 Nico Becker
Nico Becker Anja Heiner
Anja Heiner Andrea Maisner
Andrea Maisner
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol., 23 February 2022
Sec. Emerging and Reemerging Viruses
Volume 1 - 2021 | https://doi.org/10.3389/fviro.2021.821004
Nipah virus (NiV) is a BSL-4 classified zoonotic paramyxovirus that causes respiratory or encephalitic diseases. A hallmark of NiV infections, as with all cell infections caused by non-segmented negative-strand RNA viruses, is the formation of cytoplasmic inclusion bodies (IBs). We previously showed that cytosolic NiV IBs, which are formed in infected cells or in cells minimally expressing the NiV nucleocapsid proteins, are associated with the microtubule-organizing center (MTOC) marker γ-tubulin. They also recruit overexpressed cytosolic proteins that are not functionally required for viral replication in IBs and that otherwise might form toxic protein aggregates. Therefore, NiV IBs are thought to share some functional properties with cellular aggresomes. The fact that aggresomes were not found in NiV-infected cells supports the idea that NiV IBs are successfully reducing the proteotoxic stress in infected cells. Only if the proteasome-ubiquitin system is artificially blocked by inhibitors, cellular aggresomes are formed in addition to IBs, but without colocalizing. Although both structures were positive for the classical aggresome markers histone deacetylase 6 (HDAC6) and Bcl-2-associated athanogene 3 (BAG3), they clearly differed in their cellular protein compositions and recruited overexpressed proteins to different extents. The further finding that inhibition of aggresome pathways by HDAC6 or microtubule (MT) inhibitors did neither interfere with IB formation nor with protein sequestration, strengthens the idea that cytosolic NiV IBs can assume some aggresome-like functions without involving active transport processes and canonical cellular aggresome pathways.
Nipah virus (NiV) is a biosafety-level 4 (BSL-4) classified paramyxovirus that causes severe respiratory or encephalitic diseases in pigs and humans (1). The natural reservoir of NiV are Southeast Asian fruit bats, from which the virus is sporadically transmitted to humans, either by direct spillover (NiV-Bangladesh) or via pigs as intermediate hosts (NiV-Malaysia) (1–4). NiV is listed on the Blueprint list of priority pathogens by the World Health Organization, because the zoonotic infection causes highly lethal human disease that can also be transmitted from human to human and for which there is no approved therapeutic treatment available (5).
NiV, a member of the henipavirus genus within the family Paramyxoviridae, is an enveloped virus with a single-stranded negative-sense RNA genome, which encodes for six structural proteins. The two NiV surface glycoproteins, the receptor-binding G protein and the fusion protein (F), are required for virus entry into cells and support the spread of NiV in infected cells by cell-cell fusion or syncytia formation (6). In virus particles or infected cells, the viral RNA is encapsidated by the nucleocapsid protein (N), the phosphoprotein (P), and the large viral polymerase (L), which together form the so-called ribonucleoprotein complex (RNP). The NiV matrix protein (M) is located at the inner side of the viral envelope and is associated with the inner leaflet of the plasma membrane in infected cells (7). The M protein links the RNP to the NiV surface glycoproteins and is absolutely essential for the assembly and release of infectious NiV particles (8–10).
As a virus with a non-segmented negative-strand RNA genome, NiV belongs to the order Mononegavirales (MNV), whose members are characterized by inducing the formation of cytoplasmic inclusion bodies (IBs) in infected cells. These IBs are often referred to as viral factories, as they concentrate all viral nucleocapsid components and have been shown to be the main site of viral mRNA synthesis and genome replication in many MNV infections (11). In addition to viral proteins, IBs contain a variety of host cell proteins that bind either directly or indirectly to viral components. However, for most of these host proteins, the functional role in IBs is unknown (11).
The current concept describes IBs in MNV infections as proteinaceous, membrane-less compartments, which share common properties with liquid organelles or biomolecular condensates. As these, IBs represent dynamic and mobile structures which are regulated by liquid-liquid phase separation and can fuse and segregate (11, 12). Also similar to cellular liquid organelle compartments, IBs are generally heterogeneous in sizes and concentrate specific viral and cellular proteins, while others are excluded. In infected cells, MNV IBs contain viral RNAs aside of proteins. In cotransfection systems, which can recapitulate the basic properties of IBs formed in infected cells, cellular RNAs are found in IBs. In case of NiV, the minimal requirement for the formation of cytosolic IBs is the coexpression of two nucleocapsid proteins; the nucleoprotein (NiV N) and the phosphoprotein (NiV P), which form small RNP-like structures with cellular RNAs (13, 14).
Although NiV forms typical cytosolic IBs, it differs from other MNVs by additionally forming a second, independent IB population in close association with the plasma membrane (14). These so-called IB-PM likely represent platforms for NiV assembly and depend on functional NiV matrix protein at the plasma membrane. If NiV M is completely lacking or assembly defective, only cytosolic IBs and no IB-PM are formed. Consequently, NiV particle formation and release is drastically impaired, although intracellular virus RNA and protein synthesis is ongoing in infected cells (14, 15). Thus, IB-PM substantially differ from the cytosolic IBs by their strict association with the plasma membrane and, importantly, by containing the viral M protein, which is neither required for formation nor present in cytosolic NiV IBs (14) (Figure 1).
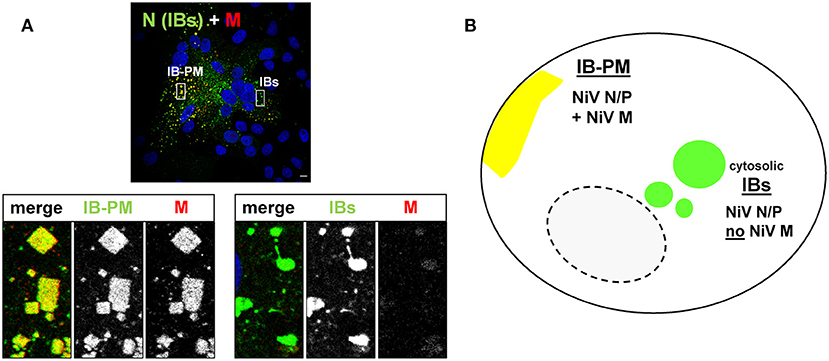
Figure 1. NiV induces the formation of two different inclusion body populations. (A) Vero76 cells were infected with NiV at an MOI of 0.05. 24 h p.i. cells were fixed with 4% PFA for 48 h and stained with a NiV N antiserum to visualize IBs (green) and with a Zenon-labeled M-specific antiserum to detect the M protein (red). Nuclei were counterstained with DAPI (blue). Images were recorded with a Leica SP5 confocal laser scanning microscope. Scale bar, 10 μm. The magnified areas in the lower panel display the two different IB types and also show the single channel greyscale images of the labeled proteins. (B) Simplified model of the two different inclusion body populations and their protein composition in NiV infected cells.
A viral infection is always a stressor for the host cell, and massive overproduction of viral proteins is one of the factors. To rapidly produce the required high levels of viral proteins, viruses hijack the cellular machinery and potentially saturate the particular folding pathways required for their maturation (16). Therefore, infection can lead to macromolecular crowding and proteotoxic stress that can overwhelm the cellular capacity of protein refolding by chaperones and the ubiquitin-proteasome degradation system (17–19). As a consequence, misfolded viral or cellular proteins adopting non-native conformations can form microaggregates in the cytoplasm, which if not degraded or sequestered, can be cytotoxic and interfere with productive virus replication. Proteotoxic stress can be restricted by forming juxtanuclear aggresomes around the microtubule organizing center (MTOC). Polyubiquitinated proteins are linked to histone deacetylase 6 (HDAC6), which harbors both an ubiquitin and dynein binding domain, and acts as an adaptor for the retrograde transport of ubiquitinated misfolded proteins along microtubules (MTs) (20). Microaggregates formed in the peripheral cytoplasm can also be actively transported to juxtanuclear aggresomes via an ubiquitin-independent pathway. This is mediated by the co-chaperone Bcl-2-associated athanogene 3 (BAG3), which can directly transfer misfolded protein substrates bound to heat-shock protein 70 (Hsp70) to the MT motor dynein (21). Aggresomes thus serve as a cytoplasmic recruitment center to deposit misfolded or overexpressed proteins. If proteotoxic stress goes on and protein aggregates sequestered to aggresomes cannot be refolded or degraded by the sequestered proteasome components, aggresomes are finally cleared by autophagic processes.
While aggresomes are induced or even adopted for replication by some, mostly DNA viruses (22–24), aggresome formation is not described in MNV infections, suggesting that these have evolved other mechanisms to interfere with aggresome formation or limit proteotoxic stress. One of those mechanisms might be the formation of cytosolic IBs, which aside of accumulating abundantly expressed viral proteins required for virus replication, can additionally sequester non-essential proteins, thereby exerting some aggresome-like function. This idea is based on our previous observation, that cytosolic NiV IBs were associated with the MTOC and aggresome marker γ-tubulin, and did not only concentrate viral nucleocapsid proteins but also sequestered unrelated cytosolic proteins such as mCherry (Figure 2) (14). Aside of unrelated proteins, some defective NiV proteins could also be sequestered to cytosolic NiV IBs. For example, a mutant NiV matrix protein that accumulates to large amounts in the nucleus (MNESmut) was found in cytosolic IBs, limiting nuclear accumulation of this non-functional M protein in infected cells (15). In view of their ability to sequester potentially proteotoxic proteins, cytosolic NiV IBs were assumed to share some functional properties with cellular aggresomes. However, it is yet unclear, whether canonical cellular aggresome pathways are involved. To address this question, this study aims to determine if cytosolic NiV-IBs hijack aggresome components and to what extent the aggresome formation machinery is required to form IBs or to sequester non-essential overabundant proteins from the cytosolic periphery.
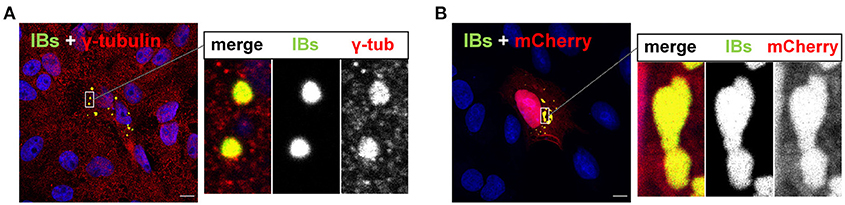
Figure 2. Cytosolic IBs formed by NiV N and P proteins contain γ-tubulin and sequester mCherry. (A) Vero76 cells were transfected with plasmids encoding NiV N and NiV PeGFP. 24 h p.t. cells were fixed with 4% PFA and permeabilized with methanol/acetone. The MTOC component and aggresome marker γ-tubulin was labeled with an anti-γ-tubulin antibody (red). IBs were detected by eGFP autofluorescence (green). (B) Cells were transfected with plasmids encoding NiV N, NiV PeGFP and mCherry. Cells were fixed with 4% PFA at 24 h p.t. and IBs (green) and mCherry (red) were detected by autofluorescence. Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. The magnified area in the right panel highlights the IBs and also shows the single channel greyscale images of the labeled proteins.
To first clarify whether the rapid and high expression of viral proteins in NiV infection triggers aggresome formation, we infected Vero76 cells with NiV at a multiplicity of infection (MOI) of 0.05 for 24 h. To detect aggresomes, cells were stained for p62, which is one of the scaffold proteins and a typical marker of cellular aggresomes (Supplementary Figure 1). Immunostaining of the NiV N protein in NiV-infected cells was used to visualize NiV IBs. As expected, NiV infection resulted in the formation of large syncytia, in which multiple IBs in perinuclear and peripheral regions were found (Figure 3A). Infection apparently did not trigger aggresome formation, as p62 was detected only dispersed in the cytoplasm and neither colocalized with IBs nor accumulated independently in the perinuclear region, where aggresome formation is normally observed. The absence of aggresomes in NiV-infected cells at such late time points of infection supports the idea that NiV has evolved alternative mechanisms to limit proteotoxic stress or inhibits aggresome pathways, thereby actively preventing aggresome formation. To test the latter, we treated infected cells with the proteasome inhibitor bortezomib, a FDA-approved drug for treating multiple myeloma (25), known to induce aggresome formation. As shown in Figure 3B, bortezomib elicited the formation of juxtanuclear aggresomes with characteristic immunoreactivity for p62 equally in uninfected and infected cells (Figure 3B). In NiV-positive cells, aggresomes neither colocalized with IBs nor blocked IB formation per se, demonstrating that the two structures can form independently and simultaneously in the same cell (boxed regions in Figures 3B,C). Since aggresomes are only formed in infected cells when the proteasome is artificially inhibited, proteotoxic stress appears to be limited, despite the strong expression of viral proteins. This fits with the idea that IBs can take over some aggresomal functions by sequestering a large fraction of cytosolic NiV proteins.
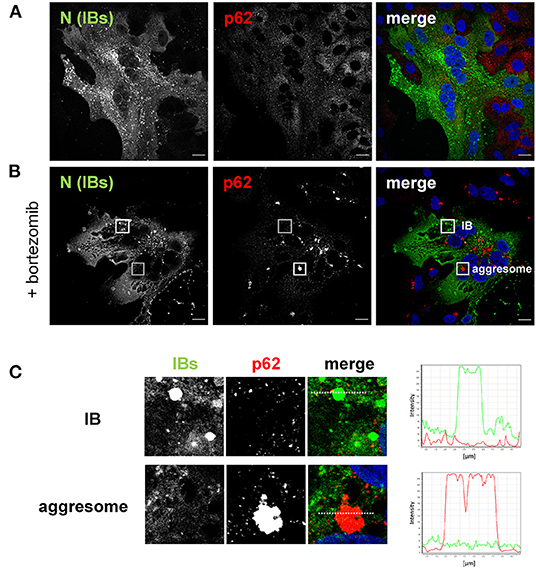
Figure 3. Aggresome formation in NiV-infected cells. Vero76 cells were infected with NiV at an MOI of 0.05 in the absence (A) or presence of 50 nM bortezomib (B). At 24 h p.i. cells were fixed with 4% PFA for 10 min and permeabilized with methanol/acetone. The aggresome marker p62 was labeled with a specific antibody (red). Then, cells were fixed with 4% PFA for further 48 h. After removal from the BSL-4 lab, cells were stained with a NiV N antiserum to visualize IBs (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (C) Enlarged views of the areas marked by the white boxes in (B). The histograms in the right panel show the fluorescence intensity profile of the respective colors along the dotted line through an aggresome and an IB.
The aggresomal scaffold protein p62 was only detected in aggresomes but not in IBs, which raised the question to what extent IBs and aggresomes differ in their protein composition. We therefore performed colocalization studies of diverse marker proteins found in cellular aggresomes (Supplementary Figure 1) and cytosolic IBs in either NiV-infected or NiV N and P coexpressing cells, which is the minimal and sufficient requirement for cytosolic IB formation (14).
IBs neither in NiV-infected cells nor in N/P-coexpressing cells did contain p62 (Figures 4A,B, top panels). We detected neither the proteasomal 26S subunit nor poly-ubiquitin, indicating that the proteasome-ubiquitin machinery is not recruited to IBs. In line with our previous report (14), IBs had no vimentin cage. So, the protein composition of IBs clearly differed from that of cellular aggresomes (Supplementary Figure 1). However, we detected the important aggresomal marker proteins, HDAC6 and Hsp70/BAG3 (Figures 4C,D). The fact that HDAC6 and Hsp70/BAG3 are involved in the MT-driven ubiquitin-dependent and ubiquitin-independent aggresomal transport pathways, together with our observation that NiV IBs contain the MTOC marker γ-tubulin (14), may indicate a functional role of aggresomal transport pathways for IB formation.
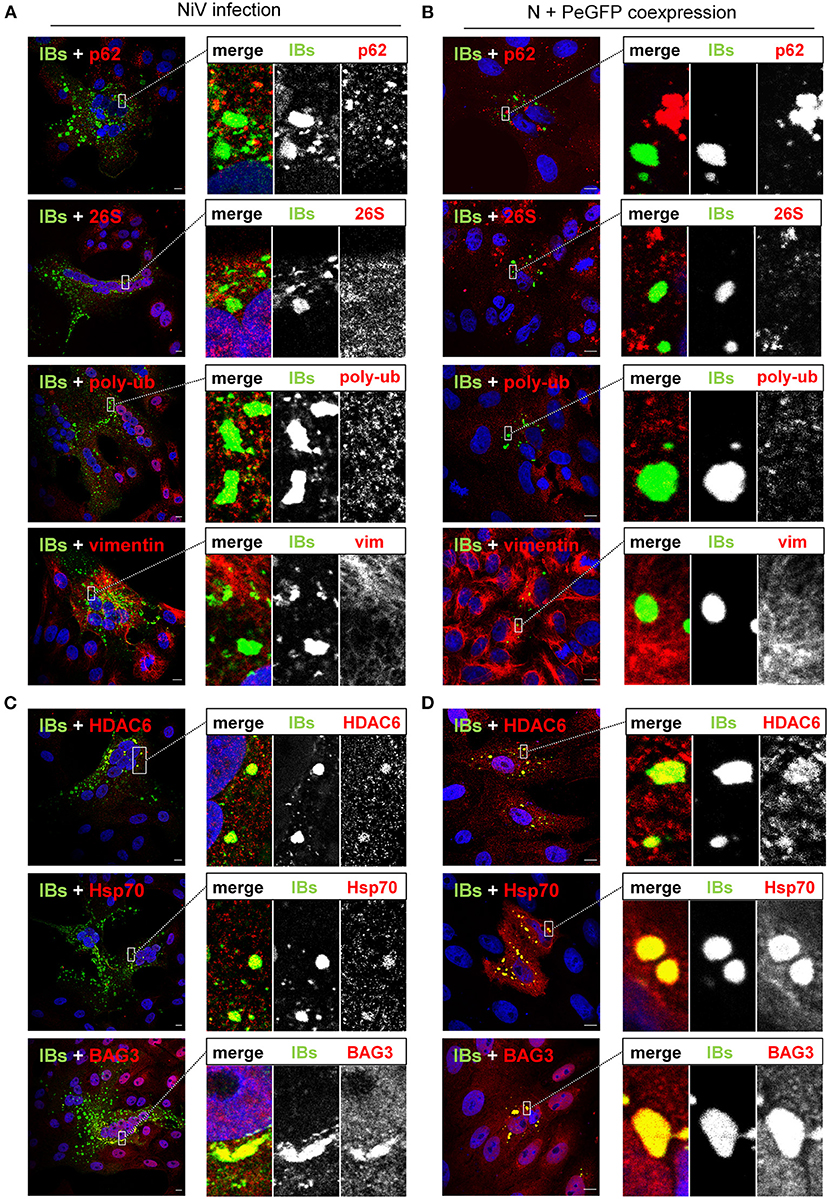
Figure 4. Colocalization of IBs and aggresome markers. (A,C) Vero76 cells were infected with NiV at an MOI of 0.05. (B,D) Vero76 cells were transfected with plasmids encoding NiV N and NiV PeGFP. 24 h later, infected or transfected cells were fixed with 4% PFA and permeabilized with Triton X-100. The indicated endogenous marker proteins were labeled with specific antibodies (red). IBs (green) were detected by a NiV N antiserum (A,C) or eGFP autofluorescence (B,D). Nuclei were counterstained with DAPI (blue). Only merged images are shown. The boxed areas are displayed in a higher magnification together with the single channel greyscale images. Scale bars, 10 μm.
The detection of HDAC6 and BAG3 raised the question whether NiV exploits aggresomal pathways to either form IBs, or to support functional virus replication. To address this, we first tested the effect of tubacin, a highly potent and cell-permeable HDAC6 inhibitor, recently shown to affect replication of some RNA viruses (26, 27). Tubacin blocks the major enzymatic function of HDAC6, which is the deacetylation of diverse cellular proteins, including α-tubulin thereby regulating MT stability and MT-dependent intracellular trafficking. The immunostaining and western blot analyses shown in Figures 5A,B clearly show that tubacin treatment of Vero76 cells for 24 h increased the cellular content of acetylated tubulin as a result of HDAC6 inhibition. Also in NiV-infected cell cultures, the amount of actetylated tubulin was increased in tubacin-treated cells compared to DMSO-treated cells (Figure 5C). However, virus replication was not affected (Figure 5D). Although tubacin has been added to the infected cells directly after virus adsorption and was therefore present from the earliest replication steps on, numerous IBs were formed and spread of infection via cell-cell fusion and syncytia formation was undisturbed, indicating a productive viral protein synthesis (Figure 5C). In line with that, viral titers in the supernatants of tubacin-treated cells were not significantly reduced (Figure 5D). It thus can be concluded that the enzymatic function of HDAC6 is neither required for IB formation nor NiV replication. This however, does not exclude the possibility that HDAC6 is required as an adaptor protein for delivering IB components. HDAC6- and Hsp70/BAG3-mediated active transport processes along MTs that deliver proteins to aggresomes can be prevented by nocodazole, a MT-interrupting inhibitor (20). To address whether such active MT-dependent transport processes might be required for IB formation or viral replication, NiV-infected cells were treated with 250 nM nocodazole, a concentration that destroyed MTs in Vero76 cells (Figure 6A) and prevented MT-dependent cellular aggresome formation with minimal cytotoxicity (Supplementary Figure 2). Despite MT disruption in NiV-infected cells, virus replication was basically unaffected. IBs were formed (Figure 6B) and viral titers in the supernatants were not significantly reduced (Figure 6C). This led us to conclude, that although IBs contain HDAC6 and Hsp70/BAG3, inhibition of the MT-dependent aggresome pathways had no impact on IB formation or viral replication.
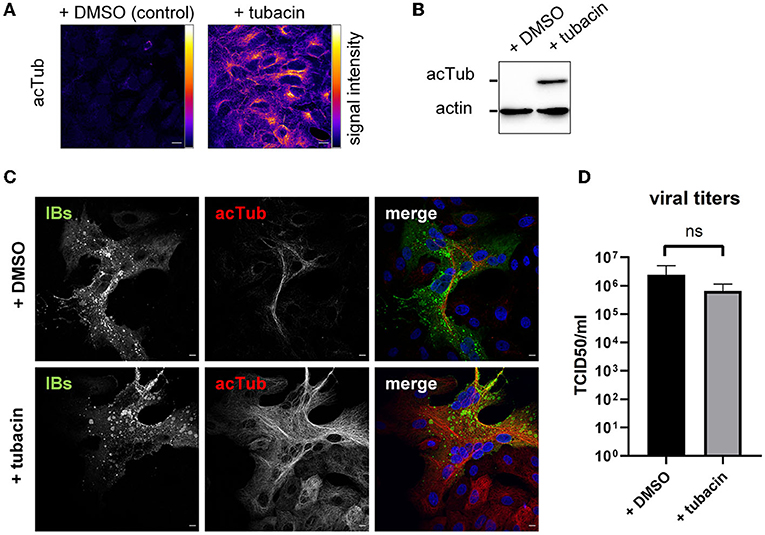
Figure 5. Influence of HDAC6 inhibition on IB formation and NiV replication. (A) Vero76 cells were treated with 10 μM of the HDAC6 inhibitor tubacin. After 24 h, the cells were fixed with 4% PFA and permeabilized with Triton X-100. Acetylated tubulin (acTub) was detected with specific antibodies and visualized with a pseudocolor-LUT to show differences in fluorescence signal intensity. (B) Vero76 cells treated with 10 μM tubacin for 24 h, were harvested, lysed and subjected to SDS-PAGE and Western blot analysis. Acetylated tubulin and the housekeeping protein actin were detected with specific primary antibodies and subsequent labeling with biotinylated secondary antibodies and Streptavidin-HRP. Chemiluminescence signal was detected using a ChemiDoc Imager (BioRad). (C) Vero76 cells were infected with NiV at an MOI of 0.05 and treated with 10 μM tubacin or DMSO. 24 h p.i. cells were fixed with 4% PFA for 10 min and permeabilized with Triton X-100. Acetylated tubulin was labeled with specific antibodies (red). Subsequently the cells were fixed for 48 h with 4% PFA to remove them from the BSL-4 lab. Then, cells were stained with a NiV N antiserum to visualize IBs (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (D) Vero76 cells were infected with NiV at an MOI of 1 and treated with 10 μM tubacin. 24 h p.i. supernatants were harvested and viral titers were determined by a TCID50 assay. Error bars indicate standard deviation (SD); n.s., not significant.
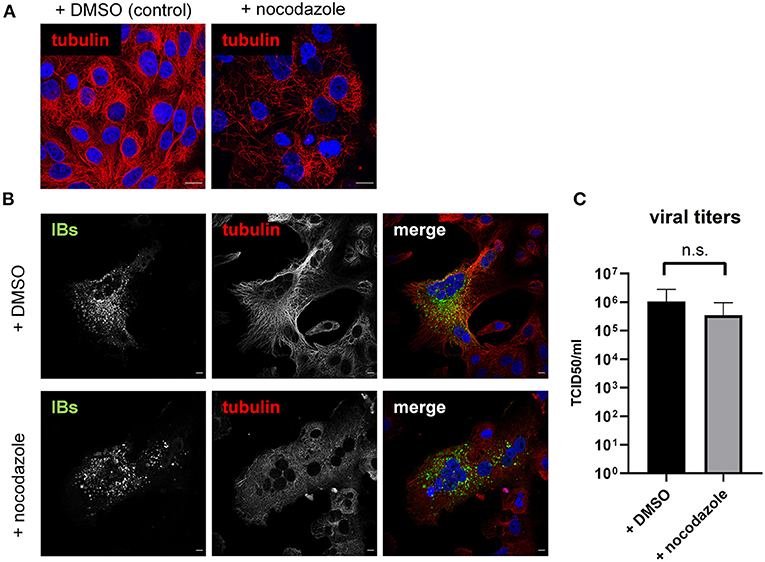
Figure 6. Influence of MT disruption on IB formation and NiV replication. (A) Vero76 cells were treated with 250 nM of nocodazole. After 18 h the cells were fixed with 4% PFA and permeabilized with Triton X-100. α-tubulin was detected with specific antibodies (red). (B) Vero76 cells were infected with NiV at an MOI of 0.05 and treated with 250 nM nocodazole. 18 h p.i. cells were fixed with 4% PFA for 10 min and permeabilized with Triton X-100. α-tubulin was labeled with specific antibodies (red). Cell were fixed with 4% PFA for 48 h and stained with a NiV N antiserum to visualize IBs (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (C) Vero76 cells were infected with NiV at an MOI of 1 and treated with 250 nM nocodazole. 18 h p.i. supernatants were harvested and viral titers were determined. Error bars indicate standard deviation (SD); n.s., not significant.
Despite the differences in marker protein compositions, IBs and aggresomes can both sequester overexpressed proteins that might otherwise accumulate widely distributed all over the cytosol. To evaluate the similarities or differences of IBs and aggresomes with regard to their capacity to recruit proteins from the cytosol, we compared their ability to sequester overexpressed cytosolic proteins using three model “guest” proteins. We analyzed soluble mCherry, we already know is recruited to IBs and two functionally defective NiV M proteins, which have not yet been tested for their recruitment to IBs. One of them is a mCherry-tagged NiV M (mCh-NiV M), which is assembly defective because it cannot interact with NiV N proteins and nucleocapsids, but undergoes a nuclear transit and is then transported to the plasma membrane like wildtype NiV M (14). The second M protein is NiV MNLSmut, which has a defective nuclear import signal and is unable to undergo nuclear transit (8). MNLSmut therefore accumulates in the cytoplasm without reaching the plasma membrane.
As shown in Figure 7A, soluble mCherry but not mCherry-tagged NiV M accumulated in IBs. Vice versa, mCh-NiV M but not mCherry was sequestered to aggresomes (Figure 7B). MNLSmut was found in IBs and aggresomes (Figures 7A,B, bottom panels). Thus, only one of the three model proteins was sequestered to both structures.
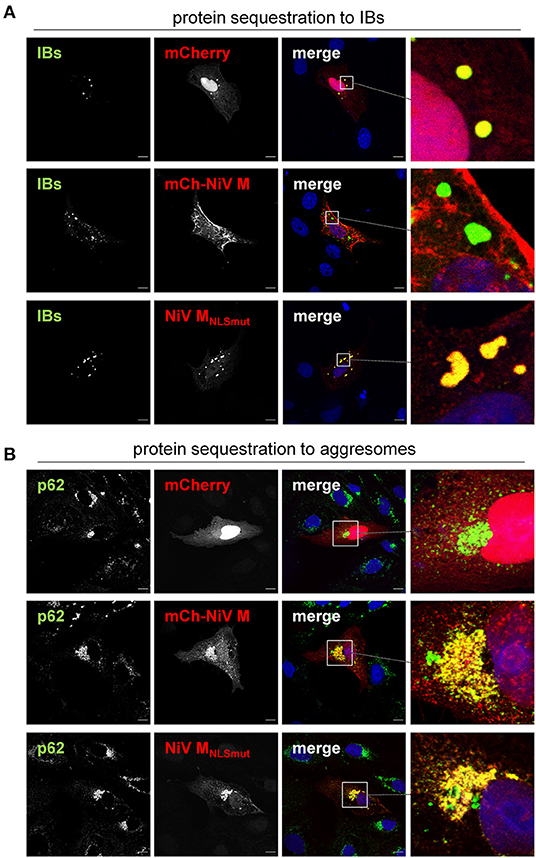
Figure 7. Sequestration of overexpressed proteins to IBs and cellular aggresomes. (A) Vero76 cells were transfected with plasmids encoding NiV N and NiV PeGFP, together with either mCherry, mCherry-NiV M or NiV MNLSmut. 24 h later, cells were fixed with 4% PFA for 15 min and permeabilized with Triton X-100. IBs were detected by eGFP autofluorescence (green). mCherry or mCherry-tagged NiV M protein (mCh-NiV M) were detected by autofluorescence (red). NiV MNLSmut was detected using an M-specific antiserum (red). Nuclei were counterstained with DAPI (blue). (B) Vero76 cells were transfected with plasmids encoding mCherry or mCherry-NiV M or NiV MNLSmut. 4 h p.t. cells were treated with 50 nM bortezomib to induce aggresome formation. 24 h after transfection, cells were fixed and permeabilized. Aggresomes were labeled with anti-p62 antibody (green). mCherry, mCh-NiV-M and NiV MNLSmut were detected as described above (red). Nuclei were counterstained with DAPI (blue). Boxed areas are shown at higher magnification on the right. Scale bars, 10 μm.
To determine if recruitment of the two “guest” proteins to IBs depends on HDAC6- or BAG3-dependent transport pathways, we assessed the sequestration mCherry and MNLSmut to IBs in the presence of tubacin or nocodazole. As shown in Figure 8, none of the inhibitors affected the sequestration to IBs. These findings clearly demonstrate that IBs not only differ structurally from aggresomes by containing only some of the major aggresomal marker proteins, but also have different “guest” protein specificities and recruit those without using active MT-dependent transport pathways.
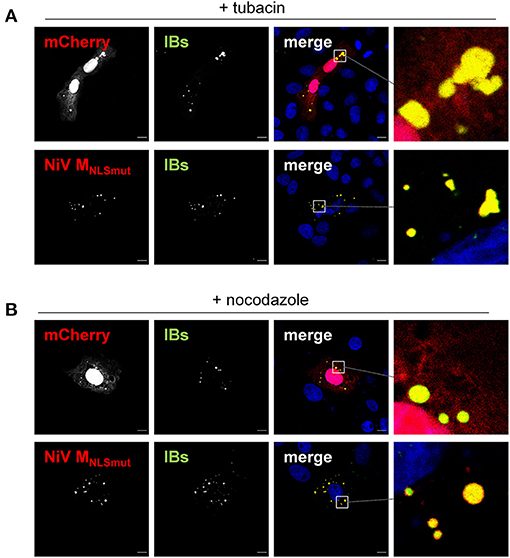
Figure 8. Influence of HDAC6 inhibition and MT disruption on protein recruitment to IBs. Vero76 cells were transfected with plasmids encoding NiV N and NiV PeGFP together with either mCherry or NiV MNLSmut. After 4 h, cells were treated with either 10 μM tubacin (A) or 250 nM nocodazole (B). 24 h post transfection, cells were fixed with 4% PFA and permeabilized with Triton X-100. mCherry and eGFP were detected by autofluorescence (green, red). NiV MNLSmut was detected using an M-specific antiserum (red). Nuclei were counterstained with DAPI (blue). Boxed areas are shown at higher magnification on the right. Scale bars, 10 μm.
A substantial number of proteins have been identified in viral IBs of MNV-infected cells (11). However, the exact role of these associations as well as the underlying sequestration mechanisms are poorly understood and are most likely specific for the individual viruses in the MNV order. Here we show that cytosolic NiV IBs and cellular aggresomes share similar intracellular locations (perinuclear), some components (γ-tubulin, HDAC6, BAG3) and can partially sequester the same cytosolic, potentially proteotoxic proteins, such as the transport-defective NiV MNLSmut. Although this suggests functional similarities, NiV IBs and aggresomes differ in many important characteristics. Neither inhibition of proteasomes, nor HDAC6 inhibitors, nor disruption of MTs had a positive or negative effect on IB formation or sequestration of proteins into IBs. Thus, the canonical transport pathways of aggresomes (MT-dependent HDAC6 and BAG3 pathways) are not required for protein accumulation in NiV IBs. In line with the differences in their formation and protein sequestration pathways, IBs did not concentrate polyubiquitinated proteins and did not contain the aggresomal marker proteins of the proteasome-ubiquitin machinery, p62 or 26S.
Supporting the idea that IBs and aggresomes are different but possibly complementary compartments, aggresomes induced by bortezomib treatment can form in NiV infected cells, but did not colocalize with IBs or affected IB formation in any way. The further finding that aggresomes, although they can be artificially induced, are not formed at any time during NiV infection supports the idea that proteotoxic stress caused by an excess of de novo synthesized cytosolic viral proteins is prevented by the concentration of these proteins in IBs. On top of concentrating viral nucleocapsid proteins needed for NiV replication, IBs might help to limit cellular stress responses by sequestering overexpressed (guest) proteins from the cytoplasm, which are not functionally needed in IBs (shown here for mCherry and transport-defective NiV MNLSmut). We suppose that NiV infection would not benefit from additional aggresome formation because this cellular stress response may trigger further processes that would ultimately counteract productive replication. Although aggresomes are supposed to reduce proteotoxic stress, they are, nevertheless, still cytotoxic. For example, the sequestration of ubiquitin to aggresomes could lead to starvation of ubiquitin, which may result in a deficient DNA damage response since ubiquitination of histones is an important factor for DNA protein localization and repair (28). Aggresomes also sequester other critical components including chaperones or proteasomes, potentially triggering a positive feedback loop increasing protein misfolding (29). Formation of aggresomes at the MTOC has also been associated with the destruction of the centrosome, which is critical for cilia formation (30). Moreover, the aggresome's perinuclear localization is supposed to cause distortions of the nuclear membrane, which might affect nucleocytoplasmic transport processes (31). As all these consequences of aggresome formation can severely affect cell homoeostasis in infected cells, the aggresome-like ability of IBs to collect overexpressed proteins might be one way to limit cytotoxic effects that may disturb virus replication, cell-cell-spread, NiV assembly and release.
The finding that neither proteasome inhibition (bortezomib), nor HDAC6 inhibition (tubacin) nor MT disruption (nocodazole) affected IB formation or protein sequestration indicates that active transport processes and canonical aggresome transport pathways are not involved. With regard to IB formation, this is in line with the concept that IBs, similar to biomolecular condensates, are formed by liquid-liquid phase separation. Based on their intrinsic properties, NiV N and P proteins trigger their preferential partitioning in the dense phase, with their intrinsically disordered protein domains as a crucial denominator that drive the liquid phase separation for cytosolic IB formation (11, 12). In line with this model, neither HDAC6 inhibition (tubacin) nor MT disruption (nocodazole) influenced IB formation in N/P-coexpressing or NiV-infected cells. Although the number of large IBs in nocodazole-treated NiV-infected cells was slightly increased (Supplementary Figure 3), none of the inhibitors had significant negative effects on NiV titers. This suggests that neither HDAC6 nor MTs play an important role in NiV replication, protein transport and assembly, which contrasts with the findings for rabies or measles viruses, where nocodazole functionally impaired IB formation and replication (32, 33).
Interestingly, liquid phase separation appears to be an efficient process not only for the formation of NiV IBs but also for the enrichment of cytosolic proteins that are not required for viral replication in IBs (mCherry, MNLSmut). Since none of the inhibitors prevented the sequestration of “guest” proteins, it can be assumed that they were not actively transported to IBs, but rather their intrinsic biophysical properties caused their preferential distribution in the dense phase and their sequestration in IBs. Protein features facilitating phase separation are for example intrinsic disorder, dynamic conformations, multivalence, nucleic acid binding or oligomeric nature (34). As mentioned above, viral nucleocapsid proteins such as NiV N and P have the required properties to drive phase separation without other components after reaching a threshold concentration (11, 12). We suppose that, as described for cellular condensates such as stress granules or P bodies (35), cytosolic NiV IBs act as scaffolds or seed IBs, which then recruit other proteins that cannot spontaneously form biomolecular condensates. Recruitment of “guest” proteins might depend on their general electrostatic properties with aromatic residues regarded as “stickers” and polar moieties as “spacers” (36). Aside of this, sequestration of “guest” proteins might need some molecular crowding leading to protein condensation, which enhances partitioning into the dense liquid phase of preformed seed IBs (37). This idea is supported by the description that normally about one-third of a cell is filled by macromolecules and that net repulsion among proteins by negative surface charges likely prevents proteins from aggregating. An increase in protein concentrations can perturb the balance, allowing proteins at the saturation concentrations to undergo condensation (37, 38). In line with this idea, only the “guest” proteins, which actually accumulated in the cytoplasm were sequestered to IBs. On one hand, transport-defective NiV MNLSmut, which cannot undergo nuclear transit required for onward transport to the plasma membrane (8), and therefore accumulates in the cytoplasm. On the other hand soluble mCherry. Although cytosolic accumulation is more limited because a substantial fraction of overexpressed mCherry resides in the nucleus, the cytosolic threshold required for partitioning into IBs is nevertheless reached over time. In contrast to the two “guest” proteins that were enriched in IBs, mCherry-labeled NiV M was not found there. The reason is most likely that mCherry-NiV M, like wild-type NiV M, is rapidly and effectively transported to the plasma membrane (8, 14) and does not accumulate sufficiently in the cytoplasm to be sequestered to IBs.
Viral IBs are known to have important functions during the viral life cycle. For many viruses these “viral factories” represent the site of replication accumulating all viral components essential for this process, and also shield viral RNA from recognition by cytosolic sensors (11). Because NiV IBs not only accumulate viral proteins but also sequester overexpressed cytosolic proteins, they may serve an additional function in limiting proteotoxic stress during infection. Although typical aggresomal marker proteins such as γ-tubulin, HDAC6, and BAG3 are present in cytosolic IBs, neither NiV nucleocapsid proteins nor “guest” proteins enriched in IBs are transported via HDAC6- or MT-dependent pathways. This independence on active transport mechanisms fits to the concept that NiV IBs are biomolecular condensates, which form by liquid-liquid phase separation induced by intrinsically disordered regions of the viral nucleocapsid proteins. Additional cellular factors are not needed. However, once formed, IBs can recruit “guest” proteins due to their biophysical properties. By separating not only viral proteins required for replication but also non-functional overexpressed proteins from the cytosol, IBs may reduce the impact of infection on cellular proteostasis, thereby improving cellular fitness and preventing cellular stress responses, which might counteract either viral replication or the survival of infected cells.
All infection experiments were carried out under biosafety level 4 (BSL-4) conditions at the Institute of Virology, Philipps University Marburg.
The NiVMalaysia (NiV) isolate used in this study has been described previously (39). Vero76 cells (CRL-1587, ATCC) were cultivated in Dulbecco's modified Eagle's medium (DMEM, Gibco) with 10% fetal calf serum (FCS), 100 U penicillin ml−1, 0.1 mg streptomycin ml−1 and 4 mM L-glutamine. Confluent Vero76 cells were infected with NiV at an MOI of 0.05 or 1 for 1 h at 37°C. After virus adsorption, cells were washed five times with DMEM 2% FCS and then incubated in DMEM 2% FCS at 37°C. For inhibitor studies, infected cells were incubated with medium containing either 250 nM nocodazole (Selleck Chemicals), 10 μM tubacin (Selleck Chemicals), 50 nM bortezomib (Selleck Chemicals) or DMSO (Wak-Chemie).
Virus titers in the supernatants of infected cells were quantified by serial dilution on Vero76 cells to determine 50% tissue culture infectious dose (TCID50/ml). Viral titers of three individual experiments are presented as mean ± SD and statistical comparison was performed using an unpaired t-test.
pCG and pCAGGS-vector based expression plasmids encoding NiV M, NiV N and NiV PeGFP, mCherry and mCherry-NiV M have been described previously (14). To generate pCG-NiV-MNLSmut, the Q5 Site-Directed Mutagenesis Kit (NEB) was used to substitute four arginines in the bipartite nuclear localization signal of NiV M at position 244 and 245 as well as position 256 and 257 by alanines (8). All transfection experiments were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Four hours post transfection (p.t.) the medium was exchanged. For inhibitor studies, transfected cells were incubated with medium containing either 250 nM nocodazole, 10 μM tubacin or DMSO.
For immunostaining of transfected cells, Vero76 cells grown on glass coverslips were fixed at 24 h p.t. with 4% paraformaldehyde (PFA, Merck) in DMEM. PFA was quenched by 0.1 M glycine in PBS supplemented with MgCl2 and CaCl2 (PBS++). Then, cells were permeabilized with 0.1% Triton X-100 (Merck) in PBS++ or ice-cold methanol/acetone (1:1) and treated with a blocking-buffer containing 2% BSA (Serva), 5% glycerol (Roth), 0.2% Tween20 (Sigma), 0.05% NaN3 (Merck). Primary antibodies were diluted in blocking-buffer and added for 1 h followed by incubation with appropriate Alexa Fluor-conjugated secondary antibodies for 45 min. A list of the different antibodies and their dilutions is provided in Supplementary Table 1. In infected cells, the immunostaining of endogenous cellular proteins was performed at 18–24 h p.i. within the BSL4 laboratory as described above using Alexa Fluor 568-conjugated secondary antibodies. After the staining of endogenous proteins, infected cells were inactivated with 4% PFA for 48 h and removed from the BSL4 laboratory. PFA was again quenched by 0.1 M glycine in PBS++. To visualize viral IBs, NiV N was stained with a guinea pig NiV antiserum (GP3) and Alexa Fluor 488-conjugated secondary antibodies. After blocking with 5% rabbit serum, NiV M was detected with a rabbit NiV M-specific antiserum (IG1321) labeled with a Zenon Alexa Fluor 647 Rabbit IgG Labeling Kit (ThermoFisher) as described previously (14). Cell nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI). Then, coverslips were mounted with mowiol (Calbiochem) and analyzed using a confocal laser scanning microscope (Leica TCS SP5 II).
To determine the total expression of acetylated α-tubulin and β-actin, cells were lysed in sample buffer containing 40% glycerol (Roth), 0.1% bromophenol blue (Merck), 200 mM Tris pH 6.8 (Roth), 8% SDS (Roth) in H2O, to which 4% β-mercaptoethanol (Sigma) was added. The proteins were separated by 10% SDS-PAGE under reducing conditions and transferred onto a nitrocellulose membrane. After blocking with 5% non-fat dry milk solution, the proteins were stained with specific primary antibodies, biotinylated secondary antibodies and peroxidase-conjugated streptavidin and detected by enhanced chemiluminescence (SuperSignal West Dura Substrate, ThermoFisher). Antibodies and dilutions are listed in Supplementary Table 1.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
NB and AM designed and conceived the study, wrote the article, and analyzed the data. NB and AH performed the experiments. All authors agreed to the final draft of the manuscript, contributed to the article, and approved the submitted version.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) to AM (Projektnummer 197785619 - SFB 1021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All infection studies were carried out under biosafety level 4 (BSL-4) conditions at the Institute of Virology, Philipps University Marburg. We thank all BSL-4 staff members for their support. We gratefully acknowledge Heinz Feldmann for providing guinea pig anti-NiV antibodies. We also thank all lab members for their critical and supportive comments on this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2021.821004/full#supplementary-material
Supplementary Figure 1. Cellular marker proteins in bortezomib-induced aggresomes. Vero76 cells were treated with 50 nM bortezomib (A) for 24 h to induce aggresome formation or with DMSO (B) as a control. Cells were fixed with 4% PFA and permeabilized with methanol/acetone or Triton X-100. Cellular proteins were labeled with specific antibodies (red). Nuclei were counterstained with DAPI (blue). Images were recorded with a Leica SP5 confocal laser scanning microscope. Aggresomes within the boxed areas are shown in higher magnification. Scale bars, 10 μm.
Supplementary Figure 2. Nocodazole prevents MT-dependent aggresome formation. (A) Vero76 cells were treated with different concentrations of nocodazole (noco) or DMSO. After 18 h cell viability was assessed with the “Ready Probes Cell Viability Imaging Kit” (Invitrogen). To determine the percentage of dead cells, 10 randomly chosen images from two individual experiments were quantified for each nocodazole concentration (>10.000 cells in total). Error bars indicate the standard deviation. Statistical analyses was performed with Prism using a one-way ANOVA with Dunnett's multiple comparisons test. ****p < 0.0001; n.s., not significant. (B) Vero76 cells were treated with either 50 nM bortezomib (BZ) or 50 nM BZ together with 250 nM nocodazole for 24 h. Cells were fixed with 4% PFA and permeabilized with Triton X-100. The aggresomal marker protein p62 was labeled with a specific antibody (green). Nuclei were counterstained with DAPI (blue). Images were recorded with a Leica SP5 confocal laser scanning microscope. Scale bars, 10 μm.
Supplementary Figure 3. Effect of inhibitor treatments on IB sizes in NiV-infected cells. Vero76 cells were infected with NiV at an MOI of 0.05 and treated with 10 μM tubacin for 24 h (A) or with 250 nM nocodazole for 18 h (B). Cells were fixed with 4% PFA for 48 h and immunostained with a NiV N antiserum to visualize IBs. To compare the IB size distribution in DMSO and inhibitor-treated cells, the numbers and areas of IBs from at least 5 different syncytia was measured using the particle analyzer function in ImageJ. Statistical analysis was performed with Prism using the Holm-Sidak t-test for multiple comparisons. Error bars indicate standard deviation (SD); *p < 0.05; n.s., not significant.
Supplementary Table 1. List of antibodies and dilutions used for immunostainings and western blots.
1. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. (2000) 288:1432–5. doi: 10.1126/science.288.5470.1432
2. Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg Infect Dis. (2001) 7:439–41. doi: 10.3201/eid0703.017312
3. Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, Gurley E, et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg Infect Dis. (2006) 12:1888–94. doi: 10.3201/eid1212.060732
4. Epstein JH, Anthony SJ, Islam A, Kilpatrick AM, Ali Khan S, Balkey MD, et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc Natl Acad Sci USA. (2020) 117:29190–201. doi: 10.1073/pnas.2000429117
5. Johnson K, Vu M, Freiberg AN. Recent advances in combating Nipah virus. Fac Rev. (2021) 10:74. doi: 10.12703/r/10-74
6. Tamin A, Harcourt BH, Ksiazek TG, Rollin PE, Bellini WJ, Rota PA. Functional properties of the fusion and attachment glycoproteins of Nipah virus. Virology. (2002) 296:190–200. doi: 10.1006/viro.2002.1418
7. Diederich S, Maisner A. Molecular characteristics of the Nipah virus glycoproteins. Ann N Y Acad Sci. (2007) 1102:39–50. doi: 10.1196/annals.1408.003
8. Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, et al. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. (2010) 6:e1001186. doi: 10.1371/journal.ppat.1001186
9. Dietzel E, Kolesnikova L, Sawatsky B, Heiner A, Weis M, Kobinger GP, et al. Nipah virus matrix protein influences fusogenicity and is essential for particle infectivity and stability. J Virol. (2015) 90:2514–22. doi: 10.1128/JVI.02920-15
10. Yun T, Park A, Hill TE, Pernet O, Beaty SM, Juelich TL, et al. Efficient reverse genetics reveals genetic determinants of budding and fusogenic differences between Nipah and Hendra viruses and enables real-time monitoring of viral spread in small animal models of henipavirus infection. J Virol. (2015) 89:1242–53. doi: 10.1128/JVI.02583-14
11. Nevers Q, Albertini AA, Lagaudrière-Gesbert C, Gaudin Y. Negri bodies and other virus membrane-less replication compartments. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118831. doi: 10.1016/j.bbamcr.2020.118831
12. Dolnik O, Gerresheim GK, Biedenkopf N. New perspectives on the biogenesis of viral inclusion bodies in negative-sense RNA virus infections. Cells. (2021) 10:1460. doi: 10.3390/cells10061460
13. Ong ST, Yusoff K, Kho CL, Abdullah JO, Tan WS. Mutagenesis of the nucleocapsid protein of Nipah virus involved in capsid assembly. J Gen Virol. (2009) 90:392–7. doi: 10.1099/vir.0.005710-0
14. Ringel M, Heiner A, Behner L, Halwe S, Sauerhering L, Becker N, et al. Nipah virus induces two inclusion body populations: identification of novel inclusions at the plasma membrane. PLoS Pathog. (2019) 15:e1007733. doi: 10.1371/journal.ppat.1007733
15. Ringel M, Behner L, Heiner A, Sauerhering L, Maisner A. Replication of a Nipah virus encoding a nuclear-retained matrix protein. J Infect Dis. (2020) 221:S389–S394. doi: 10.1093/infdis/jiz440
16. Latorre V, Mattenberger F, Geller R. Chaperoning the mononegavirales. Viruses. (2018) 10:699. doi: 10.3390/v10120699
17. Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. (2001) 26:597–604. doi: 10.1016/S0968-0004(01)01938-7
18. Marques M, Ramos B, Soares AR, Ribeiro D. Cellular proteostasis during influenza A virus infection-friend or foe? Cells. (2019) 8:228. doi: 10.3390/cells8030228
19. Aviner R, Frydman J. Proteostasis in viral infection: unfolding the complex virus-chaperone interplay. Cold Spring Harb Perspect Biol. (2020) 12:a034090. doi: 10.1101/cshperspect.a034090
20. Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. (1998) 143:1883–98. doi: 10.1083/jcb.143.7.1883
21. Gamerdinger M, Carra S, Behl C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J Mol Med. (2011) 89:1175–82. doi: 10.1007/s00109-011-0795-6
22. Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. (2006) 312:875–8. doi: 10.1126/science.1126766
23. Liu Y, Shevchenko A, Berk AJ. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J Virol. (2005) 79:14004–16. doi: 10.1128/JVI.79.22.14004-14016.2005
24. Olasunkanmi OI, Chen S, Mageto J, Zhong Z. Virus-induced cytoplasmic aggregates and inclusions are critical cellular regulatory and antiviral factors. Viruses. (2020) 12:399. doi: 10.3390/v12040399
25. Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. (2003) 8:508–13. doi: 10.1634/theoncologist.8-6-508
26. Lu CY, Chang YC, Hua CH, Chuang C, Huang SH, Kung SH, et al. Tubacin, an HDAC6 selective inhibitor, reduces the replication of the japanese encephalitis virus via the decrease of viral RNA synthesis. Int J Mol Sci. (2017) 18:954. doi: 10.3390/ijms18050954
27. Chen H, Qian Y, Chen X, Ruan Z, Ye Y, Babiuk LA, et al. HDAC6 restricts influenza A virus by deacetylation of the RNA polymerase PA subunit. J Virol. (2019) 93:e01896–18. doi: 10.1128/JVI.01896-18
28. Ben Yehuda A, Risheq M, Novoplansky O, Bersuker K, Kopito RR, Goldberg M, et al. Ubiquitin accumulation on disease associated protein aggregates is correlated with nuclear ubiquitin depletion, histone de-ubiquitination and impaired DNA damage response. PLoS ONE. (2017) 12:e0169054. doi: 10.1371/journal.pone.0169054
29. Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, et al. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell. (2018) 172:696–705.e12. doi: 10.1016/j.cell.2017.12.030
30. Iqbal A, Baldrighi M, Murdoch JN, Fleming A, Wilkinson CJ. Alpha-synuclein aggresomes inhibit ciliogenesis and multiple functions of the centrosome. Biol Open. (2020) 9:054338. doi: 10.1242/bio.054338
31. Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, et al. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington's disease. Hum Mol Genet. (2015) 24:1602–16. doi: 10.1093/hmg/ddu574
32. Nikolic J, Le Bars R, Lama Z, Scrima N, Lagaudrière-Gesbert C, Gaudin Y, et al. Negri bodies are viral factories with properties of liquid organelles. Nat Commun. (2017) 8:58. doi: 10.1038/s41467-017-00102-9
33. Zhou Y, Su JM, Samuel CE, Ma D. Measles virus forms inclusion bodies with properties of liquid organelles. J Virol. (2019) 93:e00948–19. doi: 10.1128/JVI.00948-19
34. Alberti S, Dormann D. Liquid-liquid phase separation in disease. Annu Rev Genet. (2019) 53:171–94. doi: 10.1146/annurev-genet-112618-043527
35. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. (2017) 18:285–98. doi: 10.1038/nrm.2017.7
36. Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. (2020) 367:694–9. doi: 10.1126/science.aaw8653
37. Jalihal AP, Schmidt A, Gao G, Little SR, Pitchiaya S, Walter NG. Hyperosmotic phase separation: condensates beyond inclusions, granules and organelles. J Biol Chem. (2021) 296:100044. doi: 10.1074/jbc.REV120.010899
38. Wennerström H, Vallina Estrada E, Danielsson J, Oliveberg M. Colloidal stability of the living cell. Proc Natl Acad Sci USA. (2020) 117:10113–21. doi: 10.1073/pnas.1914599117
Keywords: Nipah virus, inclusions, aggresomes, HDAC6, BAG3, microtubules
Citation: Becker N, Heiner A and Maisner A (2022) Cytosolic Nipah Virus Inclusion Bodies Recruit Proteins Without Using Canonical Aggresome Pathways. Front. Virol. 1:821004. doi: 10.3389/fviro.2021.821004
Received: 23 November 2021; Accepted: 23 December 2021;
Published: 23 February 2022.
Edited by:
Thomas Hoenen, Friedrich-Loeffler-Institut, GermanyReviewed by:
Yves Gaudin, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2022 Becker, Heiner and Maisner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Maisner, bWFpc25lckB1bmktbWFyYnVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.