 Maria Elena Turba1
Maria Elena Turba1 Domenico Mion2
Domenico Mion2 Stavros Papadimitriou3Francesca Taddei4Giorgio Dirani4
Stavros Papadimitriou3Francesca Taddei4Giorgio Dirani4 Vittorio Sambri4,5
Vittorio Sambri4,5 Fabio Gentilini2*
Fabio Gentilini2*- 1Xenturion SRL, Forlì (Forlì-Cesena), Italy
- 2Department of Veterinary Medical Sciences, (DIMEVET), University of Bologna, Bologna, Italy
- 3ADS Biotec Inc., Omaha, NE, United States
- 4Unit of Microbiology, The Great Romagna Hub Laboratory, Cesena (Forlì-Cesena), Italy
- 5Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Bologna, Italy
Mutations in the receptor binding domain (RBD) of SARS-CoV-2 alter the infectivity, pathogenicity, and transmissibility of new variants of concern (VOCs). In addition, those mutations cause immune escape, undermining the population immunity induced by ongoing mass vaccination programs. There is an urgent need for novel strategies and techniques aimed at the surveillance of the active emergence and spread of the VOCs. The aim of this study was to provide a quick, cheap and straightforward denaturing high-performance liquid chromatography (DHPLC) method for the prompt identification of the SARS-CoV-2 VOCs. Two PCRs were designed to target the RBD region, spanning residues N417 through N501 of the Spike protein. Furthermore, a DHPLC screening analysis was set up. The screening consisted of mixing the unknown sample with a standard sample of a known variant, denaturing at high temperature, renaturing at room temperature followed by a 2-minute run using the WAVE DHPLC system to detect the heteroduplexes which invariably form whenever the unknown sample has a nucleotide difference with respect to the standard used. The workflow was able to readily detect all the variants including B.1.1.7, P.1, B.1.585 B.1. 617.2 and lineages at a very affordable cost. The DHPLC analysis was robust being able to identify variants, even in the case of samples with very unbalanced target concentrations including those samples at the limit of detection. This approach has the potential of greatly expediting surveillance of the SARS-CoV-2 variants.
Introduction
The SARS-CoV-2 genome is more stable than other RNA viruses thanks to the proofreading activity operated by a 3’-to-5’ exoribonuclease (nsp14-ExoN) during replication which reduces the error rate of RNA polymerase 100–1000-fold. This confers the capacity of maintaining its 30,000 nt genome to the virus without catastrophic mutational events hampering its integrity (1–3). Nonetheless, errors still occur in the SARS-CoV-2 genome at a higher rate than in eukaryotic cells and, together with high replication rates, allow for the accumulation of mutations in the viral genome including amino acid changes, truncations, or the loss of viral proteins (4–6). These changes may impact infectivity, pathogenicity, and transmissibility, and they could lead to higher fitness and undergo positive selection (5, 7–9).
The D614G substitution of the Spike (S) protein is the first and most investigated of the positively selected mutations. This mutation occurred early in the first months of 2020 and became rapidly prevalent worldwide due to the increased infectivity of the strains carrying it (4, 10–12).
In immunologically naïve COVID-19 patients, as those prevailing at the beginning of the pandemic, immune response exerts limited selection pressure on the virus transmission. However, as the COVID-19 pandemic continues, and vaccination programs expand, it is expected that the rapid rise of population-level immunity might exert a strong positive selection pressure for those mutations responsible for immune escape or combination thereof, prompting immune escape. In other words, the speed at which resistance against acquired immunity develops in the population increases substantially as the number of infected or vaccinated individuals increases As most vaccines exploit the immunogenicity of the Spike protein, due to its pivotal role in binding to the ACE2 cell receptor and entry into the host cell, the strict monitoring of mutations in the domains targeted by the neutralising antibodies should be implemented worldwide (13–17). Substitutions in the receptor binding domain (RBD) have emerged, and they are of particular concern due to the possibility of being responsible for immune escape (17). In fact, after the D614G variant, at least five variants (B.1.1.7 also known as the Alpha variant, B.1.351 also known as the Beta variant, P.1 also known as the Gamma variant, B.1.617.2 also known as the Delta variant, and B.1.1.529 also known as the Omicron variant) carrying several mutations in the RBD of the S protein (9, 18) have emerged, impacting the transmission dynamic and causing epidemic waves which succeeded each other. More specifically, these variants carry mutations of concern or mutations of interest in the 417, 440, 452, 477 478 and 484 493, 496, 498, 501 and 505 codons of the S protein which have strong implications for infectivity and immune evasion (19, 20).
Variants of concern (VOCs) which might escape vaccine immunity are being monitored by genomic surveillance based on next generation sequencing (NGS) implemented in wealthier and many developing countries. Mass-scale sequencing has had a dramatic impact associated with the pandemic; it has enabled epidemiologic surveillance allowing the phylogenetic analysis to trace the origin and the spread, and to predict the evolution of the epidemic waves (18, 21–25). However, the sequencing of all or of even most of the isolates is still far from a reality. To date, only 21 of 160 countries have sequenced more than 1% of the confirmed cases, and 83 of the 160 have sequenced less than 0.1% of the positive cases (19, 20). Precisely those countries which have limited diagnostic capability and even more limited sequencing possibilities are those having a population which is less vaccinated and those in which it is more likely that new VOCs might emerge.
There is an urgent need for methods capable of permitting affordable genomic surveillance in the real world. Alternatives have been proposed to be integrated with sequencing in order to more effectively monitor the VOCs (26–29). Heteroduplex analysis using denaturing high-performance liquid chromatography (DHPLC) is a fast, very sensitive, low cost and reliable technique for screening nucleic acid variations
It has been used for two decades in many different applications but, for the most part, for detecting cancer somatic mutations owing to its extreme analytical sensitivity (30); DHPLC discovers DNA variations by separating heteroduplex and homoduplex DNA fragments using ion-pair reverse-phase liquid chromatography (30). This technique is based on the assumption that when two distinct PCR-amplified DNA targets containing nucleotide variations are denatured by heating and then left to renature by cooling, heteroduplexes, due to cross-hybridisation between the mismatched strands, in addition to homoduplexes, are also formed. Heteroduplexes and homoduplexes are bound to a stationary phase and eluted by a denaturing gradient at a constant slightly denaturing temperature. Since heteroduplexes have a lower stability, they are eluted before homoduplexes, and they are detected as different peaks modifying the chromatogram shape by ultraviolet absorption.
The aim of this study was to set up and evaluate a very quick, accurate and straightforward DHPLC method for scanning the RBD of SARS-CoV-2 isolates and, eventually, to readily screen the SARS-CoV-2 VOCs to undergo full genome sequencing. The proposed workflow is intended to screen all the PCR positive samples using DHPLC in addition to those randomly undergoing full genome sequencing. All positive samples are examined with two overlapping endpoint PCRs targeting the RBD. The amplicons are then mixed with the standard represented by the predominant variant and assayed using DHPLC. Those samples positive for heteroduplexes at DHPLC then undergo sequencing of amplicons and/or for full genome sequencing. The overall workflow of the screening proposed is represented in Figure 1.
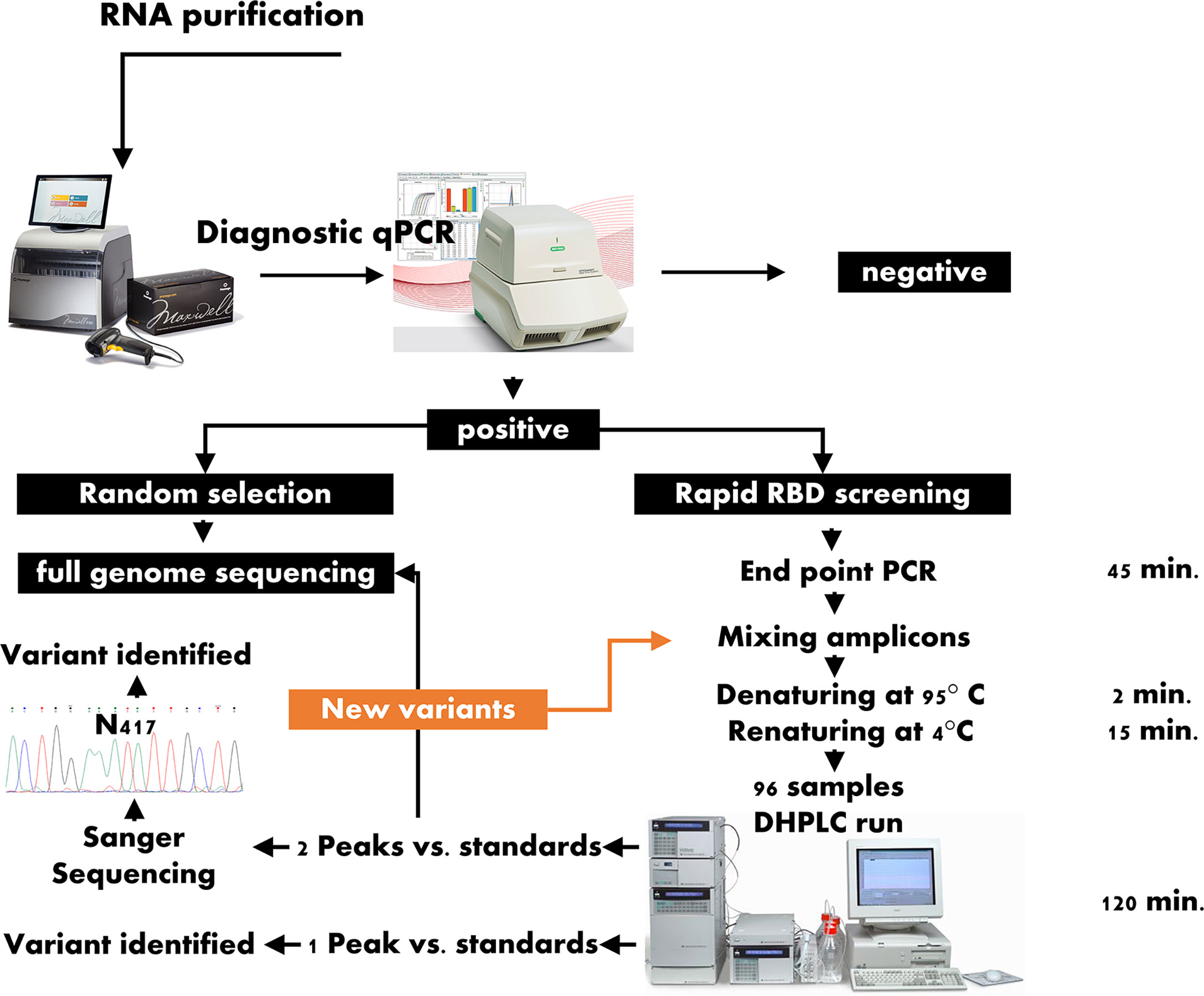
Figure 1 Workflow for the rapid screening of the variants of concern. RBD: SARS-CoV-2 Region Binding Domain; DHPLC, Denaturing high-performance liquid chromatography; contrary to the approach represented in the left branch of the scheme, which include the full genome sequencing of a random selection of positive samples, in the workflow proposed here, all samples positive to the diagnostic quantitative PCR are submitted for DHPLC screening; those samples forming heteroduplexes when mixed with a reference sample are submitted to either Sanger sequencing of the receptor binding domain or to full genome sequencing or to both.
Materials and Methods
Samples and Experimental Design
The experimental layout consisted of a preliminary setup of the DHPLC method using open-label lineage assigned SARS-CoV-2 isolates combined with a validation phase using blind-label isolates. In setting up the method, the positive samples were diagnosed with Covid-19 by means of a diagnostic molecular assay. Furthermore, some samples with lineage assigned from External Quality Assessment programs for Molecular Diagnostics were included in this step (Virus Genome Detection – SARS-CoV-2 VOC Analysis, Instand e.V.). The blind-label phase of the study used blind-label samples. These were represented by a group of 8 samples (blind group 1) assayed against the B.1.1.7 variant as a reference and an additional group of 19 samples (blind group 2) assayed against the B.1.617.2 variant as reference.
All the RT-PCR-positive samples had been lineage assigned by means of sequencing using either the Oxford Nanopore or the Illumina platform.
RNA Purification and Endpoint RT-PCR
For the DHPLC screening, RNA was re-extracted starting from 200 µL of UTM-RT (Copan) samples using a commercial kit (Maxwell® RSC Blood RNA Kit) with an automatic instrument (Maxwell RSC).
The iScript cDNA Synthesis kit (Bio-Rad) was used to reverse transcribe the RNA samples to cDNA. In brief, 10 µL of RNA were mixed with 4 µL of iScript Reaction mix (containing MMLV RNase H, dNTPs, oligo(dT)s and random primers), 1 µL of iScript Reverse Transcriptase and nuclease-free water to a final volume of 20 µL using the following protocol: 5 minutes at 25°C for priming, 20 minutes at 46°C for reverse transcription and 1 minute at 95°C for reverse transcriptase inactivation. The cDNA thereby obtained was consequently used as a template for PCR.
The PCR reactions for amplifying two subregions within the RBD of the S gene were designed using the Primer Quest web design tool (https://eu.idtdna.com/Primerquest/Home/Index). The PCR reactions were designed keeping the amplicon approximately 200-300 bp long and by including the 417 and 452 codons in the RBD_1 PCR, and the 452, 478, 484 and 501 codons in the RBD_2 PCR. The PCR primers were F_RBD1, 5’ TAAGTGTTATGGAGTGTCTCCTACTA 3’, R_RBD1, 5’ GGTTTGAGATTAGACTTCCTAAACAATC 3’, F_RBD2, 5’ TCTTGATTCTAAGGTTGGTGGTAA 3’ and R_RBD2, 5’ AGTTGCTGGTGCATGTAGAA 3’.
Briefly, the PCR reactions consisted of 1X Buffer (Phusion HF Buffer, Life Technologies), 0.2 mM MgCl2, 350 nM each of the forward and reverse primers, deoxynucleotide triphosphates (dNTPs) 250 µM, 0.4 U of Phusion Taq DNA polymerase (ThermoFisher), 2 µL of cDNA template brought up to a final volume of 20 µL with molecular biology grade water. The cycling programme for both conventional PCRs consisted of the following steps: 98°C for 30 s, 40 cycles at 98°C for 5 s, 63°C for 5 s and 72°C for 10 s, and a final elongation step at 72° C for 10 min.
DHPLC
In this study, heteroduplexes, if any, were generated by mixing the amplicons of the above-described endpoint PCRs of a standard sample (reference) and a test sample (unknown), by denaturing the mixed fragments at 95°C for 2 min, and then allowing them to renature at room temperature for 15 min. The reference samples may conveniently be the predominant variant identified in a given period. In this study, either B.1, B.1.177 or B.1.617.2 were used as standard. Each test sample was then run as such and mixed with the reference on an automated DHPLC apparatus (WAVEDHPLC system, ADS Biotec) equipped with a proprietary column (DNASep, ADS Biotec) which used alkylated non-porous polystyrene-divinylbenzene copolymer hydrophobic beads for high performance nucleic acid separations. Separation of the products was carried out by a mobile phase obtained by continuously mixing buffer B (0.1 mol/L triethylammonium acetate with 25% v/v in water acetonitrile) to buffer A (0.1 mol/L triethylammonium acetate), according to a gradient (Table 1) calculated by Navigator™ Software (ADS Biotec) and experimentally confirmed.
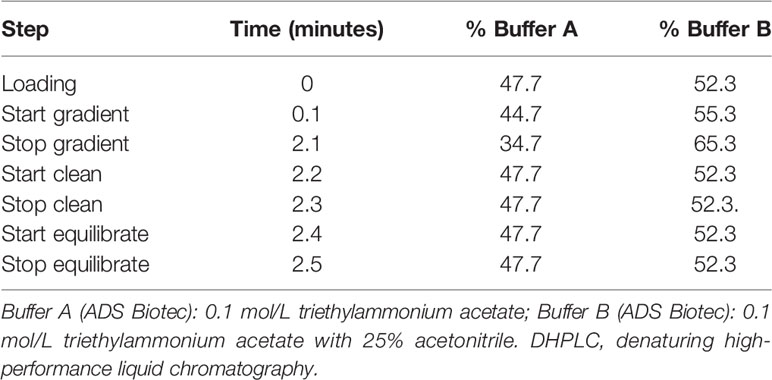
Table 1 The denaturing acetonitrile gradient used in DHPLC.
Likewise, the optimal oven temperatures for heteroduplex separation were determined using NavigatorSoftware (ADS Biotec) which gave a computer‐assisted determination of the melting profile and analytical conditions for each fragment which were then experimentally verified. In this study, partially denaturing temperatures of 55.5°C and 56.6°C were used for the DHPLC screening of both the RBD_1 and the RBD_2 amplicons.
In the DHPLC system, amplicons are screened for chromatogram shape and, in particular, for the presence or not of more than one peak with respect to the reference control. Since the method is not quantitative but qualitative, the peak height and thereafter the peak quantitation are not relevant. Conversely, the inherent impressive analytical sensitivity as low as 1%, allows for detecting a heteroduplex peak, even if the amount of mixed amplicons are not quantified and normalised.
Of course, to readily identify additional peaks unveiling a nucleotide variation with respect to the standard used, the test sample should be adequately amplified. To avoid the need for quantifying and normalising the nucleic acid of the variants and, hence, speeding up the workflow, a serial dilution experiment was carried out by spiking, in human saliva, the viral mRNA of a B1.617.2 variant sample serially diluted 1:10 in molecular biology grade water from 3.5.0x108 to 3.5x103 copies/mL. The copy number was quantified using a digital PCR method (31). The mRNA was then purified from the saliva samples spiked with the viral mRNA, and the mRNA was retrotranscribed according to the above-mentioned methods, The cDNA samples were then PCR amplified, checked for the presence of an amplification band and were then tested using DHPLC to assess the last dilution yielding a distinct heteroduplex peak.
Before each run, the column was prepared according to the manufacturer’s instructions. In particular, the Wave Low-range mutation control standard and the Wave High-range mutation control standard were used to check the apparatus before each run. One reference control was included in each assay run as well as a blank sample.
The data analysis was carried out using Navigator Software (ADS Biotec). Following the DHPLC screening, the same amplicons were purified and sequenced using an ABI310 automated sequencer (ThermoFisher).
According to the workflow in Figure 1, all positive samples should undergo the screening.
Results
The endpoint PCR assays, run by agarose gel electrophoresis, showed discrete bands without smearing or additional non-specific bands (Figure 2A).
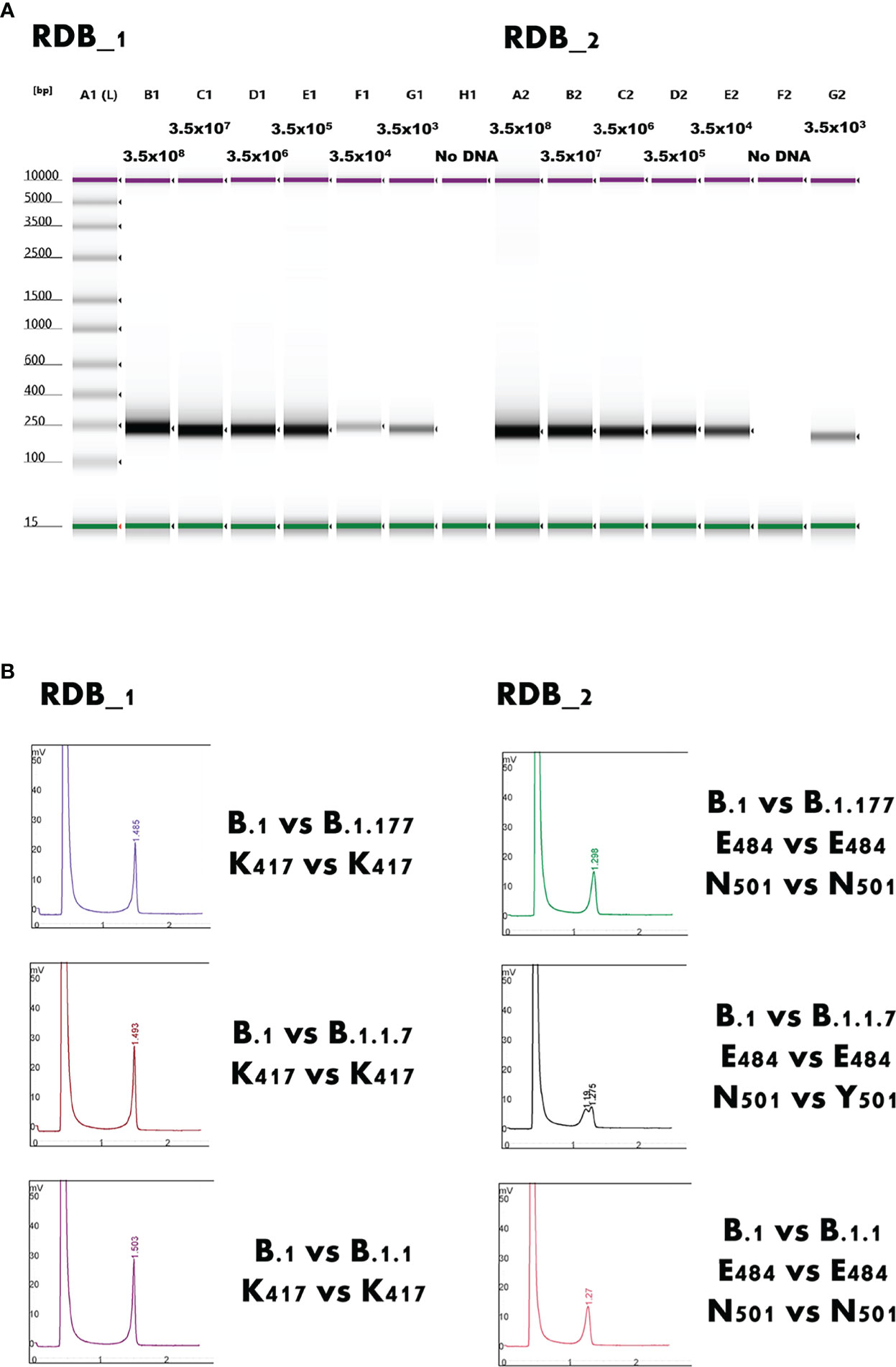
Figure 2 (A) Gel image of the electrophoresis analysis of the RBD_1 and RBD_2 end-point PCR. A1 lane shows the sizing ladder. Purple and green bands represent the upper and lower markers, respectively. The expected viral load expressed as copies/mL are indicated above each lane. Discrete sharp bands without smearing or additional non-specific bands are clearly identifiable in all cases. RBD: Receptor Binding Domain of SARS-CoV-2. (B) Detection of Y501 residue (B.1.1.7 variant) throughout heteroduplex detection between the region binding domain Target 2 (RBD_2) amplified from a B.1 strain and a B.1.1.7 strain. The mixing, denaturing at 95°C, and renaturing at room temperature between two mutations causes a heteroduplex formation detected by the Wave Denaturing high-performance liquid chromatography system, such as peak splitting N501 vs Y501. All other examples represent the typical appearance of homoduplexes.
Even samples with a very low viral load, close to the limit of detection of the diagnostic PCR, could be amplified using endpoint PCR and, notably, they could be readily screened using DHPLC (Figure 3). The method, carried out by mixing the B.1.117 and the emerging B.1.617.2 variants, even at a concentration of 7 copies/reaction, allowed identifying the heteroduplexes. In other words, DHPLC identified the heteroduplexes by mixing the D614G variant (B.1) with the B.1.1.7 variant (Alpha) as a reference (Figure 2B). Then, using the B.1.1.7 as a reference, the DHPLC assay identified the emerging VOCs P.1 (Gamma), B.1.585 (Beta) (Figure 4), and B.1.617.2 (Delta) (blind group #1; Figure 4). Finally using the dominant B.1.617.2 (Delta), DHPLC identified the most recently emerging VOC B.1.1.529 (Omicron) and the Delta sublineage B.1.617.2.39.1 (blind group #2; Figure 5).
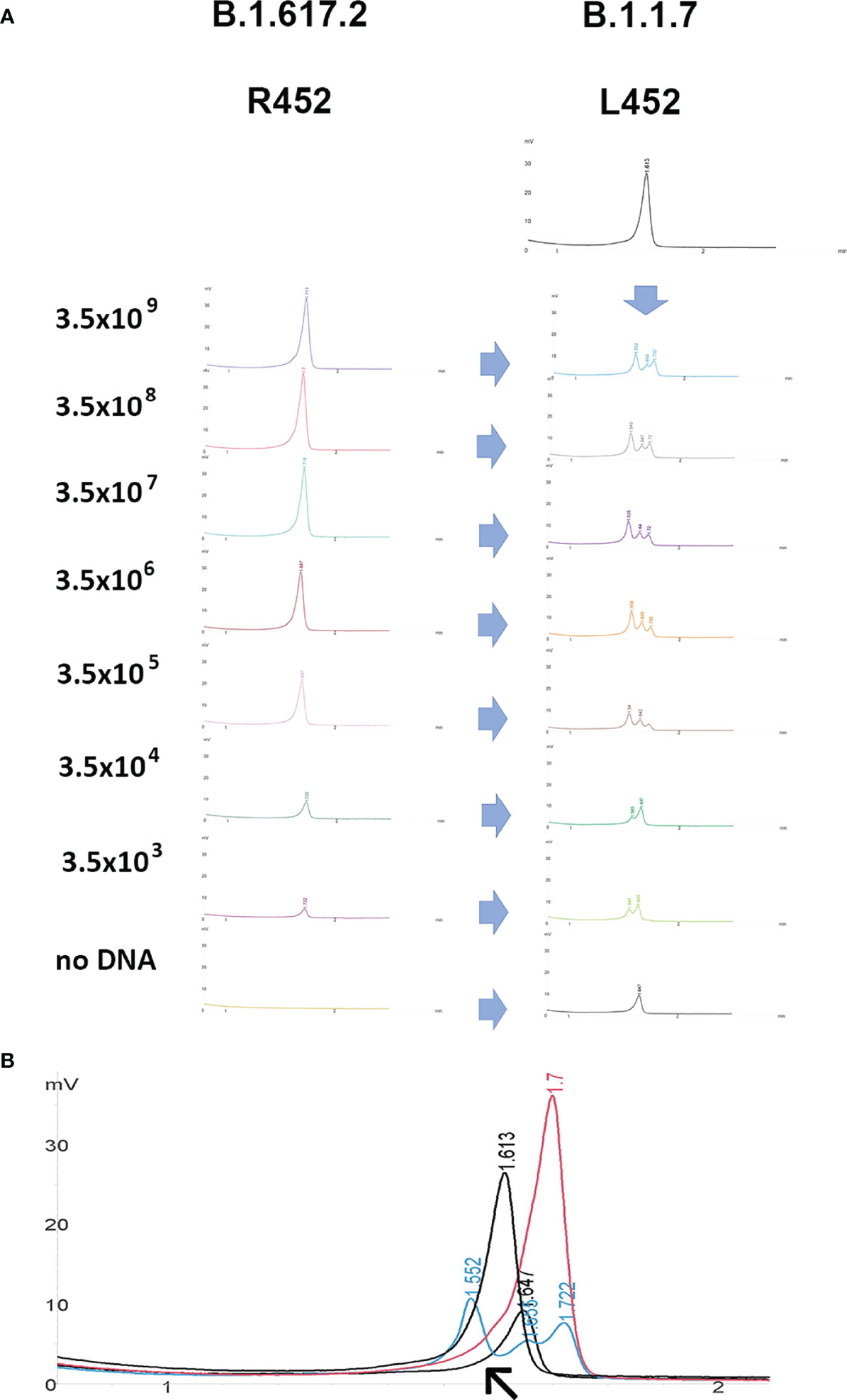
Figure 3 Denaturing high-performance liquid chromatography. (A) dilutional experiment to verify the effect of unbalanced concentration of amplicons. Example of the RBD_1 target screened by mixing amplicons obtained from samples containing different viral loads expressed as copies/mL (B.1.617.2) with the reference sample (B.1.1.7). The (B.1.1.7 and B.1.617.2) variants present a L452R amino acid substitution. The mixed samples containing heteroduplexes can be readily identified at any viral load. (B) Higher magnification of overlapping chromatograms of the reference sample (B.1.1.7; red curve) and the test (B.1.617.2; black curve) samples as such and mixed with each other to form heteroduplexes appearing as multiple peaks, modifying the chromatogram shape (cyan and grey curves). The no-DNA control mixed with the reference samples is also shown in the lower grey curve containing a single peak (arrow).
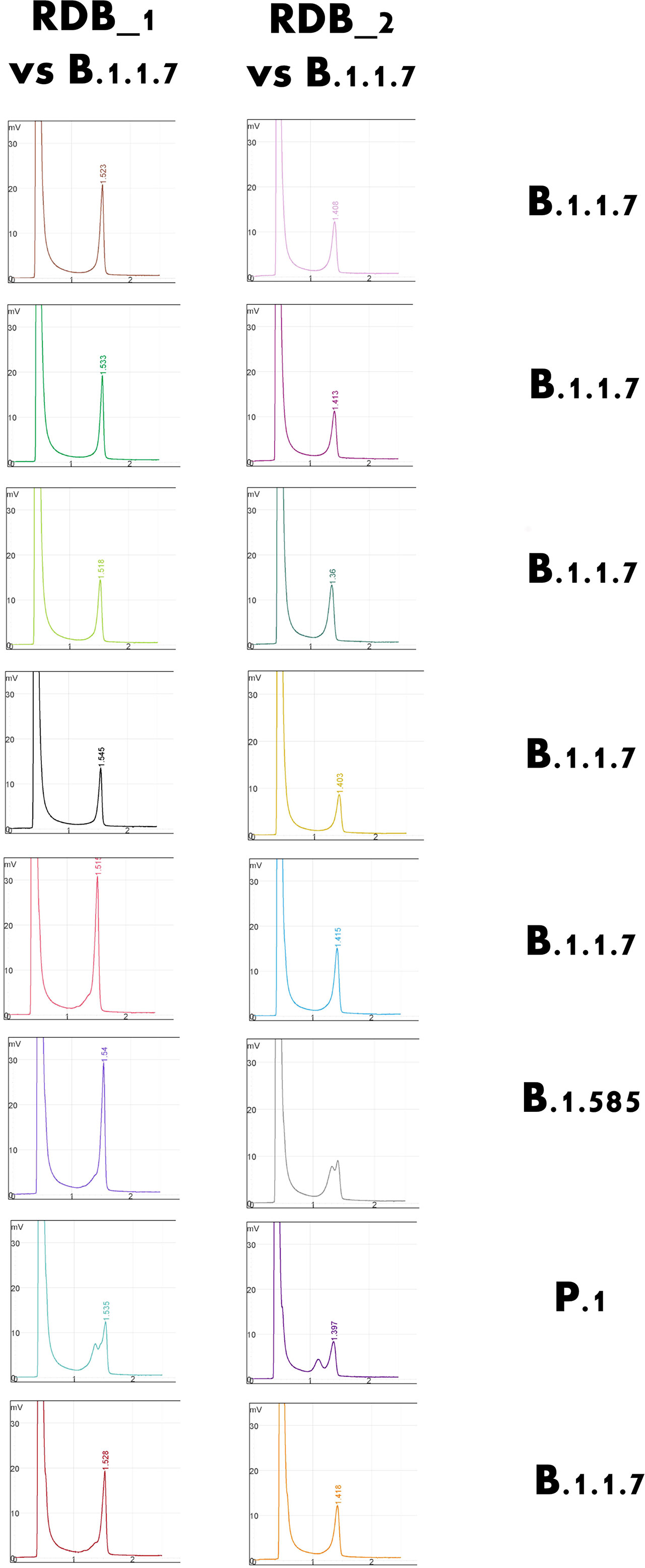
Figure 4 Selected example of the detection of rare variants of concern using the Denaturing High-Performance Liquid Chromatography system. Using the B.1.1.7 strain as a standard, the rarer B.1.585 (Nigerian) and P.1. (Brazilian) variants could be detected and confirmed.
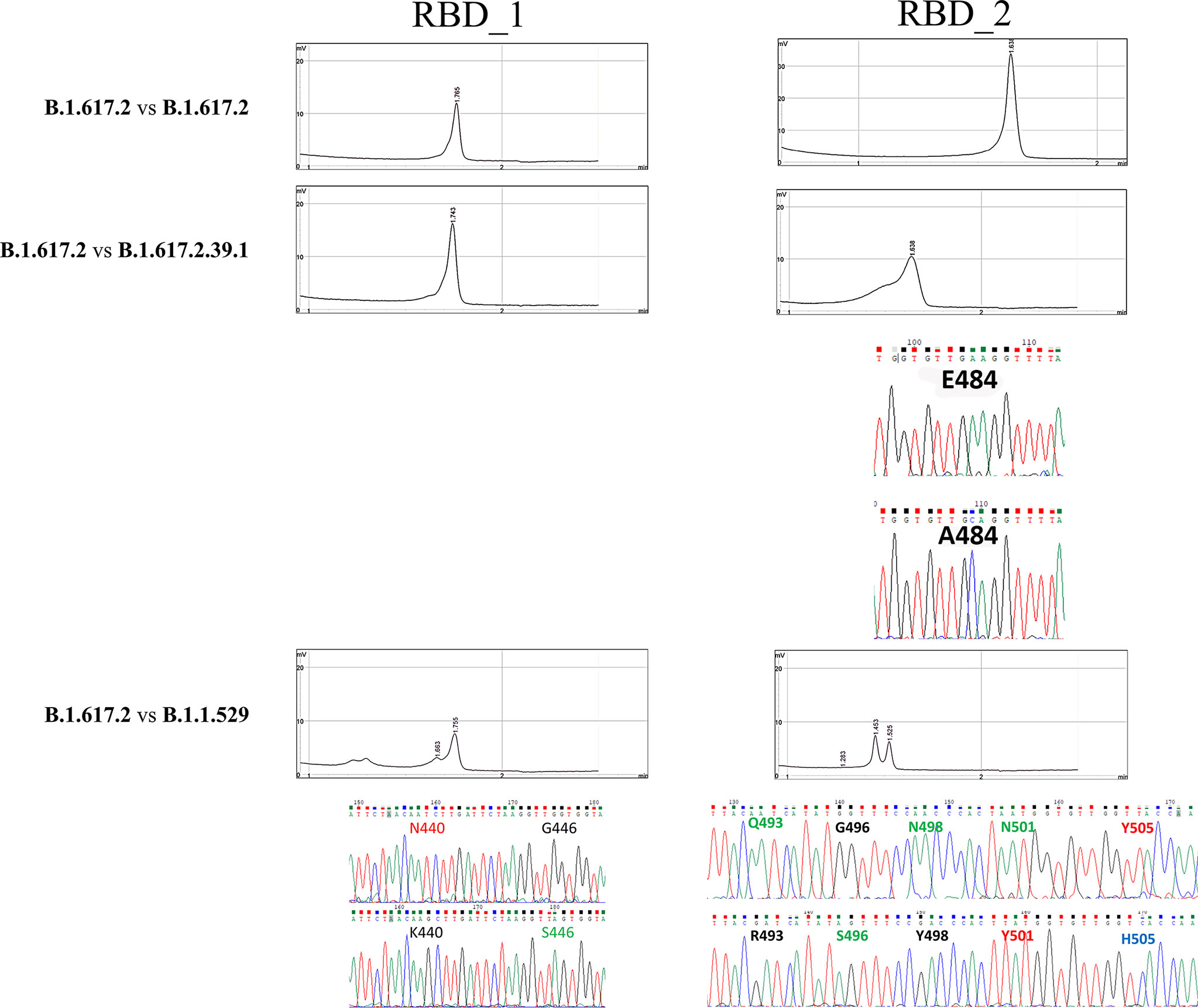
Figure 5 Denaturing High-Performance Liquid Chromatography findings in the blind-label group. Comparison of the reference used (B.1.617.2) against unknown positive samples. Whenever heteroduplexes were found, the sequenced amplicons (chromatograms) are reported, highlighting the nucleotide substitutions and the respective aminoacidic change in the S protein.
Complete findings of the latter are reported in Table 2.
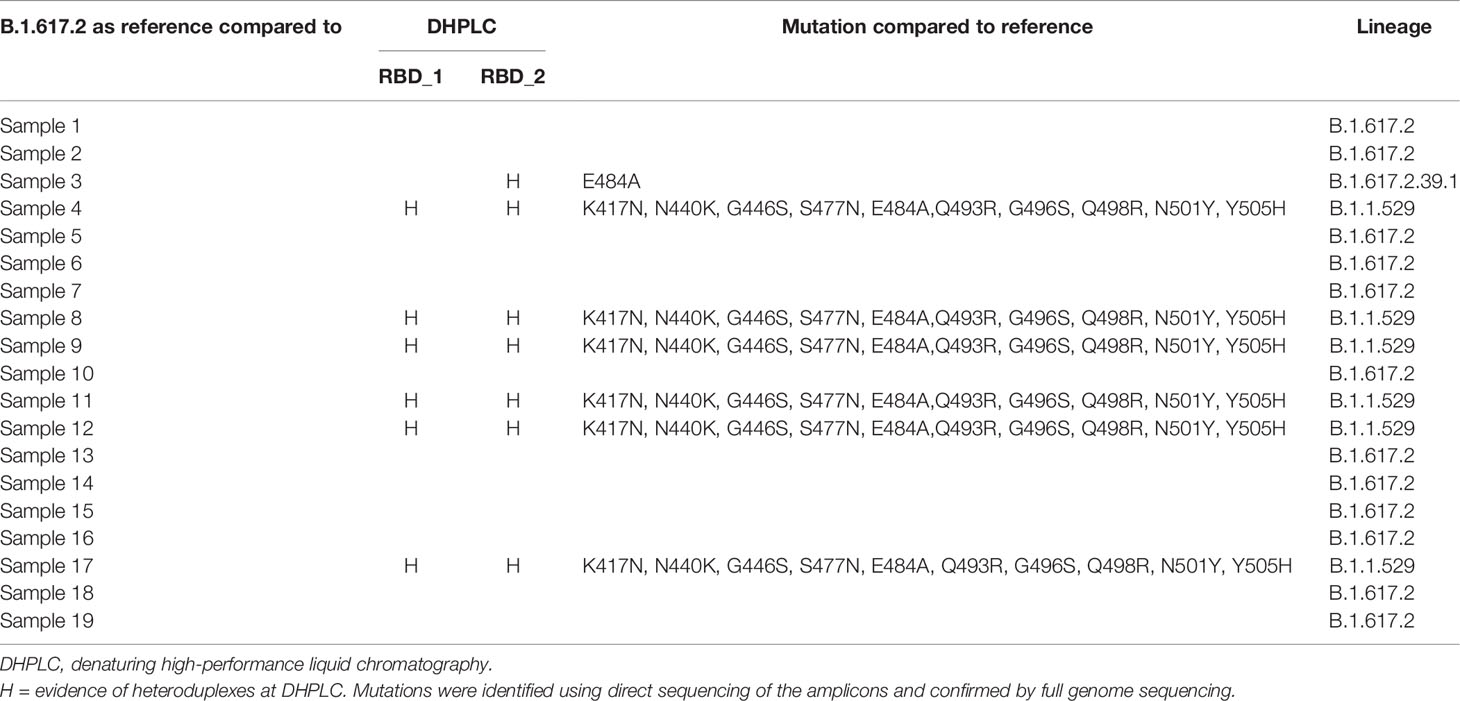
Table 2 Complete findings of the blind-label group using B.1.617.2 as reference against unknown positive samples.
Discussion
Variant surveillance is crucial for assessing whether there are emerging mutations which might make the SARS-CoV-2 more contagious, virulent, or be capable of escaping natural or vaccinal immunity. This surveillance is of paramount importance for researchers, public health authorities and policy makers to implement actions aimed at mitigating the effect of the pandemic on the healthcare system. In particular, the transmission chains and local outbreaks in hospital settings may be investigated very effectively using massive sequencing, shedding light on how to prevent transmission (22–25).
More broadly, NGS on a global scale may serve to monitor emerging variants capable of escaping immunity and, to some extent, to predicting epidemic waves or assessing pathogenicity (18, 22–25).
In this regard, an unprecedented effort to build up networks, platforms, and facilities capable of handling this great workload has been reinforced in developed and, to some extent, in developing countries. As of January 15, 2022, 7,181,951 SARS-CoV-2 full genome sequences had been filed of the 331,009,268 confirmed cases (2.1%) (19, 20). However, after two years of the pandemic and the epidemic waves caused by the spread of new more transmissible variants,the majority of healthcare systems worldwide have been overwhelmed once again by the spread of the Omicron variant (B.1.1.529 and sublineages).
Two main limitations, such as the promptness (the ability of effectively sequencing isolates in a timely manner, useful for managing single clusters of VOCs) and the comprehensiveness (the ability to sequence all the positive samples in a timely manner) of NGS may hamper the possibility of readily undertaking actions. These measures could include non-pharmaceutical interventions (NPIs) intended to put more stringent contact tracing and isolation procedures in place so as to control the outbreak of new variants as much as possible. This became dramatically evident during the spreading of the Omicron variants in Italy. In late November 2021, when the Omicron variants were beginning to emerge, Italy had a low level of SARS-CoV-2 circulation, recording fewer than 50/100,000 cases daily (32). In that critical phase, the Epidemiology and Disease Control Division (EDCD) issued a warning (33) to reinforce genomic surveillance. Although the first Omicron case identified in Italy on 26 November was promptly sequenced, and accurate contact tracing with sequencing was carried out (34), no impact of the sequencing program was observed on the epidemic curve which followed those of neighbouring countries by only a few days of delay. This led to the loss of control of the epidemic spread in only a few days as occurred during the months of December 2021 and January 2022 in Italy with millions of new cases deeply impacting the health care system (32) (Supp. Figure 1).
In Italy, a network, namely I-Co-Gen (Italian COVID-19 Genomic) commissioned to coordinate the genome sequencing surveillance, which started to operate in April 2021 was implemented by the policy maker. The consortium started operating in April 2021 and an increase in the absolute number sequenced was achieved in the following months. However, the positive isolates sequenced in Italy was still less than 1% of the number of positive cases (82,793 filed genome sequences of the 8,706,915 confirmed cases) or less than 3% of the 3,019,676 cases reported to the Surveillance system (34, 35) (Supp. Figure 1). Higher rates of sequencing were carried out with a low number of daily cases; however, eventually, the percentage dropped below 0.5% as the incidence increased (34, 35) (Supp. Figure 1).
As a matter of fact, genomic surveillance carried out at the level reported, even in developed countries, does not represent a measure which implements contact tracing and isolation procedures, and does not allow effective managing of discrete clusters. Technological advances are ongoing, arousing expectations that the above will be possible in the future (36).
However, it would be helpful to acknowledge that NGS could not address this issue until now and more practical to build up affordable platforms placed upstream from the sequencing facilities. Those platforms should be capable of investigating all positive samples in order to pre-select those samples with mutations in critical regions of the viral genome. To date, all the VOCs responsible for the epidemic waves had a mutation of concern in the RBD with no exceptions (19, 20). The above would allow focusing sequencing efforts on samples of concern to immediately establish NPI limited to specific cases.
Denaturing high-performance liquid chromatography has been demonstrated to have valuable features asscreening step in different settings. This study demonstrated that DHPLC readily detected mutations in selected SARS-CoV-2 genome regions which might be of concern. To that end, DHPLC exploits the different retention times of homoduplexes and heteroduplexes to detect their multiple retention peaks. A sample of a known sequence was used as a standard and was mixed with the unknown samples. Whenever more than one retention peak was evident at DHPLC, it indicated that at least one mismatch, existed between the standard and the sample, and hence one or more mutation/s are present in the test sample. In other words, the DHPLC is able to distinguish whether the test sample is different from the reference in the RBD. To achieve adequate separation of the elution peaks, DHPLC is suitable for screening PCR fragments with a relatively limited length ranging from 150 to 400 bp (30). To almost cover the entire RBD of the Spike protein, two PCRs were designed, and the respective amplicons were screened for mutations using DHPLC. An inherent limitation of the technique is the need for additional studies including viral targets which would require additional PCR reactions.
To obtain sharp and easily interpretable peaks at DHPLC analysis, PCR reactions should avoid the formation of non-specific amplicons and smears (Figure 2A). The high-fidelity Phusion Taq polymerase is well-suited for this purpose; furthermore, the PCR reactions using Phusion Taq are very rapid, thus reducing the duration of the PCR step. Moreover,the Phusion buffer does not contain any detergent which would be detrimental for downstream DHPLC analysis.
In this study, it was demonstrated that DHPLC analysis, by screening the presence of mutations of concerns in the RBD, would have been able to detect all the VOCs which have emerged in the two years of the pandemic. In the study presented here, we were also able to identify a Delta variant (B.1.617.2) having a rare mutation of concern in this lineage (E484A). However, sublineages assessment relies on full genome sequencing and DHPLC cannot unveil sublineages unless hallmark mutations are present in the RBD. For instance, BA.4 and BA.5 Omicron sublineage cannot be distinguished using DHPLC on the RBD since no differences exists at this level (Suppl. Figure 2).
Overall, the process was able to be completed in a few hours. Potentially, all new positives would be able to be screened on the same day as diagnosis using a dedicated thermal cycler and the DHPLC system (Figure 1). In addition to the cost of the additional PCR reaction, an additional reagent cost for each DHPLC analysis would be 1.40 Euros/amplicon. This amounts to 2.80 Euros/sample when considering both RBD_1 and RBD_2 targets. Also, DHPLC could identify the presence of a new variant even in those samples with low viral loads typical of the interwave periods (31). Our results showed that DHPLC was able to assess the VOCs with a viral load two or three order of magnitude less than the minimal amount of viral copies required for an effective amplicon-based full genome sequencing (37)
The S protein has different hotspots of mutation and deletion; the most likely candidates for immune escape are those within the RBD, such as K417N/T, E484K, and N501Y; N439K, N440K, G446S, L452R, Y453F, S477N, T478K, Q493R, G496S, N498Y, N501Y and Y505H may have a negative impact on the effectiveness of the vaccination (4, 5, 10, 19, 20, 38–40). Alarmingly, a convergent evolution has led to diverse groupings of those mutations among different clades (14). Using known sequenced strains, DHPLC is able to rapidly screen a large number of samples, establishing whether or not they share the same combination of mutations in the RBD. In the latter case, additional combinations may be rapidly ascertained in a few minutes by mixing and denaturing at high temperatures, allowing renaturing, and running in DHPLC with amplicons from known mutation combinations. Other possible targets could also be added to the analysis when their role emerges in vaccination escape in other domains (41, 42). It is important to check for the PCR amplicon presence in the test sample using a fragment analyser or an agarose gel; this step ensures that the presence of a unique peak in the DHPLC reflects the perfect sequence identity of the test sample with the standard and it is not an artifact due to the analysis of the standard sample alone.
As compared to other screening methods, such as multiplex real-time PCR genotyping (43), or multiplex amplification refractory mutation system (ARMS) PCR (44), the assay has some drawbacks and many advantages. The main drawbacks are the need for an additional instrumentation while the other methods are run on widely available qPCR thermal-cyclers, and the additional costs with respect to the use of diagnostic multiplex PCR with alerting of “suspect samples” software-based.
On the other hand, DHPLC screening may detect all the variants, either the known or the still unknown ones while these the former techniques are strictly sequence specific and can only detect known variants for whose specific probes have been designed. Only unexpected or inconsistent findings due to mismatches restricted to the probes sequence prompt the suspect of unknown variants and in-depth analysis of the sample. DHPLC. does not require complicated procedures for establishing or maintaining analytical performance as designing and validating increasing number of probes, or cumbersome technical replicatesOther sequence non-specific methods, such as High-Resolution Melting (HRM) may be valid alternatives. Nonetheless, HRM has been limited by inherently shorter amplicons and a longer running time (45–50). Furthermore, HRM approaches are greatly limited to discrete mutations targeted to achieve consistency and reliability in performance (45–50) In conclusion, the advantages for the healthcare systems of the flexible, cost-effective streamlined approach herein described, which integrates DHPLC with Sanger sequencing and, eventually, NGS should be considered as valuable alternative for variant screening in the genomic surveillance.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of AUSL Romagna under the protocol code “COVdPCR of 07/02/2020 (it includes appropriate approvals or waivers). Written informed consent for participation was not provided by the participants’ legal guardians/next of kin because it is not applicable. The samples included in this study were sent to the Unit of Micro-biology, Greater Romagna Area Hub Laboratory, Cesena, Italy, for routine diagnostic purposes, and the laboratory data results were reported as an answer to clinical suspicion. As such, informed consent from patients was not required. Each sample included was preventively anonymized.
Author Contributions
Conceptualization, FG and MT. Formal analysis, DM, FT, and GD. Data curation, FG, MT, DM, FT, and GD. Writing—original draft preparation, FG and MT. Writing—review and editing, SP and VS. Validation, DM, FT, GD, and VS. Supervision, FG and SP. Project administration, MT. Funding acquisition, VS. All authors have read and agreed to the published version of the manuscript
Funding
Partial financial support was received from AUSL Romagna under the protocol code “COVdPCR”.
Conflict of Interest
SP is an employee of ADS Biotec. ADS Biotec retains proprietary technologies for carrying out DHPLC. SP supported the study by providing technical support. No financial support that may have inappropriately influenced the study was provided by ADS Biotec. There are no patented products in development or marketed products to declare. All the authors are committed to adhering to the Journal policies regarding sharing data and materials.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Some viral strains included in the study had been investigated within the interlaboratory “INSTAND e.V. “external proficiency testing program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.889592/full#supplementary-material
References
1. Smith EC, Blanc H, Surdel MC, Vignuzzi M, Denison MR. Coronaviruses Lacking Exoribonuclease Activity are Susceptible to Lethal Mutagenesis: Evidence for Proofreading and Potential Therapeutics. PloS Pathog (2013) 9(8):e1003565. doi: 10.1371/journal.ppat.1003565
2. Sevajol M, Subissi L, Decroly E, Canard B, Imbert I. Insights Into RNA Synthesis, Capping, and Proofreading Mechanisms of SARS-Coronavirus. Virus Res (2014) 194:90–9. doi: 10.1016/j.virusres.2014.10.008
3. van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect Genet Evol (2020) 83:104351. doi: 10.1016/j.meegid.2020.104351
4. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell (2020) 182(4):812–827.e19. doi: 10.1016/j.cell.2020.06.043
5. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature (2021) 592:116–21. doi: 10.1038/s41586-020-2895-3
6. Triggle CR, Bansal D, Ding H, Islam MM, Farag EABA, Hadi HA, et al. A Comprehensive Review of Viral Characteristics, Transmission, Pathophysiology, Immune Response, and Management of SARS-CoV-2 and COVID-19 as a Basis for Controlling the Pandemic. Front Immunol (2021) 12:631139. doi: 10.3389/fimmu.2021.631139
7. Giovanetti M, Benedetti F, Campisi G, Ciccozzi A, Fabris S, Ceccarelli G, et al. Evolution Patterns of SARS-CoV-2: Snapshot on its Genome Variants. Biochem Biophys Res Commun (2021) 538:88–91. doi: 10.1016/j.bbrc.2020.10.102
8. Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell (2020) 182:1284–1294.e9. doi: 10.1016/j.cell.2020.07.012
9. Zhang L, Li Q, Liang Z, Li T, Liu S, Cui Q, et al. The Significant Immune Escape of Pseudotyped SARS-CoV-2 Variant Omicron. Emerg Microbes Infect (2022) 11(1):1–5. doi: 10.1080/22221751.2021.2017757
10. Groves DC, Rowland-Jones SL, Angyal A. The D614G Mutations in the SARS-CoV-2 Spike Protein: Implications for Viral Infectivity, Disease Severity and Vaccine Design. Biochem Biophys Res Commun (2021) 538:104–7. doi: 10.1016/j.bbrc.2020.10.109
11. Jackson CB, Zhang L, Farzan M, Choe H. Functional Importance of the D614G Mutation in the SARS-CoV-2 Spike Protein. Biochem Biophys Res Commun (2021) 538:108–15. doi: 10.1016/j.bbrc.2020.11.026
12. Plante JA, Mitchell BM, Plante KS, Debbink K, Weaver SC, Menachery VD. The Variant Gambit: COVID-19’s Next Move. Cell Host Microbe (2021) 29(4):508–15. doi: 10.1016/j.chom.2021.02.020
13. Cobey S, Larremore DB, Grad YH, Lipsitch M. Concerns About SARS-CoV-2 Evolution Should Not Hold Back Efforts to Expand Vaccination. Nat Rev Immunol (2021) 1:1–6. doi: 10.1038/s41577-021-00544-9
14. Focosi D, Maggi F. Neutralising Antibody Escape of SARS-CoV-2 Spike Protein: Risk Assessment for Antibody-Based COVID-19 Therapeutics and Vaccines. Rev Med Virol (2021) 31(6):e2231. doi: 10.1002/rmv.2231
15. Gómez CE, Perdiguero B, Esteban M. Emerging SARS-CoV-2 Variants and Impact in Global Vaccination Programs Against SARS-CoV-2/COVID-19. Vaccines (Basel) (2021) 9:243. doi: 10.3390/vaccines9030243
16. Jung J. Preparing for the Coronavirus Disease (COVID-19) Vaccination: Evidence, Plans, and Implications. J Korean Med Sci (2021) 36(7):e59. doi: 10.3346/jkms.2021.36.e59
17. Van Egeren D, Novokhodko A, Stoddard M, Tran U, Zetter B, Rogers M, et al. Risk of Rapid Evolutionary Escape From Biomedical Interventions Targeting SARS-CoV-2 Spike Protein. PloS One (2021) 16(4):e0250780. doi: 10.1371/journal.pone.025078013
18. Zella D, Giovanetti M, Benedetti F, Unali F, Spoto S, Guarino M, et al. The Variants Question: What Is the Problem? J Med Virol (2021) 93(12):6479–85. doi: 10.1002/jmv.27196
19. Gangavarapu K, Latif AA, Mullen J, Alkuzweny M, Hufbauer E, Tsueng G, et al. Outbreak.Info, >Lineage Comparison. Available at: https://outbreak.info/compare-lineages (Accessed 21 January 2022).
20. GISAID Initiative, Elbe S, Buckland-Merrett G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Global Chall (2017) 1:33–46. doi: 10.1002/gch2.1018
21. Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, et al. Coast-To-Coast Spread of SARS-CoV-2 During the Early Epidemic in the United States. Cell (2020) 181(5):990–996.e5. doi: 10.1016/j.cell.2020.04.021
22. González-Candelas F, Shaw MA, Phan T, Kulkarni-Kale U, Paraskevis D, Luciani F, et al. One Year Into the Pandemic: Short-Term Evolution of SARS-CoV-2 and Emergence of New Lineages. Infect Genet Evol (2021) 92:104869. doi: 10.1016/j.meegid.2021.104869
23. Guo S, Liu K, Zheng J. The Genetic Variant of SARS-CoV-2: Would It Matter for Controlling the Devastating Pandemic? Int J Biol Sci (2021) 17(6):1476–85. doi: 10.7150/ijbs.59137
24. Moustafa AM, Planet PJ. Jumping a Moving Train: SARS-CoV-2 Evolution in Real Time. J Pediatr Infect Dis Soc (2021) 10(Supplement_4):S96–S105. doi: 10.1093/jpids/piab051
25. Page AJ, Mather AE, Le-Viet T, Meader EJ, Alikhan NF, Kay GL, et al. The COVID-Genomics UK Cog-UK Consortium. Large-Scale Sequencing of SARS-CoV-2 Genomes From One Region Allows Detailed Epidemiology and Enables Local Outbreak Management. Microb Genom (2021) 7(6):589. doi: 10.1099/mgen.0.000589
26. Bal A, Destras G, Gaymard A, Stefic K, Marlet J, Eymieux S, et al. Two-Step Strategy for the Identification of SARS-CoV-2 Variant of Concern 202012/01 and Other Variants With Spike Deletion H69-V70, France, August to December 2020. Euro Surveill (2021) 26(3):2100008. doi: 10.2807/1560-7917.ES.2021.26.3.2100008
27. Banada P, Green R, Banik S, Chopoorian A, Streck D, Jones R, et al. A Simple Reverse Transcriptase PCR Melting-Temperature Assay To Rapidly Screen for Widely Circulating SARS-CoV-2 Variants. J Clin Microbiol (2021) 59(10):e0084521. doi: 10.1128/JCM.00845-21
28. Aoki A, Mori Y, Okamoto Y, Jinno H. Development of a Genotyping Platform for SARS-CoV-2 Variants Using High-Resolution Melting Analysis. J Infect Chemother (2021) 27(9):1336–41. doi: 10.1016/j.jiac.2021.06.007
29. Barua S, Bai J, Kelly PJ, Hanzlicek G, Noll L, Johnson C, et al. Identification of the SARS-CoV-2 Delta Variant C22995A Using a High-Resolution Melting Curve RT-FRET-PCR. Emerg Microbes Infect (2022) 11(1):14–7. doi: 10.1080/22221751.2021.2007738
30. Xiao W, Oefner PJ. Denaturing High-Performance Liquid Chromatography: A Review. Hum Mutat (2001) 17:439–74. doi: 10.1002/humu.1130
31. Gentilini F, Turba ME, Taddei F, Gritti T, Fantini M, Dirani G, et al. Modelling RT-qPCR Cycle-Threshold Using Digital PCR Data for Implementing SARS-CoV-2 Viral Load Studies. PloS One (2021) 16(12):e0260884. doi: 10.1371/journal.pone.0260884
32. Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. COVID-19 Data Repository (2020). Available at: https://github.com/CSSEGISandData/COVID-19https://github.com/CSSEGISandData/COVID-19/blob/master/README.md.
33. ECDC. Threat Assessment Brief: Implications of the Emergence and Spread of the SARS-CoV-2 B.1.1. 529 Variant of Concern (Omicron) for the EU/EEA (2021). Available at: https://www.ecdc.europa.eu/en/publications-data/threat-assessment-brief-emergence-sars-cov-2-variant-b.1.1.529.
34. Prevalenza E Distribuzione Delle Varianti Di SARS-CoV-2 Di Interesse Per La Sanità Pubblica in Italia. Rapporto N (2021). Available at: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-10-dicembre-2021.pdf.
35. Stima Della Prevalenza Delle Varianti VOC (Variants of Concern) in Italia: Beta, Gamma, Delta, Omicron E Altre Varianti Di SARS-CoV-2 (2021). Available at: https://www.iss.it/documents/20126/0/Flash_survey31dicembre.pdf/317a7725-a278-12f9-1834-a3e0bcd19557?t=1640962083615.
36. Behrmann O, Spiegel M. COVID-19: From Rapid Genome Sequencing to Fast Decisions. Lancet Infect Dis (2020) 20(11):1218. doi: 10.1016/S1473-3099(20)30580-6
37. Kubik S, Marques AC, Xing X, Silvery J, Bertelli C, De Maio F, et al. Recommendations for Accurate Genotyping of SARS-CoV-2 Using Amplicon-Based Sequencing of Clinical Samples. Clin Microbiol Infect (2021) 27(7):1036.e1–1036.e8. doi: 10.1016/j.cmi.2021.03.029
38. Garcia-Beltran WF, Lam EC, St Denis K, Nitido AD, Garcia ZH, Hauser BM, et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell (2021) 184(9):2372–83.e9. doi: 10.1016/j.cell.2021.03.013
39. Karim SSA. Vaccines and SARS-CoV-2 Variants: The Urgent Need for a Correlate of Protection. Lancet (2021) 397:1263–4. doi: 10.1016/S0140-6736(21)00468-2
40. Knoll MD, Wonodi C. Oxford-AstraZeneca COVID-19 Vaccine Efficacy. Lancet (2021) 397:72–4. doi: 10.1016/S0140-6736(20)32623-4
41. McCarthy KR, Rennick LJ, Nambulli S, Robinson-McCarthy LR, Bain WG, Haidar G, et al. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science (2021) 371(6534):1139–42. doi: 10.1126/science.abf6950
42. Pereira F. SARS-CoV-2 Variants Combining Spike Mutations and the Absence of ORF8 May Be More Transmissible and Require Close Monitoring. Biochem Biophys Res Commun (2021) 550:8–14. doi: 10.1016/j.bbrc.2021.02.080
43. Vogels CBF, Breban MI, Ott IM, Alpert T, Petrone ME, Watkins AE, et al. Multiplex qPCR Discriminates Variants of Concern to Enhance Global Surveillance of SARS-CoV-2. PloS Biol (2021) 19(5):e3001236. doi: 10.1371/journal.pbio.3001236
44. Islam MT, Alam ARU, Sakib N, Hasan MS, Chakrovarty T, Mohammad T, et al. A Rapid and Cost-Effective Multiplex ARMS-PCR Method for the Simultaneous Genotyping of the Circulating SARS-CoV-2 Phylogenetic Clades. J Med Virol (2021) 93:2962–70. doi: 10.1002/jmv.26818
45. Aoki A, Adachi H, Mori Y, Ito M, Sato K, Okuda K, et al. A Rapid Screening Assay for L452R and T478K Spike Mutations in SARS-CoV-2 Delta Variant Using High-Resolution Melting Analysis. J Toxicol Sci (2021) 46(10):471–6. doi: 10.2131/jts.46.471
46. Barua S, Hoque M, Kelly PJ, Bai J, Hanzlicek G, Noll L, et al. High-Resolution Melting Curve FRET-PCR Rapidly Identifies SARS-CoV-2 Mutations. J Med Virol (2021) 93(9):5588–93. doi: 10.1002/jmv.27139
47. Diaz-Garcia H, Guzmán-Ortiz AL, Angeles-Floriano T, Parra-Ortega I, López-Martínez B, Martínez-Saucedo M, et al. Genotyping of the Major SARS-CoV-2 Clade by Short-Amplicon High-Resolution Melting (SA-HRM) Analysis. Genes (Basel) (2021) 12(4):531. doi: 10.3390/genes12040531
48. Ferreira BIDS, da Silva-Gomes NL, Coelho WLDCNP, da Costa VD, Carneiro VCS, Kader RL, et al. 30. Validation of a Novel Molecular Assay to the Diagnostic of COVID-19 Based on Real Time PCR With High Resolution Melting. PloS One (2021) 16(11):e0260087. doi: 10.1371/journal.pone.0260087
49. Gazali FM, Nuhamunada M, Nabilla R, Supriyati E, Hakim MS, Arguni E, et al. Detection of SARS-CoV-2 Spike Protein D614G Mutation by qPCR-HRM Analysis. Heliyon (2021) 7(9):e07936. doi: 10.1016/j.heliyon.2021.e07936
Keywords: SARS-CoV-2, denaturing high-performance liquid chromatography, variant of concern, surveillance, Receptor Binding Domain
Citation: Turba ME, Mion D, Papadimitriou S, Taddei F, Dirani G, Sambri V and Gentilini F (2022) Rapid and Affordable High Throughput Screening of SARS-CoV-2 Variants Using Denaturing High-Performance Liquid Chromatography Analysis. Front. Virol. 2:889592. doi: 10.3389/fviro.2022.889592
Received: 04 March 2022; Accepted: 15 June 2022;
Published: 07 July 2022.
Edited by:
Pedro Martinez-Gomez, Center for Edaphology and Applied Biology of Segura (CSIC), SpainReviewed by:
Dominique Goedhals, Federal University of São Carlos, BrazilFarid Rahimi, Australian National University, Australia
Diana Mariani, Federal University of Rio de Janeiro, Brazil
Copyright © 2022 Turba, Mion, Papadimitriou, Taddei, Dirani, Sambri and Gentilini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Gentilini, ZmFiaW8uZ2VudGlsaW5pQHVuaWJvLml0