
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 26 November 2021
Sec. Translational Virology
Volume 1 - 2021 | https://doi.org/10.3389/fviro.2021.793320
 Paul G. Ayoub1
Paul G. Ayoub1 Arunima Purkayastha2Jason Quintos3Curtis Tam3
Arunima Purkayastha2Jason Quintos3Curtis Tam3 Lindsay Lathrop3Kevin Tam3Marlene Ruiz3Roger P. Hollis3Brigitte N. Gomperts2,4,5
Lindsay Lathrop3Kevin Tam3Marlene Ruiz3Roger P. Hollis3Brigitte N. Gomperts2,4,5 Donald B. Kohn1,2,3,4,5*
Donald B. Kohn1,2,3,4,5*The spike (S) glycoprotein of SARS-Cov-2 facilitates viral entry into target cells via the cell surface receptor angiotensin-converting enzyme 2 (ACE2). Third generation HIV-1 lentiviral vectors can be pseudotyped to replace the native CD4 tropic envelope protein of the virus and thereby either limit or expand the target cell population. We generated a modified S glycoprotein of SARS-Cov-2 to pseudotype lentiviral vectors which efficiently transduced ACE2-expressing cells with high specificity and contain minimal off-target transduction of ACE2 negative cells. By utilizing optimized codons, modifying the S cytoplasmic tail domain, and including a mutant form of the spike protein, we generated an expression plasmid encoding an optimized protein that produces S-pseudotyped lentiviral vectors at an infectious titer (TU/mL) 1000-fold higher than the unmodified S protein and 4 to 10-fold more specific than the widely used delta-19 S-pseudotyped lentiviral vectors. S-pseudotyped replication-defective lentiviral vectors eliminate the need for biosafety-level-3 laboratories required when developing therapeutics against SARS-CoV-2 with live infectious virus. Furthermore, S-pseudotyped vectors with high activity and specificity may be used as tools to understand the development of immunity against SARS-CoV-2, to develop assays of neutralizing antibodies and other agents that block viral binding, and to allow in vivo imaging studies of ACE2-expressing cells.
Human coronaviruses (CoV) are enveloped, positive stranded RNA viruses of the family of Coronaviridae (1). Three coronaviruses within the past two decades were transmitted from animals to humans to cause severe respiratory diseases in afflicted individuals: the 2002 severe acute respiratory syndrome coronavirus (SARS-CoV), the 2012 Middle East respiratory syndrome coronavirus (MERS-CoV), and most recently, the 2019 SARS-CoV-2 (1, 2). Although researchers have raced to develop safe and efficacious vaccines to prevent SARS-CoV-2 spread (3), the virus has resulted in over 4.25 million deaths worldwide since the inception of its pandemic spread.
The spike (S) glycoprotein mediates viral entry into target cells after engaging with the cell surface receptor angiotensin converting enzyme 2 (ACE2) (4). In addition to the prevalence of ACE2 receptors, various factors affect the potency of SARS-CoV-2 entry and transmission. One such factor includes the availability of proteases in target cells, such as the transmembrane protease serine 2 (TMPRSS2) or furin (4–6). TMPRSS2 facilitates S protein priming of SARS-Cov-2 to promote fusion of viral and cellular membranes to the target cell. Similarly, furin, a ubiquitous protease that activates a variety of viruses such as influenza A, HIV, Ebola, or measles (6), cleaves SARS-CoV-2 S protein at the S1/S2 site to facilitate infection. A viral factor affecting potency includes the D614G substitution in the spike glycoprotein, which has quickly become the most prevalent form of SARS-Cov-2. The dominance of the D614G variant is attributed to its increased viral infectivity and transmission (7).
Despite the progressive development of vaccines and therapeutics to treat SARS-CoV-2, there is a need for a safer alternative to infectious SARS-CoV-2 virus for research studies quantifying neutralizing antibody activity, developing high-throughput drug screens, or performing animal studies. By “pseudotyping” a virus, one can effectively replace the envelope protein of a virus with that of another virus to limit or broaden its targeting capabilities (8). Recent studies demonstrated that the spike glycoprotein from SARS-CoV-2 can be “pseudotyped” onto replication defective viral particles such as HIV-based lentiviral particles, murine leukemia virus (MLV)-based retroviral particles, or Vesicular Stomatitis Virus (VSV) (4, 9–14). As a result, generating S glycoprotein pseudotyped non-replicative viral particles capable of specifically infecting ACE2-expressing cells will avoid the need for a biosafety-level-3 facility and expand the capabilities for studies of SARS-CoV-2.
A major hurdle with this approach, however, stems from the low titer of S-pseudotyped vectors (15, 16). Previous groups have shown that modifications to the cytoplasmic tail (CT) domain of viruses can increase their lentiviral vector pseudotype efficiency (17–21). For example, researchers have attempted to increase the infectious titers of lentiviral vectors pseudotyped by the gibbon ape leukemia virus (GaLV) by adjusting its CT (18, 20, 22). The GaLV envelopes were modified to harbor the CT from MLV-A thereby increasing infectious titers by 25-fold. In the case of SARS-CoV-2, various researchers have demonstrated that simply deleting the final 19 or 21 amino acids of the cytoplasmic tail of SARS-CoV-2 (d19/d21) can substantially increase titers of SARS-CoV-2 pseudotyped lentiviral vectors (11–14). We describe a strategy to generate an S glycoprotein, with a CT replaced with that of the influenza hemagglutinin protein, capable of pseudotyping a non-replicative 3rd generation HIV-1 lentiviral vector with a titer 3-logs greater than its unmodified counterpart. Furthermore, the HA-tailed pseudotype not only maintains high levels of infectivity but also demonstrates a 4 to 10-fold greater specificity to ACE2-expressing cells than the d19 pseudotyped viruses.
Large airways and bronchial tissues were acquired from de-identified normal human donors after lung transplantations at the Ronald Reagan UCLA Medical Center. Tissues were procured under an Institutional Review Board-approved protocol at the David Geffen School of Medicine at UCLA, protocol no.16-000742. ABSCs were used from two biological replicates. One experiment was done using normal human bronchial epithelial cells (NHBE) from non-smokers obtained from Lonza and all samples were de-identified.
Human ABSCs were isolated following a previously published method (23–28). Briefly, airways were dissected, cleaned, and incubated in 16U/mL dispase for 30 min at room temperature. Tissues were then incubated in 0.5 mg/mL DNase for another 30 min at room temperature. Epithelium was stripped and incubated in 0.1% Trypsin-EDTA for 30 min shaking at 37°C to generate a single cell suspension. Isolated cells were passed through a 40 μm strainer and plated for Air Liquid Interface cultures.
24-well 6.5 mm transwells with 0.4 μm pore polyester membrane inserts were coated with collagen type I dissolved in cell culture grade water at a ratio of 1:10. 100 μl was added to each transwell and allowed to air dry. ABSCs were seeded at 100,000 cells per well directly onto collagen-coated transwells and allowed to grow in the submerged phase of culture for 4–5 days with 500 μL media in the basal chamber and 200 μL media in the apical chamber. ALI cultures were then established and transduced with equal amounts of p24 protein content. 72 h post transduction, cultures were harvested for IF studies and vector copy number analysis. Human ABSCs were grown in Pneumacult Ex serum-free media during the submerged and Pneumacult ALI media during the ALI phases of culture, respectively. Media was changed every other day and cultures were maintained at 37°C and 5% CO2.
ALI cultures were fixed in 4% paraformaldehyde for 15 min followed by permeabilization with 0.5% Triton-X for 10 min. Cells were then blocked using serum-free protein block (Dako X090930) for 1 h at room temperature and overnight for primary antibody incubation. After several washes of Tris-Buffered Saline and Tween-20 (TBST), secondary antibodies were incubated on samples for 1 h in darkness, washed, and mounted using Vectashield hardest mounting medium with DAPI (Vector Labs H-1500). The following antibodies were used for staining: Mouse Acetylated α-Tubulin (Cell Signaling Technology) and GFP antibody (Rockland Inc). IF images were obtained using an LSM700 or LSM880 Zeiss confocal microscope and composite images generated using ImageJ. Approximately equal numbers of cells (around 1,000 cells) were counted for each experimental group. All immunofluorescence images used for scoring cells consisted of a z-series of optical sections captured on the Zeiss LSM 700 or 880 confocal microscopes.
HEK-293T [#CRL-3216; American Type Culture Collection (ATCC), Manassass, VA], PKR -/- HEK 293T (in house)13, ACE-293T [ACE2-expressing 293T cells generously provided by Dr. Lili Yang (UCLA) and Dr. Pin Wang (USC)], VeroE6 [generously provided by Dr. Jocelyn Kim (UCLA)], and VeroE6/TMPRSS2 [#JCRB1819; JCRB Cell Bank (29)] cells were cultured in DMEM (#10-017-CV; Corning Inc., Corning, NY) supplemented with 10% fetal bovine serum (#100-106; GeminiBio, Calabasas, CA) and 1% penicillin/streptomycin/L-glutamine (#400-110; GeminiBio). Cell counts were measured with a Vi-CELL XR automated cell counter (Beckman Coulter, Indianapolis, IN).
The spike pseudotype backbone was generated by PCR amplification using the pMD.G-VSVG (30) and primers oPAF147 and oPAR43 (see Supplementary Materials). Ou (13) and IDT (9) codon optimized full-length spike glycoproteins were ordered as double stranded DNA fragments (Integrated DNA Technologies (IDT), Coralville, IA) with the inclusion of wildtype, HA (9), ALAYT (31), and MLV (20) cytoplasmic tails for downstream cloning. The IDT codon optimization was generated by inserting the Wuhan-Hu-1 SARS-CoV-2 spike glycoprotein sequence (Reference Sequence: NC_045512.2) into the IDT Codon Optimization Tool. The Ou codon optimization was taken directly from Supplementary Materials from a paper by Ou et al. and codon optimized from the SARS-CoV-2 S glycoprotein (QHU36824.1) (13). The codon optimized fragments were synthesized as double-stranded DNA fragments from IDT (See Supplementary Materials). The following constructs were generated using the New England Biosciences (NEB) Gibson Assembly workflow with double-stranded DNA fragments that replaced the VSV-G open reading frame from the pMD2.G plasmid (Addgene #12259): IDT (oPAG40 and oPAG41), IDT-ALAYT (oPAG40 and oPAG42), IDT-HA (oPAG40 and oPAG43), IDT-MLV (oPAG40 and oPAG44), Ou (oPAG45 and oPAG46), and Ou-MLV (oPAG45 and oPAG47). The cytoplasmic tail of the spike glycoprotein – the final 34 amino acids–were replaced with either the HA or the MLV tail. The HA tail (9) consists of the final 10 amino acids of the influenza A hemagglutinin protein (NGSLQCRICI) whereas the MLV tail consists of the final 30 amino acids of the murine leukemia virus (4070A) cytoplasmic tail (20). The Q5 Site-Directed Mutagenesis Kit (#E0554S, NEB, Ipswich, MA)–with primers oPAF176 and oPAR56 or oPAF177 and oPAR57 – was used to generate the D614G mutants from each respective plasmid. Ou-D614G-HA spike glycoprotein was PCR amplified and cloned from the Ou-D614G plasmid using primers oPAF184 and oPAR63. Primers oPAF228 and oPAR103 were used to remove the final 19 amino acids of the Ou-D614G cytoplasmic tail to generate Ou-D614G-d19 spike glycoprotein. The United Kingdom Alpha variant, the South African Beta variant, and the Indian Kappa and Delta variants were generated using the NEB Gibson Assembly workflow and the following double-stranded DNA fragments: Alpha (oPAG59 and oPAG60), Beta (oPAG61 and oPAG62), Kappa (oPAG67) and Delta (oPAG68). Any further mutations for Alpha, Beta, Kappa, and Delta were generated via the Q5 Site-Directed Mutagenisis Kit with primers (oPAF222, oPAF232-234, oPAR97, oPAR108-110, oPAF282-284, and oPAR127-129). Alpha pseudotypes contained the following mutations: Δ69-70, Δ144Y, N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H. Beta pseudotypes contained the following mutations: L18F, D80A, D215G, Δ241-243, R246I, K416N, E484K, N501Y, D614G, and A701V. Kappa pseudotypes contained the following mutations: L452R, E484Q, D614G, P681R, Q1071H. Delta pseudotypes contained the following mutations: T19R, delta157-158, L452R, T478K, D614G, P681R, D950N. The addition of the HA tail and the d19 tail were completed as described above. All plasmids were mini prepped using the PureLink™ Quick Plasmid Miniprep Kit (#K210010; Invitrogen, Carlsbad, CA). All plasmids were maxi prepped using NucleoBond Xtra Maxi Kit (#740414; Machery-Nagel Inc., Düren, Germany).
Spike-pseudotyped lentiviruses were packaged by transient transfection of PKR -/- 293T cells with fixed amounts of HIV Gag/Pol, Rev, and Lentiviral envelope (VSV-G or Spike) expression plasmids and equimolar amounts of either MNDU3-eGFP or roUBC-mCitrine transfer plasmid using TransIT-293 (Mirus Bio, Madison, WI) as described in the Supplementary Materials and Cooper et al. (32, 33). Viral supernatants were then directly used for titer determination or concentrated by tangential flow filtration, as described by Cooper et al., 2011. Briefly, ACE-293T, HEK-293T, VeroE6, or VeroE6/TMPRSS2 (29) cells were transduced at equal amounts of p24 protein content with either a 1:10 dilution of raw or a 1:10,000 dilution of concentrated vector. To calculate titers, we harvested cells and determined VCNs by ddPCR approximately 72 h post-transduction.
ACE-293T, HEK-293T, VeroE6, or VeroE6/TMPRSS2 cells, 1 x 105 per sample, were collected by trypsinization, centrifuged at 90g for 10 min and resuspended in 2 mL of culture medium for plating in a 6-well plate (#3516; Corning Inc.). 24 h after plating, cells were transduced with equal amounts of p24 protein content; culture medium was replaced with a 1:10 dilution of viral supernatant in 1 mL of culture medium. 24 h after transduction, culture medium was refreshed on all wells. 72 h after transduction, cells were harvested for downstream analyses. Cell counts were measured with a Vi-CELL XR automated cell counter. Cells were assayed for GFP expression with a BD LSRFortessa or BD LSRII flow cytometer (BD Biosciences, San Jose, CA) and analyzed with FlowJo (Tree Star, Ashland, OR).
Genomic DNA from transduced cells was extracted using PureLink Genomic DNA Mini Kit (K182002; Invitrogen). VCN was calculated by using the vector GFP gene (primers eGFP616F and eGFP705R; probe eGFP653Pr) and an endogenous human diploid gene control (SCD4 (Human Syndecan 4) primers oPAF-SDC4 and oPAR-SDC4; probe oPAP-SDC4) as a reference. Reaction mixtures of 22uL volume, comprising 1 × Digital droplet (dd)PCR Master Mix (#1863010; BioRad, Hercules, CA), 400 nmol/L primers and 100 nmol/L probe for each set, 40 U DraI (R0129S; NEB) and 30– 100 g of the gDNA to study, were prepared and incubated at 37°C for 1 h. Droplet generation was performed as described in Hindson et al. (10). with 20 L of each reaction mixture. The droplet emulsion was then transferred with a multichannel pipet to a 96-well twin.tec® real-time PCR Plates (Eppendorf, Hamburg, Germany), heat sealed with foil, and amplified in a conventional thermal cycler (T100 Thermal Cycler, Bio-Rad). Thermal cycling conditions consisted of 95°C 10 min, (94°C 30 s and 60°C 1 min) (55 cycles), 98°C 10 min (1 cycle) and 12°C hold. After PCR, the 96-well plate was transferred to a droplet reader (Bio-Rad). Acquisition and analysis of the ddPCR data was performed with the QuantaSoft software (Bio-Rad), provided with the droplet reader. Vector Titer (TU/mL) was calculated as TU=VCN x (cell count at day of transduction) x virus dilution.
p24 antigen concentration in vector supernatants were measured by the UCLA/CFAR (Center for AIDS Research) Virology Core using the Alliance HIV-1 p24 Antigen ELISA Kit (#NEK050, PerkinElmer, Waltham, MA), following the manufacturer's manual.
To achieve the most effective S-pseudotyped lentiviral vector, we modified the full-length SARS-CoV-2 S glycoprotein gene to generate a series of envelope expression plasmids for transfection experiments. The modifications included various codon optimizations (36), an amino acid substitution, and CT modifications (Figure 1A). As such, the S glycoprotein was codon optimized using the Integrated DNA Technologies algorithm, denoted “IDT,” or using the codon optimization described by a Ou et al. denoted “Ou” (13). We also modified the constructs to include an aspartic acid to glycine substitution at the 614th amino acid position of the S protein. This mutation reproduces the D614G SARS-CoV-2 variant that has rapidly become the dominant form around the world (7). Recent research has demonstrated that this strain exhibits increased competitive fitness and infectivity thereby increasing transduction of ACE2-expressing cells.
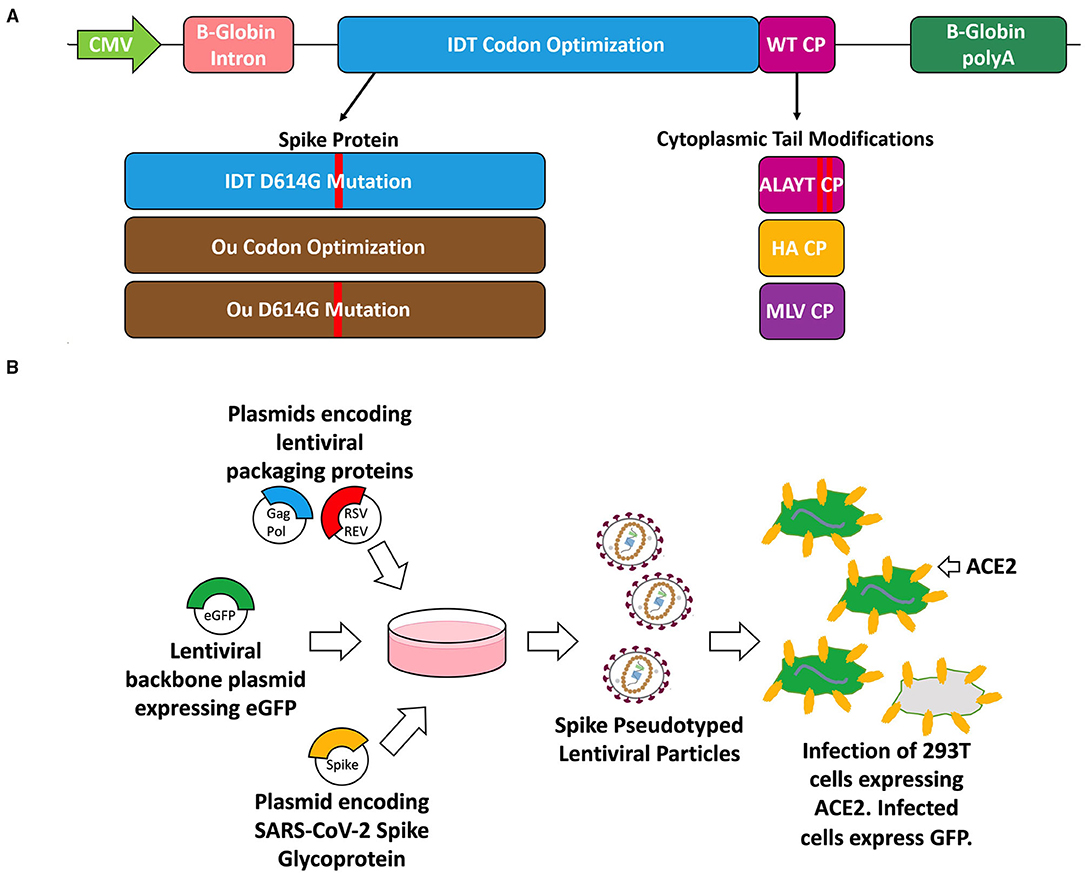
Figure 1. Schematic of the spike glycoprotein variants (A), vector packaging and transduction (B) experiments. (A) The S glycoproteins were cloned into the pMD2.G plasmid backbone (Addgene plasmid #12259) which is driven by a CMV promoter and includes a beta-globin intron and beta-globin polyA. The three variants include codon optimization (IDT or Ou), an amino acid substitution (D614G), and cytoplasmic tail modifications (wildtype tail, mutated ALAYT, influenza HA tail, and murine leukemia virus MLV tail). (B) The spike (S) glycoprotein variants were packaged in PKR-/- HEK-293T cells alongside plasmids encoding for a 3rd generation HIV-1 lentivirus and a plasmid encoding for an eGFP transgene [pCCL-MNDU3-eGFP (34, 35)].
We modified the spike CT to introduce amino acid substitutions into its C terminus or replace it with the CT of the influenza A or murine leukemia envelope glycoprotein (Figure 1A). Various groups have shown that mutating the five most C-terminal residues of the S glycoprotein eliminates the endoplasmic reticulum retention signal and improves surface expression of S-pseudovirions (9, 31). This mutant, termed “ALAYT,” replaces K1269 and H1271 with alanines (A). Additionally, we replaced the spike CT with the final 10 amino acids of the influenza A virus hemagglutinin (HA) glycoprotein (9). Given that SARS-Cov-2, influenza HA, and HIV-1 are homotrimeric class I fusion glycoproteins (1, 7, 37), we reasoned that the influenza CT may pseudotype HIV-1 lentiviral vectors more effectively than that of SARS-CoV-2. Lentiviral vectors pseudotyped with GALV, RD114, or baboon envelopes have also shown increased stability and transduction when their native CTs are replaced with the MLV cytoplasmic tail (18, 20, 22). As a result, we also replaced the spike CT with that from the murine leukemia virus (MLV) glycoprotein.
We generated the SARS-CoV-2 pseudotyped virus using a 3rd generation HIV-1 packaging system (Figure 1B). We transfected PKR -/- 293T cells with plasmids encoding for HIV-1 proteins, Gag- Pol and Rev; a plasmid encoding for the full-length spike glycoprotein envelope; and a lentiviral transfer plasmid containing an eGFP reporter cassette (33). The S glycoprotein on the produced lentivirus should improve specificity toward ACE2-expressing cells.
We first examined the effect of glycoprotein modifications on viral expression and functionality. ACE2-expressing 293T cells (ACE-293T) or parental 293T cells that do not express ACE2 (HEK-293T) were transduced with the series of lentiviral vectors. Three days post-transduction cells were harvested to quantify vector expression and infectivity. All spike-pseudotyped lentiviral vectors exhibited specificity to ACE2-expressing cells with essentially no off-target transduction of the parental HEK-293T cells that lack expression of ACE2 (Figure 2A). Furthermore, Ou codon optimized variants presented greater infectivity than their IDT counterparts. The inclusion of either the D614G mutant or the HA tail increased vector copy number (VCN) by 10-fold (Figure 2B), whereas the ALAYT amino acid modifications had no impact on pseudotyping efficiency. When combined, the D614G mutant and HA tail achieved a 50-fold synergistic effect of the percentage of cells expressing GFP (Figure 2A), a 1000-fold increase of vector copy number (Figure 2B), and a mid-107 titer (Figure 2C) −3 logs greater than its unmodified spike counterpart. In contrast, the MLV tail impeded pseudotyping efficiency. When combined with the D614G mutant, the MLV tail reduced expression and infectivity of the pseudotyped virus in comparison to the D614G mutant alone (Figures 2A,C). The S-pseudotyped vector also supports large scale concentration (Supplementary Figure 1) > 1000-fold by means of tangential flow filtration (32). Ultimately, these data demonstrate that the combination of the Ou codon optimization, the D614G mutation, and the HA cytoplasmic tail drastically increased pseudotyping efficiency of lentiviruses with the spike glycoprotein.
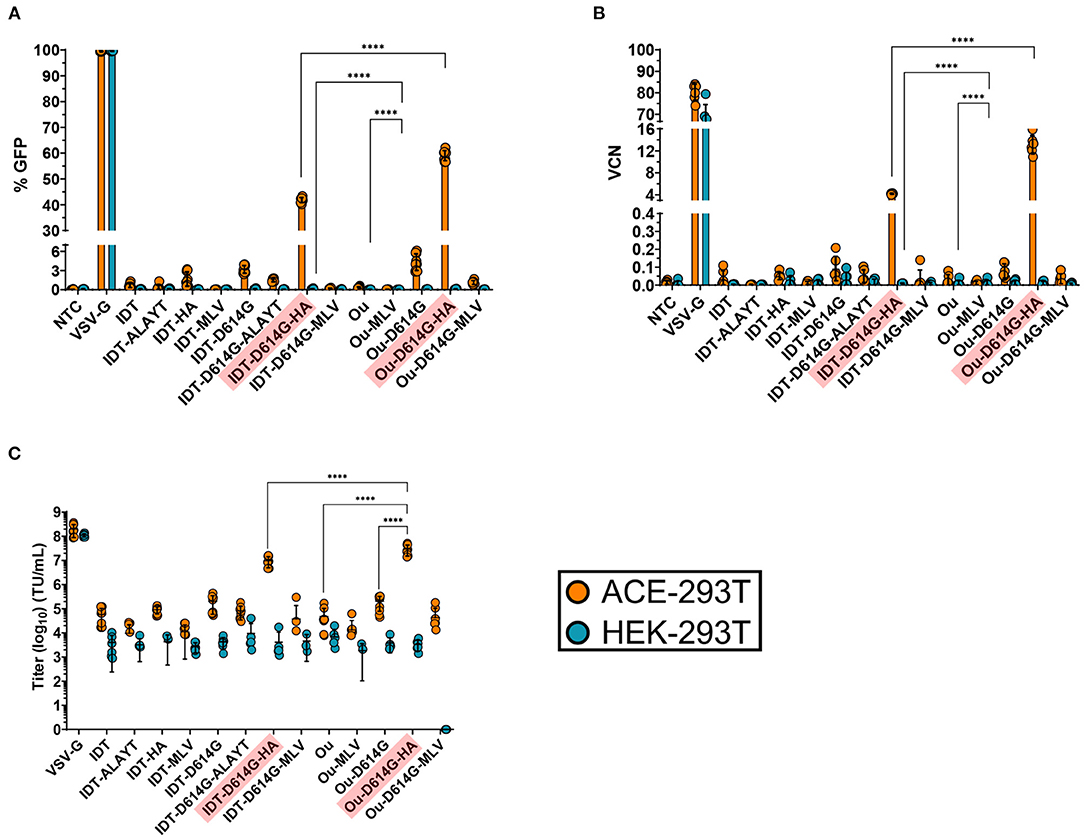
Figure 2. GFP percentage (A), vector copy number (B) and titer (C) of spike pseudotyped lentiviral vectors. Raw viral supernatants were harvested 3 days post transfection and stored at−80C for future use. (A–C) 100,000 ACE2-expressing 293T cells (ACE-293T) and HEK-293T cells were transduced with raw virus at equal amounts of p24 protein content from a series of S-pseudotyped lentiviral vectors containing an MNDU3-eGFP reporter cassette. Three days post-transduction, cells were harvested and measured for %GFP (A) via flow cytometry to assess pseudotype functionality and expression. (B,C) illustrate the infectivity of spike pseudotype vectors. To measure infectivity, genomic DNA was extracted, and vector copy number (VCN) was measured (B) by quantifying GFP integrants via QX200 Droplet Digital PCR System. The infectious titer (transducing units/mL) of each vector was determined (C) from the VCN shown in (B). All data sets were compared to a non-transduced control (NTC) and a VSV-G pseudotyped lentiviral vector containing an MNDU3-eGFP reporter cassette. Data are represented as mean ± SD of biological triplicates from n = 3 experiments. Statistical significance was analyzed using one-way ANOVA followed by multiple paired comparisons for normally distributed data (Tukey test). All statistical tests were two-tailed and a p-value of < 0.05 was deemed significant (ns non-significant, ****P < 0.0001).
Infected cells can be measured by means of the eGFP reporter driven by the MNDU3 promoter, an enhancer/synthetic promoter that contains the U3 region of the Myeloproliferative Sarcoma Virus long terminal repeat (35). It is also worth noting the reduction in transduction activity assessed by GFP expression across all spike-pseudotyped variants compared to the VSV-G control (Figure 2A). Even with a VCN of 0.1, various pseudotyped lentiviruses exhibited negligible GFP percentage (<5%) measured by flow cytometry. This reduced expression may be attributed to the MNDU3 promoter driving the GFP reporter, as it is a relatively weak expressing promoter within 293T cell lines. This is evidenced by the increase in expression when we packaged the spike-pseudotyped vectors with a GFP reporter cassette driven by the ubiquitin C promoter (Supplementary Figure 2). These UBC-mCitrine spike-pseudotyped vectors had greater GFP expression in ACE-293T cells in comparison to the MNDU3-eGFP pseudotyped variants after transduction at equal p24.
We also determined whether increasing levels of TMPRSS2 protease in target cells could affect infectivity by the S-pseudotyped lentiviral vectors. VeroE6 cells, which lack TMPRSS2 expression, or VeroE6 cells transfected to express TMPRSS2 (VeroE6/TMPRSS2 [41]) were used to titer the vectors. VeroE6 and VeroE6/TMPRSS2 cells were transduced with IDT, IDT-D614G-HA, Ou, and Ou-D614G-HA pseudotyped lentiviral vectors. Three days post-transduction, cells were harvested to quantify vector expression and infectivity. For both the IDT-D614G-HA and Ou-D614H-HA pseudotyped vectors, transduction was 5-fold higher on the VeroE6/TMPRSS2 cells, demonstrating their dependence on this protease for enhanced transduction (Figures 3A,B, Supplementary Figures 3A–C).
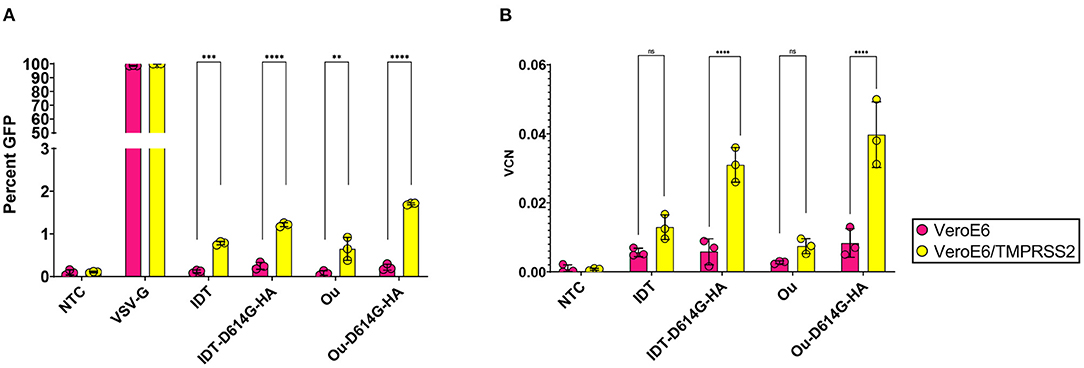
Figure 3. 100,000 VeroE6, and VeroE6/TMPRSS2 cells were transduced with raw viral supernatant containing equal amounts of p24 protein content from a series of pseudotyped lentiviral vectors containing an MNDU3-eGFP reporter cassette. (A,B) illustrate the effect of TMPRSS2 expression on spike pseudotype functionality. Three days post-transduction, cells were harvested and measured for (A) GFP percentage and (B) vector copy number (VCN). GFP percentage was measured via flow cytometry. To measure infectivity, genomic DNA was extracted to measure VCN by quantifying GFP integrants via QX200 Droplet Digital PCR System. All data sets were compared to a non-transduced control (NTC) and a VSV-G pseudotyped lentiviral vector containing an MNDU3-eGFP reporter cassette. Data are represented as mean ± SD of biological triplicates from two experiments. We analyzed statistical significance using a two-way ANOVA followed by multiple paired comparisons for normally distributed data (Tukey test). All statistical tests were two-tailed and a p-value of < 0.05 was deemed significant (ns non-significant, **P < 0.01, ***P < 0.001, ****P < 0.0001).
We next set out to compare the infectivity and specificity of the S-pseudotyped HA variant to the d19 cytoplasmic tail, the current standard for SARS-CoV-2 pseudotyping vectors (11–14). The d19 plasmid encodes for a spike envelope protein with the final 19 most C-terminal amino acid residues removed from the cytoplasmic tail. We generated two new pseudotype sets containing the Alpha and Beta SARS-CoV-2 mutations. These included the following glycoprotein envelopes: wildtype spike containing the HA or d19 tail (Ou-HA, Ou-d19), the United Kingdom spike variant (Ou-Alpha, Ou-Alpha-HA, Ou-Alpha-d19) or the South African spike variant (Ou-Beta, Ou-Beta-HA, and Ou-Beta-d19). We packaged each S-pseudotyped vector head-to-head with a GFP reporter cassette driven by a reverse oriented ubiquitin C promoter (roUBC-mCitrine) (33). ACE-293T or HEK-293T cells were transduced with the S-pseudotyped virus at equal amounts of p24 particles. Three days post-transduction, the cells were measured for their GFP percentage by flow cytometry (Figures 4A,B, Supplementary Figures 4D,E). When transducing ACE2-expressing 293Ts, Ou-Alpha-d19 and Ou-Beta-d19 variants contained a 2.5 and 1.25-fold greater GFP percentage than their HA-tail counterparts, respectively (Figure 4A, Supplementary Figure 4D). Both the HA and d19 tails produced at least 10-fold greater GFP percentages than their unmodified counterparts. When transducing non-ACE2-expressing cells, Ou-Alpha-d19 and Ou-Beta-d19 variants contained a 10-fold greater GFP percentage than their HA-tail counterparts (Figure 4B, Supplementary Figure 4E; 0.4 vs. 4% for Ou-Alpha and 0.25 vs. 3% for Ou-Beta). The infectious capability of Kappa and Delta spike-pseudotyped lentiviral vectors were also tested in ACE2 expressing and non-expressing cells (Supplementary Figure 5). Kappa-HA contained a 4-fold greater GFP percentage than Kappa-d19 in ACE2 expressing 293T cells. Further, Kappa and Delta spike-pseudotyped lentiviral vectors bearing a d19 cytoplasmic tail had 2 to 5-fold more GFP positive cells when transducing non-ACE2 expressing 293Ts (Supplementary Figure 5). These data further suggest that the HA-tail spike pseudotypes exhibit greater specificity to ACE2 expressing cells than the d19-tail spike pseudotypes.
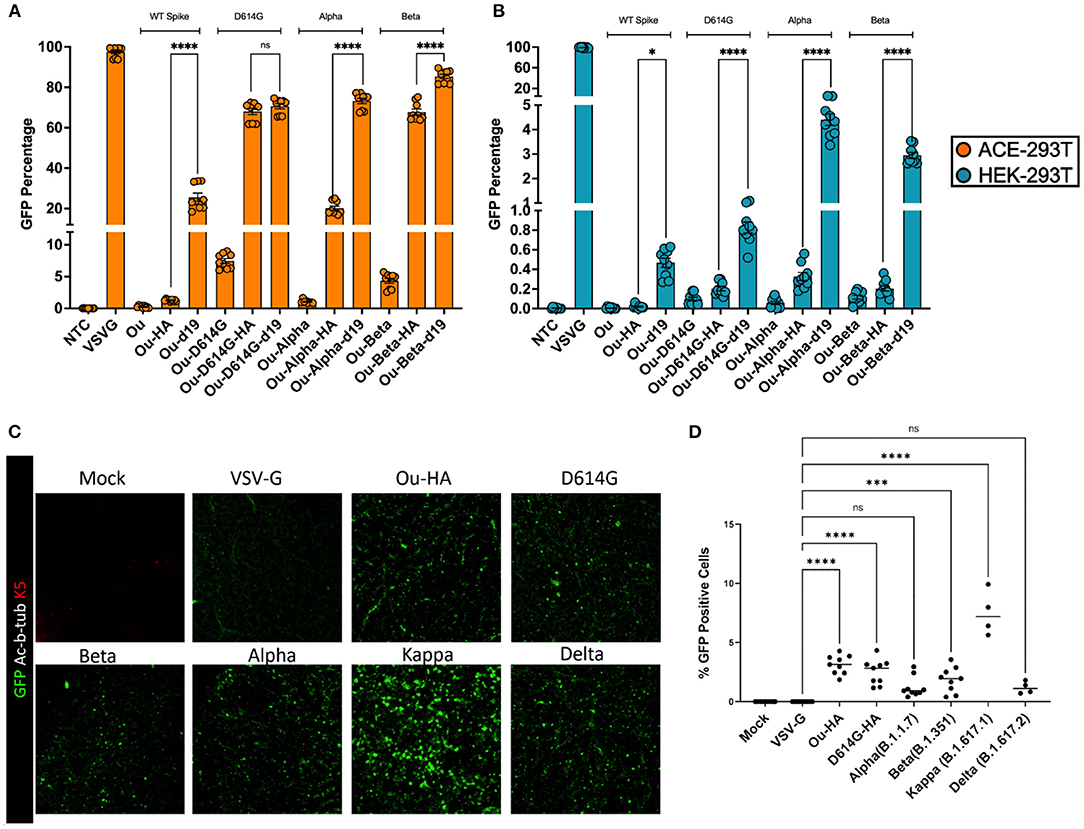
Figure 4. 100,000 ACE-293T or HEK-293T cells were transduced with raw viral supernatant containing equal amounts of p24 protein content from a series of pseudotyped lentiviral vectors containing an roUBC-mCitrine reporter cassette. Three days post-transduction, cells were harvested and measured for GFP percentage in (A) ACE2-expressing ACE-293T cells or (B) non-ACE2 expressing HEK-293T cells via flow cytometry. (C) ALI cultures were transduced with raw viral supernatant containing equal amounts of p24 protein content from a series of pseudotyped lentiviral vectors containing an roUBC-mCitrine reporter cassette. Three days post-transduction, cells were harvested and measured for immunofluorescence of GFP using confocal imaging. Nuclei were determined by DAPI staining and basal stem cells were determined by Keratin5 staining. (D) The number of infected cells in ALI cultures across the pseudotyped lentiviral vectors was quantified by counting GFP positive cells per total number of nuclei. All data sets were compared to a non-transduced control (NTC) and a VSV-G pseudotyped lentiviral vector containing an roUBC-mCitrine reporter cassette. With exception to Kappa and Delta in (C,D) (n = 1), data are represented as mean ± SEM of biological triplicates from three experiments. We analyzed statistical significance using a one-way ANOVA followed by multiple paired comparisons for normally distributed data (Tukey test). All statistical tests were two-tailed and a p-value of < 0.05 was deemed significant (ns non-significant, *P < 0.05, ***P < 0.001, ****P < 0.0001).
Finally, we investigated the ability of S-pseudotypes bearing an HA cytoplasmic tail to infect air-liquid interface (ALI) cultures derived from primary human airway basal stem cells (ABSCs). We utilized primary human ABSCs from three healthy lung transplant donors to generate the ALI cultures (23–28). The cultures were transduced with HA-tailed SARS-CoV-2 pseudotypes at equal amounts of p24 particles. Three days after infection, we examined the cultures for evidence of transduction. We validated the differentiation of the ALI cultures by immunofluorescent (IF) staining with primary antibodies for Acetylated α-Tubulin for ciliated cells for ABSCs (Supplementary Figure 6). ALI cultures were immunostained with an anti-GFP antibody and images were obtained by confocal microscopy (Figure 4C, Supplementary Figure 6). Confocal imaging demonstrated GFP expression in cells of the ALI cultures that were transduced with SARS-CoV-2 HA tail pseudotypes. However, as expected, VSV-G pseudotyped vectors failed to transduce the ALI cultures. The number of infected cells in ALI cultures across the pseudotyped lentiviral vectors was quantified by counting GFP positive cells per total number of nuclei (Figure 4D). The %GFP cells in the ALI cultures ranged from 1% (Alpha) to 7% (Kappa). No GFP positive cells were detectable in VSV-G or Mock samples. These data demonstrate that HA-tailed SARS-Cov-2 pseudotyped lentiviral vectors are capable of transduction of primary human airway cells in an epithelial barrier culture system.
Despite the continued progression of vaccines and therapeutics for the treatment of SARS-CoV-2, the methods to support high-throughput neutralization studies and drug screens remain sub-optimal. It has been shown for several envelope glycoproteins, such as those of HIV-1, GaLV, RD114, and the baboon envelope retroviral glycoprotein, that the CT domain of the envelope determines pseudotyping constraints (17–19, 21, 38). Ultimately, the CT recruits the envelope proteins for incorporation into viral particles through interactions with the Gag protein (39). As such, previous groups have attempted to pseudotype viral vectors with the S glycoprotein (4, 9, 11–15), by replacing the S cytoplasmic tail with the influenza HA cytoplasmic tail or by modifying its amino acids composition. Those pseudotyped vectors, however, failed to achieve infectious titers high enough for use in downstream assays such as in vivo studies. In this report, we incorporated these methods to generate modified S-pseudotyped lentiviral vectors capable of infecting ACE2-expressing cells at greater levels than the unmodified counterpart.
We investigated whether various CT modifications to the spike glycoprotein could affect pseudotype efficiency. We utilized previously published codon optimizations and CT modifications as well as the D614G mutant of SARS-Cov-2 (9, 13, 18, 19, 31). We showed that the influenza HA cytoplasmic tail and the D614G mutant could synergistically increase infectivity over the unmodified S counterpart and maintain the high infectivity of the widely utilized d19 pseudotype. This alternative pseudotyped lentiviral vector can provide a means for studying both the neutralizing antibodies in recovered or symptomatic patients and the potency of antibody responses from current vaccine candidates. Furthermore, the ability to concentrate the spike-pseudotyped vectors by means of tangential flow filtration (Supplementary Figure 1) should support in vivo studies and the infection of difficult to transduce cell lines and primary cells (32).
In contrast to the HA cytoplasmic tail, the murine leukemia virus (MLV) CT hampered pseudotyping efficiency. It has been shown that within the MLV cytoplasmic tail, several elements regulate the envelope's incorporation into the virion and aid with fusogenicity into host cell membranes. The C-terminus of the MLV cytoplasmic tail, known as the R peptide, includes a conserved leucine-valine dipeptide cleavage site and a tyrosine (YXXL) motif that has been implicated in promoting endocytosis of the envelope glycoprotein (39–42). These motifs contained within the MLV tail –but not the HA tail–may hinder particle formation or membrane fusion of spike-pseudotyped vectors.
Various groups have indicated the importance of the TMPRSS2 protease for S protein priming and subsequent infectivity (4, 7, 13). We demonstrated that TMPRSS2 increased the transduction of the HA tail pseudotypes suggesting that the S-pseudotyped lentiviral vectors depend on TMPRSS2 for successful transduction. As a result, they can be utilized for drug screens against SARS-CoV-2 or as models for in vivo imaging studies of ACE2 and TMPRSS2-expressing cells.
Many groups have also established that the removal of the final 19 amino acids of the spike cytoplasmic tail (d19) can significantly increase titer and infectivity of S-pseudotyped lentiviral vectors. We compared the HA tail to the d19 tail [harboring the D614G, Alpha, or Beta mutations (25–27, 43)] and demonstrated that S-pseudotyped lentiviral vectors with an HA cytoplasmic tail exhibit not only high levels of infectivity but also increased specificity over the widely used d19 S-pseudotyped vector. We speculate that the increase in specificity of the HA tail could be a result of increased binding affinity to ACE2 receptors. Previous literature indicated that although the spike proteins with the HA tail and wildtype S cytoplasmic tail are neutralized by patient sera at similar IC50s, the HA tail renders S pseudotypes more sensitive to neutralization by soluble ACE2 than the wildtype S cytoplasmic tail (9). Although it has not been further explored, an increase in binding affinity of HA-tailed S-pseudotyped vectors to ACE2 may explain both its increase in specificity and its increase in sensitivity to neutralization by soluble ACE2.
The increase in specificity of the HA S-pseudotyped vector will be important when studying the pathology of SARS-CoV-2 infection in animal models or air liquid interface cultures – including the newly dominant Delta variant (44, 45) – given it will more accurately represent the specificity to ACE2 of the SARS-CoV-2 virus. Furthermore, there is an unmet need for a lentiviral vector gene therapy to combat cystic fibrosis and other lung disorders. However, the receptors for the widely used VSV-G glycoprotein are located on the basolateral surface of epithelial cells, preventing efficient transduction of basal stem cells without disruption of the airway junctions (46). Our results demonstrated the lack of transduction of VSV-G pseudotyped lentiviral vectors in the differentiated ALI culture system. The HA tail S-pseudotyped lentiviral vectors demonstrated successful transduction of human epithelial cells within ALI cultures suggesting that ALI cultures can be utilized as tools for SARS-CoV-2 research. The lack of GFP positive cells in ALI cultures may be attributed to both donor variation and viral titer (TU/mL). Increasing the viral titer through ultracentrifugation or tangential flow filtration (Supplementary Figure 1) can increase the transduction capability of each lentivirus. It is also worth noting that the GFP positive cells in the transduced ALI cultures do not accurately depict the transmission capability of each variant, given the most transmissible variant (Delta) contained 3-fold less GFP positive cells than the less transmissible wildtype counterpart (Ou-HA). This may be due to multiple factors such as donor to donor variability, differences in cell entry mechanisms, kinetics of viral entry, expression of TMPRSS2 and furin proteases, or the requirement of other viral proteins to facilitate increased infection (47, 48) (i.e., membrane protein, or ORF1a/b accessory proteins) – all of which may be studied with the HA-pseudotypes described in this manuscript.
The HA tail S-pseudotyped lentiviral vector may provide a potential alternative to target epithelial cells expressing ACE2 and TMPRSS2 without the aberrant expression of the transgene cassette in off-target populations caused by transduction of the d19 tail. These experiments include deducing the transduction capability of single mutations within different variants (i.e., D614G, N501Y, E484K, L452R, K417N, T478K etc.) or even testing therapeutics to combat SARS-CoV-2 infection. The non-physiological expression of the truncated d19 cytoplasmic tail may also prevent the appropriate understanding of how each mutation affects the kinetics of new variants. A great example is that of the Kappa variant, where the HA-tailed pseudotype demonstrates a 3-fold greater transduction of ACE-2 expressing cells over the d19 tail (Supplementary Figure 5) and at least a 3-fold greater transduction in ALI cultures compared to other variants. This superiority in infection, however, is only a snapshot in time illustrated by the number of GFP expressing cells and not representative of viral transmission. To better understand transmission capabilities of SARS-CoV-2 variants, the HA tail S-pseudotyped vectors can be employed to study kinetics of infection without the compromised specificity of infection of the d19 tail S-pseudotypes. The SARS-CoV-2 model system described in this manuscript can help determine the physiological alterations in kinetics after mutating the receptor binding domain or even the furin cleavage site. These kinetic alterations include changes in receptor binding, enhanced cleavage of the S protein, accelerated fusion, and increased cell to cell infection. Understanding these changes, may be a causal factor for the Delta variant outcompeted previous SARS-CoV-2 variants such as D614G, Alpha, Beta, Gamma, or Kappa.
Given the non-replicative nature of pseudotyped lentiviral vectors, they have significant value in studying the biology of pathogenic viruses, such as SARS-CoV-2, due to their lower biosafety requirements. The highly infectious nature of SARS-CoV-2 requires biosafety level 3 (BSL-3) equipment within laboratories to appropriately handle and study the pathogenesis or treatment of the virus (49). By designing an efficient spike-pseudotyped HIV-1 lentiviral vector with greater potency than the unmodified S spike pseudotype, the need for BSL-3 laboratories can be avoided for many studies of SARS-CoV-2. Given the rampant, global spread of the virus, the shift from BSL-3 to BSL-2 laboratories will facilitate screening of patient's serum for neutralizing antibodies in a high-throughput fashion without risk of infection. Furthermore, this pseudotyped vector expands the capacity of research to help investigators study the effectiveness of current vaccine candidates, establish new treatments via high-throughput drug screens, examine lung pathology via infection of animal models or air-liquid interface cultures, and even explore the possible applications to gene therapy for treatment of lung diseases such as cystic fibrosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
PA, RH, and DK conceived and designed all experiments and wrote the manuscript. JQ and CT provided help on portions of the experiments. PA executed and analyzed all experiments and performed statistical analysis. AP and BG designed, performed, and analyzed all primary human ALI culture experiments and data. DK provided financial and administrative support. PA and DK approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a UCLA David Geffen School of Medicine (DGSOM) and Broad Stem Cell Research Center (BSCRC) COVID 19 Research Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Dr. Lili Yang (UCLA) and Dr. Pin Wang (USC) provided the ACE2-expressing 293T cell line (ACE-293T). Dr. Jocelyn Kim (UCLA) provided the VeroE6 cell lines. The Flow Cytometry Core of the UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, the Imaging Core of the UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research and the Virology Core of the UCLA Center for AIDS Research (CFAR) (5P30 AI028697) were used to support studies. This work was supported by UCLA David Geffen School of Medicine - Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Award Program.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2021.793320/full#supplementary-material
1. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat Rev Microbiol. (2020) 19:155–70. doi: 10.1038/s41579-020-00468-6
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. China Novel coronavirus investigating and research team, a novel coronavirus from patients with pneumonia in China 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
3. Poland GA, Ovsyannikova IG, Kennedy RB. SARS-CoV-2 immunity: review and applications to phase 3 vaccine candidates. Lancet. (2020) 396:1595–606. doi: 10.1016/S0140-6736(20)32137-1
4. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80. doi: 10.1016/j.cell.2020.02.052
5. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. (2020) 176:104742. doi: 10.1016/j.antiviral.2020.104742
6. Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulto H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life sci Alliance. (2020) 3:e202000786. doi: 10.26508/lsa.202000786
7. Yurkovetskiy L, Wang X, Pascal KE, Tomkins-Tinch C, Nyalile TP, Wang Y, et al. Structural and functional analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell. (2020) 183:739–51. doi: 10.1016/j.cell.2020.09.032
8. Verhoeyen E, Cosset FL. Surface-engineering of lentiviral vectors. J Gene Med. (2004) 6:S83–94. doi: 10.1002/jgm.494
9. Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, et al. (2020). Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses. (2020) 12:513. doi: 10.3390/v12050513
10. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. (2011) 83:8604–10. doi: 10.1021/ac202028g
11. Havranek KE, Jimenez AR, Acciani MD, Lay Mendoza MF, Reyes Ballista JM, Diaz DA, et al. SARS-CoV-2 Spike alterations enhance pseudoparticle titers and replication-competent VSV-SARS-CoV-2 virus. Viruses. (2020) 12:1465. doi: 10.3390/v12121465
12. Johnson MC, Lyddon TD, Suarez R, Salcedo B, LePique M, Graham M, et al. Optimized pseudotyping conditions for the SARS-COV-2 Spike glycoprotein. J Virol. (2020). 94:e01062. doi: 10.1128/JVI.01062-20
13. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. (2020) 11:1620. doi: 10.1038/s41467-020-15562-9
14. Schmidt F, Weisblum Y, Muecksch F, Hoffmann HH, Michailidis E, Lorenzi JCC, et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J Exp Med. (2020) 217:e20201181. doi: 10.1084/jem.20201181
15. Nie J, Li Q, Wu J, Zhao C, Hao H, Liu H, e al. Quantification of SARS-CoV-2 neutralizing antibody by a pseudotyped virus-based assay. Nat Protoc. (2020) 15:3699–715. doi: 10.1038/s41596-020-0394-5
16. Tandon R, Mitra D, Sharma P, McCandless MG, Stray SJ, Bates JT, et al. Effective screening of SARS-CoV-2 neutralizing antibodies in patient serum using lentivirus particleS-pseudotyped with SARS-CoV-2 spike glycoprotein. Sci Rep. (2020) 10:19076. doi: 10.1038/s41598-020-76135-w
17. Christodoulopoulos I, Cannon PM. Sequences in the cytoplasmic tail of the gibbon ape leukemia virus envelope protein that prevent its incorporation into lentivirus vectors. J Virol. (2001) 75:4129–38. doi: 10.1128/JVI.75.9.4129-4138.2001
18. Girard-Gagnepain A, Amirache F, Costa C, Lévy C, Frecha C, Fusil F, et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood. (2014) 124:1221–31. doi: 10.1182/blood-2014-02-558163
19. Sandrin V, Cosset FL. Intracellular versus cell surface assembly of retroviral pseudotypes is determined by the cellular localization of the viral glycoprotein, its capacity to interact with Gag, and the expression of the Nef protein. J Biol Chem. (2006) 281:528–42. doi: 10.1074/jbc.M506070200
20. Sandrin V, Boson B, Salmon P, Gay W, Nègre D, Le Grand R, et al. Lentiviral vectorS-pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. (2002) 100:823–32. doi: 10.1182/blood-2001-11-0042
21. Schnierle BS, Stitz J, Bosch V, Nocken F, Merget-Millitzer H, Engelstädter M, et al. Pseudotyping of murine leukemia virus with the envelope glycoproteins of HIV generates a retroviral vector with specificity of infection for CD4-expressing cells. Proc Natl Acad Sci U S A. (1997) 94:8640–5. doi: 10.1073/pnas.94.16.8640
22. Tomás HA, Mestre DA, Rodrigues AF, Guerreiro MR, Carrondo MJT, Coroadinha AS, et al. Improved GaLV-TR Glycoproteins to Pseudotype Lentiviral Vectors: Impact of Viral Protease Activity in the Production of LV Pseudotypes. Mol Ther Methods Clin Dev. (2019) 15:1–8. doi: 10.1016/j.omtm.2019.08.001
23. Gruenert DC, Finkbeiner WE, Widdicombe JH. Culture and transformation of human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. (1995) 268:L347–60. doi: 10.1152/ajplung.1995.268.3.l347
24. Purkayastha A, Sen C, Garcia G Jr, Langerman J, Shia DW, Meneses LK, et al. Direct exposure to SARS-CoV-2 and cigarette smoke increases infection severity and alters the stem cell-derived airway repair response. Cell Stem Cell. (2020) 27:869–75.e4. doi: 10.1016/j.stem.2020.11.010
25. Hegab AE, Ha VL, Darmawan DO, Gilbert JL, Ooi AT, Attiga YS, et al. Isolation and in vitro characterization of basal and submucosal gland duct stem/progenitor cells from human proximal airways. Stem Cells Transl Med. (2012) 1:719-24. doi: 10.5966/sctm.2012-0056
26. Hegab AE, Luan Ha V, Attiga YS, Nickerson DW, Gomperts BN. Isolation of basal cells and submucosal gland duct cells from mouse trachea. J Vis Exp. (2012) 67. doi: 10.3791/3731
27. Hegab AE, Ha VL, Bisht B, Darmawan DO, Ooi AT, Zhang KX, et al. Aldehyde dehydrogenase activity enriches for proximal airway basal stem cells and promotes their proliferation. Stem Cells Dev. (2014) 23:664–75. doi: 10.1089/scd.2013.0295
28. Paul MK, Bisht B, Darmawan DO, Chiou R, Ha VL, Wallace WD, et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent notch signaling. Cell Stem Cell. (2014) 15:199–214. doi: 10.1016/j.stem.2014.05.009
29. Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A. (2020) 117:7001–3. doi: 10.1073/pnas.2002589117
30. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. (1996) 272:263–7. doi: 10.1126/science.272.5259.263
31. McBride CE, Li J, Machamer CE. The cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds COPI and promotes interaction with membrane protein. J Virol. (2006) 81:2418–28. doi: 10.1128/JVI.02146-06
32. Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, Hollis RP, et al. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J Virol Methods. (2011) 177:1–9. doi: 10.1016/j.jviromet.2011.06.019
33. Han J, Tam K, Ma F, Tam C, Aleshe B, Wang X, et al. β-Globin Lentiviral vectors have reduced titers due to incomplete vector RNA genomes and lowered virion production. Stem Cell Reports. (2021) 16:198–211. doi: 10.1016/j.stemcr.2020.10.007
34. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Tron D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. (1998) 72:8463–71. doi: 10.1128/JVI.72.11.8463-8471.1998
35. Logan AC, Nightingale SJ, Haas DL, Cho GJ, Pepper KA, Kohn DB. Factors influencing the titer and infectivity of lentiviral vectors. Hum Gene Ther. (2004) 15:976–88. doi: 10.1089/hum.2004.15.976
36. Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. (2014) 20:604–13. doi: 10.1016/j.molmed.2014.09.003
37. Du R, Cui Q, Rong L. Competitive cooperation of hemagglutinin and neuraminidase during influenza a virus entry. Viruses. (2019) 11:458. doi: 10.3390/v11050458
38. Stitz J, Buchholz CJ, Engelstadter M, Uckert W, Bloemer U, Schmitt I, et al. Lentiviral vectorS-pseudotyped with envelope glycoproteins derived from gibbon ape leukemia virus and murine leukemia virus 10A1. Virology. (2000) 273:16–20. doi: 10.1006/viro.2000.0394
39. Duvergé A, Negroni M. Pseudotyping lentiviral vectors: when the clothes make the virus. Viruses. (2020) 12:1311. doi: 10.3390/v12111311
40. Blot V, Lopez-Vergès S, Breton M, Pique C, Berlioz-Torrent C, Grange MP. The conserved dileucine- and tyrosine-based motifs in MLV and MPMV envelope glycoproteins are both important to regulate a common Env intracellular trafficking. Retrovirology. (2006) 3:62. doi: 10.1186/1742-4690-3-62
41. Kubo Y, Tominaga C, Yoshii H, Kamiyama H, Mitani C, Amanuma H, et al. Characterization of R peptide of murine leukemia virus envelope glycoproteins in syncytium formation and entry. Arch Virol. (2007) 152:2169–82. doi: 10.1007/s00705-007-1054-6
42. Löving R, Li K, Wallin M, Sjöberg M, Garoff H. R-Peptide cleavage potentiates fusion-controlling isomerization of the intersubunit disulfide in Moloney murine leukemia virus Env. J Virol. (2008) 82:2594–7. doi: 10.1128/JVI.02039-07
43. Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, et al. SARS-CoV-2 501YV2 escapes neutralization by South African COVID-19 donor plasma. Nat Med. (2021) 27:622–5. doi: 10.1038/s41591-021-01285-x
44. Callaway E. Delta coronavirus variant: scientists brace for impact. Nature. (2021) 595:17–8. doi: 10.1038/d41586-021-01696-3
45. Bolze A, Cirulli ET, Luo S, White S, Wyman D, Dei Rossi A, et al. (2021). Rapid Displacement of SARS-CoV-2 Variant B.1.1.7 by B.1.617.2 and P.1 in the United States. Cold Spring Harbor Laboratory. doi: 10.1101/2021.06.20.21259195
46. Marquez Loza LI, Cooney AL, Dong Q, Randak CO, Rivella S, Sinn PL, et al. Increased CFTR expression and function from an optimized lentiviral vector for cystic fibrosis gene therapy. Mol Ther Methods Clin Dev. (2021) 21:94–106. doi: 10.1016/j.omtm.2021.02.020
47. Yadav R, Chaudhary JK, Jain N, Chaudhary PK, Khanra S, Dhamija P, et al. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. (2021) 10:821. doi: 10.3390/cells10040821
48. Satarker S, Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res. (2020) 51:482–91. doi: 10.1016/j.arcmed.2020.05.012
49. Bain W, Lee JS, Watson AM, Stitt-Fischer MS. Practical guidelines for collection, manipulation and inactivation of SARS-CoV-2 and COVID-19 clinical specimens. Curr Protoc Cytom. (2020) 93:e77. doi: 10.1002/cpcy.77
Resource Identification Initiative
Life Science Identifiers
Keywords: SARS-CoV-2, pseudotype, COVID-19, lentivirus, air-liquid interface
Citation: Ayoub PG, Purkayastha A, Quintos J, Tam C, Lathrop L, Tam K, Ruiz M, Hollis RP, Gomperts BN and Kohn DB (2021) Improved SARS-CoV-2 Spike Glycoproteins for Pseudotyping Lentiviral Vectors. Front. Virol. 1:793320. doi: 10.3389/fviro.2021.793320
Received: 11 October 2021; Accepted: 08 November 2021;
Published: 26 November 2021.
Edited by:
Reingard Grabherr, University of Natural Resources and Life Sciences, AustriaReviewed by:
Miriam Klausberger, University of Natural Resources and Life Sciences Vienna, AustriaCopyright © 2021 Ayoub, Purkayastha, Quintos, Tam, Lathrop, Tam, Ruiz, Hollis, Gomperts and Kohn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald B. Kohn, ZGtvaG4xQG1lZG5ldC51Y2xhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.