
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 07 February 2025
Sec. Veterinary Epidemiology and Economics
Volume 12 - 2025 | https://doi.org/10.3389/fvets.2025.1523628
 Sinegugu Kholeka Mhlophe1
Sinegugu Kholeka Mhlophe1 Charlene Clarke1
Charlene Clarke1 Giovanni Ghielmetti1,2
Giovanni Ghielmetti1,2 Megan Matthews1
Megan Matthews1 Tanya Jane Kerr1
Tanya Jane Kerr1 Michele Ann Miller1†
Michele Ann Miller1† Wynand Johan Goosen1,3*†
Wynand Johan Goosen1,3*†African buffaloes (Syncerus caffer) are wildlife maintenance hosts of Mycobacterium bovis (M. bovis), the causative agent of animal tuberculosis (aTB) in multiple ecosystems across South Africa. In addition to their role as keystone species, these animals are vital to South Africa’s economy as a highly valuable species. Controlling aTB in South Africa relies on mycobacterial culture as the gold standard for M. bovis confirmation, with the single intradermal comparative cervical test (SICCT) and Bovigam™ assays as validated cell-mediated immunological assays for detection. However, these methods are not without their shortfalls, with a suboptimal ability to discern true positive results amidst certain non-tuberculous mycobacteria (NTM) interference. This study employed a culture-independent approach using oronasal swabs collected from African buffaloes (n = 19), originating from three herds with no recorded history of M. bovis infection, to elucidate the possible cause of observed discordant immunological aTB test results. The DNA was extracted directly from the oronasal swabs, amplified using Mycobacterium genus-specific PCRs, then amplicons were pooled and sequenced using Oxford Nanopore Technologies (ONT) long-read platform. Mycobacterium tuberculosis complex DNA, along with various NTM species, were identified in 8/19 samples. The methods described support a more robust interrogation of the buffalo oronasal mycobacteriome. These findings highlight the value of accurately distinguishing between mycobacterial species in complex samples, especially in high-value animals, to facilitate accurate interpretation of immunological test results and management of aTB.
African buffaloes (Syncerus caffer) are well-known wildlife maintenance hosts for Mycobacterium bovis (M. bovis) in various national parks and game reserves across southern Africa (1, 2). In South Africa (SA), these include the Hluhluwe-iMfolozi Park (HiP) and Kruger National Park (KNP), where M. bovis is endemic (3). African buffaloes provide vital ecological functions as keystone species in these ecosystems (4–6). Additionally, they are important for ecotourism, hunting, and game sales, which are significant economic contributors to the wildlife industry in SA (4, 7). However, the presence of M. bovis in buffaloes can result in the quarantine of farms or parks, with restrictions on movement, trade, and potential culling of genetically valuable individuals (4). Early detection and control of M. bovis infections in African buffaloes can prevent its introduction through translocation, reduce inter- and intra-species transmission, and reduce economic loss associated with animal tuberculosis (aTB).
Although antemortem methods for detecting M. bovis infection have improved, mycobacterial culture and speciation, usually from postmortem tissue samples, continues to be the gold standard for diagnosing infection (8). Positive cultures require further downstream genetic speciation, commonly using PCR-based methods targeting areas such as region-of-difference (RD) or insertion elements for Mycobacterium tuberculosis complex (MTBC) identification (8, 9). However, in the absence of tissue samples, antemortem samples, such as oronasal, respiratory, or fecal samples, are often used for mycobacterial culture, especially for diagnosing high-value individuals.
Respiratory samples are typically used for antemortem direct detection of MTBC (8, 10, 11). Oronasal swabs are easier to collect, less invasive and inexpensive when compared to obtaining respiratory samples using bronchioalveolar lavage (12). However, samples from the oronasal cavity often contain a variety of microbial contaminants, including environmental non-tuberculous mycobacteria (NTM), and results may represent shedding from an infected animal as well as oronasal colonization (9, 13). Although culture can detect viable M. bovis within 6–8 weeks, rapidly growing mycobacteria often outcompete slow-growing organisms in vitro for nutrients and oxygen, subsequently impeding detection of MTBC in paucibacillary samples, leading to false-negative culture results (14, 15). Consequently, the conventional culture approach may not accurately identify all mycobacteria present in complex samples, especially in samples with low MTBC abundance and heterogeneous mycobacterial populations. Thus, accurate characterization of MTBC and NTMs in respiratory samples is crucial for a correct diagnosis of aTB (15–17). This will require improved detection and characterization techniques to identify MTBC in paucibacillary complex samples.
Conventional mycobacterial culture, followed by PCR and amplicon sequencing, have been reported for speciating mycobacteria, although there are still limitations using this approach (16–18). Recently, a culture-independent approach, using Oxford Nanopore Technologies (ONT) targeted next generation sequencing (tNGS) of DNA extracted from bronchoalveolar lavage fluid (BALF), has shown superior sensitivity for MTBC detection compared to culture (10). This method has also been used with M. bovis infected buffalo tissue, with ONT tNGS results consistent with results from culture (17). These promising results suggest that ONT tNGS could be used in time-sensitive cases or for a more detailed characterization of mycobacterial species and strains in clinical samples. Therefore, this study aimed to characterize the oronasal mycobacteriome of African buffalo culture-independently using ONT tNGS, comparing results to previously determined mycobacterial culture outcomes, to assess its potential as a complementary technique to identify MTBC, and NTMs that may lead to discordant immunological test results.
Ethical approval for African buffalo sampling was obtained from Stellenbosch University’s (SU) Animal Care and Use Committee (ACU-2019-9081 and ACU-2019-9086). Permission for animal research was granted by the South African Department of Agriculture, Land Reform, and Rural Development (DALRRD) under Section 20 of the Animal Diseases Act (12/11/1/7/2).
Two oronasal swabs were collected from each of 120 buffalo, as part of a previous study (19), to characterize the diversity of NTMs present in four African buffalo herds in South Africa with no previous herd history of M. bovis infection. In addition, blood was collected for antigen-specific cytokine release assays. Despite the herds’ TB-free status, several buffalo from three herds (Figure 1) showed immunological responses to mycobacterial antigens, based on either the Single Intradermal Comparative Cervical Tuberculin (SICCT) test, QuantiFERON®-TB Gold Plus (QFT) bovine interferon gamma (IFN-γ), or QFT bovine interferon gamma-induced protein 10 (1P-10) release assays. A subset of these individuals (n = 19) was selected to investigate the presence of MTBC and NTM associated with the immune sensitization responses (19). Briefly, mycobacterial culture as well as direct DNA extraction of oronasal swabs were performed, followed by heat shock protein of 65 kDa (hsp65) and partial RNA polymerase beta subunit (rpoB) PCR amplicon Sanger sequencing (19). Sequences with ≥90% sequence identity match were used to identify NTM species (in the absence of MTBC) in 19 oronasal swab samples (19). The oronasal swabs were further evaluated using culture-independent ONT tNGS to identify and determine the relative abundance of mycobacterial species for comparison with the results of the previous study (19). Postmortem samples were not available for the study cohort. For the current study, the previous swab culture Sanger sequences for hsp65 and rpoB were re-analyzed to apply stricter coverage and percentage identity thresholds, as described below. This was performed to ensure that the results obtained were comparable to those obtained with ONT tNGS.

Figure 1. Map of South Africa with balloons showing the locations where African buffalo (Syncerus caffer) herds were sampled for this study. Oronasal swabs were collected from immobilized animals, located in the Northern Cape, North-West, and Limpopo provinces, in wildlife reserves where Mycobacterium bovis (M. bovis) infection had not been previously detected. Image created with National Geographic MapMaker (arcgis.com).
Oronasal swab samples from African buffalo (n = 19) were subjected to DNA extraction using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany), as previously described (20). Initial screening for Mycobacterium spp. DNA was accomplished using PCR amplification of the hsp65 and rpoB genes (Table 1), as previously described (19). Briefly, each PCR reaction had a total volume of 25 μL and contained 12.5 μL of Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs, Ipswich, MA, USA), 1 μL of forward primer and 1 μL of reverse primer at 10 μM, 2 μL of extracted DNA, and 8.5 μL of nuclease-free water (Thermo Fisher Scientific™, Waltham, MA, USA). Nuclease-free water and M. bovis DNA were used as the negative and positive controls, respectively. The thermal cycling program included an initial denaturation step at 98°C for 15 min. This was followed by 35 cycles, each consisting of denaturation at 98°C for 30s, annealing at 62.5°C (for hsp65) or 64°C (for rpoB) for 30s, and extension at 72°C for 1 min. The final reaction step was performed at 72°C for 5 min. The presence of amplicons was confirmed by visualization on a 1% agarose gel, and Sanger sequencing was performed at Stellenbosch University’s Central Analytical Facility (CAF). Sanger sequences were assembled into contiguous sequences (contigs) using ApE-A Plasmid Editor version 3.1.5 (21). The resulting contigs were aligned with Mycobacterium spp. sequences, using the NCBI Nucleotide Basic Local Alignment Search Tool (BLASTn) (47). Genus was assigned to samples that had Sanger sequences with ≥90% similarity to the reference sequence, with a minimum sequence coverage of 90%. The most likely mycobacterial species identity for each sequence was selected based on ≥99% similarity to the reference sequence, with a minimum coverage of 90%. The same criteria were applied with other genera identified.
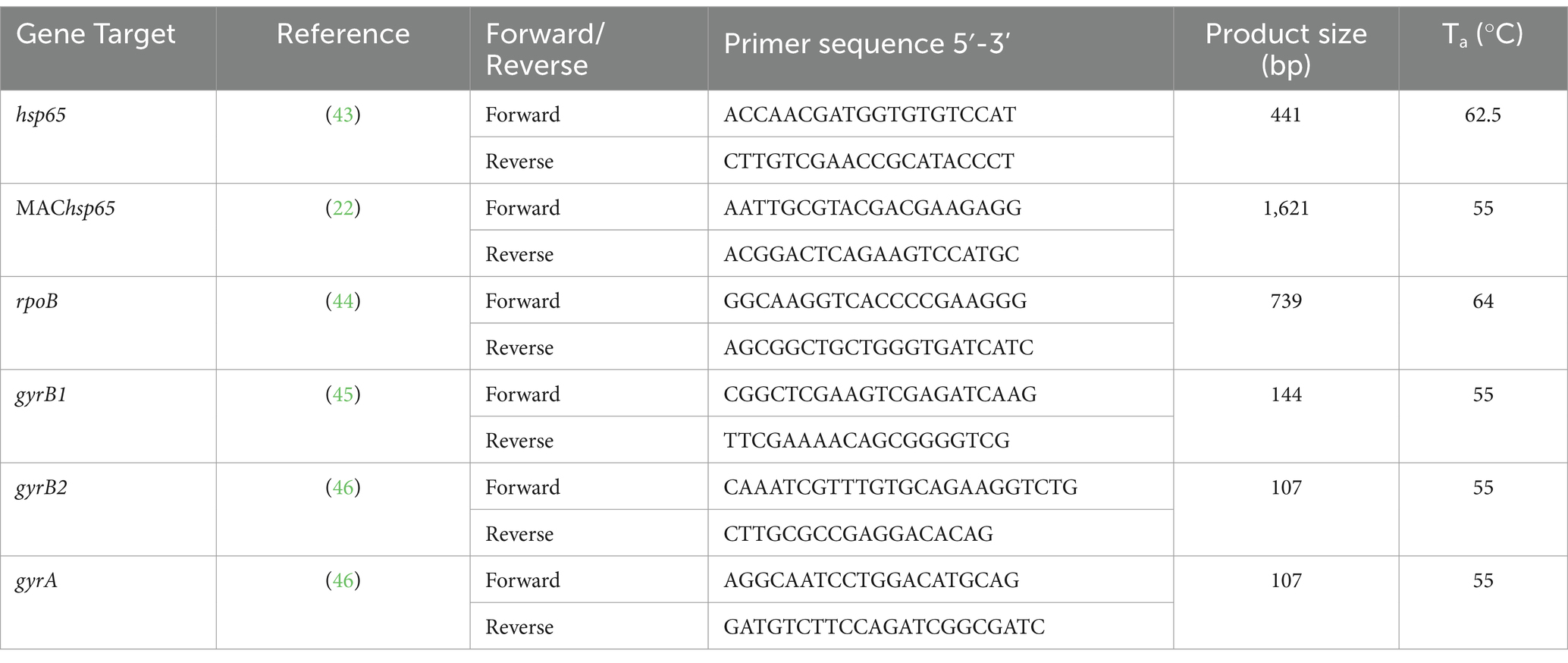
Table 1. Oligonucleotide primer sequences (Integrated DNA Technologies, Coralville, IA, USA), resulting amplicon size for Mycobacterium spp. genes, and annealing temperatures for PCR amplification of DNA extracted directly from African buffalo (Syncerus caffer) oronasal swabs.
Additional mycobacterial gene targets (Table 1) were selected, and swab DNA underwent ONT tNGS. Mycobacterium avium complex (MAC) hsp65 (MAChsp65) was used to detect the presence of MAC and other NTM (22). The target DNA gyrase subunits A and B (gyrA, gyrB1, and gyrB2) were amplified to confirm the presence of M. bovis and visualized on a 1% agarose gel, as described by Ghielmetti et al. (17). The DNA concentration of PCR amplicons for all amplified targets in each sample was quantified with the Qubit 1x dsDNA High Sensitivity (HS) Assay Kit (Thermo Fisher Scientific™). Equal amounts of 300 femtomoles of each target were pooled and prepared as a single barcoded library, using the ONT Native Barcoding Kit 96 v14 (ONT, Oxford Science Park, Oxford, UK), according to the manufacturer’s instructions. The pooled library was loaded onto primed flow cells, either R10.4.1 or Flongle (ONT), each with >1,400 and > 60 active pores, respectively. Each barcode represented a sample from one buffalo, which allowed simultaneous multigenetic sequencing using the ONT MinION Mk1C device (ONT).
For the targeted amplicon sequencing datasets, base-calling, demultiplexing, and trimming of the barcodes were performed in real time using Guppy [v6.4.6] (260 bps, high accuracy) (23). Data acquisition and base-calling were stopped after 16 h for the hsp65, rpoB, and MAChsp65 runs, using an R10.4.1 flow cell (FLO-MIN114, ONT), and after 17 h for the gyrA and gyrB runs, using a Flongle flow cell (FLO-FLG114, ONT) (Supplementary material S1). Quality control, filtering, and summary reports for Nanopore reads were generated using Nanoq v0.10.0 (24), and reads with a Q score of <12 were discarded. Following quality control checks, reference-free read sorting, based on similarity and length, was performed using Amplicon sorter (v2023-06-19) (25). A total of 200,000 randomly chosen reads with minimum and maximum lengths of 300 and 2,000 bp, respectively, were selected for each barcode generated using three target amplicons (hsp65, rpoB, and MAChsp65). Finally, ABRicate and custom databases were used to screen consensus sequences and summarize the report files, as previously described (20).
Reads of gyrA and gyrB amplicons were selected based on a minimum length of 50 bp to a maximum of 200 bp. Consensus sequences were then generated for each identified target gene and bacterial genus/species. Relative abundance was determined by analyzing a representative pool of reads. Sequence comparison with a custom database, generated with gyrA and gyrB sequences for all MTBC members, was performed as described above. For all bioinformatics tools, default settings were used unless stated otherwise.
The ONT sequencing results were interpreted based on the same criteria used for Sanger sequences: only sequences with ≥90% coverage were considered for further analysis. Sequences with a percentage identity match <90% were considered unclassified. Sequences with a percentage identity match between 90 and 99% were reported at the genus level. Sequences with a percentage identity match of ≥99% were reported as the species listed in the database. The resulting high-quality sequenced reads were reported as described above. The relative abundance of reads for each identified genus/species was graphically summarized, where the number of reads was normalized by converting the values into percentages. Samples with MTBC DNA were further investigated to detect M. bovis DNA using RD4 PCR, as previously described (26).
Sanger sequencing of hsp65 and rpoB PCR amplicons generated from DNA extracted directly from oronasal swabs or swab cultures (19) identified 13 and 12 out of 19 samples, respectively, as positive for Mycobacterium spp. at the genus level (90–99% identity match). When using a ≥ 99% identity match to assign species directly from the oronasal swabs or swab cultures, 5 and 2 out of 19 samples, respectively, could be speciated, with all species identified as M. avium (Supplementary materials S3). Bacteria other than Mycobacterium spp. with ≥99% identity match from both culture and swabs included Cellulomonas spp. (n = 2), Rhodococcus spp. (n = 1), and Cellulosimicrobium spp. (n = 1). No MTBC DNA was identified in any of the samples by PCR amplicon Sanger sequencing of the swab cultures, nor directly from the oronasal swabs.
Sequencing reads with Q scores >12 ranging from 14,310 to 262,898 per sample [mean (M) = 110,900; standard deviation (SD) = 79,866] were generated (Supplementary materials S1, S2). The number of bases ranged from 4,376,240 to 210,600,527 [M = 66,579,718; SD = 57,957,100]. The median read length of the longest contigs (N50 read length) spanned 268–881 reads. The Phred score (indicative of the average read quality) was between 13.1 and 14.8 per sample. The distribution of the number of reads per PCR target is presented in Table 2. Twelve oronasal swab samples contained Mycobacterium spp. (identity match ≥99%), based on hsp65 and rpoB ONT tNGS (Supplementary materials S3).
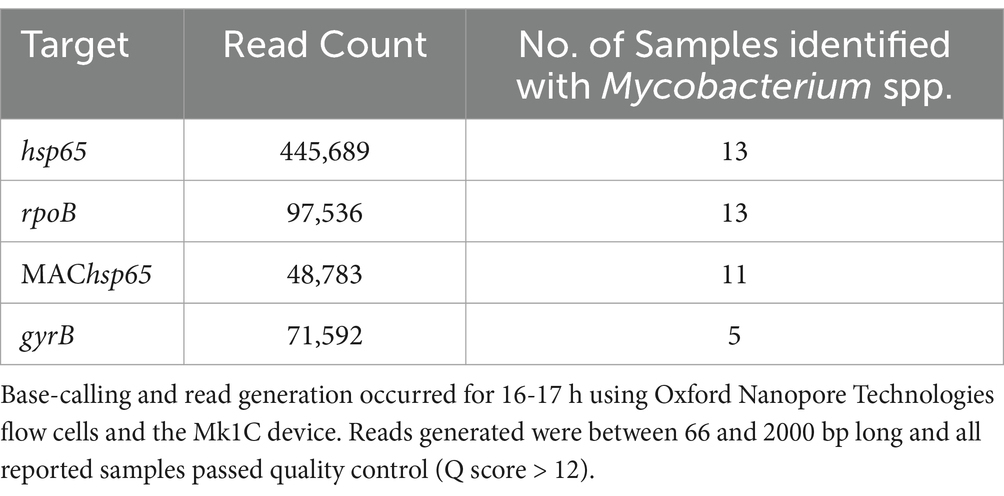
Table 2. Distribution of read counts per PCR amplification target used to identify mycobacteria in DNA extracted from African buffalo (Syncerus caffer) oronasal swabs (n = 19), based on sequence coverage and identity match at ≥90%.
The ONT tNGS results, using hsp65, rpoB, MAChsp65, and gyrB amplicon sequences with ≥99% identity match, showed that 17/19 samples contained Mycobacterium spp. DNA; 13 were identified using hsp65 and rpoB, 11 with MAChsp65, and 5 with gyrB (Table 2). Figure 2 shows the relative abundance of hsp65 and rpoB amplicon reads sequenced from 15 out of 19 DNA samples extracted from African buffalo (Syncerus caffer) oronasal swabs, using the MinION Mk1C device (ONT). Four DNA samples failed to generate amplicons from hsp65 and rpoB PCRs and were excluded from this figure. Based on the 15 hsp65 and rpoB amplicon sequences, 3 samples were identified as containing MTBC DNA (Figure 2). An additional five samples were confirmed to contain M. bovis DNA, based on gyrB sequencing, bringing the total to eight samples with MTBC DNA (Figure 3; Supplementary material S3). Speciation by RD4 PCR (based on 268 bp band on gel electrophoresis) confirmed an additional sample contained M. bovis DNA (identity assigned as MTBC by tNGS) (Figure 3; Table 3). Interestingly, DNA from 4 of the 5 gyrB positive samples could not be amplified using either hsp65, rpoB, or MAChsp65. Therefore, based on multiple amplicon sequences, a total of 8 out of 19 samples that had DNA extracted directly from oronasal swabs contained MTBC DNA, with 6 speciated to M. bovis (Figure 3).
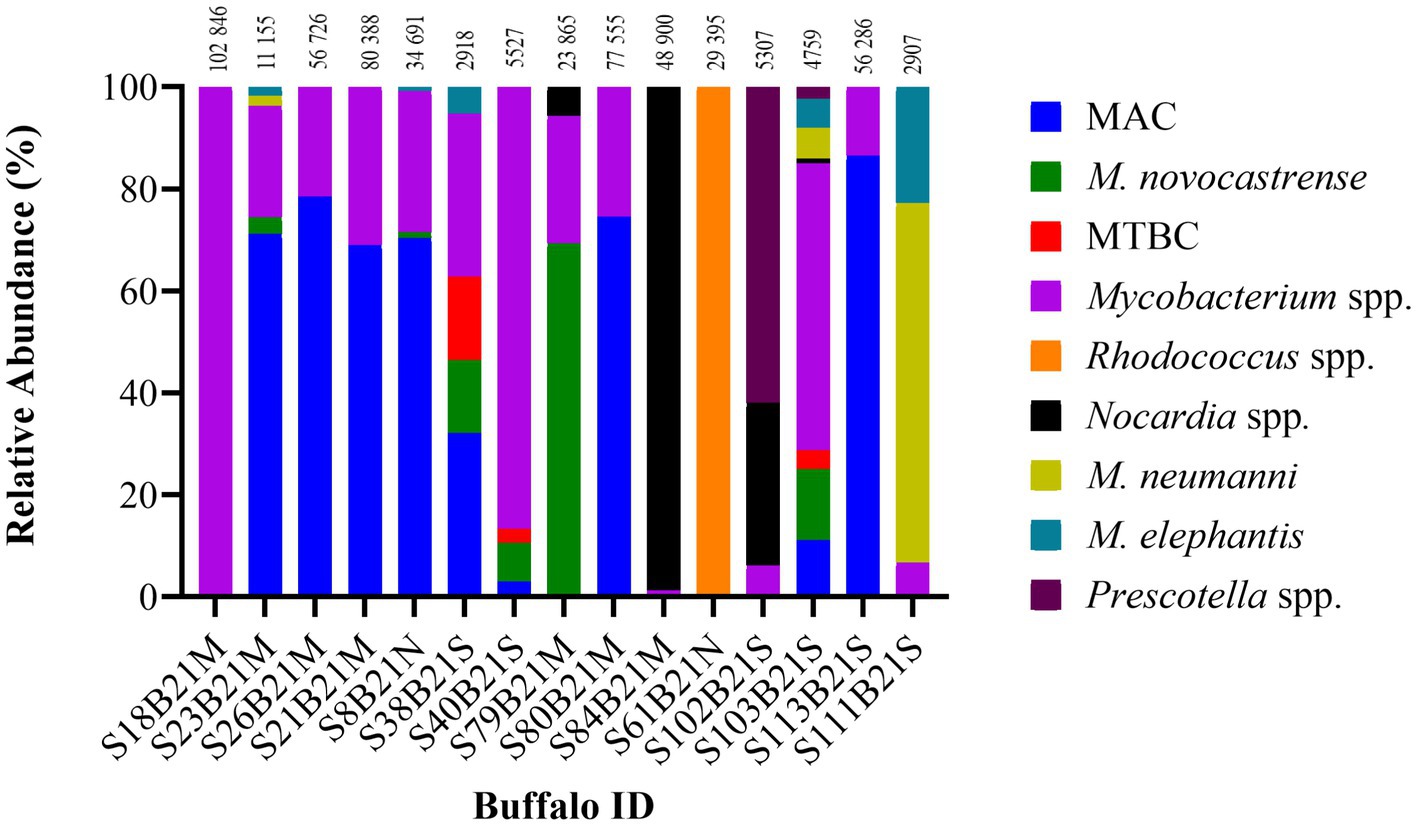
Figure 2. Distribution of the relative abundance of hsp65 and rpoB amplicon reads sequenced from 15 out of 19 DNA samples extracted from African buffalo (Syncerus caffer) oronasal swabs using the MinION Mk1C device (ONT). Four DNA samples failed to generate amplicons from hsp65 and rpoB PCRs and were excluded from the figure. Relative read abundance was based on sequences with a ≥ 90% identity match to reference sequences, with the number above each bar representing the total number of reads assigned to each sample at this threshold. The MTBC DNA in sample S40B21S was identified as M. bovis by RD4 PCR; the remaining two MTBC samples (S38B21S, S103B21S) could not be speciated. Reference-free sorting and assembly of consensus sequences were performed using a maximum of 200,000 randomly selected reads (> Q12) for each sample. Reads that could not be assigned were grouped as unclassified and excluded. MAC, Mycobacterium avium complex; MTBC, Mycobacterium tuberculosis complex.
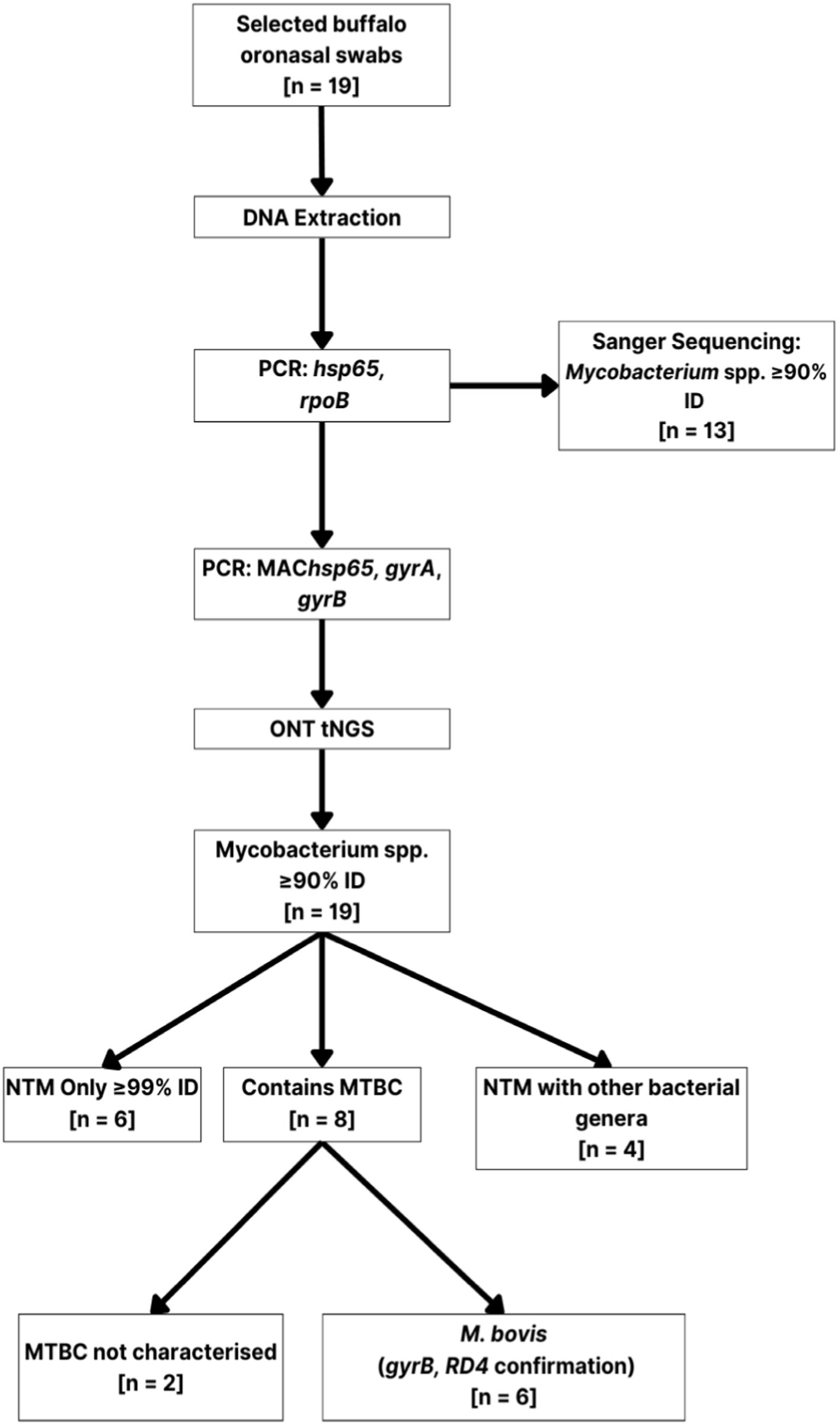
Figure 3. Branched diagram summarizing the culture-independent methods used to characterize mycobacteria present in African buffalo (Syncerus caffer) oronasal swabs (n = 19). Extracted DNA was subjected to multi-target PCR (hsp65, rpoB, MAChsp65, gyrA, gyrB, RD4), Sanger sequencing, and ONT sequencing for mycobacterial speciation. Numbers (n) in boxes represent the number of samples assigned to that category. Mycobacterium spp. DNA was detected in samples with PCR amplicon Sanger sequences having ≥90% identity match (≥ 90% ID) with reference sequences. ONS, oronasal swab; PCR, polymerase chain reaction; NTM, non-tuberculous mycobacteria; ONT, Oxford Nanopore Technologies; MTBC, Mycobacterium tuberculosis complex.
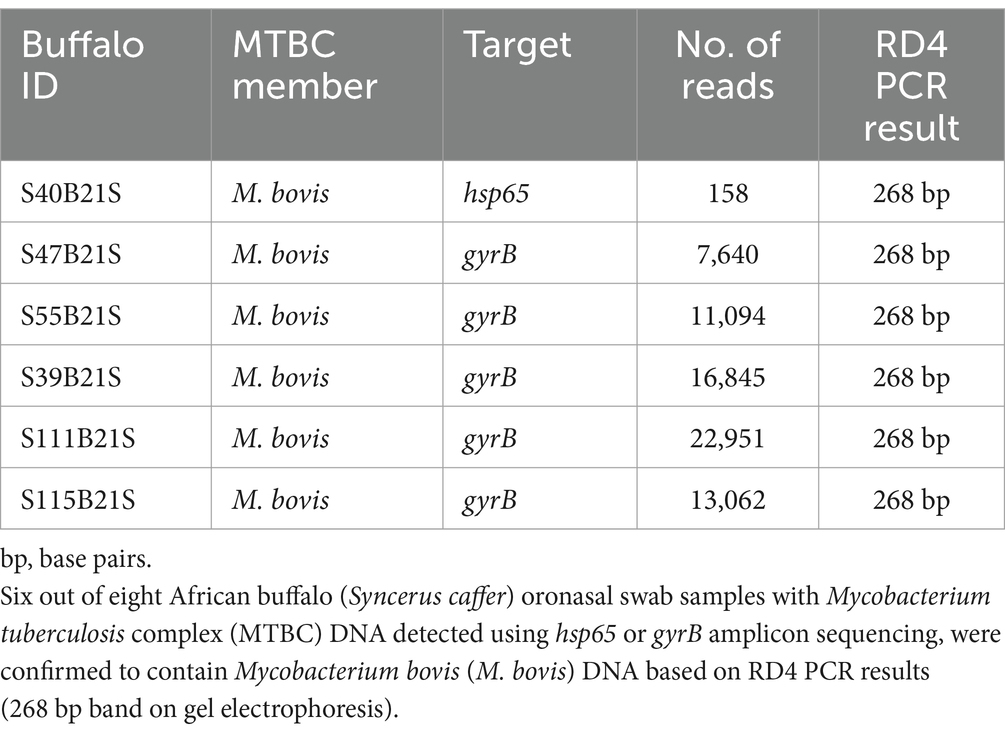
Table 3. Oxford Nanopore Technologies (ONT) targeted (hsp65, gyrB) next generation sequencing read count distribution (using the Mk1C device) and RD4 PCR results.
In addition to MTBC DNA, NTMs were identified in 13 samples using hsp65, rpoB, and MAChsp65 tNGS (≥ 99% identity match), with the results from hsp65 and rpoB shown in Figure 2. Using these three targets, oronasal swab DNA had matches with M. avium complex members (MAC) (10/13), M. novocastrense (5/13), M. neumanni (4/13), and M. elephantis (6/13), with variable abundance. In addition to NTMs (with or without MTBC), Rhodococcus spp. (n = 1) and Nocardia spp. (n = 1) were also identified at a ≥ 99% identity match to the reference sequence. These genera, alongside Precotella spp. were also identified at varying percentage identities between 90 and 99%. Four of the eight samples identified with MTBC DNA also contained NTMs identified at the species level (heterogeneous mycobacterial population).
Mycobacterial species identified from culture, followed by PCR speciation using Sanger sequencing, were compared to those obtained from culture-independent ONT tNGS. A total of 12/19 samples showed concordant results for the presence of mycobacterial DNA (Supplementary materials S3). Of these 12 samples, 2 were concordant for the presence of M. avium based on ≥99% identity match (S8B21N and S21B21M). In addition, ONT tNGS identified M. novocastrense, M. neumannii, and M. elephantis DNA alongside M. avium in one of these samples (S8B21N), while the other sample (S21B21M) also contained unspecified Mycobacterium spp. DNA that was not detected by Sanger sequencing. While Sanger sequencing of hsp65 and rpoB amplicons was utilized to speciate mycobacteria in oronasal swab cultures, this method was unable to detect heterogeneous mycobacterial populations (Supplementary material S3). In contrast, the ONT tNGS of DNA extracted from oronasal swabs using the same targets, detected multiple samples with heterogeneous mycobacterial populations (Figure 2; Supplementary material S3). None of the 8 samples, which contained MTBC DNA based on ONT tNGS, had MTBC detected in swab cultures using Sanger sequencing. It is important to note that four culture sample DNA Sanger sequences were below the 90% coverage and identity match threshold for mycobacterial speciation, however, these were later found to contain mycobacteria with ONT tNGS.
This study aimed to culture-independently detect and characterize the mycobacterial species present in 19 African buffalo oronasal swabs collected from individuals with unexpected immune sensitization to mycobacterial antigens from historically M. bovis-free herds. These samples were selected based on identification of NTM DNA in oronasal swabs from an earlier study (19). In the current study, a panel of mycobacterial gene targets was used for PCR amplification, followed by long-read ONT amplicon sequencing to characterize the mycobacteriome. In order to identify all mycobacteria present in buffalo oronasal cavities, PCR targets were based on mycobacterial housekeeping genes (rpoB, hsp65, gyrB), which have been shown to be generally conserved across species but contain unique sequences that allow differentiation of Mycobacterium spp. (27). These results were compared to identified species generated from swab mycobacterial culture and speciation.
Conventional mycobacterial culture with speciation is the gold standard for detecting M. bovis infection in African buffalo and other species (8). However, this method has limitations, as shown by the lack of MTBC detection in buffalo oronasal swabs from a previous study (19). A potential explanation is the harsh decontamination processing of samples prior to culture, which could result in lower numbers of viable MTBC (12, 28, 29). Additionally, mycobacterial culture may change the microbial composition especially with long incubation times, favoring the more abundant, resilient, and often faster growing mycobacteria (15). This may lead to the underrepresentation or loss of mycobacteria that were initially present in lower numbers and slow growing, when characterizing microbial populations from culture (19, 28, 30). Therefore, in this study, culture-independent characterization of buffalo oronasal mycobacteriome was compared with mycobacterial species generated previously from paired buffalo oronasal swab cultures (19). The culture sequences were re-analyzed with stricter thresholds to ensure that the results obtained used the same criteria as the culture-independent sequences generated in this study. Sanger sequencing of hsp65 and rpoB amplicons, using DNA extracted directly from the swabs, showed slightly higher numbers (n = 13) of samples identified with Mycobacterium spp. DNA compared to culture (n = 12), although no MTBC DNA was detected using either approach. The Sanger sequencing results identified the majority of Mycobacterium spp. as M. avium. This is not surprising since M. avium is ubiquitous in the environment, and likely contaminated the buffalo oronasal cavities.
An intriguing finding in this study was the identification of MTBC DNA in 8 of the 19 buffalo oronasal samples, 6 of which were speciated as M. bovis. None of the other previous analyses, using Sanger sequencing of PCR amplicons, indicated the presence of MTBC in these samples (19). Notably, in four of these samples, both MTBC and NTM DNA were present, which suggests that earlier results based on Sanger sequences missed paucibacillary MTBC. The low relative abundance of MTBC in these samples was not surprising, since these animals originated from historically M. bovis free herds. However, these results highlight the limitation of using Sanger sequencing for paucibacillary respiratory samples, due to its low coverage, and the inherent complexity of the sample type. In addition, Sanger amplicon sequencing may fail to identify co-occurring species, since strict criterion must be applied, which further impedes accurate speciation (31).
To overcome these disadvantages, ONT tNGS was employed to investigate the presence of mixed mycobacterial species, in DNA extracted directly from the oronasal swabs. One advantage of ONT tNGS is that it generates higher numbers and quality of reads, compared to Sanger sequencing (20). The increased depth of sequencing afforded by ONT tNGS facilitated a more thorough analysis of the mycobacterial species present in the samples (10, 11). This approach demonstrated that the majority of oronasal swabs contained multiple species of mycobacteria, including MTBC in eight cases, which is consistent with the complex nature of the oronasal mycobacteriome. Although MTBC DNA was identified using ONT tNGS, it is important to consider the historical M. bovis negative status of the herds, as well as the clinical assessment of the individuals when interpreting these findings. None of the animals showed clinical signs consistent with TB and initial testing was undertaken to meet regulatory requirements for transporting buffalo (48). The 19 buffalo swabs selected for further investigation were based on evidence of immunological responses to mycobacteria, which may be sensitization to MTBC or cross-reactivity to NTMs (19). Therefore, without evidence that the MTBC were viable, it is unclear whether the detection of DNA in eight oronasal swabs represented infection and shedding, residual MTBC DNA after clearing infection, or environmental contamination. Since cultures were negative for MTBC, the bacteria could have been non-replicating, or were present in too few numbers, which might have been eliminated during harsh decontamination steps prior to culture (12). When Cooke et al. (28) applied a similar approach to goat nasal swabs for MTBC detection, the culture-independent detection was higher than from cultures.
The source of MTBC in this study was unknown. Interestingly, the eight buffalo with MTBC DNA detected were from the same herd. Transmission of M. bovis in buffalo is believed to be primarily via infectious aerosols from shedding animals (6, 32). However, MTBC shed into the environment can remain viable for variable durations (33, 34). As animals graze, wallow, and create environmental aerosols with movement, exposure to environmental MTBC may occur (35, 36). There is growing evidence for indirect transmission of M. bovis between cattle and badgers sharing a contaminated environment as well as indirect interspecies spread in other ecosystems (36, 37). Therefore, one possible source of MTBC may have been previous environmental contamination associated with the historical presence of infected livestock, or current wildlife sharing the game reserve, although further investigation of environmental samples would be needed to determine epidemiological links to MTBC isolated from animals. In addition, culture would be required to confirm the presence of viable MTBC in buffalo and provide a source for whole genome sequencing to epidemiologically link cases.
This study had several limitations. The inability to acquire postmortem samples for gold standard confirmation of mycobacterial infection led to the alternative use of microbially complex antemortem samples (oronasal swabs). Previous reports suggest that MTBC shedding occurs sporadically from infected buffalo, and only some animals will be positive when using direct detection; thus, the use of respiratory samples could lead to false negative results (9). Buffalo in this study were suspected to be infected, based on immunological test results and presence of MTBC DNA in oronasal swabs. However, without evidence of mycobacterial viability, it was not possible to differentiate contamination or colonization, from infection. Importantly, it was not possible to rule-out false positive immunological results due to cross-reactivity to NTMs (19, 38). The paucibacillary nature of the samples may also indicate that sufficient antigen load may not have been able to cause immunosensitization, although low doses of M. bovis (1 cfu) can result in interferon gamma expression (39). Another limitation was the lack of PCR target amplification in a number of samples, especially for hsp65, rpoB and gyrB. The optimization of PCR for use with paucibacillary and complex samples remains a challenge. Alternative approaches, such as16S rRNA (16S) and shotgun metagenomic (SMg) sequencing, could have been employed. However, the amount of host DNA found in the directly extracted sample would have posed a challenge during SMg, sequestering the majority of much-needed reads (40). The speciation thresholds for 16S sequencing in mycobacteria have previously underperformed, given the genomic similarity found within the genus (41). Despite the higher resolution of ONT tNGS, a potential limitation is the reported error rates of this technology (42), although only high-quality sequences that could be assigned with high identity match with listed mycobacterial species were included in the analyses. The challenges of characterizing the diversity of microbial communities in complex samples suggests that new approaches are needed to interpret results when pathogenic mycobacteria are identified (17, 20).
In summary, the findings of this study demonstrate the utility of ONT tNGS to identify MTBC DNA in complex antemortem samples as an ancillary tool to conventional mycobacterial culture, especially in high value animals. The higher resolution and discriminatory power of ONT tNGS was used to investigate the apparent mycobacterial sensitization of individual African buffalo, based on diagnostic test results. Immunosensitization was hypothesized to have been due to low levels of MTBC, which conventional mycobacterial culture and subsequent PCR speciation did not detect. Culture-independent PCR and ONT tNGS, using DNA extracted from oronasal swabs, facilitated antemortem detection of MTBC in several African buffalo with evidence of immune sensitization. When used alongside mycobacterial culture, this technology may provide a comprehensive approach to characterizing the mycobacteriome in animals. Although methods may yield discordant results, their combined use allows for a more thorough understanding of the mycobacterial species present in a population, which may inform disease surveillance and control efforts.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was approved by Stellenbosch University’s (SU) Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
SM: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. CC: Methodology, Writing – review & editing. GG: Formal analysis, Writing – original draft, Writing – review & editing. MM: Methodology, Writing – review & editing. TK: Methodology, Project administration, Writing – review & editing. MAM: Supervision, Writing – original draft, Writing – review & editing. WG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the National Research Foundation South African Research Chair Initiative (grant #86949), Wellcome Trust Foundation (222941/Z/21/Z), European Union supported by the Global Health EDCTP3 Joint Undertaking and its members [Project 101103171], and American Association of Zoo Veterinarians Wild Animal Health Fund (S005651). The content is the sole responsibility of the authors and does not necessarily represent the official views of the funders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1523628/full#supplementary-material
1. Hlokwe, TM, and Mogano, RM. Utility of xpert® MTB/RIF ultra assay in the rapid diagnosis of bovine tuberculosis in wildlife and livestock animals from South Africa. Prev Vet Med. (2020) 177:104980. doi: 10.1016/j.prevetmed.2020.104980
2. Sichewo, PR, Vander Kelen, C, Thys, S, and Michel, AL. Risk practices for bovine tuberculosis transmission to cattle and livestock farming communities living at wildlife-livestock-human interface in northern KwaZulu Natal. South Africa PLoS Negl Trop Dis. (2020) 14:e0007618. doi: 10.1371/journal.pntd.0007618
3. Michel, AL, Coetzee, ML, Keet, DF, Maré, L, Warren, R, Cooper, D, et al. Molecular epidemiology of Mycobacterium bovis isolates from free-ranging wildlife in south African game reserves. Vet Microbiol. (2009) 133:335. doi: 10.1016/j.vetmic.2008.07.023
4. Arnot, LF, and Michel, A. Challenges for controlling bovine tuberculosis in South Africa. Onderstepoort J Vet Res. (2020) 87:e1. doi: 10.4102/ojvr.v87i1.1690
5. Caron, A, Cross, PC, and Du Toit, JT. Ecological implications of bovine tuberculosis in African buffalo herds. Ecol Appl. (2003) 13:1338–45. doi: 10.1890/02-5266
6. Michel, AL, and Bengis, RG. The African buffalo: a villain for inter-species spread of infectious diseases in southern Africa. Onderstepoort J Vet Res. (2012) 79:2. doi: 10.4102/ojvr.v79i2.453
7. de Macedo Couto, R, Ranzani, OT, and Alves Waldman, E. Zoonotic tuberculosis in humans: control, surveillance, and the one health approach. Epidemiol Rev. (2019) 41:130–44. doi: 10.1093/epirev/mxz002
8. Bernitz, N, Kerr, TJ, Goosen, WJ, Chileshe, J, Higgitt, RL, Roos, EO, et al. Review of diagnostic tests for detection of Mycobacterium bovis infection in south African wildlife. Front Vet Sci. (2021) 8, 1–11. doi: 10.3389/fvets.2021.588697
9. Clarke, C, Smith, K, Goldswain, SJ, Helm, C, Cooper, DV, Kerr, TJ, et al. Novel molecular transport medium used in combination with Xpert MTB/RIF ultra provides rapid detection of Mycobacterium bovis in African buffaloes. Sci Rep. (2021) 11:7061. doi: 10.1038/s41598-021-86682-5
10. Ye, J, Huang, K, Xu, Y, Chen, N, Tu, Y, Huang, J, et al. Clinical application of nanopore-targeted sequencing technology in bronchoalveolar lavage fluid from patients with pulmonary infections. Microbiol Spectr. (2024) 12:e0002624. doi: 10.1128/spectrum.00026-24
11. Yu, G, Shen, Y, Zhong, F, Zhou, L, Chen, G, Fang, L, et al. Diagnostic accuracy of nanopore sequencing using respiratory specimens in the diagnosis of pulmonary tuberculosis. Int J Infect Dis. (2022) 122:237–43. doi: 10.1016/j.ijid.2022.06.001
12. Clarke, C, Cooper, DV, Miller, MA, and Goosen, WJ. Detection of Mycobacterium tuberculosis complex DNA in oronasal swabs from infected African buffaloes (Syncerus caffer). Sci Rep. (2022) 12:1834. doi: 10.1038/s41598-022-05982-6
13. Goosen, WJ, Clarke, C, Kleynhans, L, Kerr, TJ, Buss, P, and Miller, MA. Culture-independent PCR detection and differentiation of mycobacteria spp. in antemortem respiratory samples from African elephants (Loxodonta africana) and Rhinoceros (Ceratotherium simum, Diceros bicornis) in South Africa. Pathogens. (2022) 11:6. doi: 10.3390/pathogens11060709
14. Hamed, KA, and Tillotson, G. A narrative review of nontuberculous mycobacterial pulmonary disease: microbiology, epidemiology, diagnosis, and management challenges. Expert Rev Respir Med. (2023) 17:973–88. doi: 10.1080/17476348.2023.2283135
15. Liang, Q, Shang, Y, Huo, F, Xue, Y, Li, Y, Dong, L, et al. Assessment of current diagnostic algorithm for detection of mixed infection with Mycobacterium tuberculosis and nontuberculous mycobacteria. J Infect Public Health. (2020) 13:1967–71. doi: 10.1016/j.jiph.2020.03.017
16. Byrne, AS, Goudreau, A, Bissonnette, N, Shamputa, IC, and Tahlan, K. Methods for detecting mycobacterial mixed strain infections–a systematic review. Front Genet. (2020) 11, 1–20. doi: 10.3389/fgene.2020.600692
17. Ghielmetti, G, Kerr, TJ, Bernitz, N, Mhlophe, SK, Streicher, E, Loxton, AG, et al. Insights into mycobacteriome composition in Mycobacterium bovis-infected African buffalo (Syncerus caffer) tissue samples. Sci Rep. (2024) 14:17537. doi: 10.1038/s41598-024-68189-x
18. Gcebe, N, and Hlokwe, TM. Non-tuberculous mycobacteria in south African wildlife: neglected pathogens and potential impediments for bovine tuberculosis diagnosis. Front Cell Infect Microbiol. (2017) 7, 1–15. doi: 10.3389/fcimb.2017.00015
19. Clarke, C, Kerr, TJ, Warren, RM, Kleynhans, L, Miller, MA, and Goosen, WJ. Identification and characterisation of nontuberculous mycobacteria in African buffaloes (Syncerus caffer), South Africa. Microorganisms. (2022) 10:9. doi: 10.3390/microorganisms10091861
20. Ghielmetti, G, Loubser, J, Kerr, TJ, Stuber, T, Thacker, T, Martin, LC, et al. Advancing animal tuberculosis surveillance using culture-independent long-read whole-genome sequencing. Front Microbiol. (2023) 14, 1–13. doi: 10.3389/fmicb.2023.1307440
21. Davis, MW, and Jorgensen, EM. ApE, a plasmid editor: a freely available dna manipulation and visualization program. Front Bioinform. (2022) 2, 1–15. doi: 10.3389/fbinf.2022.818619
22. Turenne, CY, Semret, M, Cousins, DV, Collins, DM, and Behr, MA. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J Clin Microbiol. (2006) 44:433. doi: 10.1128/JCM.44.2.433-440.2006
23. Wick, RR, Judd, LM, and Holt, KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. (2019) 20:129. doi: 10.1186/s13059-019-1727-y
24. Steinig, E, and Coin, L. Nanoq: ultra-fast quality control for nanopore reads. J Open Source Softw. (2022) 7:69. doi: 10.21105/joss.02991
25. Vierstraete, AR, and Braeckman, BP. Amplicon_sorter: a tool for reference-free amplicon sorting based on sequence similarity and for building consensus sequences. Ecol Evol. (2022) 12:e8603. doi: 10.1002/ece3.8603
26. Warren, RM, Gey van Pittius, NC, Barnard, M, Hesseling, A, Engelke, E, de Kock, M, et al. Differentiation of Mycobacterium tuberculosis complex by PCR amplification of genomic regions of difference. Int J Tuberc Lung Dis. (2006) 10:818–22.
27. Devulder, G, de Montclos, MP, and Flandrois, JP. A multigene approach to phylogenetic analysis using the genus Mycobacterium as a model. Int J Syst Evol Microbiol. (2005) 55:293–302. doi: 10.1099/ijs.0.63222-0
28. Cooke, DM, Clarke, C, Kerr, TJ, Warren, RM, Witte, C, Miller, MA, et al. Detection of Mycobacterium bovis in nasal swabs from communal goats (Capra hircus) in rural KwaZulu-Natal, South Africa. Front Microbiol. (2024) 15, 1–11. doi: 10.3389/fmicb.2024.1349163
29. Goosen, WJ, Kleynhans, L, Kerr, TJ, van Helden, PD, Buss, P, Warren, RM, et al. Improved detection of Mycobacterium tuberculosis and M. bovis in African wildlife samples using cationic peptide decontamination and mycobacterial culture supplementation. J Vet Diagnost Invest. (2022) 34:61. doi: 10.1177/10406387211044192
30. Goosen, WJ, Moodley, S, Ghielmetti, G, Moosa, Y, Zulu, T, Smit, T, et al. Identification and molecular characterization of Mycobacterium bovis DNA in GeneXpert® MTB/RIF ultra-positive, culture-negative sputum from a rural community in South Africa. One Health. (2024) 18:100702. doi: 10.1016/j.onehlt.2024.100702
31. Bunnik, EM, and Le Roch, KG. An introduction to functional genomics and systems biology. Adv Wound Care. (2013) 2:490–8. doi: 10.1089/wound.2012.0379
32. Lakin, HA, Tavalire, H, Sakamoto, K, Buss, P, Miller, M, Budischak, SA, et al. Bovine tuberculosis in African buffalo (Syncerus caffer): progression of pathology during infection. PLoS Negl Trop Dis. (2022) 16:e0010906. doi: 10.1371/journal.pntd.0010906
33. Fine, AE, Bolin, CA, Gardiner, JC, and Kaneene, JB. A study of the persistence of Mycobacterium bovis in the environment under natural weather conditions in Michigan, USA. Vet Med Int. (2011) 2011:4. doi: 10.4061/2011/765430
34. Ghodbane, R, Medie, FM, Lepidi, H, Nappez, C, and Drancourt, M. Long-term survival of tuberculosis complex mycobacteria in soil. Microbiology. (2014) 160:496–501. doi: 10.1099/mic.0.073379-0
35. Carneiro, PAM, Takatani, H, Pasquatti, TN, Silva, CBDG, Norby, B, Wilkins, MJ, et al. Epidemiological study of Mycobacterium bovis infection in buffalo and cattle in Amazonas. Brazil Front Vet Sci. (2019) 6, 1–9. doi: 10.3389/fvets.2019.00434
36. Dwyer, RA, Witte, C, Buss, P, Goosen, WJ, and Miller, M. Epidemiology of tuberculosis in multi-host wildlife systems: implications for black (Diceros bicornis) and white (Ceratotherium simum) rhinoceros. Front. Vet. Sci. (2020) 7, 1–15. doi: 10.3389/fvets.2020.580476
37. Allen, AR, Ford, T, and Skuce, RA. Does Mycobacterium tuberculosis var. bovis survival in the environment confound bovine tuberculosis control and eradication? A literature review. Vet. Med. Int. (2021) 2021:1–19. doi: 10.1155/2021/8812898
38. Krasinilkov, I, Lehnherr-Ilyina, T, Djonovic, M, Artamonova, I, Nikitin, M, and Kislichkin, N. Cracking the antigenic code of mycobacteria: CFP-10/ESAT-6 tuberculosis skin test and misleading results. J Clin Tuberc Other Mycobact Dis. (2024) 36:100436. doi: 10.1016/j.jctube.2024.100436
39. Johnson, L, Dean, G, Rhodes, S, Hewinson, G, Vordermeier, M, and Wangoo, A. Low-dose Mycobacterium bovis infection in cattle results in pathology indistinguishable from that of high-dose infection. Tuberculosis. (2007) 87:71–6. doi: 10.1016/j.tube.2006.04.002
40. Kok, NA, Peker, N, Schuele, L, de Beer, JL, Rossen, JWA, Sinha, B, et al. Host DNA depletion can increase the sensitivity of Mycobacterium spp. detection through shotgun metagenomics in sputum. Front Microbiol. (2022) 13, 1–11. doi: 10.3389/fmicb.2022.949328
41. Beye, M, Fahsi, N, Raoult, D, and Fournier, PE. Careful use of 16S rRNA gene sequence similarity values for the identification of Mycobacterium species. New Microbes New Infect. (2018) 22:24–9. doi: 10.1016/j.nmni.2017.12.009
42. Gómez-González, PJ, Campino, S, Phelan, JE, and Clark, TG. Portable sequencing of Mycobacterium tuberculosis for clinical and epidemiological applications. Brief Bioinform. (2022) 23:5. doi: 10.1093/bib/bbac256
43. Telenti, A, Marchesi, F, Balz, M, Bally, F, Botrger, EC, and Bodmer, T. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J Clin Microbiol. (1993) 31:175. doi: 10.1128/jcm.31.2.175-178.1993
44. Adékambi, T, Colson, P, and Drancourt, M. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol. (2003) 41:5699. doi: 10.1128/JCM.41.12.5699-5708.2003
45. Landolt, P, Stephan, R, and Scherrer, S. Development of a new high-resolution melting (HRM) assay for identification and differentiation of Mycobacterium tuberculosis complex samples. Sci Rep. (2019) 9:1850. doi: 10.1038/s41598-018-38243-6
46. Landolt, P, Stephan, R, Stevens, MJA, and Scherrer, S. Three-reaction high-resolution melting assay for rapid differentiation of Mycobacterium tuberculosis complex members. Microbiology Open. (2019) 8:12. doi: 10.1002/mbo3.919
47. National Center for Biotechnology Information (NCBI). Nucleotide Basic Local Alignment Search Tool (BLASTn). (2024) Available at: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome (Accessed September, 2024).
48. Department of Agriculture Land Reform and Rural Development (DALRRD) Buffalo disease risk management. (2017) Availble at: https://www.dalrrd.gov.za/images/Branches/AgricProducHealthFoodSafety/animal-health/disease-control/information/dah-policy/buffalo-disease-risk-management-vpn-addendums-march-2017.pdf (Accessed September 2024).
Keywords: African buffaloes, culture-independent detection, Mycobacterium tuberculosis complex, oronasal swabs, Oxford Nanopore Technologies, targeted next generation sequencing
Citation: Mhlophe SK, Clarke C, Ghielmetti G, Matthews M, Kerr TJ, Miller MA and Goosen WJ (2025) Culture-independent detection of Mycobacterium tuberculosis complex DNA using targeted next generation sequencing in African buffalo (Syncerus caffer) oronasal swabs in South Africa. Front. Vet. Sci. 12:1523628. doi: 10.3389/fvets.2025.1523628
Edited by:
Javier Bezos, Complutense University of Madrid, SpainReviewed by:
Susana Flores-Villalva, National Institute of Forestry and Agricultural Research (INIFAP), MexicoCopyright © 2025 Mhlophe, Clarke, Ghielmetti, Matthews, Kerr, Miller and Goosen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wynand Johan Goosen, d2pnb29zZW5Ac3VuLmFjLnph
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.