
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Virol. , 04 February 2022
Sec. Viral Diversification and Evolution
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.758191
This article is part of the Research Topic Insights in Viral Diversification and Evolution: 2021 View all 5 articles
 Angelica Bianco1†
Angelica Bianco1† Loredana Capozzi1†
Loredana Capozzi1† Laura Del Sambro1
Laura Del Sambro1 Domenico Simone1
Domenico Simone1 Lorenzo Pace1Valeria Rondinone1Laura M. Difato1Angela Miccolupo1Caterina Manzari2Alberto Fedele3
Lorenzo Pace1Valeria Rondinone1Laura M. Difato1Angela Miccolupo1Caterina Manzari2Alberto Fedele3 Antonio Parisi1*
Antonio Parisi1*The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a pandemic, threatening global public health. Several cases of persistent infection have been described, but there are few reports that compared the genetic variability among samples collected from the patient during infection. In the current study, we reported a viral genetic analysis of a diabetic male patient with Non-Hodgkin Lymphoma affected by persistent SARS-CoV-2 infection. We sequenced the patient-derived viral isolated both from oro/nasopharyngeal swab and VeroE6 cell line, collected from the same patient at different points of the infection. Due to the insufficient material of the second swab received, in order to obtain a complete coverage of the viral genome, it was convenient to perform a virus isolation after cell culture. Both genomes belonged to Pangolin Lineage B.1, Nextstrain clade 20A and GISAID clade G. The mutation spectrum predicted for the two viral genomes reveal three additionally mutations in the sequence of second sample when compared with mutations set identified in the first sample. Our findings show the evolution of the intra-host variability during the course of a long-lasting infection.
The novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a pandemic, threatening global public health. SARS-CoV-2 is the seventh member of the Coronaviridae family known to infect humans and it is responsible for respiratory illness of varying severity (1). Among the clinical symptoms, the most common are fever and cough (2). The mean duration of the virus infection has been estimated to be about 14 days (3). However, the persistence of the virus for more than 30 days has been described (4). Previous studies have found correlation between infection duration and presence of pre-existing diseases (3) or with severe COVID-19 illness (5); conversely, individuals that can remain positive to virus also after symptom resolution were described (6–8). Thus, the factors that impact the persistence of the virus are still uncertain. This report describes the case of a diabetic man affected by Non-Hodgkin Lymphoma with persistent SARS-CoV-2 infection. Whole genome sequence of SARS-CoV-2 was performed on the first oro/nasopharyngeal swab and on a later one collected after more than 1-month, following isolation from VeroE6 cell cultures. The intra-host variability of the viral genome was also assessed.
On 7 April 2020, a man was admitted to hospital of Lecce, in Apulia Region, with diagnosis of acute respiratory failure and bilateral pneumonia caused by SARS-CoV-2 virus infection. The patient was also affected by hypertension, obesity, diabetic and follicular Non-Hodgkin Lymphoma (NHL). During hospitalization, he showed fever and he needed moderate support of oxygen. No additional complain of any significant disorders was referred. After about 5 weeks, the patient did not show fever and after 11 weeks he became asymptomatic. One oro/nasopharyngeal swab every week from April to July 2020, for a total of 13 samples, was performed and processed in hospital laboratory. SARS-CoV-2 biomolecular tests performed on the oro/nasopharyngeal swabs always showed a positive outcome, while the specific serological tests (IgG and IgM) were negative. The first positive oro/nasopharyngeal swab performed on April 7th 2020 (named IZSPB_141) was sent to Istituto Zooprofilattico of Puglia and Basilicata to perform whole genome sequencing (Figure 1). During hospitalization the patient received infusion therapy with (High-dose Intravenous Immunoglobulin) IVIg (two administrations: June 22nd and 29th 2020). Outpatient nasopharyngeal RT-PCR SARS-CoV-2 amplification testing was persistently positive and on June 30th 2020 a new positive oro/nasopharyngeal swab (named IZSPB_123), was sent to our institute to understand the infectivity potential of this asymptomatic patient, in order to perform the viral isolation in cell culture. No direct sequencing of the virus was performed on the last swab specimen due to insufficient material. On July 2nd, the asymptomatic patient was dismissed. On July 20th 2020 the patient tested negative to the virus and, following another negative test, he was declared healed.
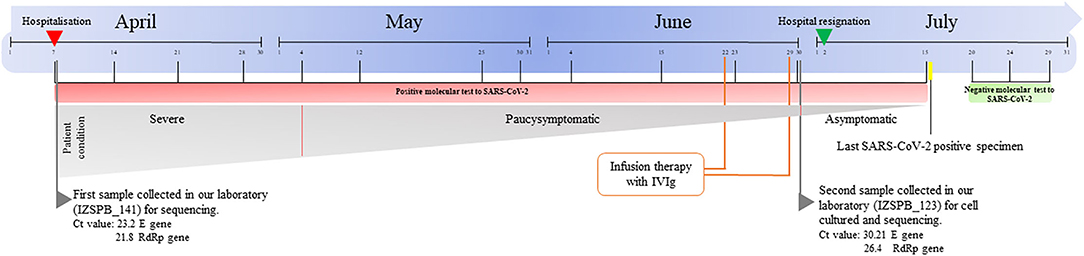
Figure 1. Timeline of symptom onset, molecular diagnosis, and sequencing of specimens.
Samples IZSPB_141 and IZSPB_123 were collected from the patient to detect SARS-CoV-2 by real-time reverse transcriptase rtPCR. RNA was extracted from clinical samples with QIAamp viral RNA mini kit (Qiagen, Germany) following the manufacturer's instructions. The specimen was handled in a biosafety level 3 laboratory at the Istituto Zooprofilattico of Puglia and Basilicata (Italy), under a class II type A 2 biosafety cabinet. The SARS-CoV-2 RNA was detected by a One-step real-time reverse transcription (RT-PCR) multiplex assay (DIATHEVA, Italy) by amplification of RdRp and E genes. The assay also included the amplification of RNase P gene as internal positive control. PCR amplification was performed at 48 °C for 30 min for reverse transcription, followed by 95 °C for 10 min and then 50 cycles of 95 °C for 15 s, 58 °C for 30 s, using CFX Connect Real-Time PCR Detection System (Biorad).
For SARS-CoV-2 isolation, the Vero E6 cell line (African green monkey kidney cells) was used as previously described (9).
For virus isolation from swab, cells were plated into a 25 cm2 cell culture flasks (Corning, CLS430168) at a confluency of 70–80% in 6 mL EMEM medium with 6% fetal bovine serum (FBS) and incubated overnight at 37°C. The following day, 1,500μL of the swab medium was incubated with 500 μL of an antibiotic solution (2,000 U/mL of penicillin/streptomycin) for 1 h at room temperature. Two mL of suspension was inoculated on the monolayer of the VeroE6 cells and incubated at 37°C for 1 h. Then, 4 mL of EMEM with 6% FBS was added and incubated at 37°C for 72 h. Virus replication and isolation were confirmed through cytopathic effect and gene detection.
Total RNA was used to perform cDNA synthesis by LunaScript RT SuperMix Kit (New England Biolabs) and then amplified with SARS-CoV-2 primers panel ARTIC nCOV-2019 V3 (© 2021 Integrated DNA Technologies, Inc.). The library preparation was performed according to Illumina Illumina DNA Prep kit protocol (Illumina) and sequenced on MiSeq Sequencing Platform (Illumina). For each sample, the sequence analysis was performed as previously described (10). Specifically, the paired-end raw reads were quality-filtered and trimmed using Trimmomatic v0.38 (11). De novo genome assembly and scaffolding were executed with SPAdes v. 3.12 (12). Bowtie2 v2.3.4 software (13) was used to performed the quality check of genome assemblies by aligning the filtered reads to the SARS-CoV-2 reference genome NC_045512.2. Sequence alignments were converted to binary alignments (BAM format) and sorted using SAM-tools version 1.3.1. Read alignments were evaluated with QualiMap (14). Two hundred and ninety-eight consensus genome sequences passed the quality control assessment by Nextclade (https://clades.nextstrain.org/), whose parameters include missing data, mixed sites, private mutations, and mutation clusters, and were deposited in the GISAID database (https://www.gisaid.org/) with the following accession ID: EPI_ISL_525555 (IZSPB_141); EPI_ISL_525554 (IZSPB_123). Additionally, single nucleotide variant (SNVs) and insertions/deletions (InDels) with minor allelic frequencies (MAF) were identified using LoFreq (Version 2.1.3.1) (15) with default parameters.
RT-PCR performed on the two oro/nasopharyngeal swabs confirmed the presence of SARS-CoV-2: IZSPB_141 showed the amplification of E gene with a Ct value of 23.2 and RdRp gene with a ct value of 21.8; IZSPB_123 showed the amplification of E gene with a Ct value of 30.21 and RdRp gene with a Ct value of 26.4. The sample oro/nasopharyngeal swab collected on June 30th 2020 (IZSPB_123) was inoculated in VeroE6 cells, and then incubation was observed at 24 h intervals. The cytopathic effects were observed 72 h after inoculation. Virus replication was confirmed using real-time RT-PCR with RNA extracted from the cell culture medium. To assess the variational spectrum of the oro/nasopharyngeal swab specimen (IZSPB_141) and viral isolates by VeroE6 cell culture (IZSPB_123), whole genome sequencing of the SARS-CoV-2 samples was performed, generating 887.638 and 881.046 number of reads, respectively, of which more than 99% resulted as mapped reads. We obtain the mean coverage of 5.763x for IZSPB_141 sample and 5.807x for IZSP_123 (Supplementary Table 1). Both SARS-CoV-2 sequences belonged to Nextstrain clade 20A, GISAID clade G and Pangolin lineage B.1 (Supplementary Table 2). We detected a total of nine mutations, of which six were shared between the two reconstructed viral sequences (Figure 2). Among the six mutations, two did not result in any amino acid change, while the remaining mutations were non-synonymous variations. Interestingly, the sequence of SARS-CoV-2 isolated from VeroE6 cells harbored three additional mutations, namely C24117T – Spike (S) A852V; C17326T – ORF1b P1287S; C27710T - ORF7a A106V. In particular, mutation (S A852V) was detected in other 84 SARS-CoV-2 sequences available in GISAID database, as per July 28th 2021. Additionally, searching among available sequencing in GISAID database we detected four additional amino acid changes in the same protein position (i.e. A852G; A852S; A852T; A852E). All the nucleotide variants described were predicted by Nextclade tool, thus present in the consensus sequence of the viral genome analyzed and were detected with a frequency >61.9% by LoFreq. When we considered all the variants predicted with a lower frequency than 61.9%, we detected a total of 46 additional variants in IZSPB_141 and 32 additional variants in IZSPB_123 (Supplementary Table 3). Among these, only 8 variants were common in the two sampling; six variants were reported in Figure 2, while two variants were detected with very low frequency between 1.4 and 0.7%.
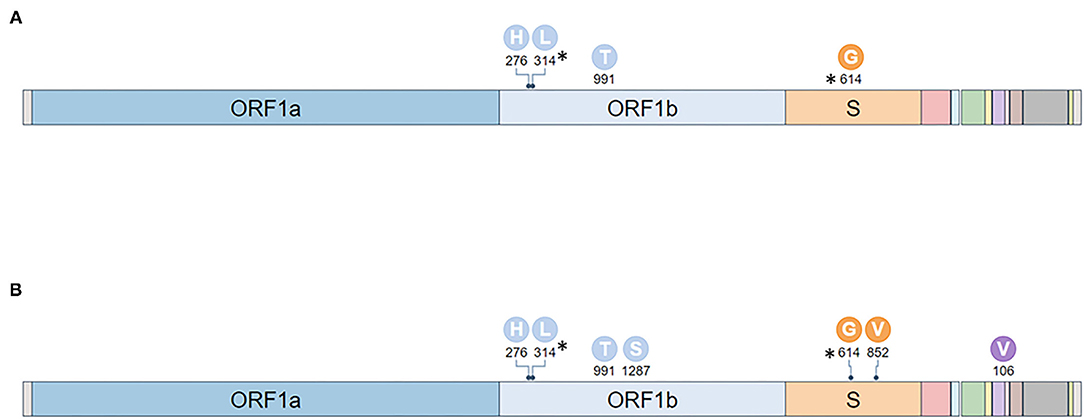
Figure 2. Mutations identified in two specimens analyzed. The mutations identified with a frequency >61.9% in IZSPB_141 (A) and in IZSPB_123 (B) were reported in the figure. The characteristic mutations of B.1 (identified in at least 75% sequences) were marked with asterisk.
We reported the case of long persistent SARS-CoV-2 infection in a diabetic patient affected by NHL. The patient initially showed modest typical COVID19 symptoms (i.e., acute respiratory failure) and later became asymptomatic and finally his swab resulted negative after 3 months of infection. Although, the serological assay resulted negative, the clinical condition of the patient was not severe. This finding disagreed with previous studies that reported an increased severity and mortality SARS-CoV-2 infection in adult immunosuppressed patients compared with immunocompetent adults (16, 17). Two infusions of IVIg were administered 2 months after the start of infection. SARS-CoV-2 genome analysis showed that the virus isolated from the oro/nasopharyngeal swab specimen and viral cell line isolates, collected in different times, belong both to pangolin lineage B.1. The two genomes analyzed shared six mutations compared to the reference genome NC_045512.2; however the strain isolated from cell culture harbored three additional mutations. Among these, we noticed a non-synonymous mutation (S A852V) in the gene encoding the Spike protein (S gene), that mediates receptor binding and fusion of the viral and cellular membrane (18), thus determining the ability of the virus to infect the cells and its transmissibility in the host (19). To date, several mutations in the S gene were reported, but it is still unclear whether these variations could influence viral infectivity, transmissibility or immune response (20). The mutation described here (S A852V) was searched in all SARS-CoV-2 genomes available in GISAID database. We detected the same amino acid change in a total of 84 strains. Interestingly, this locus is a polymorphic site, since we found other four additional amino acid changes: hcov S A852G (13 strains); hcov S A852S (186 strains); hcov S A852T (13 strains); hcov S A852E (5 strains). Other two mutations in the same strain, isolated from cell culture were found in genomes already collected in GISAID: specifically, ORF1b P1287S was identified in 83 samples, while ORFa A106V was identified in 21 samples. Nevertheless, the co-segregation of the three variations was found for the first time in our sample. The investigation of the occurrence of these mutations among our two oro/nasopharyngeal samples was not straightforward, since the cultured strain (IZSPB_123) derived from a different oro/nasopharyngeal swab and was sampled about 2 months after the first one (IZSPB_141). Thus, we cannot assess whether these mutation events occurred before or after cell culture, but certainly following the first sequenced virus sample. Viral population diversity was influenced by several factors, e.g., replication fidelity (21, 22). The mutation frequency in SARS-CoV-2 populations is strongly influenced by negative and positive selection pressures (23). Virus replication rates directly impact the accumulation of mutations in the viral genome, enabling the existence of a viral quasispecies (a swarm of different, yet highly related, viral entities) within an infected host (24–26). Thereby, a single swab obtained from an individual or patient can contain different molecules of viral RNA, thus a different level of diversity. This scenario can exert a range of effects on viral fitness (27, 28). Based on this concept, we investigated the presence of mutations with a low frequency in our strains. The aim of this analysis was to determine whether the additional variants identified in sample IZSPB_123 were already present in the previously sequenced genome IZSPB_141 with a frequency <2%. Among the tally of identified mutations, only two were carried in both strains with an allele frequency <1.5%. However, we could not detect in IZSPB_141 the three variations identified in the IZSPB_123 strain. This result suggests two hypotheses: a) the three mutations identified in IZSPB_123 but not in IZSPB_141, may have arisen as a consequence of the in vitro growth of the virus; b) the mutations identified in IZSPB_123 may have arisen de novo after the first diagnosis of SARS-CoV-2 infection. According to the first hypothesis there is evidence to indicate the occurrence of new variations during virus passage in VeroE6 cells (29). The three mutations identified in IZSPB_123 could not be found in any viral genome from Apulia or Basilicata available in the GISAID database, whose sequencing was carried out in our Laboratory following the isolation in VeroE6 cells. Conversely, the latter hypothesis is supported by the finding of the same mutations in other viral samples (none isolated from cells), suggesting that these variations did not are due to amplification errors in sequencing. Thus, these mutations may be the expected result of the constant mutation of SARS-CoV-2, which might be more noticeable in cases of persistent infection. Further studies are required to understand the role of these mutations in COVID-19 ethology and spread.
In conclusion, our findings support the concept that intra-host genetic variations occurs in SARS-CoV-2, especially when the infection is persistent, suggesting that these mutations, in combination with the clinical phenotype of the host, can potentially influence the natural history of the viral infection.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.gisaid.org/, EPI_ISL_525555; EPI_ISL_525554.
Ethical approval was not provided for this study on human participants. After diagnostic routine, strains resulting from the biological material were stored to be processed for further analysis. No personal data or any other information than the type of material and the result of routine analysis were collected from each specimen, inhibiting any correlations of these fully anonymized samples with the respective patients. Thus, according to national regulations and the institutional rules for Good Scientific Practice, the requirement for submission to an ethical committee and for obtaining patients' informed consent was waived. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. In addition, the document “Deontological rules for processing for statistical or scientific research purposes published according to Article 20, paragraph 4, of Legislative Decree Number 101 of 10 August 2018 - 19 December 2018”, in Article 2 mentions: “These deontological rules do not apply to processing for statistical and scientific purposes related to health protection activities carried out by health professions or health bodies, or with comparable activities in terms of significant personalized impact on the data subject, which shall continue to be governed by the relevant provisions”.
AP, AB, and LC provided substantial contributions to the conception and design of the work and the acquisition, analysis, and interpretation of data and drafting the work. AB, LC, LDe, DS, LP, VR, CM, LDi, AF, and AM performed the acquisition, analysis, and interpretation of data and drafting the work. AB and LC performed the acquisition, analysis, and interpretation of data and critical revising of the work for important intellectual content. All authors have approved the final version to be published and accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
This research was funded by Project COVID-19: epidemiological, clinical, genetic, and social determinants of infection and disease progression (Project Code: COVID-2020-12371675).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Anna Giannico and Donato Ridolfi for technical support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.758191/full#supplementary-material
1. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet Lond Engl. (2020) 395:565–74. doi: 10.1016/S0140-6736(20)30251-8
2. Zapor M. Persistent detection and infectious potential of SARS-CoV-2 virus in clinical specimens from COVID-19 patients. Viruses. (2020) 12:1384. doi: 10.3390/v12121384
3. Zheng S, Fan J, Yu F, Feng B, Lou B, Zou Q, et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. (2020) 369:m1443. doi: 10.1136/bmj.m1443
4. Zhou B, She J, Wang Y, Ma X. Duration of viral shedding of discharged patients with severe COVID-19. Clin Infect Dis Off Publ Infect Dis Soc Am. (2020) 71:2240–2. doi: 10.1093/cid/ciaa451
5. Fang Z, Zhang Y, Hang C, Ai J, Li S, Zhang W. Comparisons of viral shedding time of SARS-CoV-2 of different samples in ICU and non-ICU patients. J Infect. (2020) 81:147–78. doi: 10.1016/j.jinf.2020.03.013
6. Dou P, Zhang S, Wang C, Cai L, Liu Z, Xu Q, et al. Serial CT features in discharged COVID-19 patients with positive RT-PCR re-test. Eur J Radiol. (2020) 127:109010. doi: 10.1016/j.ejrad.2020.109010
7. Lan L, Xu D, Ye G, Xia C, Wang S, Li Y, et al. Positive RT-PCR test results in patients recovered from COVID-19. JAMA. (2020) 323:1502–3. doi: 10.1001/jama.2020.2783
8. Li XJ, Zhang ZW, Zong ZY. A case of a readmitted patient who recovered from COVID-19 in Chengdu, China. Crit Care Lond Engl. (2020) 24:152. doi: 10.1186/s13054-020-02877-8
9. Manzulli V, Scioscia G, Giganti G, Capobianchi MR, Lacedonia D, Pace L, et al. Real time PCR and culture-based virus isolation test in clinically recovered patients: is the subject still infectious for SARS-CoV2? J Clin Med. (2021) 10:20309. doi: 10.3390/jcm10020309
10. Capozzi L, Bianco A, Del Sambro L, Simone D, Lippolis A, Notarnicola M, et al. Genomic surveillance of circulating SARS-CoV-2 in South East Italy: a one-year retrospective genetic study. Viruses. (2021) 13:50731. doi: 10.3390/v13050731
11. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma Oxf Engl. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
12. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
13. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
14. García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. (2012) 28:2678–9. doi: 10.1093/bioinformatics/bts503
15. Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. (2012) 40:11189–201. doi: 10.1093/nar/gks918
16. Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med. (2020) 383:2291–3. doi: 10.1056/NEJMc2031364
17. Orner EP, Rodgers MA, Hock K, Tang MS, Taylor R, Gardiner M, et al. Comparison of SARS-CoV-2 IgM and IgG seroconversion profiles among hospitalized patients in two US cities. Diagn Microbiol Infect Dis. (2021) 99:115300. doi: 10.1016/j.diagmicrobio.2020.115300
18. Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun. (2020) 11:19808. doi: 10.1038/s41467-020-19808-4
19. Hulswit RJG, de Haan CM, Bosch BJ. Coronavirus spike protein and tropism changes. Adv Virus Res. (2016) 96:29–57. doi: 10.1016/bs.aivir.2016.08.004
20. Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. (2020) 182, 1284–94.e9. doi: 10.1016/j.cell.2020.07.012
21. Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. (2008) 9:267–76. doi: 10.1038/nrg2323
22. Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. (2020) 369:1014–8. doi: 10.1126/science.abd0831
23. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182, 812–27.e19. doi: 10.1016/j.cell.2020.06.043
24. Jary A, Leducq V, Malet I, Marot S, Klement-Frutos E, Teyssou E, et al. Evolution of viral quasispecies during SARS-CoV-2 infection. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis. (2020) 26:1560.e1-1560.e4. doi: 10.1016/j.cmi.2020.07.032
25. Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L, et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin Infect Dis Off Publ Infect Dis Soc Am. (2020) 71:713–20. doi: 10.1093/cid/ciaa203
26. Siqueira JD, Goes LR, Alves BM, de Carvalho PS, Cicala C, Arthos J, et al. SARS-CoV-2 genomic and quasispecies analyses in cancer patients reveal relaxed intrahost virus evolution. bioRxiv. (2020). [Preprint]. doi: 10.1101/2020.08.26.267831
27. Andino R, Domingo E. Viral quasispecies. Virology. (2015) 479–80:46–51. doi: 10.1016/j.virol.2015.03.022
28. Dolan PT, Whitfield ZJ, Andino R. Mapping the evolutionary potential of RNA viruses. Cell Host Microbe. (2018) 23:435–46. doi: 10.1016/j.chom.2018.03.012
Keywords: SARS-CoV-2, virus culture, cancer, quasispecies, WGS
Citation: Bianco A, Capozzi L, Del Sambro L, Simone D, Pace L, Rondinone V, Difato LM, Miccolupo A, Manzari C, Fedele A and Parisi A (2022) Persistent SARS-CoV-2 Infection in a Patient With Non-hodgkin Lymphoma: Intra-Host Genomic Diversity Analysis. Front. Virol. 2:758191. doi: 10.3389/fviro.2022.758191
Received: 15 September 2021; Accepted: 03 January 2022;
Published: 04 February 2022.
Edited by:
Rafael Sanjuán, University of Valencia, SpainReviewed by:
Judd Hultquist, Northwestern University, United StatesCopyright © 2022 Bianco, Capozzi, Del Sambro, Simone, Pace, Rondinone, Difato, Miccolupo, Manzari, Fedele and Parisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Parisi, YW50b25pby5wYXJpc2lAaXpzcGIuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.