 Abdulkarim Fahad Alhetheel
Abdulkarim Fahad Alhetheel
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Virol. , 20 January 2022
Sec. Virus and Host Immunity
Volume 1 - 2021 | https://doi.org/10.3389/fviro.2021.810231
Hepatitis C is a worldwide liver disease caused by hepatitis C virus (HCV) infection. The virus causes acute and chronic liver inflammation, and it is transmitted mainly by exposure to contaminated blood. HCV is capable of infecting hepatocytes and peripheral blood mononuclear cells, causing complications and disease progression. This mini review provides an overview of HCV infection, including details on the virological aspects, infection of the immune cells, and its impact on the immune system.
Over 30 years have passed since the discovery of hepatitis C virus (HCV) infection in 1989, which is one of the most common causes of liver disease worldwide (1). HCV infection causes acute and chronic hepatitis and is the main cause of post-transfusion hepatitis. It is transmitted mainly by exposure to contaminated blood, contaminated needles or razors, hemodialysis, organ transplantation, and, to a lesser extent, by sexual intercourse (1, 2). HCV is highly heterogeneous and is susceptible to ongoing mutation. It has been estimated that over 170 million people are currently infected with HCV, and over three million people are estimated to be infected annually. Moreover, ~85 % of newly infected individuals develop chronic hepatitis, which may lead to further complications such as liver cirrhosis and hepatocellular carcinoma. To date, seven main HCV genotypes (genotypes 1–7) and many other subtypes have been identified. The sequence diversity among the genotypes is approximately 30%. Genotypes 1 and 2 are the most prevalent HCV genotypes. HCV genotypes 1, 2, and 3 are found in the USA, Europe, and Japan, while genotypes 4 and 5 are found in Africa. Genotypes 3 and 6 are endemic to Asia, whereas genotype 7 is endemic to central Africa and has not been fully evaluated (1, 2). This article discusses recent published literature on antiviral therapy for HCV infection, focusing on infection of the peripheral blood mononuclear cells (PBMCs), and the effects of HCV infection on the immune system.
HCV belongs to the genus Hepacivirus, a member of the Flaviviridae family. It is a small spherical enveloped virus with a molecular size of 50–80 nm in diameter (Figure 1A). The virus has a 9.6 kb positive-sense single-stranded RNA (ssRNA) genome, which has a long open reading frame between the 5' and 3' untranslated regions (1, 2). It encodes a large polypeptide chain consisting of 3,010 amino acids that are processed to produce 10 different proteins known as the HCV structural and non-structural (NS) proteins. The structural proteins include the core capsid (C) and envelope subunit glycoproteins (E1 and E2). The NS proteins include NS1, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Figure 1B). The HCV core is a highly conserved protein that forms the viral nucleocapsid and is involved in HCV pathogenesis, including steatosis hepatitis, hepatocellular carcinoma, and alteration of different cellular signaling pathways and functions. The envelope is composed of two subunit glycoproteins, E1 and E2, and both play a vital role in viral entry (Figure 1A). The NS proteins are essential at different stages of HCV replication (1, 2).
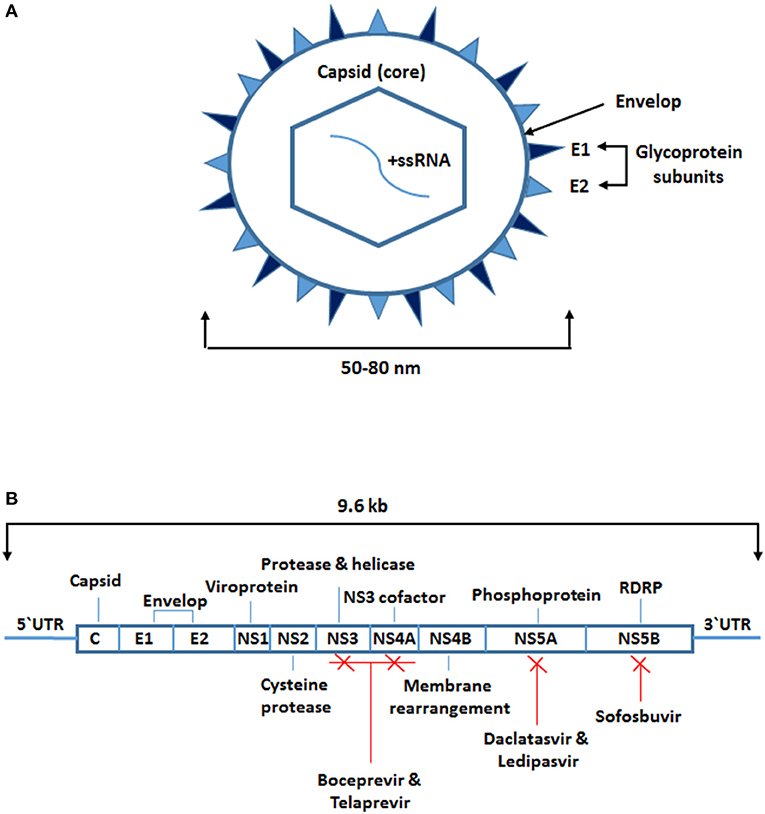
Figure 1. Structure of hepatitis C virus and sites targeted by antiviral therapy. (A) A small spherical enveloped virus with molecular size between 50 and 80 nm in diameter. The genome is a positive-sense single-stranded RNA molecule. The structural proteins include the capsid which is the core (C) protein and the envelope subunits glycoproteins (E1 and E2). (B) The viral long open reading frame in-between 5‘ and 3‘ untranslated regions. It encodes for 10 different proteins known as the HCV structural proteins the core (C) and the envelope subunits glycoproteins (E1 and E2) and the non-structural (NS) proteins (NS1, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Some of the NS proteins are targeted by the protease and RNA dependent RNA polymerase inhibitors (boceprevir, telaprevir, daclatasvir, ledipasvir, sofosbuvir).
The first approved treatment for HCV infection was ribavirin in combination with PEGylated interferon-alpha (IFN-α). This combined therapy inhibits HCV replication non-specifically by general suppression of protein synthesis, inhibition of HCV polymerase, or mutagenesis of newly manufactures HCV RNA. The effectiveness of this therapy in adherent patients is ~50%. However, the development of direct antiviral therapy and the addition of a protease inhibitor such as boceprevir or telaprevir has increased the effectiveness of the treatment to about 70%. Advances in the development of other proteases and RNA-dependent RNA polymerase inhibitors such as daclatasvir, ledipasvir, and sofosbuvir have drastically improved the clinical outcomes (1, 2). Moreover, other antiviral drugs which target some of the host key factors involved in HCV replication such as microRNA (miR)-122 and cyclophilin are currently under development (1).
The immune system is a diverse and complex network. Based on the immune response to a foreign antigen, it can be broadly categorized into innate and adaptive immune components (Figure 2) (3–6). Innate immunity is mediated by cellular components of natural killer (NK) cells, dendritic cells, macrophages, and granulocytes, whereas, adaptive immunity is mediated by B and T lymphocytes. On exposure to foreign antigens, both components act together and in parallel with regulatory soluble mediators known as cytokines and chemokines, in order to eradicate, and protect the body from, microbial pathogens (Figure 2) (3–6).
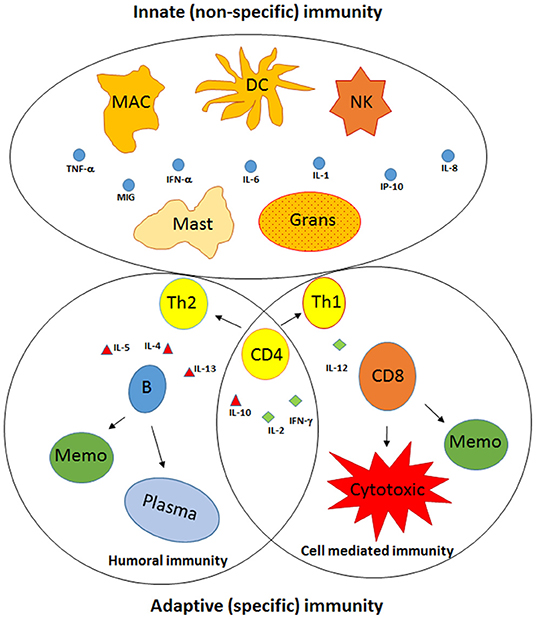
Figure 2. Components of the innate and adaptive immunity. The immune system is broadly categorized into innate and adaptive immune components. Innate immunity is recognized by cellular components, including natural killer (NK), dendritic cells (DC), macrophages (MAC), mast cells, and granulocytes (Grans), whereas adaptive immunity is designated by B lymphocytes and T lymphocytes. Following activation, the components of both branches act together and in parallel with regulatory soluble cytokines (IL-1, IL-6, IL-12, IL-10, IL-4) and chemokines (IL-8, IP-10, and MIG). Subsequently, CD4+ T lymphocytes become activated and differentiate into T helper (Th)-1 and Th-2 cells, leading to the activation and differentiation of T lymphocytes into cytotoxic CD8+ T cells and memory cells (Memo), while the B lymphocytes differentiate into plasma cells, which are antibody-specific secreted cells, and Memo, respectively.
NK cells, dendritic cells, macrophages, and granulocytes, as part of the innate immune system, and are the first line of defense against invading pathogens. NK cells kill virus-infected cells by cell lysis and perforin secretion. Dendritic cells, macrophages, and granulocytes possess antimicrobial, antifungal, and antiparasitic characteristics through their phagocytic and endocytic activity (3–6). Further, they function as antigen-presenting cells by uptake, processing, and presenting antigens linked to major histocompatibility complex class II to CD4+ T cells. Furthermore, they secrete proinflammatory cytokines such as interleukin (IL)-1, IL-6, IL-12, interferon type-1 (IFN-α/β), and chemokines such as IL-8 (3–6). This stimulates the adaptive immune system through the activation of CD4+ T lymphocytes, which differentiate into T helper (Th)-1 and Th-2 cells and leads to the activation and differentiation of T lymphocytes into cytotoxic T cells (cytotoxic CD8+ T cells) and the B lymphocytes into plasma cells, the antibody-specific cells (Figure 2) (3–6). These essential cellular activities are mostly mediated and regulated by the responsiveness of these cells to different cytokines such as IFN-γ, tumor necrosis factor (TNF)-α, IL-10, and signals delivered via the toll-like receptor (TLR) family through recognition of various microbial proteins such as bacterial lipopolysaccharides, viral proteins, and nucleic acids, including those of HCV (3–6).
Unlike human cellular RNA molecules, which are short and incomplete stems with secondary structures, RNA viruses usually produce long and perfect double-stranded RNA (dsRNA) molecules during replication in infected cells. Therefore, long dsRNA molecules are recognized as foreign molecules and trigger both cellular and humoral innate immune responses (7). There are two characteristic mechanisms by which a cell recognizes viral antigens. Extracellular viruses are recognized by the different TLRs expressed on the cell surface based on the type of their genome. For example, ssRNA, dsRNA, or CpG-rich DNA viruses are recognized by TLR7/8, TLR3, and TLR9, respectively (8). On the other hand, the intracellular replicating viruses are recognized by RNA helicases which encoded by the retinoic acid-inducible gene I (RIG-I) and/or melanoma differentiation-associated gene 5 (MDA5) (9). Following viral recognition, the activation and translocation of nuclear factor κB (NFκB) and interferon-regulatory factor 3 (IRF-3) to the nucleus occurs and promotes the transcription of IFN type-1 (10). Production of IFN type-1 triggers the neighboring cells to produce different antiviral proteins, including 2'-5' oligoadenylate synthase/RNase L, protein kinase R (PKR), myxovirus resistance factor, and dsRNA adenosine deaminase 1, which results in the activation of eukaryotic initiation factor (eIF-2), and translation inhibition of both host and viral mRNA (11).
PBMCs, which include monocytes, the precursor of macrophages, and lymphocytes (CD4+T cells, CD8+T cells, NK cells, and B cells) play a major role in clearing microbial pathogens. Studies have shown that HCV can infect and replicate in hepatocytes and other cell types, including bone marrow cells and PBMCs (12–16) by cell-to-cell infection (1). Despite the disappearance of HCV RNA in the serum of sustained responders, viral RNA can remain detectable for up to 5 years in the PBMCs of these individuals (12, 17). Persistent infection of PBMCs by HCV has been proposed to be an important extrahepatic source of reinfection and is believed to be an important site for HCV replication (18–21). HCV infects PBMCs and other cells through interaction with CD81 molecules on cell surface (22) and subsequently replication of HCV in extrahepatic tissues, is facilitated by expression of miR-122 (23). Moreover, transducing miR122 in B lymphocytes found to stabilize and enhance HCV replication in these cells (24). Direct and prolonged interaction of HCV with lymphoid tissue has been implicated in the induction of malignant lymphoma (25). Little is known about the biological significance of lymphotropic HCV but evidence suggests that it might possibly be a reservoir that contributes to recurrence of HCV infection (26). Peripheral blood B lymphocytes particularly CD27+ memory B cells not only can harbor higher HCV viral loads but also resist apoptosis thus may serve as a candidate subset for HCV reservoir (27). In addition to B lymphocytes lymphotropic HCV has been shown to be capable of infecting T cell lines and primary naïve CD4+ lymphocytes (5). Similarly, it has also been reported that HCV can infect monocytes that are CD14+/CD16+ but not CD14+/CD16- (28). Despite the existence of convincing data supporting PBMCs serving as HCV reservoir there are reports suggesting that the presence of HCV sequences in PBMCs is due to passive virus adsorption (29) or contamination by circulating virus and not due to replication of HCV (30). Because of the conflicting data the exact mechanism of HCV infection of PBMCs in-vivo remains unclear.
Infection of PBMCs results in persistence of infection, chronic immune activation, and exhaustion of the immune system (17, 31, 32). Similar to other viral infections, HCV infection often induces an IFN response which is recognized by the production and secretion of proinflammatory cytokines, including IL-6 and IFN type-1, which have antimicrobial and antiproliferative characteristics, and activate the adaptive immune responses. PBMCs play a critical role in the elimination of pathogens via phagocytic and cytolytic activities. Such PBMC functions are mediated through the regulation of different signaling pathways, including the Janus kinase/signal transducer and activator of transcription (JAK-STAT) pathway (14, 33, 34). HCV and its proteins, such as the core, E2, and NS5A polypeptides, are capable of affecting several signaling pathways, including JAK-STAT, phosphatidylinositol-3 kinase (PI3K), and mitogen-activated protein kinase pathways in macrophages and hepatocytes. HCV core protein has been shown to induce STAT-3 and PI3K activation in monocytes and macrophages through the release of IL-6 (35). Another study demonstrated up-regulation of STAT-1 expression in hepatocytes from individuals with chronic HCV viremia (36). In contrast, a significant down-regulation of STAT-1 and IRF-1 expression has been observed in PBMCs of individuals with untreated HCV infection and sustained responders (12). Other reports have shown that HCV and its proteins repress STAT-1 and IRF-1 expression in human hepatocellular carcinoma cell-line (Huh7 cells) and other cell types (37–41). STAT-1 and IRF-1 are transcription factors involved in different immune functions, and dysregulation in the expression of these proteins may compromise the immune response (14, 33, 34, 42). Down-regulation of STAT-1 and IRF-1 expression in HCV-transfected cell lines has been shown to promote viral replication (43). However, IFN-α and IFN-γ co-treatment in human hepatoma cells has been shown to up-regulate the expression of STAT-1 and IRF-1, leading to enhanced interferon stimulated gene (ISG) expression with robust antiviral activity (43). It is therefore possible that persistent HCV infection is attributable to virus-induced suppression of IFN expression. Moreover, as IFN therapy up-regulates the expression of STAT-1 and IRF-1, monitoring the expression of these two genes may be used to assess the response to treatment.
One of the main side effects of viral infections, including HCV infection, is the alteration in cytokine and chemokine secretion. Studies have shown alterations in the serum levels of cytokines and chemokines such as IL-8, IL-6, IL-10, and IL-12p70 in individuals with untreated HCV infection and non-responders (44–46). IL-8, which is also known as CXCL8, is a CXC chemokine that plays a major role in neutrophil and macrophage trafficking toward the site of tissue injury (47). It is produced by different types of cells expressing TLR, especially macrophages and monocytes (48, 49). IL-8 binds to the chemokine receptors, CXCR1 and CXCR2, which are both expressed by neutrophils, monocytes, and macrophages (50, 51). It has been suggested that in chronic HCV infection, elevated levels of IL-8 in the intrahepatic tissues and peripheral blood and its interaction with CXCR1 resulted in increased tissue infiltration and activation of hepatic macrophages (52). In addition, increased levels of IL-8 in individuals with HCV infection are associated with disease progression, resulting in chronic infection (45). The decreased level of IL-8 after antiviral therapy among sustained responders indicates that increases in serum IL-8 levels may be associated with enhanced viral activity (44). Thus, the assessment of serum IL-8 levels in HCV patients may be used to monitor disease activity and response to treatment.
It has been reported that serum IL-6 levels are elevated in individuals with untreated HCV infection, but that sustained responder HCV patients have similar serum IL-6 levels to healthy controls (44). IL-6 is a proinflammatory cytokine that is produced by different types of cells, including macrophages and T cells, and is involved in the regulation of differentiation of many cell types (53). An increased IL-6 levels in the serum of individuals with HCV infection have been shown to be linked to disease severity (46). HCV infection has been shown to be directly linked to high serum IL-6 levels. A recent animal study showed that inoculation of mice with recombinant HCV core protein resulted in increased production of IL-6 (54). Thus, it is possible that the decreased IL-6 levels in HCV patients serum after successful antiviral therapy may indicate clearance of the infection (44).
It has been found that serum IL-10 levels are elevated in individuals with untreated HCV infection, but that IL-10 levels decrease and serum IL-12p70 levels increase following successful antiviral treatment in sustained responders (44). IL-10 is a Th2 cytokine, whereas IL-12 is a Th1 cytokine (55). Increased levels of IL-10 in the serum of individuals with untreated HCV infection suggests Th2 polarization of the immune response in individuals with HCV infection. However, the immune response switches to a Th1 immune response after antiviral treatment, as evidenced by an increase in IL-12 levels and a decrease in IL-10 levels (44). Several studies have shown that HCV infection is associated with a Th2-skewed immune response (56, 57). Increased serum IL-12 levels after treatment with ribavirin/PEGylated IFN-α2A have been associated with a sustained viral suppression, whereas increased serum IL-10 levels may indicate late viral activation (58). These results suggest that successful antiviral treatment is linked to a modification in the pretreatment Th2 cytokine immune response to the Th1 response.
There has been considerable interest in the persistence of low-level HCV RNA in PBMCs after completing a course of treatment in individuals with chronic HCV infection. HCV RNA has been shown to remain detectable 5 years after sustained viral suppression following treatment with ribavirin/PEGylated IFN-α2A, along with up-regulation of IL-6, IL-8, IL-12, TNF-α, and macrophage inflammatory protein (MICP)-1β expression (17, 44). PBMCs appear to be infected with HCV that leads to an increase in cytokine expression due to persistent antigen stimulation (29). IL-6, a proinflammatory cytokine associated with the innate immune response, promotes T- and B-cell activation, proliferation, differentiation, and survival (59), whereas IL-8, a chemokine, is involved in neutrophil and macrophage infiltration and activation (52). Therefore, it is possible that both cytokines involve in both the innate and the adaptive immune response to HCV. However, increased IL-12 levels in the serum of HCV patients after antiviral treatment suggest its key role in Th1 polarization of the immune response and increasing antiviral and cytotoxic activity (44, 60).
Programmed cell death (PCD) through apoptosis or autophagy plays a major role in the maintenance of immune regulation, cellular homeostasis, and normal cell death. Thus, alterations in PCD processes may result in malignancies and/or immune dysfunction (61–63). The apoptosis signaling cascade is initiated either through cell surface death receptors such as Fas, TNF-αR, TRAILR, and APO3-R (the extrinsic pathway), which results in activation of caspase 8 and caspase 10 or disturbance of the mitochondrial membrane potential, leading to cytochrome C release and the activation of caspase 9 (the intrinsic pathway). As a result, the downstream effector caspases 3, 6, and 7 are activated, leading to cleavage of nuclear poly (ADP-ribose) polymerase, DNA fragmentation, and cell death (61, 62). However, autophagy is triggered by metabolic stress such as nutrient starvation or by pharmaceutical or chemotherapeutic agents (such as rapamycin and etoposide, respectively). The process starts by the sequestration of a portion of cytoplasmic materials, which include proteins and organelles, into a double-membrane vesicle known as an autophagosome. Thereafter, the autophagosome vesicles fuse with cellular endosomes and lysosomes to form autolysosomes. Lastly, the cytoplasmic contents within the autolysosome breaks down, leading to cell death or cell survival (61, 63).
A number of reports have elucidated the role of PCD in the pathogenesis of chronic HCV infection through the course of hepatitis and fibrosis, or its progression to hepatocellular carcinoma (64–67). PBMCs, through their phagocytic and cytolytic activities, are critical for microbial pathogen clearance. After activation of PBMCs, several cytokines are produced, such as L-1 α and β, IL-2, IL-6, IL-10, IL-12, and IL-18, leading to the activation of the adaptive immune response. It has been proposed that the PBMCs of individuals with HCV infection are more susceptible to PCD and are thus not capable of clearing HCV. The PBMCs of individuals with untreated HCV infection and sustained responders have been shown to exhibit significantly increased spontaneous PCD compared to healthy controls (68). The increased level of PCD has been shown to be associated with significant up-regulation of caspase 3 expression in the PBMCs of individuals with untreated HCV infection and sustained responders compared to healthy controls (12). Several other studies have shown similar results (69–71). For example, increased PCD rates have been shown to be associated with the up-regulation of the apoptotic genes, such as Fas, caspase 8, caspase 9, and caspase 3, in the PBMCs of patients with HCV infection, (69). In addition, on starvation, the cell death rates in T cells and monocytes of patients with chronic HCV infection, with or without liver cirrhosis or hepatocellular carcinoma, were significantly higher than in healthy controls (70). Moreover, several reports have shown that caspase 3 plays a role in PBMC cell death and dysfunction (64, 70, 71).
It has been shown that increased susceptibility of PBMCs to spontaneous PCD is associated with decreased levels of proinflammatory cytokines, including IL-6, IL-8, and IL-10, in the supernatants of PBMC cultures of individuals with untreated HCV infection and sustained responders (68). These findings indicate that the PBMCs of individuals with HCV infection are dysfunctional. Although low levels of supernatant proinflammatory cytokines could indicate defective PBMCs, they may have an adverse effect on PBMC PCD. These cytokines may have a role in PBMC survival as recombinant IL-8 inhibits apoptosis of endothelial cells by enhancing the expression of anti-apoptotic genes such as B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma extra-large (Bcl-xL) (72). IL-10 has also been shown to inhibit spontaneous T cell apoptosis in patients with infectious mononucleosis (73). Furthermore, IL-6 prevents cluster of differentiation (CD) 3-induced T cell death and hydrogen peroxide-induced PCD in alveolar epithelial cells (74, 75). These results indicate that despite increased blood levels of antiapoptotic cytokines such as IL-8, IL-10, and IL-6, the PBMCs of individuals with HCV infection are defective and more prone to PCD. Thus, it is possible that factors related to the presence of HCV within PBMCs are critical for enhanced susceptibility to PCD.
PBMCs, which include monocytes, CD4+T cells, CD8+T cells, NK cells, and B cells, play an essential role in eradicating microbial pathogens. Infection of PBMCs with HCV results in dysregulation of the signaling pathway mediators such as STAT-1 and IRF-1, and alterations in cytokine and chemokine production, including IL-1, IL-6, IL-10, and IL-8. In individuals with chronic HCV infection, persistent HCV RNA and its antigens, combined with chronic immune activation, lead to PBMCs becoming exhausted, defective, and more prone to PCD (Figure 3).
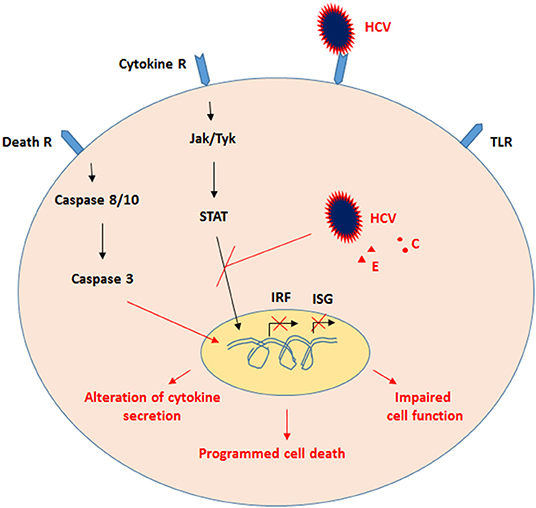
Figure 3. Proposed model for the effects of HCV on PBMCs. Infection of PBMCs, which include monocytes, CD4+T cells, CD8+T cells, NK cells, and B cells, with HCV resulted in dysregulation of signaling-pathway mediators such as STAT-1 and IRF-1, as well as alteration in cytokine and chemokine production. Subsequently, persistent HCV RNA and its antigens, along with chronic immune activation, the PBMCs become exhausted, impaired, and more prone to PCD.
AA collecting and reviewing the published literature and writing and revising the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The author would like to thank Editage (www.editage.com) for English language editing.
1. Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. (2013) 19:837–49. doi: 10.1038/nm.3248
2. Preciado MV, Valva P, Escobar-Gutierrez A, Rahal P, Ruiz-Tovar K, Yamasaki L, et al. Hepatitis C virus molecular evolution: transmission, disease progression and antiviral therapy. World J Gastroenterol. (2014) 20:15992–6013. doi: 10.3748/wjg.v20.i43.15992
3. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. (2003) 111(2 Suppl):S442–59. doi: 10.1067/mai.2003.125
4. Delves PJ, Roitt IM. The immune system. Second of two parts. N Engl J Med. (2000) 343:108–17. doi: 10.1056/NEJM200007133430207
5. Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med. (2000) 343:37–49. doi: 10.1056/NEJM200007063430107
6. Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. (2000) 343:338–44. doi: 10.1056/NEJM200008033430506
7. Cullen BR. Is RNA interference involved in intrinsic antiviral immunity in mammals? Nat Immunol. (2006) 7:563–7. doi: 10.1038/ni1352
8. Uematsu S, Akira S. The role of Toll-like receptors in immune disorders. Expert Opin Biol Ther. (2006) 6:203–14. doi: 10.1517/14712598.6.3.203
9. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. (2006) 441:101–5. doi: 10.1038/nature04734
10. Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. (2005) 11:263–70. doi: 10.1038/nm1191
11. Schlee M, Hornung V, Hartmann G. siRNA and isRNA: two edges of one sword. Mol Ther. (2006) 14:463–70. doi: 10.1016/j.ymthe.2006.06.001
12. Alhetheel A, Albarrag A, Hakami A, Shakoor Z, Alswat K, Abdo A, et al. In the peripheral blood mononuclear cells (PBMCs) of HCV infected patients the expression of STAT1 and IRF-1 is downregulated while that of caspase-3 upregulated. Acta Virol. (2020) 64:352–8. doi: 10.4149/av_2020_313
13. Bare P. Hepatitis C virus and peripheral blood mononuclear cell reservoirs Patricia Bare. World J Hepatol. (2009) 1:67–71. doi: 10.4254/wjh.v1.i1.67
14. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
15. Manzin A, Candela M, Paolucci S, Caniglia ML, Gabrielli A, Clementi M. Presence of hepatitis C virus (HCV) genomic RNA and viral replicative intermediates in bone marrow and peripheral blood mononuclear cells from HCV-infected patients. Clin Diagn Lab Immunol. (1994) 1:160–3. doi: 10.1128/cdli.1.2.160-163.1994
16. Revie D, Salahuddin SZ. Human cell types important for hepatitis C virus replication in vivo and in vitro: old assertions and current evidence. Virol J. (2011) 8:346. doi: 10.1186/1743-422X-8-346
17. Radkowski M, Opoka-Kegler J, Cortes KC, Bukowska-Osko I, Perlejewski K, Pawelczyk A, et al. Evidence for immune activation in patients with residual hepatitis C virus RNA long after successful treatment with IFN and ribavirin. J Gen Virol. (2014) 95:2004–9. doi: 10.1099/vir.0.064709-0
18. Laskus T, Radkowski M, Piasek A, Nowicki M, Horban A, Cianciara J, et al. Hepatitis C virus in lymphoid cells of patients coinfected with human immunodeficiency virus type 1: evidence of active replication in monocytes/macrophages and lymphocytes. J Infect Dis. (2000) 181:442–8. doi: 10.1086/315283
19. Lerat H, Rumin S, Habersetzer F, Berby F, Trabaud MA, Trepo C, et al. In vivo tropism of hepatitis C virus genomic sequences in hematopoietic cells: influence of viral load, viral genotype, and cell phenotype. Blood. (1998) 91:3841–9. doi: 10.1182/blood.V91.10.3841
20. Muller HM, Pfaff E, Goeser T, Kallinowski B, Solbach C, Theilmann, et al. Peripheral blood leukocytes serve as a possible extrahepatic site for hepatitis C virus replication. J Gen Virol. (1993) 74:669–76. doi: 10.1099/0022-1317-74-4-669
21. Willems M, Peerlinck K, Moshage H, Deleu I, Van den Eynde C, Vermylen J, et al. Hepatitis C virus-RNAs in plasma and in peripheral blood mononuclear cells of hemophiliacs with chronic hepatitis C: evidence for viral replication in peripheral blood mononuclear cells. J Med Virol. (1994) 42:272–8. doi: 10.1002/jmv.1890420314
22. Kondo Y, Ueno Y, Kakazu E, Kobayashi K, Shiina M, Tamai K, et al. Lymphotropic HCV strain can infect human primary naive CD4+ cells and affect their proliferation and IFN-gamma secretion activity. J Gastroenterol. (2011) 46:232–41. doi: 10.1007/s00535-010-0297-2
23. Fukuhara T, Kambara H, Shiokawa M, Ono C, Katoh H, Morita E, et al. Expression of microRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol. (2012) 86:7918–33. doi: 10.1128/JVI.00567-12
24. Chen CL, Huang JY, Wang CH, Tahara SM, Zhou L, Kondo Y, et al. Hepatitis C virus has a genetically determined lymphotropism through co-receptor B72. Nat Commun. (2017) 8. doi: 10.1038/ncomms13882
25. De Vita S, Sansonno D, Dolcetti R, Ferraccioli G, Carbone A, Cornacchiulo V, et al. Hepatitis C virus within a malignant lymphoma lesion in the course of type II mixed cryoglobulinemia. Blood. (1995) 86:1887–92. doi: 10.1182/blood.V86.5.1887.bloodjournal8651887
26. Pal S, Sullivan DG, Kim S, Lai KK, Kae J, Cotler SJ, et al. Productive replication of hepatitis C virus in perihepatic lymph nodes in vivo: implications of HCV lymphotropism. Gastroenterology. (2006) 130:1107–16. doi: 10.1053/j.gastro.2005.12.039
27. Ito M, Masumi A, Mochida K, Kukihara H, Moriishi K, Matsuura Y, et al. Peripheral B cells may serve as a reservoir for persistent hepatitis C virus infection. J Innate Immun. (2010) 2:607–17. doi: 10.1159/000317690
28. Coquillard G, Patterson BK. Determination of hepatitis C virus-infected, monocyte lineage reservoirs in individuals with or without HIV coinfection. J Infect Dis. (2009) 200:947–54. doi: 10.1086/605476
29. Laskus T, Radkowski M, Wang LF, Nowicki M, Rakela J. Uneven distribution of hepatitis C virus quasispecies in tissues from subjects with end-stage liver disease: confounding effect of viral adsorption and mounting evidence for the presence of low-level extrahepatic replication. J Virol. (2000) 74:1014–7. doi: 10.1128/JVI.74.2.1014-1017.2000
30. Laskus T, Radkowski M, Wang LF, Cianciara J, Vargas H, Rakela, et al. Hepatitis C virus negative strand RNA is not detected in peripheral blood mononuclear cells and viral sequences are identical to those in serum: a case against extrahepatic replication. J Gen Virol. (1997) 78:2747–50. doi: 10.1099/0022-1317-78-11-2747
31. Barathan M, Mohamed R, Yong YK, Kannan M, Vadivelu J, Saeidi A, et al. Viral Persistence and Chronicity in Hepatitis C Virus Infection: Role of T-Cell Apoptosis, Senescence and Exhaustion. Cells. (2018) 7. doi: 10.3390/cells7100165
32. Nguyen LN, Nguyen LNT, Zhao J, Schank M, Dang X, Cao D, et al. Immune activation induces telomeric DNA damage and promotes short-lived effector T cell differentiation in chronic HCV infection. Hepatology. (2021) 74:2380–94. doi: 10.1002/hep.32008
33. Imada K, Leonard WJ. The Jak-STAT pathway. Mol Immunol. (2000) 37:1–11. doi: 10.1016/S0161-5890(00)00018-3
34. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
35. Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. (2011) 286:10847–55. doi: 10.1074/jbc.M110.217653
36. El-Saadany S, Ziada DH, El Bassat H, Farrag W, El-Serogy H, Eid M, et al. The role of hepatic expression of STAT1, SOCS3 and PIAS1 in the response of chronic hepatitis C patients to therapy. Can J Gastroenterol. (2013) 27:e13–7. doi: 10.1155/2013/562765
37. Ciccaglione AR, Stellacci E, Marcantonio C, Muto V, Equestre M, Marsili G, et al. Repression of interferon regulatory factor 1 by hepatitis C virus core protein results in inhibition of antiviral and immunomodulatory genes. J Virol. (2007) 81:202–14. doi: 10.1128/JVI.01011-06
38. Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol. (1999) 73:8469–75. doi: 10.1128/JVI.73.10.8469-8475.1999
39. Kanazawa N, Kurosaki M, Sakamoto N, Enomoto N, Itsui Y, Yamashiro T, et al. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J Virol. (2004) 78:9713–20. doi: 10.1128/JVI.78.18.9713-9720.2004
40. Kumthip K, Chusri P, Jilg N, Zhao L, Fusco DN, Zhao H, et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J Virol. (2012) 86:8581–91. doi: 10.1128/JVI.00533-12
41. Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. (2005) 128:1034–41. doi: 10.1053/j.gastro.2005.02.006
42. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. (2001) 19:623–55. doi: 10.1146/annurev.immunol.19.1.623
43. Zhang XN, Liu JX, Hu YW, Chen H, Yuan ZH. Hyper-activated IRF-1 and STAT1 contribute to enhanced interferon stimulated gene (ISG) expression by interferon alpha and gamma co-treatment in human hepatoma cells. Biochim Biophys Acta. (2006) 1759:417–25. doi: 10.1016/j.bbaexp.2006.08.003
44. Alhetheel A, Albarrag A, Shakoor Z, Alswat K, Abdo A, Al-Hamoudi W. Assessment of pro-inflammatory cytokines in sera of patients with hepatitis C virus infection before and after anti-viral therapy. J Infect Dev Ctries. (2016) 10:1093–8. doi: 10.3855/jidc.7595
45. Polyak SJ, Khabar KS, Rezeiq M, Gretch DR. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J Virol. (2001) 75:6209–11. doi: 10.1128/JVI.75.13.6209-6211.2001
46. Zhang L, Hao CQ, Miao L, Dou XG. Role of Th1/Th2 cytokines in serum on the pathogenesis of chronic hepatitis C and the outcome of interferon therapy. Genet Mol Res. (2014) 13:9747–55. doi: 10.4238/2014.November.27.2
47. Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. (2006) 26:912–9. doi: 10.1111/j.1478-3231.2006.01327.x
48. Hashimoto S, Yoda M, Yamada M, Yanai N, Kawashima T, Motoyoshi K. Macrophage colony-stimulating factor induces interleukin-8 production in human monocytes. Exp Hematol. (1996) 24:123–8.
49. Kaplanski G, Farnarier C, Kaplanski S, Porat R, Shapiro L, Bongrand P, et al. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood. (1994) 84:4242–8. doi: 10.1182/blood.V84.12.4242.bloodjournal84124242
50. Morohashi H, Miyawaki T, Nomura H, Kuno K, Murakami S, Matsushima K, et al. Expression of both types of human interleukin-8 receptors on mature neutrophils, monocytes, and natural killer cells. J Leukoc Biol. (1995) 57:180–7. doi: 10.1002/jlb.57.1.180
51. Murphy PM. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin Hematol. (1997) 34:311–8.
52. Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE. (2011) 6:e21381. doi: 10.1371/journal.pone.0021381
53. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374:1–20. doi: 10.1042/bj20030407
54. Torbati E, Ghassab RK, Davachi ND. Recombinant HCV core protein and the secretion of associated cytokines (IL-6, TNF-alpha and IFN-gamma) in immunized mice. Pak J Biol Sci. (2013) 16:2041–5. doi: 10.3923/pjbs.2013.2041.2045
55. Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol. (2006) 80:1197–213. doi: 10.1189/jlb.0506297
56. Darling JM, Wright TL. Immune responses in hepatitis C: is virus or host the problem? Curr Opin Infect Dis. (2004) 17:193–8. doi: 10.1097/00001432-200406000-00004
57. Gramenzi A, Andreone P, Loggi E, Foschi FG, Cursaro C, Margotti M, et al. Cytokine profile of peripheral blood mononuclear cells from patients with different outcomes of hepatitis C virus infection. J Viral Hepat. (2005) 12:525–30. doi: 10.1111/j.1365-2893.2005.00634.x
58. Araujo AR, Peruhype-Magalhaes V, Coelho-dos-Reis JG, Chaves LP, de Lima TA, Pimentel JP, et al. Dual role of IL-12 in the therapeutic efficacy or failure during combined PEG-Interferon-alpha2A and ribavirin therapy in patients with chronic hepatitis C. Immunol Lett. (2013) 154:61–9. doi: 10.1016/j.imlet.2013.07.010
59. Hirano T, Taga T, Nakano N, Yasukawa K, Kashiwamura S, Shimizu K, et al. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc Natl Acad Sci U S A. (1985) 82:5490–4. doi: 10.1073/pnas.82.16.5490
60. Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, Gomez E, et al. Interleukins, from 1 to 37, and interferon-gamma: receptors, functions, and roles in diseases. J Allergy Clin Immunol. (2011) 127:701–21.e1-70. doi: 10.1016/j.jaci.2010.11.050
61. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. (2004) 116:205–19. doi: 10.1016/S0092-8674(04)00046-7
62. Gupta S. Molecular mechanisms of apoptosis in the cells of the immune system in human aging. Immunol Rev. (2005) 205:114–29. doi: 10.1111/j.0105-2896.2005.00261.x
63. Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. (2007) 6:1837–49. doi: 10.4161/cc.6.15.4511
64. Bantel H, Schulze-Osthoff K. Apoptosis in hepatitis C virus infection. Cell Death Differ. (2003) 10 Suppl 1:S48–58. doi: 10.1038/sj.cdd.4401119
65. Calabrese F, Pontisso P, Pettenazzo E, Benvegnu L, Vario A, Chemello L, et al. Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology. (2000) 31:1153–9. doi: 10.1053/he.2000.7123
66. El-Domyati M, Abo-Elenin M, El-Din WH, Abdel-Wahab H, Abdel-Raouf H, El-Amawy T, et al. Expression of apoptosis regulatory markers in the skin of advanced hepatitis-C virus liver patients. Indian J Dermatol. (2012) 57:187–93. doi: 10.4103/0019-5154.96189
67. Otsuka M, Kato N, Taniguchi H, Yoshida H, Goto T, Shiratori Y, et al. Hepatitis C virus core protein inhibits apoptosis via enhanced Bcl-xL expression. Virology. (2002) 296:84–93. doi: 10.1006/viro.2002.1371
68. Alhetheel A, Albarrag A, Shakoor Z, Alswat K, Abdo A, Al-Hamoudi W, et al. Increased Spontaneous Programmed Cell Death Is Associated with Impaired Cytokine Secretion in Peripheral Blood Mononuclear Cells from Hepatitis C Virus-Positive Patients. Viral Immunol. (2017) 30:283–7. doi: 10.1089/vim.2016.0166
69. Albertoni G, Arnoni CP, Latini FR, Andrade SS, Araujo PR, Rodrigues FK, et al. Altered of apoptotic markers of both extrinsic and intrinsic pathways induced by hepatitis C virus infection in peripheral blood mononuclear cells. Virol J. (2012) 9:314. doi: 10.1186/1743-422X-9-314
70. Nakamoto Y, Kaneko S, Kobayashi K. Increased susceptibility to apoptosis and attenuated Bcl-2 expression in T lymphocytes and monocytes from patients with advanced chronic hepatitis C. J Leukoc Biol. (2002) 72:49–55. doi: 10.1189/jlb.72.1.49
71. Taya N, Torimoto Y, Shindo M, Hirai K, Hasebe C, Kohgo Y. Fas-mediated apoptosis of peripheral blood mononuclear cells in patients with hepatitis C. Br J Haematol. (2000) 110:89–97. doi: 10.1046/j.1365-2141.2000.01945.x
72. Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. (2003) 170:3369–76. doi: 10.4049/jimmunol.170.6.3369
73. Taga K, Chretien J, Cherney B, Diaz L, Brown M, Tosato G. Interleukin-10 inhibits apoptotic cell death in infectious mononucleosis T cells. J Clin Invest. (1994) 94:251–60. doi: 10.1172/JCI117315
74. Ayroldi E, Zollo O, Cannarile L, D' Adamio F, Grohmann U, Delfino DV, et al. Interleukin-6 (IL-6) prevents activation-induced cell death: IL-2-independent inhibition of Fas/fasL expression and cell death. Blood. (1998) 92:4212–9. doi: 10.1182/blood.V92.11.4212
Keywords: hepatitis C virus, peripheral blood mononuclear cells, hepatitis C virus infection, immune response, programmed cell death
Citation: Alhetheel AF (2022) Impact of Hepatitis C Virus Infection of Peripheral Blood Mononuclear Cells on the Immune System. Front. Virol. 1:810231. doi: 10.3389/fviro.2021.810231
Received: 06 November 2021; Accepted: 16 December 2021;
Published: 20 January 2022.
Edited by:
Brian Flanagan, University of Liverpool, United KingdomReviewed by:
Hussein Hassan Aly, National Institute of Infectious Diseases (NIID), JapanCopyright © 2022 Alhetheel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdulkarim Fahad Alhetheel, YWFsaGV0aGVlbEBrc3UuZWR1LnNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.