: for DNA-A, a general overrepresentation of nnt codons and underrepresentation of nna codons in the virion-sense, and relatively even RSCU except for broad underrepresentation of nng codons across sequences in anti-sense (Figure S1C). The ORFs on the DNA-B segment display high nnt and low nnc RSCU values in the virion-sense and increased relative usage of nna codons in the anti-sense (Figure S1D). PCA based on the RSCU values show that PC1 separates Rep and CP sequences into distinct clusters (Figure S2A), while it took both axes to separate the DNA-B genes into discrete clusters (Figure S2B). While PC1 and PC2 explain similar proportions of variance in DNA-A and DNA-B gene RSCU, the PCA loadings plots (Figures S2C, D) show that codons contribute to the principal components in different ways, which supports the notion that codon usage is evolving differently in each segment.
The correlation analyses show that there is a strong negative relationship between nnc-nnt content in both virion-sense sequences (CP r= -0.86, NSP r= -0.84, Figures S3A, B). Nc and PC1 did not have strong correlations with any codon type in DNA-A, implying that base constraints are not the main determinants of CUB for either Rep or CP. Conversely, both Nc and PC1 had strong negative correlations with nnt and strong positive correlations with nnc in NSP, suggesting c→t is a main factor influencing codon usage for this gene (Figure S3C). On the other hand, MP nnt and nnc content showed the same relationships as NSP with PC1 but not with Nc (Figure S3D).
Mastreviruses constitute the second largest genus within Geminiviridae and are monopartite, leafhopper-transmitted viruses that are predominantly associated with disease in monocotyledonous (or monocot) plants in the Old World (47). Mastreviruses have two genes in the virion-sense encoding CP and a movement protein. In the anti-sense, they encode Rep, which is expressed from two genes by transcript splicing, and an additional overlapping gene that supports viral replication (48). We provide results of analyses performed on CP and the spliced version of Rep sequences.
A great majority of mastrevirus CP sequences (92.3%) showed relative overrepresentation of nng codons while most Rep sequences (73.1%) revealed an nna codon overrepresentation (Figure 7C). This is consistent with codon composition analyses showing nng with the highest median content in the virion-sense (Figure S4A; Supplemental Table 1). Interestingly, while nnt is not relatively enriched in the CP sequences, there were seven sequences exhibiting very high nnt content (> 37%) (Figure S4A; Supplemental File 3). They all correspond to dicot-infecting mastreviruses while all the other sequences closer to the median correspond to monocot-infecting viruses (Supplemental File 1). In the anti-sense, instead of an increase in nna content, nnc and nnt show the highest median values. The relative nna overrepresentation in Rep sequences (Figure 7C) is therefore likely supplemented by the depletion of nna nucleotides in the virion-sense (Figure S4A).
RSCU values show a relatively uniform overrepresentation of nng codons and underrepresentation of nna codons in the virion-sense CP, with anti-sense Reps having a corresponding underrepresentation of nng codons (Figures S4B). The RSCU PCA does not show clear Rep and CP clusters along PC1 (Figure S4C). However, a more well-defined cluster would form for CP if we removed sequences corresponding to dicot-infecting mastreviruses, which account for seven of the eight CP sequences with negative PC1 values (Supplemental Files 1, 3). PC1 seems to separate sequences by nna/nnt and nng/nnc usage while PC2 is separating according to nng/nnt and nna/nnc usage (Figure S4D).
The strongest correlation in the virion-sense sequences in terms of codon composition is between nnc-nnt (r= -0.88) (Figure S4E). Anti-sense Rep sequences show similar negative correlations between nnc-nnt (r= -0.71) and nng-nna (r= -0.72). PC1 correlates positively with nng and nnc, and negatively with nna and nnt for both Rep and CP, which is consistent with the codon contributions observed in the PCA loadings plot (Figure S4D). Additionally, PC1 correlates somewhat with AROMA, which indicates a potential selective constraint imposed by amino acid composition on the RSCU of mastrevirus CP sequences.
Porprismacoviruses (family Smacoviridae) are a recently discovered group of CRESS DNA viruses that, although prevalent in vertebrate feces (including human), are yet to be cultured (49, 50). Their ambisense genomes contain only two major ORFs, Rep and CP. Although gut-associated methanogenic archaea have been implicated as potential smacovirus hosts based on the presence of CRISPR spacers that match regions of the smacovirus genome (51), no definitive host-range has been established. We performed analyses on porprismacovirus CP and Rep sequences.
Almost all porprismacoviruses exhibit nnt overrepresentation in the virion-sense and nna overrepresentation in the anti-sense (Figure 7D). The codon composition of virion-sense CP sequences displays very high median levels of nnt (39.8%) and strikingly low median levels of nna (9.7%) (Figure S5A and Supplemental Table 1). In complement, anti-sense Rep sequences show very low median levels of nnt (10.8%), yet all other codons have comparable distributions. RSCU heatmaps broadly corroborate codon compositions for both ORFs (Figure S5B). Rep and CP are separated well by PC1, which explains over a third of the variance in RSCU values (Figure S5C, D).
Codon composition correlation analysis shows that nnc-nnt is strongly correlated the virion-sense (r= -0.79) and mildly correlated in the anti-sense (r= -0.6) (Figure S5E). PC1 is most strongly correlated with nnt in both senses, indicating that the separation of Rep and CP sequences is largely due to the high levels of thymine in the virion-sense and low levels of adenine in the anti-sense. These codon usage analyses indicate that there is a uniform and strong bias against adenines at the third position in porprismacovirus genomes.
Gemycircularviruses belong to the Genomoviridae family, the sister clade to Geminiviridae that includes viruses with fungal and insect host-ranges (52–54). Genomoviruses have the same genome orientation as geminiviruses but encode only CP (virion-sense) and Rep (anti-sense) in their ssDNA genomes. Here we present results from CUB analyses performed on the two coding sequences of Gemycircularvirus species exemplars.
Virion-sense CP sequences consistently have a relative nnc overrepresentation (Figure 7C). In contrast to all other CRESS DNA virus genera examined, the anti-sense sequences often show an nnt overrepresentation, observed in 40.7% of viruses (Figure 7E). Codon composition supports these trends since nnc and nnt have the highest median content in the virion- and anti-sense strands, respectively (Figure S6A and Supplemental Table 1). Both Rep and CP show a depletion of nna content. RSCU analyses show uniform nnc codon overrepresentation and general patterns of underrepresentation across several nna codons in the virion-sense (Figure S6B). In the anti-sense Reps, some nnt codons are overrepresented across many sequences, and select nna codons (e.g., gga, aga, cga) are strongly overrepresented (Figure S6B and Supplemental File 2). The RSCU PCA mostly separates Rep and CP into individual clusters but PC1 and PC2 only account for 23.4% of the variance seen in the RSCU profiles, indicating complex and largely unexplained patterns across genes (Figure S6C). PCA loadings show separation along PC1 is mostly due to nnc usage (Figure S6D).
Despite differing patterns from other members of Cressdnaviricota, the strongest correlations between codon types are seen between nnc-nnt (r=-0.61) in the virion-sense and the complementary nng-nna (r= -0.65) in the anti-sense (Figure S6E). PC1 was most strongly correlated with nnc (r= 0.84) and nnt (r= -0.6) in Rep (Figure S6E) and nnc (r= 0.9) and nnt (r= -0.72) in CP (Figure S6F).
The examination of relative codon overrepresentation across Cressdnaviricota showed that nna codons were always the most overrepresented across anti-sense sequences in all genera (excepting Gemycircularvirus, Figure 7E). To a lesser degree, nnt codons are overrepresented in the virion-sense sequences of most CRESS DNA virus genera (Table 2). Additionally, except for gemycircularviruses, there is a consistent lack of relative nna codon overrepresentation in the virion-sense and a lack or relative nnt codon overrepresentation in the anti-sense strands of these viruses. Given these observations, we decided to conduct hypergeometric tests on Rep and CP pairs of sequences from unclassified CRESS DNA viruses with ambisense genomes to assess their virion-sense strandedness.
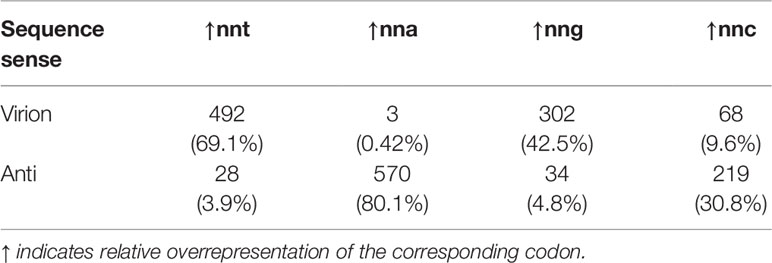
Table 2 Number of classified CRESS DNA virus sequences (n=712) that show relative overrepresentation of the corresponding codon (percentages in parentheses).
First, genome orientation for all unclassified viruses was verified by identifying the direction of putative origin of replication motifs found in stem loops predicted by the StemLoop Finder tool. We found 48 instances where the submitted unclassified CRESS DNA viruses in GenBank were potentially in the reverse complement (Supplemental File 1). After correcting the orientation of these 48 genomes, there were 35 unclassified viruses predicted to code Rep in the virion-sense and CP in the anti-sense, sharing a strand orientation that is unique to circoviruses. Hypergeometric tests of relative codon overrepresentation in Rep and CP pairs revealed that nearly half of virion- and anti-sense sequences display overrepresentation of nnt and nna codons, respectively (Figure 8). These results agree with the typical patterns observed for the established Cressdnaviricota genera. Around 12% of virion-sense and anti-sense sequences display the opposite pattern: nna codon overrepresentation in the virion-sense and nnt codon overrepresentation in the anti-sense.
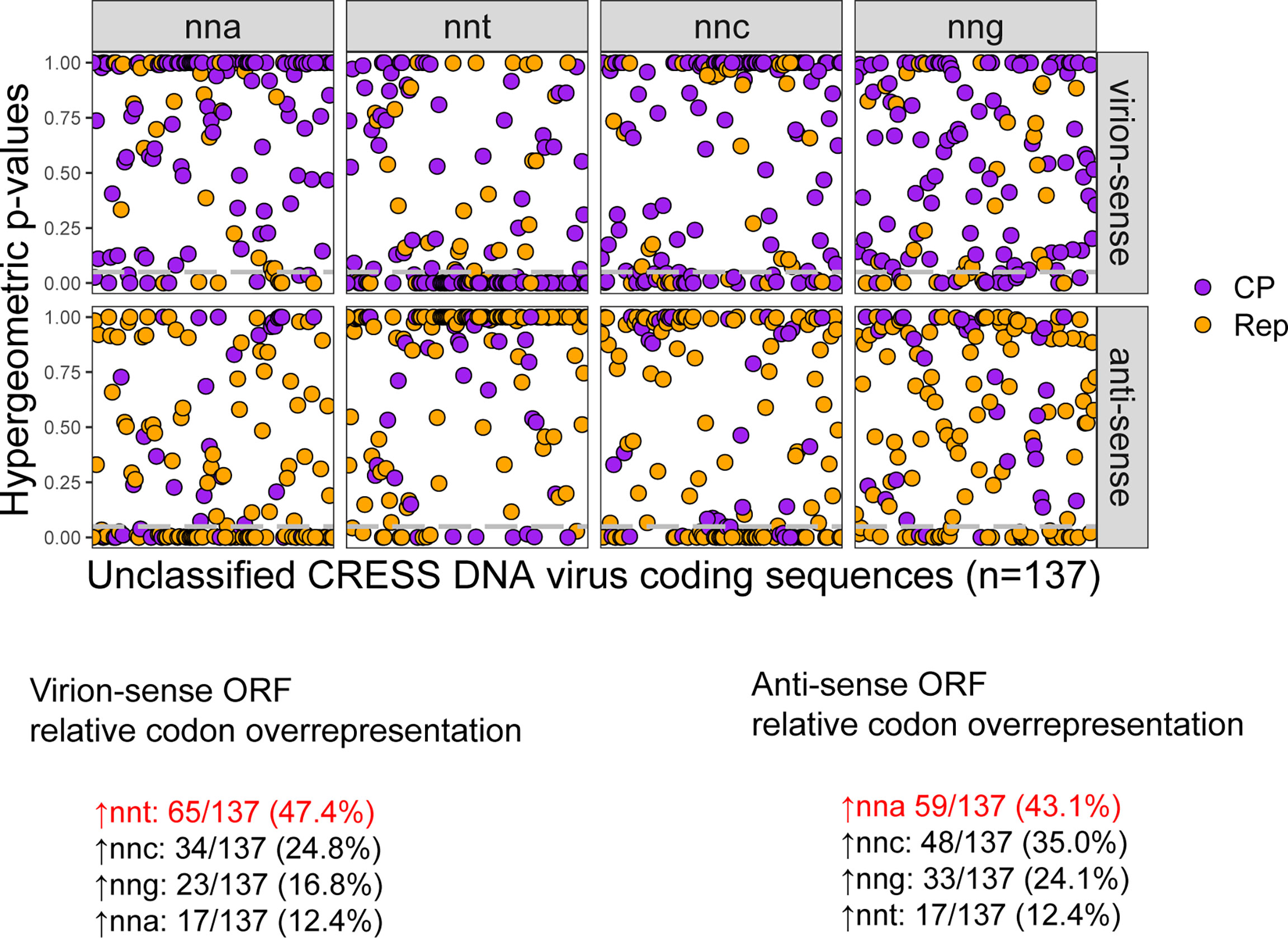
Figure 8 Hypergeometric test p-values for relative codon overrepresentation of unclassified CRESS-DNA virus coding sequences and the number of virion-sense and anti-sense sequences with relative overrepresentation of nnt, nna, nng and nnc codons. Grey dashed lines indicate a hypergeometric p-value cutoff of 0.05. Percentages indicate the number of sequences in the data set that show relative overrepresentation of the corresponding codon. The codons in red are the ones expected to be most overrepresented in each strand if c→t mutational bias is the predominant process shaping codon usage.
Out of the 35 CRESS DNA viruses with predicted circovirus-like orientation, 8 virion-sense Rep and 10 anti-sense CP sequences across 13 different viruses display the expected pattern of nnt codon overrepresentation in the virion-sense and nna codon overrepresentation in the anti-sense, supporting their circovirus-like organization. These 13 unclassified viruses were sampled from many sources including fish tissue, bird cloacal swabs, plants, and mammalian feces and meat (Supplemental File 5). We assessed the evolutionary relationships between the homologous Rep sequences of the 13 viruses by performing a pairwise nucleotide identity analysis with SDT v1.2. We found that, on average, the Rep sequences share 57.6% nucleotide identity to each other and the highest pairwise identity between two samples is just 63.6% (Supplemental File 5). Given the low pairwise percent nucleotide identity between the most well-conserved gene shared by these samples, it is likely that these viruses represent members of multiple undetermined families that possess the strand organization shared with only circoviruses among established Cressdnaviricota families.
Cressdnaviricota is a widespread and rapidly expanding phylum of small ssDNA viruses with highly diverse host ranges. As the number of sequenced CRESS DNA virus representatives grows exponentially through genomic and metagenomic surveillance, it is necessary to refine sequenced-based approaches to characterize viruses in the absence of our ability to culture them. To this end, we decided to explore trends in codon usage bias of the two major ORFs, Rep and CP, of species representatives across the entire phylum. Based on the extensive literature from experimental and comparative analyses of ssDNA bacteriophages and eukaryotic viruses (9, 10, 29, 55–60), we hypothesized that the strong bias towards c→t transitions exhibited by ssDNA viruses would result in an overrepresentation of nnt codons in virion-sense and, in complement, nna codons in anti-sense coding sequences. Results showed that most sequences, across genera, shared the predicted over- and underrepresented codons. While in members of Circoviridae these statistical over- and under-representations are complementary to each other (Figures 2, 3), we found most genera have idiosyncratic preferences. This suggests that forces shaping codon usage differ across Cressdnaviricota, but they often act in a strand-specific manner. Virion-sense genes had a high amount of nnt codons, often being the most overrepresented kind of codon in these ORFs by median value (excepting circoviruses and mastreviruses where nnt codons are second most prevalent to nng codons, and gemycircularviruses, where nnt comes in second to nnc). For anti-sense ORFs, while nna codons were usually overrepresented relative to the virion sense ORF, only two groups had median nna content that exceeded that of the other kinds of codons: cycloviruses and begomovirus DNA-B segments. Consequently, codon content indicates that nnt codons are not incredibly enriched in the virion sense and nna codons are not in anti-sense, contrary to the expectation from a persistent and quantifiable c→t substitution bias. Regardless, we observed that most virion-sense sequences across the classified genera of Cressdnaviricota do have a relative nnt overrepresentation (69.1%) while a majority of anti-sense sequences have a relative nna overrepresentation (80.1%, Table 2), which conforms to our expectations. Moreover, only a very small fraction of sequences displays the opposite pattern (i.e., virion-sense enriched for nna- 0.42% - and anti-sense enriched for nnt- 3.9%), indicating that the hypergeometric test can serve as a tool to reliably corroborate the strandedness of a CRESS DNA virus genome.
When examining unclassified CRESS DNA viruses, establishing gene orientations based on the position of the origin of replication can be difficult since the nonanucleotide varies between families and we do not have a clear sense of the unsampled motif diversity out there. Additionally, multiple candidate stem loop structures are often found throughout CRESS DNA virus genomes, the nonanucleotides where replication starts can be palindromic, and we lack experimental data revealing origin function for many of these viruses. Based on our best origin predictions aided by the StemLoop Finder tool, we applied hypergeometric tests to ambisense coding sequence pairs of unclassified CRESS DNA viruses and observed that only a little less than half of all sequences follow the expected patterns for nnt and nna relative overrepresentation (Figure 8). However, nnt overrepresentation in the virion-sense and nna overrepresentation in anti-sense are the most common patterns of overrepresentation. Around 12% of unclassified sequences in both the virion-sense and anti-sense data sets have inverse relative codon overrepresentation. It is possible that strand assignments for these annotations are incorrect despite our best efforts to correct them by predicting the direction of putative origin of replication motifs. Alternatively, they can be properly assigned, and their codon usage is a result of distinct factors from those acting at third codon positions across Cressdnaviricota. Nonetheless, results are generally consistent with those observed for established CRESS DNA virus genera. We found 13 unclassified CRESS DNA viruses with circovirus-like genome organization based on both origin predictions, and nnt/nna relative codon overrepresentation profiles. As there was substantial nucleotide divergence among the Rep gene sequences for the 13 viruses, they likely represent candidate members of more than one novel family in Cressdnaviricota. Therefore, it appears that viruses encoding Rep in the virion-sense and CP in the anti-sense are more common than what our current taxonomy suggests, where of more than 30 genera only Circovirus uses this genomic orientation.
Our results suggest that codon preference across Cressdnaviricota cannot be solely attributed to c→t mutation bias. We explore some of the factors that could potentially influence CUB in viruses of Cressdnaviricota.
Mutational biases implied by significant negative correlations between codon types are present in all data sets, suggesting base composition constraints play an integral role in shaping codon usage for all genera. A c→t bias is suggested to be present across all data sets based on negative correlations between nnc and nnt in virion-sense and nng and nna in anti-sense for all genera. Yet, there are often other codon types that are negatively correlated with each other, indicating that multiple mutational biases might be acting simultaneously. While c→t transitions represent by far the most common type of mutation in single-stranded DNA (31, 61), evolution experiments with geminiviruses have revealed that, after c→t, g→t substitutions occur more frequently than others (56–59). Interestingly, van der Walt et al. (56) and Monjane etal. (57) showed that these mutations occur in a strand-specific manner in both mastrevirus and begomovirus species, with CP exhibiting a distinct overrepresentation of g→t substitutions, which may also be caused by oxidative damage to nucleotides. While long-term g→t bias will act synergistically with c→t to enrich nnt codons in the virion-sense CP, our results show that mastrevirus CPs are enriched in nng and depleted in nna. Since no mutational bias can explain the observed pattern, we hypothesize that some form of selection accounts for the codon usage of mastrevirus CP sequences (discussed in the next section). In turn, the mastrevirus CUB signatures indicate that measured substitution biases from previous short-term evolution experiments (56, 57) do not necessarily reflect long-term evolution of synonymous sites in geminivirus genomes.
Viruses rely on the host tRNA machinery for the synthesis of viral proteins and are subjected to different selective pressures imposed by the intracellular environment as well as a variety of antiviral immune systems (62–64). Selection pressures might act in synergy or antagonistically with mutational biases to shape codon usage and could partially explain the genus and strand-specific patterns observed across the diverse host range of members of Cressdnaviricota. Since host ranges are not well-defined for most CRESS DNA viruses, it is hard to make interpretations from in silico, comparative analyses such as this one. However, we provide some hypotheses based on the existing literature for CRESS DNA viruses.
PCA analysis of RSCU in circovirus genes revealed that some variation in codon usage is host-dependent, since PC2 produced clusters based on host source and separated avian circoviruses from fish and mammalian circoviruses. A previous CUB study also identified distinct patterns of codon usage between avian and mammalian circoviruses, citing a strong deviation from mutational patterns in mammalian viruses as the determinant of the differences between the two groups (65). A more recent study revealed that tRNA genes are drastically reduced in number and complexity in birds when compared to other vertebrates, but they exhibit overall similar tRNA usage patterns (66). This suggests that birds have evolved to use their limited tRNA inventory more efficiently, and this optimization may in turn exert selective pressures on codon usage for avian viruses. Overall, selection acting in different directions between avian and mammalian circoviruses might partially explain the diverged codon usage patterns. A more detailed, host-based examination of codon usage with available circoviruses might provide further insights into factors responsible for this demarcation.
Translational selection can promote optimal viral codon usage and translation through assimilation to the host tRNA pool (67, 68), and may partially explain the strand-specific differences observed in the geminiviruses in this study. Begomo- and mastreviruses are both geminiviruses and infect plants, yet they have distinct host ranges: begomoviruses infect dicots, and mastreviruses are largely restricted to monocots. Both are similarly biased against nna codons in the virion-sense CP and against nng in the anti-sense Rep sequences (Figures S1, S4). However, they do show one notable distinction in CP: begomoviruses are biased in favor of nnt while mastreviruses are enriched for nng codons. Previous codon usage analyses revealed that mastreviruses’ monocot hosts prefer nng and nnc codons while begomoviruses’ dicot hosts prefer nna and nnt codons in highly expressed genes (30, 69, 70). Cardinale etal. (30) showed that the RSCU values of begomo- and mastrevirus CP sequences are well-correlated to their respective hosts’ RSCU while Rep sequences are not. Since likeness to host codon usage is not consistent throughout a virus genome and is generally stronger in structural proteins (23), it is plausible to suggest that there is host-imposed translational selection acting mostly on CP, leading to an increase in nnt content in begomoviruses and nng content in mastreviruses. This is further supported by the fact that we observed a much higher nnt content in dicot-infecting mastreviruses than in monocot-infecting ones (Figure S4A; Supplemental File 3).
Gemycircularviruses showed a complicated pattern of codon usage that largely did not conform to results observed for the rest of the examined genera (Figure 7). Principal components from our RSCU PCA jointly accounted for only 23.4% of the variance in the data, which points to very divergent RSCU profiles between samples. The most well-characterized gemycircularvirus replicates in a fungal host, a mycophagous insect, the cells of a distantly related moth and potentially a variety of other hosts (53). It is possible that gemycircularviruses are generalists which have not adapted to any one specific host codon usage, as viruses with a narrow host spectrum match their hosts’ tRNA pools better and exhibit stronger CUB than those with wide host ranges (71). Due to their putative ability to infect such a broad diversity of hosts, gemycircularviruses present a potential system to explore host-specific factors influencing codon usage in ssDNA viruses.
Nucleic acid secondary structures can be functionally important in virus genomes, which means that they can be targeted by selection to preserve structure integrity (72–75). Mutations disrupting predicted secondary structures have been found to reliably revert and restabilize base pairings in mastreviruses (76). Additionally, median substitutions rates of paired 3rd codon position nucleotides within predicted highly conserved secondary structures are significantly lower than for unpaired nucleotides in circoviruses, begomoviruses, mastreviruses and capulaviruses (77–80). Muhire etal. (77) also observed reduced mutation frequencies for paired sites in short term evolution experiments for mastreviruses. These results indicate that selection acts to conserve secondary structures in CRESS DNA viruses, even in extremely short time scales, and that perhaps secondary structures can protect paired nucleotides from oxidative damage. Correlating secondary structure predictions in coding regions with nucleotide frequencies in silent codon sites may inform whether secondary structures play a significant role in shaping global codon usage for CRESS DNA viruses.
Two outliers were detected within Cyclovirus in our hypergeometric test analyses: a Rep sequence from Spider associated cyclovirus 1 (SpACyV-1) and the Rep and CP sequences for Duck associated cyclovirus 1 (DuACyV-1). Rep phylogenetic analyses reveal SpACyV-1 as an intermediate between circo- and cycloviruses and its inclusion into Cyclovirus is not phylogenetics-based but rather informed by its inferred genome orientation (81). The DuACyV-1 CP shows no significant evolutionary relationship to other cyclovirus CPs and clusters with unclassified CRESS DNA viruses (82). The Rep, however, shares a 98% nucleotide sequence identity with a partial Rep sequence (~470 nucleotides in length) isolated from human faeces (83), which groups closely with circoviruses rather than other cycloviruses (84). These results suggest that DuACyV-1 might be a recombinant involving a circovirus-like Rep and a CP diverged from other cycloviruses both phylogenetically and in terms of RSCU. Potentially, both DuACyV-1 and SpACyV-1 warrant investigation into new genera within Circoviridae.
Ambisense genomes of viruses classified within Cressdnavircota display genus- and strand-specific patterns of codon usage. While influenced by different factors in each genus, hypergeometric tests reveal consistent relative nna codon overrepresentation in anti-sense sequences and, to a lesser extent, nnt codon overrepresentation in the virion-sense, suggesting it could potentially serve as a complementary tool to verify genome organization of novel sequences. Additionally, there is an even more constant absence of relative nna codon overrepresentation in the virion-sense and a lack or relative nnt codon overrepresentation in the anti-sense strands of these viruses (except for gemycircularviruses), which indicates that there is a low probability of incorrect assignment. When applied to unclassified CRESS DNA viruses, hypergeometric test results provide support for the notion that, while CP is more commonly encoded in the virion-sense strand across Cressdnaviricota, more viral genera with circovirus-like genomes (i.e., harboring a virion-sense Rep ORF) must exist. We hope that this comprehensive analysis of codon composition across Cressdnaviricota can provide a framework for more fine-scale, experimental approaches to understand the evolution of codon preference for such a diverse group of viruses.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
AC-B and SD conceived the project, AC-B conducted the analyses, AC-B and SD wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by an HHMI Gilliam Fellowship to AC-B and SD (#GT13634) and NSF OIA 1545553 to SD. AC-B was also supported by a Rutgers SUPERGrad fellowship and the Rutgers Initiative for Maximizing Student Development (NIH R25GM055145).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Lele Zhao, Erik Lavington, J Steen Hoyer and Linda Hanley-Bowdoin for useful discussions and Hector Loyola Irizarry and Lauren Chukrallah for assistance with preliminary analyses.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.899608/full#supplementary-material
1. Zhang YZ, Chen YM, Wang W, Qin XC, Holmes EC. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu Rev Virol (2019) 6:119–39. doi: 10.1146/annurev-virology-092818-015851
2. Zhao L, Rosario K, Breitbart M, Duffy S. Eukaryotic Circular Rep-Encoding Single-Stranded DNA (CRESS DNA) Viruses: Ubiquitous Viruses With Small Genomes and a Diverse Host Range. Adv Virus Res (2019) 103:71–133. doi: 10.1016/bs.aivir.2018.10.001
3. Rosario K, Duffy S, Breitbart M. A Field Guide to Eukaryotic Circular Single-Stranded DNA Viruses: Insights Gained From Metagenomics. Arch Virol (2012) 157:1851–71. doi: 10.1007/s00705-012-1391-y
4. Rosario K, Schenck RO, Harbeitner RC, Lawler SN, Breitbart M. Novel Circular Single-Stranded DNA Viruses Identified in Marine Invertebrates Reveal High Sequence Diversity and Consistent Predicted Intrinsic Disorder Patterns Within Putative Structural Proteins. Front Microbiol (2015) 6:696. doi: 10.3389/fmicb.2015.00696
5. Tisza MJ, Pastrana DV, Welch NL, Stewart B, Peretti A, Starrett GJ, et al. Discovery of Several Thousand Highly Diverse Circular DNA Viruses. Elife (2020) 9:1–26. doi: 10.7554/eLife.51971
6. Simmonds P, Adams MJ, Benko M, Breitbart M, Brister JR, Carstens EB, et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat Rev Microbiol (2017) 15:161–8. doi: 10.1038/nrmicro.2016.177
7. Aiewsakun P, Simmonds P. The Genomic Underpinnings of Eukaryotic Virus Taxonomy: Creating a Sequence-Based Framework for Family-Level Virus Classification. Microbiome (2018) 6:38. doi: 10.1186/s40168-018-0422-7
8. Krupovic M, Varsani A, Kazlauskas D, Breitbart M, Delwart E, Rosario K, et al. Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses With Single-Stranded, Circular DNA Genomes. J Virol (2020) 94:1–14. doi: 10.1128/JVI.00582-20
9. Duffy S, Holmes EC. Phylogenetic Evidence for Rapid Rates of Molecular Evolution in the Single-Stranded DNA Begomovirus Tomato Yellow Leaf Curl Virus. J Virol (2008) 82:957–65. doi: 10.1128/JVI.01929-07
10. Duffy S, Holmes EC. Validation of High Rates of Nucleotide Substitution in Geminiviruses: Phylogenetic Evidence From East African Cassava Mosaic Viruses. J Gen Virol (2009) 90:1539–47. doi: 10.1099/vir.0.009266-0
11. Firth C, Charleston MA, Duffy S, Shapiro B, Holmes EC. Insights Into the Evolutionary History of an Emerging Livestock Pathogen: Porcine Circovirus 2. J Virol (2009) 83:12813–21. doi: 10.1128/JVI.01719-09
12. Grigoras I, Timchenko T, Grande-Perez A, Katul L, Vetten HJ, Gronenborn B. High Variability and Rapid Evolution of a Nanovirus. J Virol (2010) 84:9105–17. doi: 10.1128/JVI.00607-10
13. Lefeuvre P, Lett JM, Varsani A, Martin DP. Widely Conserved Recombination Patterns Among Single-Stranded DNA Viruses. J Virol (2009) 83:2697–707. doi: 10.1128/JVI.02152-08
14. Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A. Recombination in Eukaryotic Single Stranded DNA Viruses. Viruses (2011) 3(9):1699–738. doi: 10.3390/v3091699
15. Roux S, Enault F, Bronner G, Vaulot D, Forterre P, Krupovic M. Chimeric Viruses Blur the Borders Between the Major Groups of Eukaryotic Single-Stranded DNA Viruses. Nat Commun (2013) 4:2700. doi: 10.1038/ncomms3700
16. Kazlauskas D, Varsani A, Krupovic M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses (2018) 10:1–11. doi: 10.3390/v10040187
17. Rosario K, Breitbart M, Harrach B, Segales J, Delwart E, Biagini P, et al. Revisiting the Taxonomy of the Family Circoviridae: Establishment of the Genus Cyclovirus and Removal of the Genus Gyrovirus. Arch Virol (2017) 162:1447–63. doi: 10.1007/s00705-017-3247-y
18. Hershberg R, Petrov DA. Selection on Codon Bias. Annu Rev Genet (2008) 42:287–99. doi: 10.1146/annurev.genet.42.110807.091442
19. Sharp PM, Emery LR, Zeng K. Forces That Influence the Evolution of Codon Bias. Philos Trans R Soc Lond B Biol Sci (2010) 365:1203–12. doi: 10.1098/rstb.2009.0305
20. Shah P, Gilchrist MA. Explaining Complex Codon Usage Patterns With Selection for Translational Efficiency, Mutation Bias, and Genetic Drift. Proc Natl Acad Sci U.S.A. (2011) 108(25):10231–6. doi: 10.1073/pnas.1016719108
21. Ata G, Wang H, Bai H, Yao X, Tao S. Edging on Mutational Bias, Induced Natural Selection From Host and Natural Reservoirs Predominates Codon Usage Evolution in Hantaan Virus. Front Microbiol (2021) 12:699788. doi: 10.3389/fmicb.2021.699788
22. Lucks JB, Nelson DR, Kudla GR, Plotkin JB. Genome Landscapes and Bacteriophage Codon Usage. PloS Comput Biol (2008) 4:e1000001. doi: 10.1371/journal.pcbi.1000001
23. Bahir I, Fromer M, Prat Y, Linial M. Viral Adaptation to Host: A Proteome-Based Analysis of Codon Usage and Amino Acid Preferences. Mol Syst Biol (2009) 5:311. doi: 10.1038/msb.2009.71
24. Takata MA, Goncalves-Carneiro D, Zang TM, Soll SJ, York A, Blanco-Melo D, et al. CG Dinucleotide Suppression Enables Antiviral Defence Targeting non-Self RNA. Nature (2017) 550:124–7. doi: 10.1038/nature24039
25. Lin YT, Chiweshe S, Mccormick D, Raper A, Wickenhagen A, Defillipis V, et al. Human Cytomegalovirus Evades ZAP Detection by Suppressing CpG Dinucleotides in the Major Immediate Early 1 Gene. PloS Pathog (2020) 16:e1008844. doi: 10.1371/journal.ppat.1008844
26. Jenkins GM, Holmes EC. The Extent of Codon Usage Bias in Human RNA Viruses and its Evolutionary Origin. Virus Res (2003) 92:1–7. doi: 10.1016/S0168-1702(02)00309-X
27. Deb B, Uddin A, Chakraborty S. Codon Usage Pattern and its Influencing Factors in Different Genomes of Hepadnaviruses. Arch Virol (2020) 165:557–70. doi: 10.1007/s00705-020-04533-6
28. Cardinale DJ, Duffy S. Single-Stranded Genomic Architecture Constrains Optimal Codon Usage. Bacteriophage (2011) 1:219–24. doi: 10.4161/bact.1.4.18496
29. Chithambaram S, Prabhakaran R, Xia X. The Effect of Mutation and Selection on Codon Adaptation in Escherichia Coli Bacteriophage. Genetics (2014) 197:301–15. doi: 10.1534/genetics.114.162842
30. Cardinale DJ, Derosa K, Duffy S. Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses. Viruses (2013) 5:162–81. doi: 10.3390/v5010162
31. Frederico LA, Kunkel TA, Shaw BR. A Sensitive Genetic Assay for the Detection of Cytosine Deamination: Determination of Rate Constants and the Activation Energy. Biochemistry (1990) 29:2532–7. doi: 10.1021/bi00462a015
32. Li W, Godzik A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics (2006) 22:1658–9. doi: 10.1093/bioinformatics/btl158
33. Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics (2010) 26:680–2. doi: 10.1093/bioinformatics/btq003
34. Pratt AA, Torrance EL, Kasun GW, Stedman KM, de la Higuera I. StemLoop-Finder: A Tool for the Detection of DNA Hairpins With Conserved Motifs. Microbiol Resour Announc (2021) 10:e0042421. doi: 10.1128/MRA.00424-21
35. R Devleopment Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2020).
36. Xia X. DAMBE6: New Tools for Microbial Genomics, Phylogenetics, and Molecular Evolution. J Hered (2017) 108:431–7. doi: 10.1093/jhered/esx033
37. Sharp PM, Tuohy TM, Mosurski KR. Codon Usage in Yeast: Cluster Analysis Clearly Differentiates Highly and Lowly Expressed Genes. Nucleic Acids Res (1986) 14:5125–43. doi: 10.1093/nar/14.13.5125
38. Gu Z, Eils R, Schlesner M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics (2016) 32:2847–9. doi: 10.1093/bioinformatics/btw313
39. Wright F. The 'Effective Number of Codons' Used in a Gene. Gene (1990) 87:23–9. doi: 10.1016/0378-1119(90)90491-9
40. Sun X, Yang Q, Xia X. An Improved Implementation of Effective Number of Codons (Nc). Mol Biol Evol (2013) 30:191–6. doi: 10.1093/molbev/mss201
42. Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. J Mol Biol (1982) 157:105–32. doi: 10.1016/0022-2836(82)90515-0
43. Lobry JR, Gautier C. Hydrophobicity, Expressivity and Aromaticity are the Major Trends of Amino-Acid Usage in 999 Escherichia Coli Chromosome-Encoded Genes. Nucleic Acids Res (1994) 22:3174–80. doi: 10.1093/nar/22.15.3174
44. Muhire BM, Varsani A, Martin DP. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PloS One (2014) 9:1–8. doi: 10.1371/journal.pone.0108277
45. Dennis TPW, Flynn PJ, De Souza WM, Singer JB, Moreau CS, Wilson SJ, et al. Insights Into Circovirus Host Range From the Genomic Fossil Record. J Virol (2018) 92:1–9. doi: 10.1128/JVI.00145-18
46. Ellis J. Porcine Circovirus: A Historical Perspective. Vet Pathol (2014) 51:315–27. doi: 10.1177/0300985814521245
47. Rojas MR, Macedo MA, Maliano MR, Soto-Aguilar M, Souza JO, Briddon RW, et al. World Management of Geminiviruses. Annu Rev Phytopathol (2018) 56:637–77. doi: 10.1146/annurev-phyto-080615-100327
48. Fondong VN. Geminivirus Protein Structure and Function. Mol Plant Pathol (2013) 14:635–49. doi: 10.1111/mpp.12032
49. Varsani A, Krupovic M. Smacoviridae: A New Family of Animal-Associated Single-Stranded DNA Viruses. Arch Virol (2018) 163:2005–15. doi: 10.1007/s00705-018-3820-z
50. Krupovic M, Varsani A. A 2021 Taxonomy Update for the Family Smacoviridae. Arch Virol (2021) 166:3245–53. doi: 10.1007/s00705-021-05224-6
51. Diez-Villasenor C, Rodriguez-Valera F. CRISPR Analysis Suggests That Small Circular Single-Stranded DNA Smacoviruses Infect Archaea Instead of Humans. Nat Commun (2019) 10:294. doi: 10.1038/s41467-018-08167-w
52. Krupovic M, Ghabrial SA, Jiang D, Varsani A. Genomoviridae: A New Family of Widespread Single-Stranded DNA Viruses. Arch Virol (2016) 161:2633–43. doi: 10.1007/s00705-016-2943-3
53. Liu S, Xie J, Cheng J, Li B, Chen T, Fu Y, et al. Fungal DNA Virus Infects a Mycophagous Insect and Utilizes it as a Transmission Vector. Proc Natl Acad Sci USA (2016) 113:12803–8. doi: 10.1073/pnas.1608013113
54. Zhao L, Lavington E, Duffy S. Truly Ubiquitous CRESS DNA Viruses Scattered Across the Eukaryotic Tree of Life. J Evol Biol (2021) 34:1901–16. doi: 10.1111/jeb.13927
55. Rokyta DR, Joyce P, Caudle SB, Wichman HA. An Empirical Test of the Mutational Landscape Model of Adaptation Using a Single-Stranded DNA Virus. Nat Genet (2005) 37:441–4. doi: 10.1038/ng1535
56. Van Der Walt E, Martin DP, Varsani A, Polston JE, Rybicki EP. Experimental Observations of Rapid Maize Streak Virus Evolution Reveal a Strand-Specific Nucleotide Substitution Bias. Virol J (2008) 5:104. doi: 10.1186/1743-422X-5-104
57. Monjane AL, Pande D, Lakay F, Shepherd DN, van der Walt E, Lefeuvre P, et al. Adaptive Evolution by Recombination is Not Associated With Increased Mutation Rates in Maize Streak Virus. BMC Evol Biol (2012) 12:252. doi: 10.1186/1471-2148-12-252
58. Sanchez-Campos S, Dominguez-Huerta G, Diaz-Martinez L, Tomas DM, Navas-Castillo J, Moriones E, et al. Differential Shape of Geminivirus Mutant Spectra Across Cultivated and Wild Hosts With Invariant Viral Consensus Sequences. Front Plant Sci (2018) 9:932. doi: 10.3389/fpls.2018.00932
59. Aimone CD, Lavington E, Hoyer JS, Deppong DO, Mickelson-Young L, Jacobson A, et al. Population Diversity of Cassava Mosaic Begomoviruses Increases Over the Course of Serial Vegetative Propagation. J Gen Virol (2021) 102:1–17. doi: 10.1099/jgv.0.001622
60. Ortega-Del Campo S, Grigoras I, Timchenko T, Gronenborn B, Grande-Perez A. Twenty Years of Evolution and Diversification of Digitaria Streak Virus in Digitaria Setigera. Virus Evol (2021) 7:veab083. doi: 10.1093/ve/veab083
61. Lindahl T. Instability and Decay of the Primary Structure of DNA. Nature (1993) 362:709–15. doi: 10.1038/362709a0
62. Walsh D, Mohr I. Viral Subversion of the Host Protein Synthesis Machinery. Nat Rev Microbiol (2011) 9:860–75. doi: 10.1038/nrmicro2655
63. Callens M, Pradier L, Finnegan M, Rose C, Bedhomme S. Read Between the Lines: Diversity of Nontranslational Selection Pressures on Local Codon Usage. Genome Biol Evol (2021) 13:1–14. doi: 10.1093/gbe/evab097
64. Pinto RM, Bosch A. The Codon Usage Code for Cotranslational Folding of Viral Capsids. Genome Biol Evol (2021) 13:1–5. doi: 10.1093/gbe/evab089
65. Franzo G, Segales J, Tucciarone CM, Cecchinato M, Drigo M. The Analysis of Genome Composition and Codon Bias Reveals Distinctive Patterns Between Avian and Mammalian Circoviruses Which Suggest a Potential Recombinant Origin for Porcine Circovirus 3. PloS One (2018) 13:e0199950. doi: 10.1371/journal.pone.0199950
66. Ottenburghs J, Geng K, Suh A, Kutter C. Genome Size Reduction and Transposon Activity Impact tRNA Gene Diversity While Ensuring Translational Stability in Birds. Genome Biol Evol (2021) 13:1–16. doi: 10.1093/gbe/evab016
67. Zhi N, Wan Z, Liu X, Wong S, Kim DJ, Young NS, et al. Codon Optimization of Human Parvovirus B19 Capsid Genes Greatly Increases Their Expression in Nonpermissive Cells. J Virol (2010) 84:13059–62. doi: 10.1128/JVI.00912-10
68. Albers S, Czech A. Exploiting tRNAs to Boost Virulence. Life (Basel) (2016) 6:1–15. doi: 10.3390/life6010004
69. Campbell WH, Gowri G. Codon Usage in Higher Plants, Green Algae, and Cyanobacteria. Plant Physiol (1990) 92:1–11. doi: 10.1104/pp.92.1.1
70. Mazumdar P, Binti Othman R, Mebus K, Ramakrishnan N, Ann Harikrishna J. Codon Usage and Codon Pair Patterns in non-Grass Monocot Genomes. Ann Bot (2017) 120:893–909. doi: 10.1093/aob/mcx112
71. Tian L, Shen X, Murphy RW, Shen Y. The Adaptation of Codon Usage of +ssRNA Viruses to Their Hosts. Infect Genet Evol (2018) 63:175–9. doi: 10.1016/j.meegid.2018.05.034
72. Orozco BM, Hanley-Bowdoin L. A DNA Structure is Required for Geminivirus Replication Origin Function. J Virol (1996) 70:148–58. doi: 10.1128/jvi.70.1.148-158.1996
73. Zanini F, Neher RA. Quantifying Selection Against Synonymous Mutations in HIV-1 Env Evolution. J Virol (2013) 87:11843–50. doi: 10.1128/JVI.01529-13
74. Shimoike T, Hayashi T, Oka T, Muramatsu M. The Predicted Stem-Loop Structure in the 3'-End of the Human Norovirus Antigenomic Sequence is Required for its Genomic RNA Synthesis by its RdRp. J Biol Chem (2021) 297:101225. doi: 10.1016/j.jbc.2021.101225
75. Yan ZY, Fang L, Xu XJ, Cheng DJ, Yu CM, Wang DY, et al. A Predicted Stem Loop in Coat Protein-Coding Sequence of Tobacco Vein Banding Mosaic Virus Is Required for Efficient Replication. Phytopathology (2022) 112(2):441–51. doi: 10.1094/PHYTO-10-20-0463-R
76. Shepherd DN, Martin DP, Varsani A, Thomson JA, Rybicki EP, Klump HH. Restoration of Native Folding of Single-Stranded DNA Sequences Through Reverse Mutations: An Indication of a New Epigenetic Mechanism. Arch Biochem Biophys (2006) 453:108–22. doi: 10.1016/j.abb.2005.12.009
77. Muhire BM, Golden M, Murrell B, Lefeuvre P, Lett JM, Gray A, et al. Evidence of Pervasive Biologically Functional Secondary Structures Within the Genomes of Eukaryotic Single-Stranded DNA Viruses. J Virol (2014) 88:1972–89. doi: 10.1128/JVI.03031-13
78. Stenzel T, Piasecki T, Chrzastek K, Julian L, Muhire BM, Golden M, et al. Pigeon Circoviruses Display Patterns of Recombination, Genomic Secondary Structure and Selection Similar to Those of Beak and Feather Disease Viruses. J Gen Virol (2014) 95:1338–51. doi: 10.1099/vir.0.063917-0
79. Bernardo P, Muhire B, Francois S, Deshoux M, Hartnady P, Farkas K, et al. Molecular Characterization and Prevalence of Two Capulaviruses: Alfalfa Leaf Curl Virus From France and Euphorbia Caput-Medusae Latent Virus From South Africa. Virology (2016) 493:142–53. doi: 10.1016/j.virol.2016.03.016
80. Stenzel T, Dziewulska D, Muhire BM, Hartnady P, Kraberger S, Martin DP, et al. Recombinant Goose Circoviruses Circulating in Domesticated and Wild Geese in Poland. Viruses (2018) 10:1–13. doi: 10.3390/v10030107
81. Rosario K, Mettel KA, Benner BE, Johnson R, Scott C, Yusseff-Vanegas SZ, et al. Virus Discovery in All Three Major Lineages of Terrestrial Arthropods Highlights the Diversity of Single-Stranded DNA Viruses Associated With Invertebrates. PeerJ (2018) 6:e5761. doi: 10.7717/peerj.5761
82. Kaszab E, Lengyel G, Marton S, Dan A, Banyai K, Feher E. Occurrence and Genetic Diversity of CRESS DNA Viruses in Wild Birds: A Hungarian Study. Sci Rep (2020) 10:7036. doi: 10.1038/s41598-020-63795-x
83. Feher E, Kaszab E, Forro B, Bali K, Marton S, Lengyel G, et al. Genome Sequence of a Mallard Duck Origin Cyclovirus, DuACyV-1. Arch Virol (2017) 162:3925–9. doi: 10.1007/s00705-017-3566-z
Keywords: mutational bias, single-stranded DNA virus, translational selection, CRESS DNA viruses, strand specificity, hypergeometric test
Citation: Crespo-Bellido A and Duffy S (2022) Strand-Specific Patterns of Codon Usage Bias Across Cressdnaviricota. Front.Virol. 2:899608. doi: 10.3389/fviro.2022.899608
Received: 18 March 2022; Accepted: 20 May 2022;
Published: 17 June 2022.
Edited by:
Rafael Sanjuán, University of Valencia, SpainReviewed by:
F. Murilo Zerbini, Universidade Federal de Viçosa, BrazilCopyright © 2022 Crespo-Bellido and Duffy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siobain Duffy, ZHVmZnlAc2Vicy5ydXRnZXJzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.