 Bailey S. Mosher
Bailey S. Mosher Timothy F. Kowalik5
Timothy F. Kowalik5 Andrew D. Yurochko
Andrew D. Yurochko- 1Department of Microbiology & Immunology, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 2Center for Molecular & Tumor Virology, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 3Center for Applied Immunology and Pathological Processes, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 4Center of Excellence for Emerging Viral Threats, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 5Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, United States
- 6Center for Cardiovascular Diseases & Sciences, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 7Feist-Weiller Cancer Center, Louisiana State University Health Shreveport, Shreveport, LA, United States
- 8Center for Excellence in Arthritis and Rheumatology, Louisiana State University Health Shreveport, Shreveport, LA, United States
Human cytomegalovirus (HCMV) is a significant cause of morbidity and mortality in the immunocompromised and developing fetuses. Infection has also been linked to chronic inflammatory diseases, cardiovascular disease, and the development of certain cancers. The wide range of pathologies associated with HCMV infection is attributable to the broad cellular tropism of the virus where infection affects every organ system. Like other viruses, HCMV must tailor host cells to support productive infection. In particular, HCMV dedicates many resources and various strategies to manipulate host intracellular trafficking networks to facilitate various aspects of infection across all infected cell types. The dysregulation of host intracellular trafficking networks allows the virus to translocate to the host cell nucleus for genome replication, facilitate nuclear import/export of viral proteins and immature virions, subvert the host immune response, form new organelles for progeny virion assembly, maturation and egress, and promote cellular migration and viral spread. However, due to their complex nature, many aspects of these processes are not well-studied. New research and omics-based technologies have recently begun to elucidate the extent to which HCMV dysregulates host cell trafficking machinery. Here we review the variety of strategies HCMV utilizes to dysregulate intracellular trafficking networks to promote productive infection.
Introduction
Eukaryotic cells maintain complex and dynamic intracellular trafficking networks vital for cellular processes because of their role in maintaining the balanced flow of membrane and cargo between cellular compartments (1). As obligate intracellular parasites, viruses have evolved to effectively manipulate these intracellular transport systems to facilitate their intracellular life cycles, infection, replication and eventual release (1, 2). For example, viruses can alter endocytic properties of cellular plasma membrane proteins, which in turn disrupts the physiological trafficking of these proteins, thus causing their abnormal intracellular routing and downmodulation of their surface expression (1, 3). Additionally, viral proteins can induce the formation of entirely new intracellular compartments, such as viral assembly compartments (1, 3). Viral perturbations of host cell intracellular trafficking networks can be the consequence of viral proteins directly targeting intracellular trafficking proteins or their regulators, or through an indirect mechanism such as virus-induced signaling events that regulate formation of trafficking protein complexes (1). Although the knowledge of how viruses interact with the cellular machinery involved with intracellular trafficking components to promote infection remains limited, deregulation of these intracellular pathways do appear to be unique and/or distinct in different cell types and under different viral infection systems (1, 4).
While intracellular trafficking interference is common during infection with different viral families (1, 5), herpesvirus infections in general show extensive intracellular remodeling of host trafficking systems (1–3, 6). Of note, within the herpesviruses, intracellular remodeling is especially extensive following human cytomegalovirus (HCMV) infection, particularly within the vesicular transport system (1, 3, 5). HCMV is a betaherpesvirus with high seroprevalence (60-90%) worldwide. HCMV infection poses a public health threat, as infection is a significant cause of morbidity and mortality in immunocompromised populations such as in AIDS and cancer patients, and transplant recipients. Additionally, HCMV infection can result in neurological damage in developing fetuses and can often lead to fetal demise (5, 7–9). Because HCMV has been shown to manipulate the host intracellular trafficking system in all infected cell types (4), interference with intracellular trafficking is likely directly linked to viral pathogenesis.
Unlike other viruses, HCMV does not induce a global block on intracellular trafficking systems such as in the secretory pathway (1). Rather, the virus seems to have evolved a targeted approach to disrupt specific aspects of intracellular trafficking as a mechanism to optimize the successful establishment and maintenance of both lytic and latent infections (1, 4). The HCMV genome encodes a variety of tools such as viral tegument and non-structural proteins, noncoding RNAs, and glycoproteins to manipulate the host intracellular trafficking systems (3, 10, 11). However, the information available on how HCMV manipulates intracellular trafficking is unresolved, largely due to the complex and unknown nature of the many intracellular trafficking networks themselves – including both the endosomal and endocytic trafficking networks (3). Recent data, however, has begun to shed light on how HCMV commandeers the intracellular networks of host cells during infection. Here, we discuss the broad impact HCMV infection has on intracellular trafficking networks and review the major areas in which HCMV is best known to modulate intracellular trafficking dynamics to support productive viral infection (Table 1). We will discuss these categories in the order by which the most in-depth research has been performed: 1) formation of the viral assembly compartment (VAC) and viral egress, 2) immune evasion, 3) cellular migration and viral spread, 4) viral nuclear translocation, and 5) nuclear import and export.

Table 1 Overview of HCMV-Induced Intracellular Trafficking Network Changes. Overview of the various methodologies by which HCMV manipulates intracellular trafficking networks to promote productive infection as discussed throughout the review.
HCMV infection causes broad changes to host cell intracellular trafficking networks
The advent of ‘omics and screening-based approaches has allowed for the detailed investigation of molecular changes within cells on a broad scale. These high throughput experiments facilitate the cultivation of large-scale data sets, which can be used to study general trends and inform more specific research projects. We and others have used ‘omics-based approaches to demonstrate broad changes in cellular biology, including changes in intracellular trafficking systems, upon HCMV infection of various cell types.
Our laboratory has used a transcriptomic approach to examine HCMV-induced changes in cellular gene expression prior to nuclear translocation in primary human monocytes. We have published that HCMV infection causes a sweeping remodel of these cells to create an intracellular environment conducive to productive infection (12, 13). In monocytes, HCMV infection significantly alters the transcript level of several cytoskeletal regulators (i.e. Myosin X, Kif5), membrane trafficking regulators (i.e. small GTPases of the Rab family such as Rab13, Rab7), as well as endosomal trafficking regulators (i.e. Stx6, VAMP5), all of which are crucial to normal physiological intracellular trafficking and may play a role during viral entry in these cells (12–14).
Studies in HCMV-infected fibroblasts have shown that infection leads to significant enrichment of cellular transcripts related to intracellular trafficking late during infection (3, 6, 15). Recently, a unique combination transcriptomic/proteomic approach revealed that after infection, nearly 1000 annotated genes mapped to the gene ontology (GO) term “Transport” (6). Specifically, annotated transcript and protein-associated genes mapped to “Secretion” (269 transcript/291 protein), “Intracellular transport” (305/296), and “Vesicle-mediated transport” (421/453) (6). Overall, the exocytic pathway appeared to be the most profoundly impacted following infection, particularly those components related to the endocytic recycling compartment (ERC) (6). For example, ERC pathways involving early or recycling endosomes were negatively impacted by infection. However, components of sorting vesicle-associated exocytosis experienced increased gene expression, potentially indicating a mechanism for virion egress. In this system (of fibroblast infection by HCMV), the endocytic pathway was not noted to be greatly impacted by infection (6).
Proteomic analyses have also contributed to our understanding of the broad mechanisms HCMV utilizes to manipulate intracellular trafficking pathways of host cells late during infection. A hybrid quantitative MS approach measuring both protein localization and abundance within HCMV-infected fibroblasts during late infection revealed that a global reorganization of proteins occurs within the cell (11). This reorganization stretched across major cellular systems including the secretory pathway and the plasma membrane. Additionally, many proteins were translocated between cellular compartments at specific time points during infection (11). It was also noted that fold changes in proteins within individual cellular compartments increased with the progression of infection, suggesting differential regulation of these proteins from early to late-stage infection. For example, proteins within the Golgi assigned function in “intracellular transport” and “membrane organization” experienced upregulation at late time points during infection, while proteins with assigned function in “organic substance transport” were downregulated over the same time frame (11). These changes are reflective of pathways important to viral intracellular transport, assembly complex formation, and egress. A new LC-MS-based proteomic study also demonstrated that HCMV infection of ARPE-19 epithelial cells and MRC5 fibroblasts results in significant manipulation of the cellular exosome pathway for viral egress (16). Surprisingly, viral manipulation of this pathway was shown to occur via a novel non-classical nexus of effectors including VAMP3, a trafficking protein important to recycling endosome fusion with the plasma membrane (16).
While HCMV infection is generally not associated with inhibition of cellular protein synthesis as found during infection with other viruses, such as poxvirus infections (17, 18), polysome profiling studies have shed light on how HCMV infection impacts global translation. For example, not only does HCMV infection extensively reprogram host cell translation, but infection also significantly increases translation of cellular mRNAs critical for vital cellular functions, including vesicular transport and cytoskeletal organization (4, 17). Additionally, infection causes a repression of translation for other mRNAs, including those involved in lipid transport and antigen presentation (17).
HCMV itself encodes multiple miRNAs that interfere with intracellular trafficking on a broad scale. It has been demonstrated that HCMV-encoded miRNAs such as miR-UL112-1 and miR-US5-1 target the endocytic pathway (19). These miRNAs function broadly in at least 2 capacities. One function is to mediate the host cell immune response by interfering with inflammatory cytokine trafficking and secretion, including that of TNF-α and IL-6 (19, 20). The other function is to remodel secretory pathway components, predominantly in the Golgi and endocytic compartments, to help in the formation of the VAC and increase production of infectious virus (11, 19). Mutations in the coding regions of these miRNAs have been shown to restore secretory and cytokine trafficking pathways, which results in malformation of the VAC and appropriate release of inflammatory cytokines (19). Different aspects of these HCMV-encoded miRNAs were observed in fibroblasts, epithelial, and monocytic cell lines. Further investigation into how HCMV-encoded miRNAs function to manipulate intracellular trafficking across all cell types would be beneficial to understanding HCMV infection and pathogenesis.
HCMV also manipulates the physiological trafficking of signaling receptors, and thus broadly influences downstream signaling outcomes from those receptors. HCMV infection can result in inhibition of receptor trafficking, which causes retention of receptors in various cellular compartments or enforces trafficking of receptors to lysosomes for degradation. For example, it has been demonstrated that HCMV-encoded proteins such as UL135 actively regulate the trafficking and turnover of cellular epidermal growth factor receptor (EGFR) (21). In infected fibroblasts, UL135 enforces trafficking of EGFR from early endosomes to lysosomes for degradation, while the absence of UL135 restores accumulation of EGFR in Rab5+ early endosomes and its subsequent recycling to the plasma membrane. Additionally, active EGFR is retained in the VAC during lytic infection. However, during HCMV latency in CD34+ HPCs, EGFR is increased on the surface of the cells by HCMV UL138 (21). Control of intracellular signaling is important for viral infection. In fact, over 100 cellular kinases are predicted to be important to viral infection and progeny virion production (4). It is important to note that intracellular signaling is a primary source of intracellular trafficking regulation. For example, EGFR is upstream of many kinases, such as protein kinase C (PKC), that regulate intracellular trafficking networks by activation of SNAP receptor (SNARE) complexes and other proteins that facilitate intracellular trafficking (22, 23). Disruption of appropriate receptor trafficking can in turn disrupt intracellular signaling, which can cause the dysregulation of entire trafficking networks within the cell.
Viral regulation of host cell cytoplasmic trafficking is a critical point that can significantly impact infection outcomes. Overall, these examples of the global effects that HCMV infection has on host cell intracellular trafficking networks demonstrates on a large scale the multi-faceted approach HCMV uses to shape intracellular trafficking dynamics to facilitate productive infection. Although the changes discussed above are specific to HCMV infection, other viruses target these broad pathways. For example, Epstein-Barr Virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) also encode miRNAs and proteins that target vesicular trafficking networks to achieve productive infection (3, 19). Therefore, the study of how HCMV manipulates intracellular trafficking components may have broad implications for the understanding of these processes and their contribution to viral pathogenesis across the herpesvirus family.
HCMV manipulates intracellular trafficking components to facilitate VAC formation, viral assembly, and egress
HCMV lytic infection of fibroblasts, endothelial cells, epithelial cells, smooth muscle cells, and potentially macrophages and dendritic cells, causes the formation of a large structure within the cell known as the viral assembly compartment (VAC) which functions as a space for viral tegument acquisition and envelopment (1, 6, 7, 19, 24–31). Infection causes extensive remodeling of nearly all secretory pathway components including the ER, Golgi complex, and the endosomal system to form the VAC, complete morphogenesis of progeny virions, and facilitate egress of the viral particle from the host cell (1, 6, 7, 19, 24, 25, 27, 32). Due to the complexity of these events, definition of the specific HCMV-driven molecular processes involved has proven to be a challenging area of research. Newer studies, however, have untangled more details on the mechanisms by which HCMV manipulates intracellular trafficking of the host cell following infection (1, 6, 16, 19, 25–27, 32, 33).
During infection, both the endosomal and secretory networks are disrupted to facilitate the stepwise remodeling and rearrangement of organelles and their regulatory components to form the VAC (Figure 1A). Early and recycling endosomes, as well as trans-Golgi network (TGN)-derived vesicles, are recruited to the perinuclear MTOC within the infected cell to form the many cylindrical, nested layers that make up the core of the VAC. The core is then surrounded by the rearranged Golgi complex and lysosomes (1–3, 11, 24, 25, 32–34, 36). Formation of the VAC is dependent on the host cell microtubule network, as well as the associated dynein motors (1, 2, 36). In addition to these organelles, other cellular components have been found to be associated with the VAC, including members of the SNARE family, ESCRT machinery, and Rab GTPases, all of which are important factors for intracellular trafficking. ESCRT machinery regulates multi-vesicular body (MVB) cargo incorporation, thus its association with the multi-vesicular VAC indicates that ESCRT machinery may facilitate formation of the VAC compartments (3, 24–26, 32).
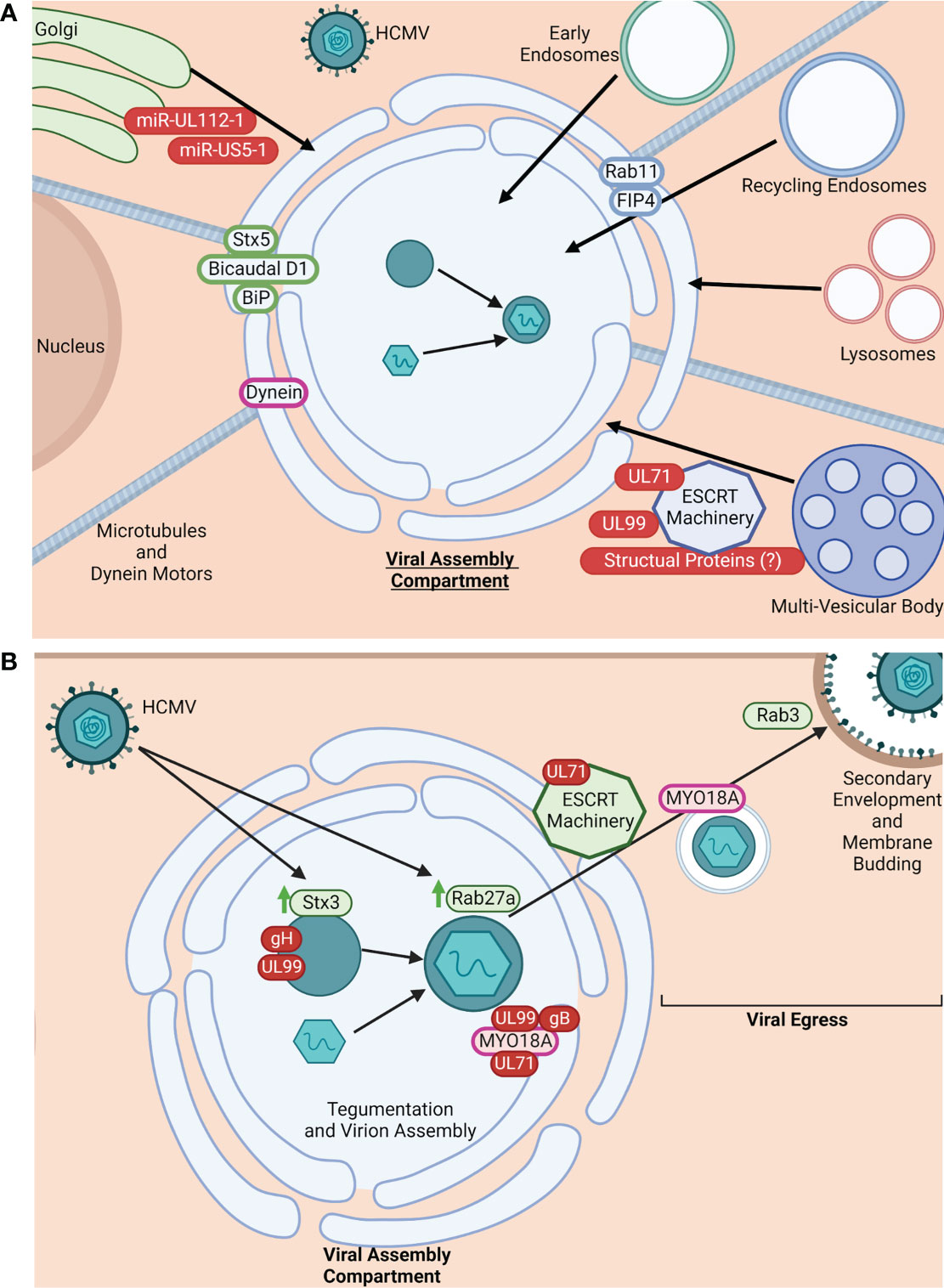
Figure 1 HCMV manipulates intracellular trafficking components for VAC formation and viral egress. (A) HCMV manipulates intracellular trafficking machinery for VAC formation. HCMV restructures the TGN, early endosomes, recycling endosomes, lysosomes, and MVBs to form the VAC (1–3, 11, 24, 25, 32–34). VAC formation is dependent on microtubules and dynein motors (1, 2, 34). Additionally, HCMV commandeers trafficking proteins to maintain the VAC and facilitate virion maturation. Some known trafficking proteins utilized in this process are: Stx5, Bicaudal D1, BiP, Dynein, Rab11, and FIP4 (1, 2, 6, 11, 16, 24, 26, 32, 34). While exact mechanisms for HCMV-induced organelle restructuring and protein recruitment are unclear, there is evidence implicating roles for HCMV-encoded miRNAs (miR-UL112-1 and miR-US5-1), UL71, UL99, and potentially structural proteins (4, 25, 26). Viral proteins/miRNA are shown in red. Golgi-associated proteins are shown in green. Recycling endosome-associated proteins are shown in blue. Molecular motors shown in purple. Arrows (black) indicate the general location of organelles within the VAC (core or periphery). Made with BioRender.com. (B). HCMV manipulates intracellular trafficking machinery for assembly and egress. It is known that cellular trafficking proteins are commandeered for virion tegumentation and assembly; for example, HCMV infection causes the upregulation (green arrows) of Stx3 and Rab27a, both of which are thought to be important for virion production/envelopment and exit from the VAC. Stx3 is found in areas of gH and UL99 accumulation within the VAC (3, 6, 25, 34, 35). Additionally, the unconventional myosin motor MYO18A is recruited to the VAC, typically associated with gB, UL99, and UL71, implicating these proteins in MYO18A regulation and potential involvement in virion assembly and egress from the VAC (11). Viral egress is also an active event mediated by viral proteins hijacking intracellular trafficking mechanisms. Evidence suggests that UL71 modulates the function of ESCRT machinery to facilitate viral egress from the VAC and the infected cell (33). MYO18A has also been implicated as the molecular motor responsible for carrying mature viral particles from the VAC to the plasma membrane (11). Further, Rab3 is also re-distributed during infection and thought to be important for secondary envelopment, implicating a Rab3/Rab27a linked trafficking pathway for HCMV egress (6). Viral proteins are shown in red, cellular trafficking proteins in green, and molecular motors in pink. Made with BioRender.com.
The assembly of the VAC and formation of the progeny virions within the VAC have little homology with normal cellular processes. That is, the regulation of formation, size, and complexity of the VAC is distinct to HCMV infection (26, 27). Because the formation and function of the VAC is thought to be largely dependent on viral proteins, VAC formation and subsequent virion assembly is an active process facilitated by viral manipulation of host cell trafficking machinery. A number of specific cellular trafficking proteins have been shown to be needed for VAC formation and maintenance, the most well-studied being the small Rab GTPase Rab11, Syntaxin 5, Bicaudal D1, FIP4, BiP, and dynein (1, 2, 6, 11, 16, 24, 26, 32, 33, 36) (Figure 1A). Endosomal recycling depends on proteins such as Rab11 as well as their effectors, and interference can significantly alter endosomal function and localization within the cell, facilitating formation of the VAC. For example, interference with Rab11 and/or its effectors leads to re-routing of cargo, disruption of recycling endosome localization to intended cellular locations, and malformation of the VAC (1, 6, 19, 37–40). HCMV viral products are known to actively recruit these cellular trafficking proteins and other components to the VAC. For example, the viral microRNAs (miR-UL112-1 and miR-US5-1) previously described to target the mRNA of trafficking proteins also disrupt physiological Golgi morphology and restructure the organelle to help form the VAC (Figure 1A). Additionally it has been shown that HCMV UL71 plays a role in multi-vesicular body (MVB) formation. For example, in the absence of UL71, MVB diameters are significantly larger during infection and exhibit enlarged vesicular and tubular morphology unlike that seen in cells infected by UL71-containing viruses. Because the formation of MVBs is predominantly regulated by endosomal sorting complexes required for transport (ESCRT) machinery, it is thought that UL71 may interfere with or manipulate ESCRT function, which has been reported to be important for HCMV maturation and VAC formation. (Figure 1A) (25, 34). UL99 may also aid in utilizing ESCRT machinery for VAC formation (4, 25, 26). The viral exploitation and recruitment of ESCRT components to the VAC has also been proposed to be a product of specific domains (conserved amino acid motifs PTAP, YXXL, or PPXY) within the HCMV tegument and/or glycoproteins (1, 19, 34, 35). However, data on the requirement of ESCRT machinery for HCMV infection remains conflicted.
Virion assembly within the VAC is also dependent on interactions with host cell trafficking proteins and membrane systems (6, 36, 41). A few cellular trafficking proteins have been identified to be important in HCMV assembly. HCMV infection causes the upregulation of Rab27a, a small GTPase of the Rab family that regulates Rab11+ membrane fusion within the recycling route, as well as late endosome and lysosome fusion events within the secretory pathway. During infection, Rab27a can be found localized within the center of the VAC and is requisite for efficient virion production and egress, suggesting the exploitation of its function in endolysosomal secretion by HCMV for viral assembly (Figure 1B) (3, 6, 36). Rab3, another member of the Rab family that regulates - dependent exocytosis, is an important component of secondary envelopment of progeny virions (Figure 1B) (6). The SNARE Syntaxin 3 (Stx3), a regulator of membrane fusion events within the exocytosis trafficking pathway, is also upregulated during HCMV infection at both the level of transcript and protein (6). In infected cells, Stx3 is found within the VAC, particularly at sites of viral glycoprotein gH and tegument protein UL99 accumulation, where it is shown to be important in the tegumentation and secondary envelopment process within the VAC, as well as in the production of infectious HCMV particles (Figure 1B) (6, 25, 36, 41). Mass spectrometry analysis of progeny virion proteomes found the presence of: MHC-I, Rab12, Rab 23, Rab32, SNX2, SNX3, VAMP2, VAMP3, STX12, VTI1A, and PIK3C2A, suggesting that membrane components for virion envelopment are derived from the tubular portions of early endosomes that participate in Arf6/Rab8-dependent cargo recycling (32). Newer data also supports that the envelopment and egress of HCMV is likely via a Rab3/Rab27a-linked lysosome or sorting vesicle-like trafficking pathway (Figure 1B) (6).
Egress of the progeny HCMV particles from the VAC through the cytoplasm, to the plasma membrane, and out of the cell is also thought to be an active process where the virus commandeers intracellular trafficking proteins, molecular motors, and the microtubule network surrounding the VAC to exit the infected cell (11, 24, 25). Secondary envelopment and viral egress have been recently reviewed by Lucin et al. (32). Physiological membrane budding typical of endosomal cargo sorting, as well as MVB formation, is controlled by the ESCRT complex (1, 34, 35). As previously mentioned, the viral protein UL71 plays an important role in VAC formation due to its manipulation of ESCRT machinery (4, 25, 26, 34). There is also evidence that UL71 plays a role in the budding of the viral progeny from the infected cell during the final steps of viral egress as ESCRT machinery is thought to be involved in the final maturation steps of the virus (Figure 1B) (34). The unconventional myosin motor, MYO18A, has also been found to be associated within and directly outside of the VAC during infection, along with the HCMV glycoprotein gB of the viral envelope, UL71, and the tegument protein UL99 (11). Along with its association with these proteins involved in assembly and egress of the viral particle, there is evidence that MYO18A functions late in infection during viral production and release, where it likely associates with virus-containing vesicles exiting the VAC and is involved in their egress and final envelopment (Figure 1B) (11).
In summary, the complex nature of HCMV assembly and egress has made research in these areas challenging, but continued study and advancing technology have begun to unravel the intricate virus-host interactions involved in these processes. The limited knowledge to date gained from these studies is predominantly derived from fibroblast cell lines. Because the processes of VAC formation, virion assembly, and viral egress are critical for the HCMV life cycle, and because HCMV exhibits broad cellular tropism, further study of these processes in other cell types is needed to solidify and expand existing knowledge. As an enveloped virus, HCMV must interact with host membranes and other membrane components to produce infectious viral progeny and successfully complete its life cycle. Because assembly and egress of new viral particles does not overlap with normal cellular function, the virus actively commandeers host resources to facilitate the latter portion of its intracellular life cycle through various means such as viral effector proteins and miRNAs. Thus, HCMV facilitates the formation of the viral assembly compartment, virion assembly, and egress of viral progeny through extensive multi-faceted manipulation and remodeling of the host intracellular trafficking system. Requisition of host trafficking resources for assembly, envelopment, and egress is a common feature for herpesviruses. For example, Herpes Simplex Virus 1 (HSV-1) also commandeers host trafficking proteins such as Rab6, Rab5, and Rab11 for progeny virion envelopment and egress (2). Thus, further study of how HCMV utilizes cellular trafficking components for assembly and egress may be beneficial for investigating these processes within the herpesvirus family as well as other viral families, and thereby contributing to greater understanding of intracellular viral life cycles.
HCMV manipulates intracellular trafficking to promote immune evasion
Evading recognition by the host immune system is an important facet of viral infections. To infect hosts and successfully produce viral progeny, viruses must have strategies to antagonize, blunt, or evade host immune function. Relative to other viruses, HCMV has a high coding capacity and employs various tools to prevent immune recognition. A large portion these strategies involve manipulating the host intracellular trafficking network to prevent antigen presentation, NK cell recognition, and inflammatory cytokine secretion.
Antigen presentation is a crucial immunologic process for the detection and elimination of infected cells by both the innate and adaptive branches of the host immune system. HCMV utilizes several mechanisms to interfere with the intracellular trafficking of MHC I and MHC II molecules, thereby diminishing the antigen presentation ability of the infected host cell. While HCMV infection perturbs both MHC I and MHC II intracellular trafficking, the strategies applied to MHC I trafficking are more well studied (3, 42, 43). Several viral effectors (US2, US3, US6, US10, US11, and UL82) that are expressed differentially throughout infection specifically target the secretory pathway to disrupt the trafficking of the MHC I complex or its assembly components (Figure 2A) (1, 3, 11, 43–47). HCMV US2 targets MHC I at the early stages of the biosynthetic secretory pathway by destabilizing and dislocating free membrane-inserted MHC I heavy chains from the ER and promoting their translocation into the cytosol, where they are then degraded by the proteasome (Figure 2A) (1, 43–48). Prior to peptide loading in the ER, HCMV US3 complexes with the MHC I heavy chain/β2m heterodimer, causing retention of the immature complex in the ER and its degradation, thereby inhibiting trafficking of the complex to the cell surface (Figure 2A) (3, 43, 45–47). HCMV US6 targets the TAP complex by inhibiting the binding of ATP to TAP1. This inhibition impairs TAP complex function, thus preventing viral cytosolic peptides from entering the ER and being loaded onto MHC I complexes, in turn causing retention of the immature MHC I complexes within the ER (Figure 2A) (3, 43, 45–47). HCMV US10 has been shown to bind the MHC I heavy chain. However, this event only delays trafficking of MHC I from the ER to the cell surface rather than preventing it. Additionally, US10 can act similarly to US11 in promoting MHC I degradation (Figure 2A) (1, 43, 45–47). Similarly, UL82 has been documented to delay the transport of peptide-loaded MHC I from both the Golgi and the ER (47). Acting in concert with US2, US3, or cellular proteins such as Derlin-1 of the UPR, the HCMV protein US11 also functions in the rapid dislocation of MHC I from the ER to the cytosol for proteasomal degradation (3, 44, 45, 47). However, US11 can also interact with free MHC I heavy chains or immature MHC I heterodimers, as well as cause the dislocation of misfolded MHC I complexes from the ER (Figure 2A) (1, 3, 43–45, 47). While there is significant overlap in function between US2, US3, US6, US10, US11, and UL82, this overlap has likely evolved to combat both MHC I allelic and cell type-specific differences in antigen presentation processes (45, 47). In addition to HCMV, HSV-1, EBV, and several other viruses (such as HIV) can prevent the translocation of MHC I molecules to the cell surface or their internalization (3). Prevention of antigen presentation is a crucial facet of most productive viral infections. Further research into how HCMV and other viruses modulate the intracellular trafficking of immune recognition molecules like MHC I may provide key insight into viral pathogenesis.
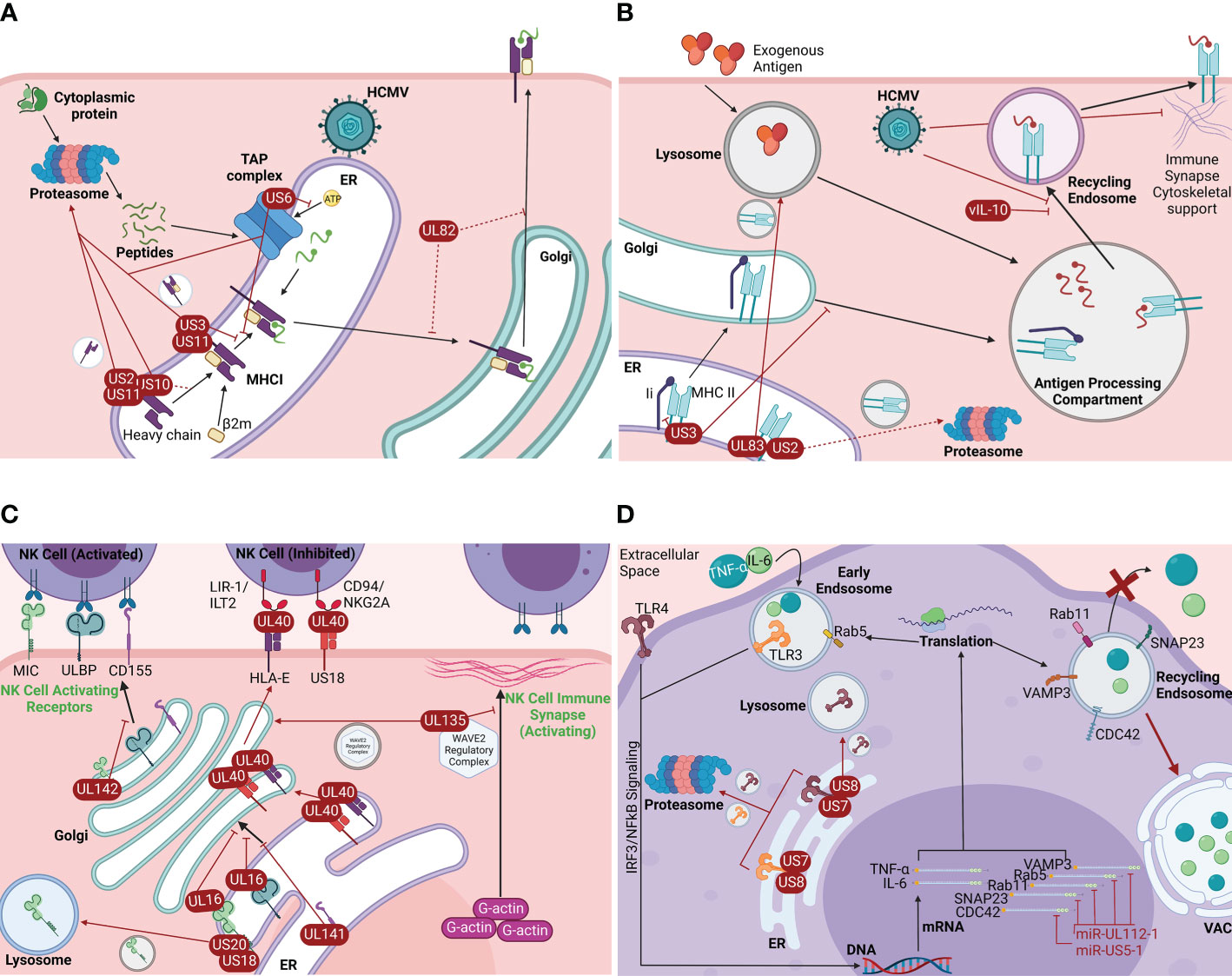
Figure 2 HCMV manipulates intracellular trafficking components to promote immune evasion. (A) HCMV-induced MHC I trafficking disruption. HCMV encodes viral proteins that disrupt the intracellular trafficking of the MHC I complex or its components early in the biosynthetic pathway: US2, US3, US6, US10, US11, and UL82 (1, 3, 11, 42–45). US2 destabilizes and dislocates free MHC I heavy chains from the ER and promotes their translocation to the cytosol and subsequent proteasomal degradation (1, 42–46). US3 binds the immature MHC I complex in the ER and causes its retention in the ER and subsequent proteasomal degradation (3, 42, 44, 45). US6 prevents TAP ATP-binding, which prevents the transport of cytosolic peptides into the ER for loading onto MHC I, in turn causing MHC I retention in the ER and ultimately proteasomal degradation (3, 42, 44, 45). US10 binds the MHC I heavy chain and delays MHC I translocation to the cell surface. US10 can also act similarly to US11 (1, 42, 44, 45). US11 acts in concert with US2, US3, and/or cellular proteins such as UPR-associated Derlin-1 to rapidly dislocate the immature MHC I complex from the ER to the cytosol for degradation. US11 can also interact with individual MHC I heavy chains or immature heterodimers and promote the dislocation of misfolded MHC I complexes (1, 3, 42–45). UL82 delays transport of mature MHC I from both the Golgi and ER (45). Viral proteins are shown in red. Dotted lines indicate delayed transport. Made with BioRender.com. (B) HCMV-induced MHC II trafficking disruption. HCMV causes the disruption of MHC II trafficking. Infection results in the retention of MHC II in an enlarged peptide loading compartment and/or its degradation (3). Some of the viral effectors involved in MHC I trafficking disruption are thought to play a role in MHC II trafficking disruption. US2 rapidly dislocates MHC II subunits from the ER, though this event is possibly cell-type specific (red dotted line) (42, 47, 48). US3 inhibits MHC II binding to the variant chain (Ii), which inhibits efficient trafficking to the antigen processing compartment and leads to MHC II lysosomal degradation (42, 48, 49). Similar to US2, UL83 binds and dislocates MHC II subunits from the ER to the lysosome for degradation (42, 47). HCMV also encodes a viral homolog of IL-10, vIL-10, which prevents MHC II trafficking to the surface after acquiring antigen (48, 50). Further, HCMV infection causes destabilization of dendrite cytoskeletal components, inhibiting formation of the immune synapse (3). Made with BioRender.com. (C) HCMV-induced disruption of NK cell receptor trafficking. HCMV dedicates several viral proteins to dysregulate the trafficking of NK cell receptors: US18, US20, UL16, UL135, UL141, UL142, and UL40 (3, 41, 50–53). The effect of HCMV proteins on NK cell-activating receptors is more well-studied. UL16, UL141, and UL142 inhibit the trafficking NK cell-activating receptors of the ULBP and MIC families, as well as CD155, by binding the receptors and causing the retention within the ER and Golgi (3, 41, 50–58). US18 and US20 promote MICA transport to the lysosome (1, 3, 52, 53). The leader sequence of UL40 functions as a peptide for HLA-E and the HCMV-encoded MHC I homolog US18, facilitating the transport of HLA-E and US18 to the cell surface where they act as ligands for the NK cell inhibitory receptors CD94/NKG2A and LIR1/ILT2, respectively (3, 42, 50, 52, 57, 59, 60). UL135 modulates the actin cytoskeleton necessary for immune synapse formation by commandeering the WAVE2 regulatory complex (WRC), a key regulator of actin polymerization. UL135 causes WRC components to traffic to the Golgi for retention which inhibits F-actin polymerization, a cytoskeleton characteristic of NK cell inhibitory receptors (52). Viral proteins are shown in red. Made with BioRender.com. (D) HCMV disrupts inflammatory cytokine trafficking. HCMV seems to rely on manipulation of intracellular trafficking networks to inhibit proinflammatory cytokine secretion (19, 61). The HCMV-encoded miRNAs miR-UL112-1 and miRNA-US5-1 target the mRNA of the major intracellular trafficking proteins CDC42, SNAP23, Rab11, Rab5, and VAMP3, preventing their translation (6, 19, 61–68). These trafficking proteins have major roles in the secretion of important inflammatory cytokines such as IL-6 and TNF-α. Lack of these trafficking proteins causes a significant reduction in the secretion of IL-6 and TNF-α (red x), as well as retention of IL-6 and TNF-α in the VAC (6, 19). Additionally, the HCMV-encoded proteins US7 and US8 each bind both TLR3 and TLR4, major components of the interferon signaling pathway (69). US7 and US8 binding of TLR3 and TLR4 dislocate the receptors from the ER to the proteasome and/or lysosome for degradation, inhibiting the production and secretion of proinflammatory cytokines downstream of the IRF3 and NFκB transcription factors, as well as other proinflammatory factors (69). Made with BioRender.com.
The viral mechanisms underlying MHC II trafficking disruption are not as well understood as those affecting MHC I trafficking. Unlike the early disruptions seen with MHC I intracellular trafficking, HCMV targets MHC II both early and late in the physiological trafficking route (3). HCMV infection results in the reduction of surface MHC II from monocytes, macrophages, dendritic cells, and mature Langerhans cells where the complex is found either retained in an enlarged peptide loading compartment with mature peptide-loaded MHC II at the periphery or degraded via lysosome or proteasome. A similar phenomenon also occurs in infected hematopoietic progenitor cells that serve as the latency compartment for HCMV, potentially by viral miRNA (3). It is known, however, that some viral effectors involved in the trafficking disruption of MHC I are also involved with trafficking disruption of MHC II (3, 43, 45, 47, 49). US2 has been shown to cause rapid dislocation of MHC II subunits from the ER, causing their transport to the proteasome for degradation (Figure 2B). However, this effect is potentially cell type-specific (43, 46, 49). There is some evidence that US3 acts earlier in the MHC II trafficking pathway. US3 is reported to bind MHC II in the ER, which reduces the ability of the complex to bind with the invariant chain (Ii/CD74), a protein that facilitates appropriate trafficking of the complex to the endosomal compartment for peptide loading (43). While US3-bound MHC II complexes traffic normally from ER to Golgi, they are unable to be sorted efficiently to peptide loading endosomes, resulting in MHC II complex accumulation and degradation (Figure 2B) (43, 49, 50). Cytoskeletal remodeling may also be associated with the viral MHC II immune evasion strategy (3). For example, it has been shown that in HCMV-infected mature Langerhans cells, loss of mature MHC II from the plasma membrane is associated with the loss of cytoskeleton components in the cellular dendrites that support the formation of the immune synapse, reducing the ability to activate T cells (Figure 2B) (3). HCMV UL83 downregulates MHC II surface expression by causing MHC II accumulation in lysosomes and subsequent degradation (43, 46). Additionally, HCMV encodes a viral homologue of interleukin 10 (vIL-10), which is known to downregulate MHC II surface expression by preventing trafficking of the complex to the cell surface after peptide loading (Figure 2B) (49, 51). Though more research is necessary to understand how HCMV modulates MHC II transport, overall, HCMV is documented to disrupt MHC I and MHC II trafficking likely to allow infected cells to resist CD8+ T cell and natural killer (NK) cell recognition and lysis (3, 42, 43).
It is currently known that HCMV dedicates at least 5 viral effectors to either increase NK cell inhibitory receptors or suppress activating receptors to modulate NK cell responses (3, 42, 51–54). Most of these viral effectors target NK cell-activating receptors by inhibiting their intracellular trafficking through the endosomal and secretory pathways. HCMV proteins UL16, UL141, and UL142 operate within the secretory pathway to inhibit the cell surface localization of NK cell-activating receptors in the ULBP family (1, 2/3, and 6) and the MIC family (A and B), as well as CD155 by causing their intracellular retention in the ER and Golgi and reducing their turnover (Figure 2C) (3, 42, 51–56). UL16 binds members of the MIC and ULBP families similarly to their ligand NKG2D, causing their retention in the ER/cis-Golgi (3, 57). Similarly, UL142 binds MICA and causes retention of the activating receptor in the Golgi while UL141 retains the immature form of the NK cell-activating receptor CD155 within the ER (3, 58, 59). HCMV effectors US18 and US20 manipulate the intracellular trafficking of NK cell-activating receptors by promoting the transport of MICA to the lysosome for degradation (Figure 2C). While there is less known about the viral mechanisms underlying the disruption of NK cell inhibitory receptor intracellular trafficking, it has been demonstrated that the leader sequence of UL40 functions as a peptide for the MHC I isotype HLA-E, facilitating MHC I antigen loading independent of US6-mediated inhibition of the TAP complex. Similarly, the US40 leader sequence serves as a peptide for US18, an HCMV-encoded MHC I homolog. By functioning as an MHC I peptide, UL40 promotes the trafficking of US40-bound HLA-E and US18 to the cell surface where they bind the NK cell inhibitory receptors CD94/NKG2A and LIR1/ILT2, respectively, to block NK cell-mediated lysis (3, 43, 51, 53, 58, 60, 70). Additionally, the HCMV effector UL135 disrupts the formation of the underlying F-actin ring necessary for the proper alignment of the NK cell recognition synapse. UL135 commandeers the WAVE2 regulatory complex (WRC), a key regulator of actin polymerization for immunological synapses, and causes the trafficking of WRC components to the Golgi (53). In this way, UL135 prevents F-actin polymerization, destabilizing the NK cell immune synapse to a state similar to that observed underlying NK cell inhibitory receptors (Figure 2C). Thereby, UL135-mediated disruption of F-actin formation reduces the recruitment of both NK and T cells to infected target cells (53). Mechanisms by which NK cell receptors are modulated by viral infection have been extensively studied following HCMV infection (3, 54–56). KSHV has also been documented to interfere with MICA, redistributing and retaining the ligand within infected cells (3, 56, 61). This is a product of convergent evolution between the viruses and an example of how important NK cell evasion is to the herpesvirus family.
Inhibition of inflammatory cytokine secretion is also important for HCMV immune temperance. HCMV has evolved methods to prevent the secretion of inflammatory cytokines from infected cells, in part by altering their intracellular trafficking (19, 62). Secretion of IL-6 and TNF-α, the central inflammatory cytokines that stimulate the innate immune response, is facilitated by a network of trafficking proteins within the endocytic and secretory pathway including VAMP3, SNAP23 and various members of the Rab family including Rab11 and Rab5 (19, 62, 63). As previously mentioned, HCMV encodes several microRNAs, such as miR-UL112-1. miR-UL112-1 targets the mRNA of VAMP3, a major trafficking component involved in the docking and fusion of vesicles with the plasma membrane. It also targets the mRNA of Rab5, a major trafficking component for intracellular vesicle trafficking, and the mRNA of Rab11, a key requisite GTPase that regulates recycling endosome trafficking. Further, miR-UL112-1 targets the mRNA of SNAP23, a key trafficking component for secretory cargo transport to the plasma membrane (Figure 2D) (6, 19, 62–69). Additional evidence demonstrates that HCMV miR-US5-1 also targets these components, as well as the mRNA of CDC42, a critical protein in actin nucleation and the retrograde transport of recycling endosomes involved in the secretory pathway (Figure 2D) (6, 19, 65). As a result of HCMV miR-UL112-1 and miR-US5-1 activity, the intracellular trafficking and secretion of TNF- α and IL-6 is significantly impaired in infected host cells, where these inflammatory cytokines are instead retained in the VAC (6, 19). HCMV also inhibits the production of inflammatory gene products, such as TNF-α and IL-6, by dislocating Toll-like receptors (TLRs) from the ER. Specifically, the HCMV protein US7 targets both TLR3 and TLR4 for ubiquitination and subsequent proteasomal degradation via potential association with the ERAD components Sec61β or Derlin-1. US8 also targets TLR3 and TLR4 for degradation, although by different mechanisms than UL7. US8 binds TLR4, destabilizing the protein and causing its ubiquitination and subsequent transport to both the lysosome and the proteasome (71). Additionally, US8 binds UNC93B1, the protein that stabilizes TLR3 and facilitates trafficking of TLR3 to endolysosomes where it functions as an interferon regulatory factor. US8-bound UNC93B1 is not only targeted to the lysosome for degradation but is also unable to bind TLR3. TLR3 unable to bind UNC93B1 is consequently destabilized, ubiquitinated, and subsequently trafficked to the proteasome for degradation (Figure 2D) (71).
Regulation of other immunologically relevant intracellular trafficking pathways, such as the ARF6 clathrin-independent endocytosis (CIE) pathway, can also be influenced by HCMV infection. The CIE pathway is a trafficking route that regulates MHC I, MHC II, ILR2-α, and CD59 endocytosis and recycling (72). ARF6, a GTP binding protein, regulates CIE by modulating CIE cargo endocytosis, deposition of the cargo into sorting endosomes, and recycling of the cargo to the plasma membrane. However, during HCMV infection, GTP-bound ARF6 protein levels fall while GDP-bound ARF6 protein levels increase, inhibiting ARF6 recycling and causing an accumulation of ARF6 in sorting endosomes (72). Importantly, the depletion of ARF6-GTP reroutes CIE cargo to EEA1+ membranes, resulting in the retention of CIE cargo - including important immunologic proteins such as MHC I, CD59, CD147, and CD98 – in enlarged sorting endosomes. Not only does HCMV-induced dysregulation of the CIE pathway restrict antigen presentation, but it may also affect the signaling that occurs from intracellular vesicles (72, 73).
The immune response of a host needs to be actively overcome, or at least dampened/delayed, by pathogens to efficiently infect a host. HCMV utilizes a variety of strategies to mitigate the host immune response with a variety of tools including viral effector proteins and viral microRNAs. While the entirety of the immune evasion tools utilized by HCMV are important for productive infection, here, we focus on the tools directly related to intracellular trafficking. As demonstrated by the above examples, a key strategy of HCMV to overcome the hurdles of the host innate and adaptive immune response is to actively target and dysregulate the intracellular trafficking of immunologic components such as MHC I and MHC II, NK cell receptors and inflammatory cytokines, as well as pathways that regulate the physiological trafficking of these components. Disruption of the appropriate intracellular trafficking of these immune response regulators restricts antigen presentation, NK and T cell activation, and virus-induced innate immune activation, facilitating an intracellular environment conducive to infection. Although discussed in other reviews, it is important to note that there is overlap of these features with other herpesviruses such as HSV-1/2, KSHV and EBV (3, 55, 56, 61), demonstrating how critical modulation of immune molecule trafficking is to not only HCMV, but the entire herpesvirus family (α, β, and γ herpesviruses).
HCMV manipulates intracellular trafficking networks to promote host cell migration and viral spread
HCMV spread from blood to various host organ systems during acute infection and reactivation from latency is predominantly facilitated by CD14+ circulating peripheral blood monocytes. Once within the tissues, HCMV is typically spread from epithelial cells, endothelial cells, fibroblasts, and smooth muscles cells in a cell-to-cell manner following lytic infection (7, 9, 74, 75). Thus, the virus must also actively modulate host trafficking systems within the cells of these separate compartments (solid tissue and blood) to promote cellular migration and viral spread during lytic and latent infections.
HCMV infection alters migration of infected cells in solid tissue. We have previously published that HCMV infection of endothelial cells triggers depolymerization of the intracellular actin network (76), causing a loss of the actin stress fibers that maintain the cell-cell junctions of the endothelial barrier (Figure 3A). The virus-induced destabilization of actin filaments, which also serve as a principal pillar of intracellular trafficking systems, increases endothelial layer permeability. This in turn increases migration of leukocytes through the endothelium, facilitating bidirectional viral spread between tissues and the blood stream (Figure 3A) (76). In fibroblasts, it has been shown that the HCMV-encoded GPCR US27 increases the amount of the cellular chemokine receptor CXCR4, co-localizes with CXCR4 at the cell surface, and is likely endocytosed in the same vesicle as CXCR4 after ligand-binding (Figure 3A) (77, 78). Additionally, US27 enhances the internalization of CXCR4 after ligand stimulation and significantly slows the recycling of the receptor back to the plasma membrane (Figure 3A) (77). This viral manipulation of CXCR4 intracellular trafficking potentiates CXCR4 signaling events, which greatly increases the migration of these cells to CXCL12, the ligand of both CXCR4 and US27 (Figure 3A). As CXCL12 is expressed in the bone marrow, the primary site of latency for HCMV, the increased migration of these cells may expand the latency pool for long term persistence (9, 74, 77, 78). It has also recently been shown that HCMV manipulates intracellular trafficking machinery to increase extracellular vesicle biogenesis in fibroblasts. Specifically, the ESCRT-III complex, a member of the ESCRT machinery and a mechanism for extracellular vesicle generation, is increased during infection and correlates with an increase in extracellular vesicle release from infected cells (80). While morphologically similar to uninfected extracellular vesicles, the extracellular vesicles generated during HCMV infection are modified in both size and content. Infection-generated extracellular vesicles are smaller and contain specific late viral proteins (gB, gH, UL46, UL82, and UL85). These late protein-containing vesicles have been shown to enhance viral spread to uninfected cells and increase the efficiency of infection, potentially by priming uninfected cells into a pro-viral environment (Figure 3A) (80). Importantly, inhibition of extracellular vesicle biogenesis pathways results in slowed and inefficient viral spread (80).
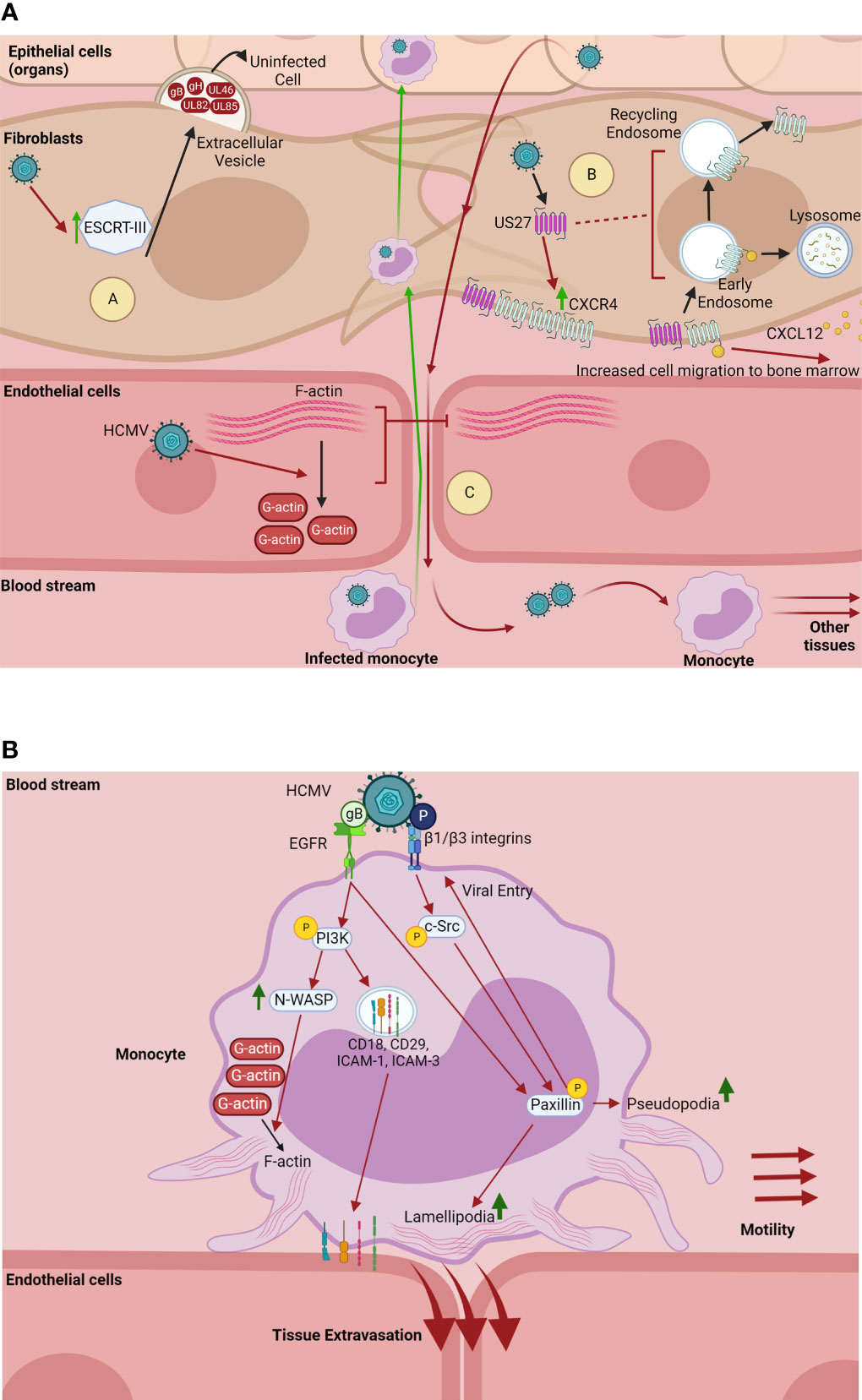
Figure 3 HCMV manipulates intracellular trafficking components to promote cell migration and viral spread. (A) HCMV dysregulates trafficking components to promote viral spread. (A) In fibroblasts, HCMV upregulates ESCRT-III, a component of the ESCRT complex and mechanism for extracellular vesicle biogenesis (77). HCMV-induced upregulation of ESCRT-III correlates with an increase in extracellular vesicle generation and release into the extracellular space. Extracellular vesicles generated through HCMV infection contain the late proteins gB, gH, UL46, UL82, and UL85. These vesicles enhance viral spread and the efficiency of infection, potentially by priming uninfected cells for a proviral environment (77). (B) HCMV-encoded US27 increases the amount of the chemokine receptor CXCR4 in fibroblasts, co-localizes with CXCR4 at the cell surface, and is likely endocytosed along with CXCR4 after ligand (CXCL12)-binding (76, 78). US27 also enhances the rapid internalization of CXCR4 after CXCL12 stimulation and significantly slows the receptor recycling back to the plasma membrane, causing a potentiation of CXCR4 chemotactic signaling. The US27-mediated potentiation of CXCR4 signaling greatly increases the chemotaxis of infected cells towards the ligand CXCL12, which is expressed in the bone marrow that serves as the HCMV latency compartment (9, 73, 76, 78). (C) HCMV infection of endothelial cells causes the depolymerization of actin which results in the loss of F-actin stress fibers that maintain the integrity of cellular junctions between endothelial cells (75). The breakdown of actin stress fibers increases the permeability of the endothelial cell layers, allowing HCMV-infected peripheral blood monocytes through to carry HCMV into the fibroblasts and epithelial cells of organs. Conversely, HCMV-induced endothelial layer permeability allows for newly produced virus to be released into the bloodstream, where circulating monocytes can then be infected, facilitating viral dissemination to other tissues (75). Made with BioRender.com. (B) HCMV enhances monocyte motility and tissue extravasation. On monocytes, HCMV binds and activates EGFR and β1/β3 integrins via its glycoproteins gB and pentamer complex (P), respectively (79). This receptor-ligand engagement activates the PI3K and c-Src signaling cascades, which in turn chronically activates the downstream cytoskeletal and focal adhesion regulator paxillin (80). Activation of PI3K by gB also upregulates the highly active actin nucleator N-WASP, which significantly upregulates the formation of F-actin (81). Further, HCMV activation of PI3K redistributes intracellular stores of adhesion molecules (CD18, CD29, ICAM-1, and ICAM-3) to the cell surface (82). Together, the HCMV-induced upregulation of N-WASP and paxillin, as well as increased transport of adhesion receptors, significantly increase the formation of monocyte lamellipodia and pseudopodia, creating a “hyper” adhesive and motile monocyte, which enhances monocyte motility and tissue extravasation (9, 80–82). Further, activation of paxillin is required for efficient viral entry into monocytes (80). Made with BioRender.com.
HCMV infection also increases migration of infected circulating cells. Circulating peripheral blood monocytes are initially non-adherent cells and cannot usually enter tissue unless stimulated by external sources. Yet in these cells that are utilized by HCMV for hematogenous dissemination to organ tissues (9, 79, 82). Our laboratory has published that HCMV manipulates the intracellular trafficking components of monocytes to increase both cellular motility and extravasation to the tissues. HCMV glycoprotein complexes gB and gH/gL/UL128-131 (pentamer) bind and activate cellular EGFR and β1/β3 integrins respectively on the surface of the monocyte (14, 81, 83). Both of these receptor-ligand engagement events cause induction of genes associated with cellular motility and adhesion. Specifically, HCMV gB and pentamer-induced signaling through EGFR and β1/β3 integrins activate the PI3K and c-Src signaling cascades respectively, and chronically activate the downstream cytoskeletal and focal adhesion regulator paxillin (Figure 3B) (82). Viral activation of PI3K signaling also increases transport of intracellular stores of adhesion molecules such as CD29, CD18, and ICAM-3 to the monocyte plasma membrane. Similarly, virus activation of NF-κB redistributes ICAM-1. Together, these signaling events alter the trafficking of cellular adhesion molecules to promote firm adhesion of infected monocytes to epithelial and endothelial barriers (79). Additionally, the virus-induced EGFR signaling upregulates the highly active actin nucleator N-WASP, which significantly enhances the formation of F-actin (Figure 3B) (84). The activation of paxillin and N-WASP in infected monocytes leads to significant increases in both pseudopodia and lamellipodia formation, allowing for enhanced cellular motility and monocyte dissemination across both epithelial and endothelial cell barriers (9, 79, 82, 84). Thus, HCMV-induced signaling cascades significantly increase the diapedesis of infected monocytes through epithelial cell barriers, as well as through endothelial cell barriers associated with the brain and umbilical cord microvasculature (9). Additionally, HCMV modulation of paxillin activity is also required for efficient viral entry into monocytes (82). Together, the modulation of intracellular trafficking components via HCMV-induced signaling events pushes infected monocytes into a distinct state of “hyper” motility and adhesion, which ultimately increases monocyte extravasation to the tissues and bone marrow for viral dissemination (9, 79, 82, 84–86).
In solid tissues and circulating blood, HCMV has developed various strategies to promote cellular migration and thus viral spread throughout the host. HCMV effector proteins as well as envelope glycoproteins directly and indirectly manipulate intracellular trafficking systems to help promote dissemination of the virus and long-term persistence.
HCMV manipulates intracellular trafficking for nuclear translocation
HCMV is a DNA virus, and like most DNA viruses, must translocate to the host cell nucleus for viral genome replication. HCMV must therefore not only orchestrate translocation of the viral genome to the nucleus, but must also coordinate the nuclear translocation of both tegument and de novo viral proteins, which are necessary for viral genome replication and appropriate expression of the viral gene cascade. After the completion of viral DNA replication, the newly synthesized genomes are encapsidated and must egress out of the nucleus to the VAC for virion maturation (as discussed earlier in this report) (7).
While precise mechanisms remain unclear, nuclear translocation of the viral genome in fibroblasts, epithelial cells, and endothelial cells is rapid (within approximately 30 minutes) and has been shown to be dependent on microtubules, actin cytoskeleton rearrangement, and the intermediate filament protein vimentin (7, 10, 83, 87, 88). HCMV nuclear translocation process in monocytes, however, is temporally extended (~3 days), and exhibits complex retrograde trafficking dynamics (Figure 4). Our laboratory has shown that on monocytes, gB and the pentamer complex bind cell surface EGFR and β1/β3 integrins, respectively. The engagement and activation of these receptors causes virus internalization into the cell where the process of nuclear translocation begins. In these cells, HCMV nuclear translocation follows a distinct retrograde transport-like pathway. HCMV is first trafficked to EEA1+ vesicles (15 – 45 mpi) (81, 83). While the initial trafficking of the viral particle to early endosomes prior to nuclear translocation is also common to epithelial cells (88–90), and potentially fibroblasts (88, 91), de-envelopment occurs at this stage, releasing tegument proteins and the genome-containing capsid into the cytosol for the completion of the nuclear translocation process (7, 88–92). However, after trafficked to early endosomes in monocytes, HCMV must then necessarily traffic through the TGN (45 mpi – 1 dpi), and subsequently to Rab11+ recycling endosomes (2 hpi – 5 dpi) wherein de-envelopment occurs approximately 1 – 2 dpi. After de-envelopment, viral DNA can first be detected in the nucleus around 3 dpi (81, 83). We have shown that the intracellular trafficking events of this distinct nuclear translocation pathway are primarily manipulated by HCMV-induced signaling. Pentamer-driven c-Src signaling prevents viral trafficking to late endosomes for degradation and promotes appropriate endocytic sorting of the viral particle to EEA1+ vesicles, thereby pushing viral trafficking to the TGN (83). Trafficking from endosomes to the TGN is characteristic of retrograde trafficking, a trafficking route uncommon in normal cellular physiology and rarely utilized by viruses for entry and nuclear transport (81, 93, 94). As previously mentioned, we have published that HCMV infection significantly alters the transcript levels of several endosomal trafficking regulators in monocytes, including the major retrograde trafficking protein Stx6 (12, 13). Further, we have recently published that not only does HCMV infection significantly upregulate Stx6 protein in these cells, but also that HCMV-driven Stx6 upregulation is an important regulator of HCMV trafficking to the TGN and nuclear translocation in monocytes (14). Simultaneously, gB-driven EGFR signaling is required for the extended trafficking pathway to the nucleus (Figure 4). Trafficking of EGFR itself is ligand-dependent, where certain ligands such as EGF induce EGFR degradation and others such as TGF-α cause EGFR recycling (81, 95). HCMV acts as a distinct activating ligand for EGFR and remains engaged with the receptor throughout nuclear translocation in monocytes. This chronic engagement and subsequent signaling not only manipulates endocytic processes within the cell to promote viral nuclear translocation, but also manipulates the trafficking of the EGFR itself into a recycling-like trafficking pathway (81). EGFR is traditionally recycled through the ERC, however, EGFR travels through the TGN with HCMV in monocytes. While the trafficking of EGFR through the TGN has been documented to occur naturally, this trafficking event occurs in the context of EGFR nuclear translocation and is characteristic of a cellular stress response (81, 96). It is believed that the viral manipulation of receptor trafficking in combination with distinct virus-driven intracellular signaling events modulate the intracellular trafficking network in monocytes to facilitate HCMV nuclear translocation.
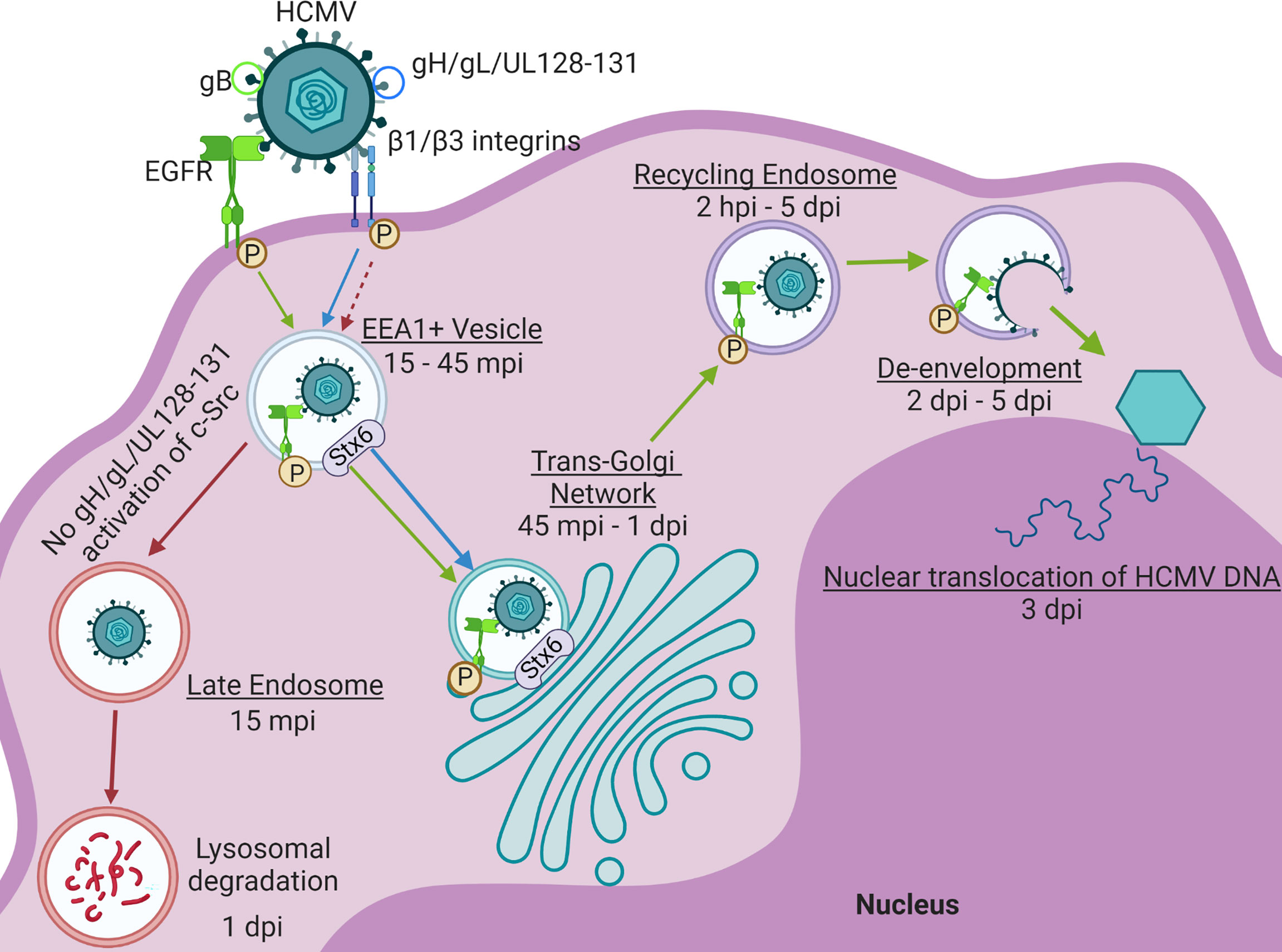
Figure 4 HCMV drives distinct Nuclear Translocation in Primary Human Monocytes. HCMV glycoproteins gB and the pentamer complex (gH/gL/UL128-131) engage and activate EGFR and β1/β3 integrins, respectively. Viral activation of these signaling cascades facilitate the internalization of the viral particle. After internalization, the virus is trafficked to EEA1+ vesicles along with phosphorylated EGFR (p-EGFR) (15 – 45 mpi) (79, 83). Lack of pentamer-activated signaling alternatively results in the sorting of virus to late endosomes and subsequently, viral degradation within 24 hpi. From the EEA1+ vesicle, both HCMV and p-EGFR traffic to the TGN (45 mpi – 1 dpi). The virus and p-EGFR are then subsequently taken to Rab11+ recycling endosomes (2 hpi – 5 dpi) where de-envelopment occurs approximately 2 – 5 dpi. Viral and p-EGFR transport to both the TGN and recycling endosome are regulated by Syntaxin 6 (Stx6) (14). After de-envelopment, the genome-containing capsid is free to dock at the nucleus and inject the HCMV genome beginning around 3 dpi (79, 83). Made with BioRender.com.
While complex navigation of intracellular trafficking networks by HCMV may be distinct to nuclear translocation in monocytes, the virus has also been documented to manipulate cellular receptor trafficking and signaling for entry and nuclear translocation in other cell types. Engagement of cellular receptors for viral entry in different cell types is partly dependent on the specific HCMV glycoprotein complexes gH/gL/gO (trimer) and pentamer. Trimer interactions with cellular receptors/co-receptors lead to virion entry via plasma membrane fusion whereas pentamer engagement with cellular receptors results in viral trafficking to endosomes for entry. Because pentamer-mediated viral entry defines HCMV tropism for several clinically significant cell types including epithelial cells, endothelial cells, and monocytes, recent efforts have been directed toward identifying cellular receptors for the pentamer complex and determining how the virus manipulates their trafficking and function to facilitate viral entry and nuclear translocation (in contrast to more well-developed trimer-receptor studies). We have shown that on monocytes, the pentamer complex engages and activates β1 and β3 integrins simultaneously, an event required for efficient viral entry into these cells and for subsequent nuclear translocation. We have further shown that HCMV infection of monocytes redistributes β1/β3 integrins away from the secretory vesicles that serve as the primary location of integrins in leukocytes and increases β1/β3 integrin presence on the cell surface within 24 hpi. The increase of integrin cell surface expression is dependent on HCMV-induced PI3K and NF-kB signaling (79, 83, 97). However, others have reported that in fibroblasts, HCMV infection re-directs β1 integrins from traditional recycling transport routes to instead be degraded to facilitate viral entry (98). Recently, an in vitro screen of all known single-pass human transmembrane proteins identified neuropilin 2 (NRP2) as a bona fide pentamer receptor (99). Additionally, our group performed a CRISPR/Cas9 screen of host genes required for HCMV infection of epithelial cells and identified the orphan olfactory receptor OR14I1 as a determinant of HCMV epithelial cell tropism (90). Specifically, OR14I1 was found to be essential for virion entry through the endosome. Thrombomodulin (THBD) was also recently identified as a pentamer receptor (100). These findings suggest a need for further study on pentamer-associated receptors and how the virus impacts their transport and function to facilitate viral entry.
Host cell signaling cascades are also critical for virion entry and trafficking, especially for endosome-mediated entry. For example, pentamer engagement of OR14I1, a GPCR (G protein-coupled receptor), results in activation of AKT signaling in epithelial cells, which was found to regulate virion entry (90). As previously mentioned, in monocytes, HCMV engagement and activation of EGFR causes chronic signaling from various endosomal compartments to facilitate viral nuclear translocation (81). HCMV signaling via the engagement of at least one host receptor contributes to viral entry across all cell types, however many unanswered questions remain. Do HCMV receptors, particularly pentamer-associated receptors, signal when engaged and if so, does the consequent signaling direct viral entry and transport? Further, is this signaling coordinated between GPCRs (i.e. OR14I1) and receptor tyrosine kinases (i.e. EGFR)? Most studies of HCMV attachment and entry associated with the pentamer have been confined to epithelial cells (89, 98, 99). However, the pentamer has been shown to be necessary for HCMV infection of cells of the monocytic lineage (83, 97) and is likely also important for viral entry in other cell types. Given the varied cell types and growing list pentamer receptors, it is possible that HCMV tropism, signaling, and entry are driven by combinations of cellular receptors available to a virion in a given cell type, thus giving potential for a wide variety of strategies for HCMV-associated disruption of receptor transport to facilitate viral entry and productive infection.
HCMV manipulates nuclear machinery for nuclear import and egress
Nuclear pore complexes within the nuclear envelope regulate traffic into and out of the nucleus. The nuclear pore complexes only allow passage to proteins and molecules of a certain size, or with specific nuclear localization signal (NLS) or nuclear export signal (NES) sequences (23). HCMV infection does not cause the breakdown of the nuclear envelope like other viruses (24). Therefore, HCMV must have more complex methods of shuttling the viral proteins required for genome replication and encapsidation into and out of the nucleus (101). Due to the limited coding capacity of most viruses, molecular mimicry of cellular NLS sequences is a common strategy used to facilitate nuclear entry of viral proteins. Indeed, some HCMV proteins such as UL44 contain a putative NLS (Figure 5) (105). However, several HCMV proteins targeted to the nucleus encode distinct, non-conventional NLS sequences such as a PY-NLS. These proteins, such as HCMV UL79 and UL84 can then interact directly with components of the nuclear import machinery such as Imp-β, thereby manipulating this restrictive trafficking machinery for nuclear entry (Figure 5) (101, 102).
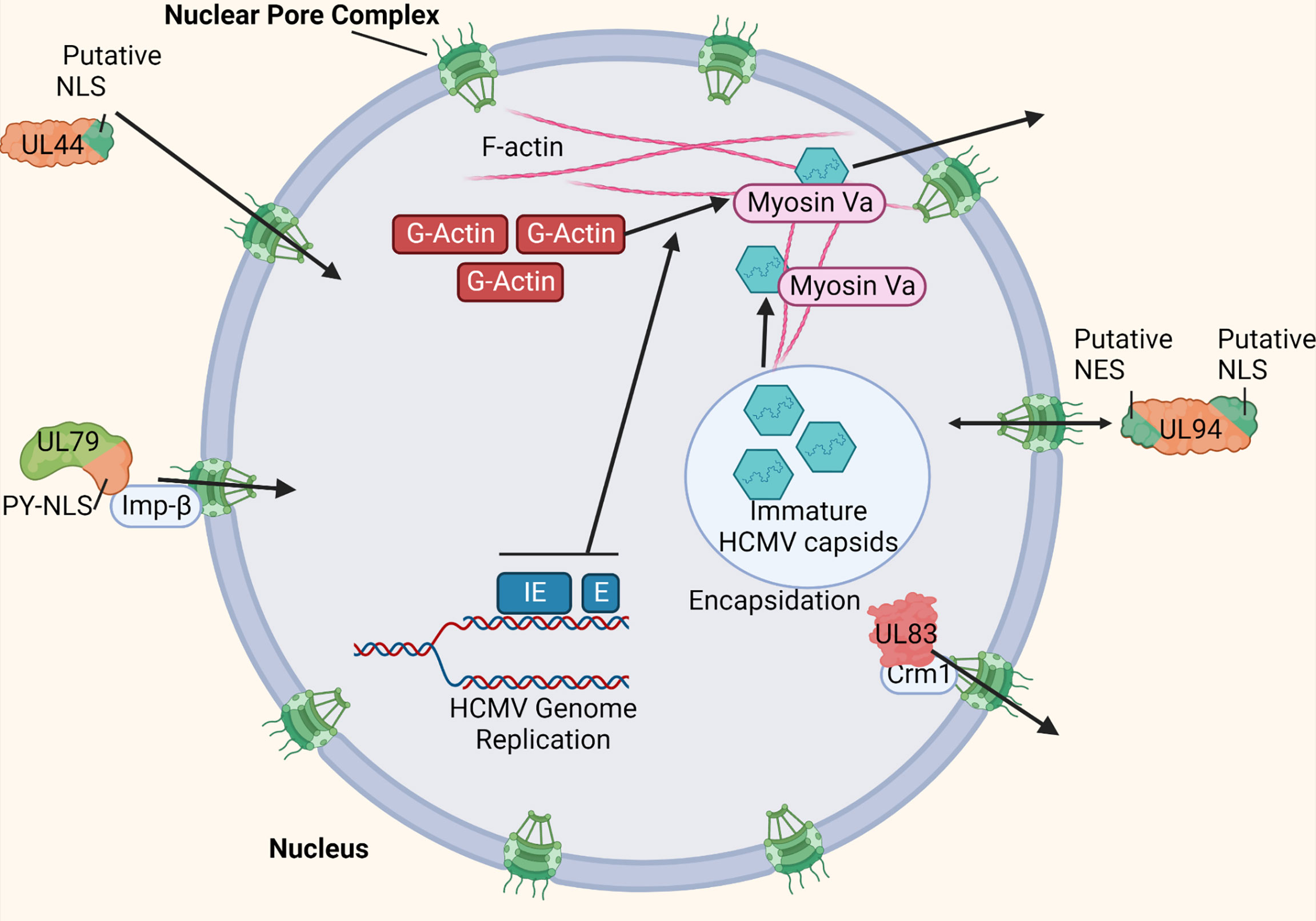
Figure 5 HCMV manipulates nuclear import and export machinery. HCMV uses various strategies to manipulate nuclear trafficking machinery for import and export. Some HCMV-encoded proteins such as UL44 mimic cellular NLS sites to achieve nuclear entry (101). Other HCMV-encoded proteins such as UL79 and UL84 contain a non-conventional NLS site (PY-NLS) that allow direct interaction with nuclear import machinery components such as Imp-β (100, 101). HCMV-encoded UL94 contains both a putative NLS an NES to manage efficient bidirectional nuclear import and export (100). HCMV proteins such as UL83 has been shown to interact directly with nuclear export machinery (such as Crm1) to manage its export from the nucleus (100, 102). HCMV IE and E gene expression has been shown to induce the formation of F-actin within the nucleus (103). Additionally, myosin Va is recruited to the replication compartment during infection. Both myosin Va and nuclear actin filaments are trafficking components required for nuclear egress of HCMV genome-containing capsids (103, 104). Made with BioRender.com.
HCMV also manipulates nuclear export. For example, HCMV UL83 has been shown to indirectly interact with Crm1, a component of the nuclear export machinery, to manage its exit from the nucleus (Figure 5) (101, 103). Additionally, HCMV encodes proteins such as UL94, which contain at least one NLS and NES each (Figure 5). This allows proteins such as UL94 to efficiently manipulate nuclear trafficking machinery for both import and export. However, because these proteins can bidirectionally cross the nuclear envelope, more complex regulation of these nuclear transport signals is required (101). HCMV-associated modulation of nuclear export machinery is not limited to viral proteins. HCMV UL69 has been shown to bind both Spt6 of RNAPII, as well as UAP56/URH49 to mediate targeting of viral transcripts to the cellular mRNA export pathway (104).
Because primary HCMV encapsidation occurs within the nucleus, the virus must also manage trafficking machinery within the nucleus to facilitate nuclear egress of the viral genome-containing capsids. Nuclear actin filaments (F-actin) are a mechanism by which the viral capsids can move around within the host cell nucleus. Indeed, HCMV infection has been shown to induce the formation of F-actin within the nucleus, most likely from a cellular pool of G-actin monomeric subunits (106). The induction of nuclear F-actin is dependent on the expression of viral immediate early (IE), and potentially early (E) gene products (Figure 5). By the late stages of infection in fibroblasts (48-72 hpi), these filaments extend from the replication compartment periphery outward to the rim of the nucleus, allowing the newly formed viral capsids a trafficking mechanism for nuclear egress (Figure 5). Additionally, the molecular motor protein myosin Va colocalizes with both the replication compartment and nuclear F-actin filaments during HCMV infection (Figure 5). Both nuclear F-actin filaments and myosin Va are cellular trafficking components required for efficient nuclear egress of HCMV, likely at least functioning in the movement of the viral genome-containing capsids from the replication compartment to the nuclear rim (106, 107).
These strategies allow for viral proteins to successfully shuttle in and out of the nucleus, facilitating viral genome replication and encapsidation, and are critical to facilitate the efficient egress of the encapsidated viral particles from the nucleus to the VAC for virion maturation.
Summary/discussion
In this review we discuss some reported approaches HCMV takes to manipulate host intracellular trafficking networks to facilitate productive infection. As obligate intracellular parasites, viruses must necessarily alter host cell physiology to create an environment suitable for productive infection. Of particular importance to this process is the ability of the virus to manipulate the host intracellular trafficking network. Of the human viruses, HCMV seems to have a profound impact on the host intracellular trafficking network (11). With its large coding capacity, HCMV has many tools at its disposal to cause sweeping alterations to the intracellular trafficking of host cells. These tools – ranging from viral miRNA and effector proteins to indirect signaling outcomes from receptor-ligand interactions – target many different components of both the endocytic and secretory trafficking pathways to different ends. Dysregulation of appropriate receptor trafficking, microtubule and actin polymerization, molecular motor function and trafficking protein dynamics, as well as the remodeling of major trafficking compartments, are all part of an extensive and targeted multi-faceted strategy by HCMV to hijack existing host intracellular trafficking pathways to facilitate productive infection of host cells. The disruption of the host intracellular trafficking network allows the virus to successfully navigate nuclear translocation, evade immune system recognition, develop a unique viral assembly compartment to generate progeny virus, and to successfully spread new virus throughout the host. However, the full extent by which HCMV remodels the host intracellular trafficking system remains to be resolved. The complex nature of these processes, as well as the many factors involved by which HCMV manipulates these networks, creates a challenging area of study. Nonetheless, many of the discussed intracellular trafficking processes, when disrupted, have been shown to be detrimental to the HCMV life cycle, and thereby have potential to generate new targets for therapeutic interventions.
Author contributions
BM, TK, and AY all helped write the review. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the (NIH) Grants AI056077, AI127335, R56-AI159672, P20GM134974, P20GM121307, 3P20GM121307-05W1, and P30GM110703.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cruz L, Buchkovich NJ. Rerouting the traffic from a virus perspective. Front Biosci (Landmark Ed) (2017) 22:1845–66. doi: 10.2741/4575
2. Spearman P. Viral interactions with host cell rab GTPases. Small GTPases (2018) 9:192–201. doi: 10.1080/21541248.2017.1346552
3. Lucin P, Mahmutefendic H, Blagojevic Zagorac G, Ilic Tomas M. Cytomegalovirus immune evasion by perturbation of endosomal trafficking. Cell Mol Immunol (2015) 12:154–69. doi: 10.1038/cmi.2014.85
4. Cohen Y, Stern-Ginossar N. Manipulation of host pathways by human cytomegalovirus: insights from genome-wide studies. Semin Immunopathol (2014) 36:651–8. doi: 10.1007/s00281-014-0443-7
5. Beltran Jean PM, Cristea ,IM. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev Proteomics (2014) 11:697–711. doi: 10.1586/14789450.2014.971116:697-711
6. Close WL, Glassbrook JE, Gurczynski SJ, Pellett PE. Infection-induced changes within the endocytic recycling compartment suggest a roadmap of human cytomegalovirus egress. Front Microbiol (2018) 9:1888. doi: 10.3389/fmicb.2018.01888
7. Mocarski ES Jr, Griffiths PD, Pass RF. Cytomegaloviruses. In: David M, Knipe PMH, editors. Fields virology, 6 ed, vol. 2. Philadelphia, PA, USA: Lippincott Williams & Wilkins (2013). p. 1961–2010.
8. Azevedo LS, Pierrotti LC, Abdala E, Costa SF, Strabelli TM, Campos SV, et al. Cytomegalovirus infection in transplant recipients. Clinics (Sao Paulo) (2015) 70:515–23. doi: 10.6061/clinics/2015(07)09
9. Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol (2004) 78:4444–53. doi: 10.1128/JVI.78.9.4444-4453.2004
10. Ogawa-Goto K, Tanaka K, Gibson W, Moriishi E, Miura Y, Kurata T, et al. Microtubule network facilitates nuclear targeting of human cytomegalovirus capsid. J Virol (2003) 77:8541–7. doi: 10.1128/JVI.77.15.8541-8547.2003
11. Beltran Jean PM, Mathias RA, Cristea IM. A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst (2016) 3:361–73. doi: 10.1016/j.cels.2016.08.012
12. Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J Immunol (2008) 181:698–711. doi: 10.4049/jimmunol.181.1.698
13. Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. Transcriptome analysis of NF-kappaB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J Virol (2008) 82:1040–6. doi: 10.1128/JVI.00864-07
14. Mosher BS, Fulkerson HL, Boyle T, Chesnokova LS, Cieply SJ, Yurochko AD. Human cytomegalovirus manipulates syntaxin 6 for successful trafficking and subsequent infection of monocytes. J Virol (2022) 96:e0081922. doi: 10.1128/jvi.00819-22
15. Hertel L, Mocarski ES. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J Virol (2004) 78:11988–2011. doi: 10.1128/JVI.78.21.11988-12011.2004
16. Turner DL, Korneev DV, Purdy JG, de Marco A, Mathias RA. The host exosome pathway underpins biogenesis of the human cytomegalovirus virion. Elife (2020) 9:e58288. doi: 10.7554/eLife.58288
17. McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep (2014) 6:1175. doi: 10.1016/j.celrep.2014.03.002
18. Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol (2011) 9:860–75. doi: 10.1038/nrmicro2655
19. Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, et al. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe (2014) 15:363–73. doi: 10.1016/j.chom.2014.02.004
20. Naqvi AR, Shango J, Seal A, Shukla D, Nares S. Viral miRNAs alter host cell miRNA profiles and modulate innate immune responses. Front Immunol (2018) 9:433. doi: 10.3389/fimmu.2018.00433
21. Rak MA, Buehler J, Zeltzer S, Reitsma J, Molina B, Terhune S, et al. Human cytomegalovirus UL135 interacts with host adaptor proteins to regulate epidermal growth factor receptor and reactivation from latency. J Virol (2018) 92:e00919–18. doi: 10.1128/JVI.00919-18
22. Snyder DA, Kelly ML, Woodbury DJ. SNARE complex regulation by phosphorylation. Cell Biochem Biophys (2006) 45:111–23. doi: 10.1385/CBB:45:1:111
23. Molenberghs F, Bogers JJ, De Vos WH. Confined no more: Viral mechanisms of nuclear entry and egress. Int J Biochem Cell Biol (2020) 129:105875. doi: 10.1016/j.biocel.2020.105875
24. Alwine JC. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PloS Pathog (2012) 8:e1002878. doi: 10.1371/journal.ppat.1002878
25. Tandon R, Mocarski ES. Viral and host control of cytomegalovirus maturation. Trends Microbiol (2012) 20:392–401. doi: 10.1016/j.tim.2012.04.008
26. Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics (2010) 9:851–60. doi: 10.1074/mcp.M900485-MCP200
27. Procter DJ, Banerjee A, Nukui M, Kruse K, Gaponenko V, Murphy EA, et al. The HCMV assembly compartment is a dynamic golgi-derived MTOC that controls nuclear rotation and virus spread. Dev Cell (2018) 45:83–100 e7. doi: 10.1016/j.devcel.2018.03.010
28. Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C. Envelopment of human cytomegalovirus occurs by budding into golgi-derived vacuole compartments positive for gB, rab 3, trans-golgi network 46, and mannosidase II. J Virol (2003) 77:3191–203. doi: 10.1128/JVI.77.5.3191-3203.2003
29. Ruzic T, Juranic Lisnic V, Mahmutefendic Lucin H, Lenac Rovis T, Zeleznjak J, Cokaric Brdovcak M, et al. Characterization of M116.1p, a murine cytomegalovirus protein required for efficient infection of mononuclear phagocytes. J Virol (2022) 96:e0087621. doi: 10.1128/JVI.00876-21
30. Lucin P, Jug Vucko N, Karleusa L, Mahmutefendic Lucin H, Blagojevic Zagorac G, Lisnic B, et al. Cytomegalovirus generates assembly compartment in the early phase of infection by perturbation of host-cell factors recruitment at the early Endosome/Endosomal recycling Compartment/Trans-golgi interface. Front Cell Dev Biol (2020) 8:563607. doi: 10.3389/fcell.2020.563607
31. Garcia-Rios E, Rodriguez MJ, Terron MC, Luque D, Perez-Romero P. Identification and characterization of epithelial cell-derived dense bodies produced upon cytomegalovirus infection. Vaccines (2022) 10:1308. doi: 10.3390/vaccines10081308
32. Mahmutefendic Lucin H, Blagojevic Zagorac G, Marcelic M, Lucin P. Host cell signatures of the envelopment site within beta-herpes virions. Int J Mol Sci (2022) 23:9994. doi: 10.3390/ijms23179994
33. Archer MA, Brechtel TM, Davis LE, Parmar RC, Hasan MH, Tandon R. Inhibition of endocytic pathways impacts cytomegalovirus maturation. Sci Rep (2017) 7:46069. doi: 10.1038/srep46069
34. Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, et al. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol (2011) 85:3821–32. doi: 10.1128/JVI.01540-10
35. Tandon R, AuCoin DP, Mocarski ES. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J Virol (2009) 83:10797–807. doi: 10.1128/JVI.01093-09
36. Das S, Pellett PE. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol (2011) 85:5864–79. doi: 10.1128/JVI.00155-11
37. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol (2009) 10:597–608. doi: 10.1038/nrm2755
38. Tomas MI, Kucic N, Mahmutefendic H, Blagojevic G, Lucin P. Murine cytomegalovirus perturbs endosomal trafficking of major histocompatibility complex class I molecules in the early phase of infection. J Virol (2010) 84:11101–12. doi: 10.1128/JVI.00988-10
39. Wilson GM, Fielding AB, Simon GC, Yu X, Andrews PD, Hames RS, et al. The FIP3-Rab11 protein complex regulates recycling endosome targeting to the cleavage furrow during late cytokinesis. Mol Biol Cell (2005) 16:849–60. doi: 10.1091/mbc.e04-10-0927
40. Krzyzaniak MA, Mach M, Britt WJ. HCMV-encoded glycoprotein m (UL100) interacts with Rab11 effector protein FIP4. Traffic (2009) 10:1439–57. doi: 10.1111/j.1600-0854.2009.00967.x
41. Cepeda V, Fraile-Ramos A. A role for the SNARE protein syntaxin 3 in human cytomegalovirus morphogenesis. Cell Microbiol (2011) 13:846–58. doi: 10.1111/j.1462-5822.2011.01583.x
42. Bennett NJ, Ashiru O, Morgan FJ, Pang Y, Okecha G, Eagle RA, et al. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J Immunol (2010) 185:1093–102. doi: 10.4049/jimmunol.1000789
43. Lin A, Xu H, Yan W. Modulation of HLA expression in human cytomegalovirus immune evasion. Cell Mol Immunol (2007) 4:91–8.
44. Lilley BN, Ploegh HL. Viral modulation of antigen presentation: manipulation of cellular targets in the ER and beyond. Immunol Rev (2005) 207:126–44. doi: 10.1111/j.0105-2896.2005.00318.x
45. Gabor F, Jahn G, Sedmak DD, Sinzger C. In vivo downregulation of MHC class I molecules by HCMV occurs during all phases of viral replication but is not always complete. Front Cell Infect Microbiol (2020) 10:283. doi: 10.3389/fcimb.2020.00283
46. Johnson DC, Hegde NR. Inhibition of the MHC class II antigen presentation pathway by human cytomegalovirus. Curr Top Microbiol Immunol (2002) 269:101–15. doi: 10.1007/978-3-642-59421-2_7
47. Noriega V, Redmann V, Gardner T, Tortorella D. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res (2012) 54:140–51. doi: 10.1007/s12026-012-8304-8
48. Machold RP, Wiertz EJ, Jones TR, Ploegh HL. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med (1997) 185:363–6. doi: 10.1084/jem.185.2.363
49. Zuo J, Rowe M. Herpesviruses placating the unwilling host: manipulation of the MHC class II antigen presentation pathway. Viruses (2012) 4:1335–53. doi: 10.3390/v4081335
50. Schroder B. The multifaceted roles of the invariant chain CD74–more than just a chaperone. Biochim Biophys Acta (2016) 1863:1269–81. doi: 10.1016/j.bbamcr.2016.03.026
51. Miller-Kittrell M, Sparer TE. Feeling manipulated: cytomegalovirus immune manipulation. Virol J (2009) 6:4. doi: 10.1186/1743-422X-6-4
52. Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, et al. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med (2003) 197:1427–39. doi: 10.1084/jem.20022059
53. Stanton RJ, Prod’homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe (2014) 16:201–14. doi: 10.1016/j.chom.2014.07.005
54. Lodoen MB, Lanier LL. Viral modulation of NK cell immunity. Nat Rev Microbiol (2005) 3:59–69. doi: 10.1038/nrmicro1066
55. Lisnic VJ, Krmpotic A, Jonjic S. Modulation of natural killer cell activity by viruses. Curr Opin Microbiol (2010) 13:530–9. doi: 10.1016/j.mib.2010.05.011
56. De Pelsmaeker S, Romero N, Vitale M, Favoreel HW. Herpesvirus evasion of natural killer cells. J Virol (2018) 92:e02105–17. doi: 10.1128/JVI.02105-17
57. Muller S, Zocher G, Steinle A, Stehle T. Structure of the HCMV UL16-MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PloS Pathog (2010) 6:e1000723. doi: 10.1371/journal.ppat.1000723
58. Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR. NKG2D ligand MICA is retained in the cis-golgi apparatus by human cytomegalovirus protein UL142. J Virol (2009) 83:12345–54. doi: 10.1128/JVI.01175-09
59. Tomasec P, Wang EC, Davison AJ, Vojtesek B, Armstrong M, Griffin C, et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol (2005) 6:181–8. doi: 10.1038/ni1156
60. Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod’homme V, Aicheler R, et al. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol (2008) 41:206–12. doi: 10.1016/j.jcv.2007.10.027
61. Fielding CA, Weekes MP, Nobre LV, Ruckova E, Wilkie GS, Paulo JA, et al. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. Elife (2017) 6:e22206. doi: 10.7554/eLife.22206
62. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol (2014) 5:491. doi: 10.3389/fimmu.2014.00491
63. Murray RZ, Stow JL. Cytokine secretion in macrophages: SNAREs, rabs, and membrane trafficking. Front Immunol (2014) 5:538. doi: 10.3389/fimmu.2014.00538
64. Simonsen A, Gaullier JM, D’Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. J Biol Chem (1999) 274:28857–60. doi: 10.1074/jbc.274.41.28857
65. Cruz L, Streck NT, Ferguson K, Desai T, Desai DH, Amin SG, et al. Potent inhibition of human cytomegalovirus by modulation of cellular SNARE syntaxin 5. J Virol (2017) 91:e02044–20. doi: 10.1128/JVI.01637-16
66. Personnic N, Barlocher K, Finsel I, Hilbi H. Subversion of retrograde trafficking by translocated pathogen effectors. Trends Microbiol (2016) 24:450–62. doi: 10.1016/j.tim.2016.02.003
67. Enrich C, Rentero C, Hierro A, Grewal T. Role of cholesterol in SNARE-mediated trafficking on intracellular membranes. J Cell Sci (2015) 128:1071–81. doi: 10.1242/jcs.164459
68. Laufman O, Hong W, Lev S. The COG complex interacts directly with syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J Cell Biol (2011) 194:459–72. doi: 10.1083/jcb.201102045
69. Riggs KA, Hasan N, Humphrey D, Raleigh C, Nevitt C, Corbin D, et al. Regulation of integrin endocytic recycling and chemotactic cell migration by syntaxin 6 and VAMP3 interaction. J Cell Sci (2012) 125:3827–39. doi: 10.1242/jcs.102566
70. Halenius A, Gerke C, Hengel H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets-but how many arrows in the quiver? Cell Mol Immunol (2015) 12:139–53. doi: 10.1038/cmi.2014.105
71. Park A, Ra EA, Lee TA, Choi HJ, Lee E, Kang S, et al. HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat Commun (2019) 10:4670. doi: 10.1038/s41467-019-12641-4
72. Zeltzer S, Zeltzer CA, Igarashi S, Wilson J, Donaldson JG, Goodrum F. Virus control of trafficking from sorting endosomes. mBio (2018) 9:e00683–18. doi: 10.1128/mBio.00683-18
73. Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: location makes a difference. Exp Cell Res (2009) 315:1601–9. doi: 10.1016/j.yexcr.2008.09.021
74. Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F. Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses (2018) 10:444. doi: 10.3390/v10080444
75. Collins-McMillen D, Chesnokova L, Lee BJ, Fulkerson HL, Brooks R, Mosher BS, et al. HCMV infection and apoptosis: How do monocytes survive HCMV infection? Viruses (2018) 10:533. doi: 10.3390/v10100533
76. Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol (2006) 80:11539–55. doi: 10.1128/JVI.01016-06
77. Boeck JM, Spencer JV. Effect of human cytomegalovirus (HCMV) US27 on CXCR4 receptor internalization measured by fluorogen-activating protein (FAP) biosensors. PloS One (2017) 12:e0172042. doi: 10.1371/journal.pone.0172042
78. Arnolds KL, Lares AP, Spencer JV. The US27 gene product of human cytomegalovirus enhances signaling of host chemokine receptor CXCR4. Virology (2013) 439:122–31. doi: 10.1016/j.virol.2013.02.006
79. Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, et al. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol (2007) 81:7683–94. doi: 10.1128/JVI.02839-06
80. Streck NT, Zhao Y, Sundstrom JM, Buchkovich NJ. Human cytomegalovirus utilizes extracellular vesicles to enhance virus spread. J Virol (2020) 94:e00609–20. doi: 10.1128/JVI.00609-20
81. Fulkerson HL CL, Kim JH, Mahmud J, Frazier LE, Chan GC, Yurochko AD. HCMV-induced signaling through gB–EGFR engagement is required for viral trafficking and nuclear translocation in primary human monocytes. PNAS (2020) 117:19507–16. doi: 10.1073/pnas.2003549117
82. Nogalski MT, Chan G, Stevenson EV, Gray S, Yurochko AD. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J Virol (2011) 85:1360–9. doi: 10.1128/JVI.02090-10
83. Kim JH, Collins-McMillen D, Caposio P, Yurochko AD. Viral binding-induced signaling drives a unique and extended intracellular trafficking pattern during infection of primary monocytes. Proc Natl Acad Sci U.S.A. (2016) 113:8819–24. doi: 10.1073/pnas.1604317113
84. Chan G, Nogalski MT, Yurochko AD. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U.S.A. (2009) 106:22369–74. doi: 10.1073/pnas.0908787106
85. Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene (2001) 20:6459–72. doi: 10.1038/sj.onc.1204786
86. Smith MS, Bentz GL, Smith PM, Bivins ER, Yurochko AD. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J Leukoc Biol (2004) 76:65–76. doi: 10.1189/jlb.1203621
87. Miller MS, Hertel L. Onset of human cytomegalovirus replication in fibroblasts requires the presence of an intact vimentin cytoskeleton. J Virol (2009) 83:7015–28. doi: 10.1128/JVI.00398-09
88. Cohen T, Schwarz TM, Vigant F, Gardner TJ, Hernandez RE, Lee B, et al. The microtubule inhibitor podofilox inhibits an early entry step of human cytomegalovirus. Viruses (2016) 8:295. doi: 10.3390/v8100295
89. Vanarsdall AL, Johnson DC. Human cytomegalovirus entry into cells. Curr Opin Virol (2012) 2:37–42. doi: 10.1016/j.coviro.2012.01.001
90. E X, Meraner P, Lu P, Perreira JM, Aker AM, McDougall WM, et al. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc Natl Acad Sci U.S.A. (2019) 116:7043–52. doi: 10.1073/pnas.1814850116
91. Hetzenecker S, Helenius A, Krzyzaniak MA. HCMV induces macropinocytosis for host cell entry in fibroblasts. Traffic (2016) 17:351–68. doi: 10.1111/tra.12355
92. Nguyen CC, Kamil JP. Pathogen at the gates: Human cytomegalovirus entry and cell tropism. Viruses (2018) 10:704. doi: 10.3390/v10120704
93. Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell (2008) 135:1175–87. doi: 10.1016/j.cell.2008.12.009
94. Sivan G, Weisberg AS, Americo JL, Moss B. Retrograde transport from early endosomes to the trans-golgi network enables membrane wrapping and egress of vaccinia virus virions. J Virol (2016) 90:8891–905. doi: 10.1128/JVI.01114-16
95. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol (2014) 24:26–34. doi: 10.1016/j.tcb.2013.11.002
96. Du Y, Shen J, Hsu JL, Han Z, Hsu MC, Yang CC, et al. Syntaxin 6-mediated golgi translocation plays an important role in nuclear functions of EGFR through microtubule-dependent trafficking. Oncogene (2014) 33:756–70. doi: 10.1038/onc.2013.1
97. Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PloS Pathog (2013) 9:e1003463. doi: 10.1371/journal.ppat.1003463
98. Hashimoto Y, Sheng X, Murray-Nerger LA, Cristea IM. Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat Commun (2020) 11:806. doi: 10.1038/s41467-020-14586-5
99. Martinez-Martin N, Marcandalli J, Huang CS, Arthur CP, Perotti M, Foglierini M, et al. An unbiased screen for human cytomegalovirus identifies neuropilin-2 as a central viral receptor. Cell (2018) 174:1158–1171 e19. doi: 10.1016/j.cell.2018.06.028
100. Kschonsak M, Johnson MC, Schelling R, Green EM, Rouge L, Ho H, et al. Structural basis for HCMV pentamer receptor recognition and antibody neutralization. Sci Adv (2022) 8:eabm2536. doi: 10.1126/sciadv.abm2536
101. Tessier TM, Dodge MJ, Prusinkiewicz MA, Mymryk JS. Viral appropriation: Laying claim to host nuclear transport machinery. Cells (2019) 8:559. doi: 10.3390/cells8060559
102. Lischka P, Rauh C, Mueller R, Stamminger T. Human cytomegalovirus UL84 protein contains two nuclear export signals and shuttles between the nucleus and the cytoplasm. J Virol (2006) 80:10274–80. doi: 10.1128/JVI.00995-06
103. Sanchez V, Mahr JA, Orazio NI, Spector DH. Nuclear export of the human cytomegalovirus tegument protein pp65 requires cyclin-dependent kinase activity and the Crm1 exporter. J Virol (2007) 81:11730–6. doi: 10.1128/JVI.02760-06
104. Toth Z, Stamminger T. The human cytomegalovirus regulatory protein UL69 and its effect on mRNA export. Front Biosci (2008) 13:2939–49. doi: 10.2741/2899
105. Alvisi G, Jans DA, Guo J, Pinna LA, Ripalti A. A protein kinase CK2 site flanking the nuclear targeting signal enhances nuclear transport of human cytomegalovirus ppUL44. Traffic (2005) 6:1002–13. doi: 10.1111/j.1600-0854.2005.00331.x
106. Wilkie AR, Lawler JL, Coen DM. A role for nuclear f-actin induction in human cytomegalovirus nuclear egress. mBio (2016) 7:e01254–16. doi: 10.1128/mBio.01254-16
Keywords: HCMV, monocytes, intracellular trafficking, signaling, SNAREs, vesicular transport, immune evasion, viral infection
Citation: Mosher BS, Kowalik TF and Yurochko AD (2022) Overview of how HCMV manipulation of host cell intracellular trafficking networks can promote productive infection. Front. Virol. 2:1026452. doi: 10.3389/fviro.2022.1026452
Received: 23 August 2022; Accepted: 09 November 2022;
Published: 28 November 2022.
Edited by:
Binod Kumar, Institute of Advanced Virology (IAV), IndiaReviewed by:
Thomas Stamminger, Ulm University Medical Center, GermanyPhilip E. Pellett, Wayne State University, United States
Reinhild Prange, Johannes Gutenberg University Mainz, Germany
Copyright © 2022 Mosher, Kowalik and Yurochko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew D. Yurochko, YW5kcmV3Lnl1cm9jaGtvQGxzdWhzLmVkdQ==