 Pablo Tamiozzo
Pablo Tamiozzo Virginia García
Virginia García Raúl E. González-Ittig
Raúl E. González-Ittig Maria Pieters
Maria Pieters- 1Departamento de Patología Animal, Facultad de Agronomía y Veterinaria, Universidad Nacional de Río Cuarto, Córdoba, Argentina
- 2Cátedra de Genética de Poblaciones y Evolución, Facultad de Ciencias Exactas, Físicas y Naturales, Universidad Nacional de Córdoba, Córdoba, Argentina
- 3Instituto de Diversidad y Ecología Animal (IDEA; CONICET-UNC), Córdoba, Argentina
- 4Department of Veterinary Population Medicine, College of Veterinary Medicine, University of Minnesota, Saint Paul, MN, United States
- 5Veterinary Diagnostic Laboratory, College of Veterinary Medicine, University of Minnesota, Saint Paul, MN, United States
- 6Swine Disease Eradication Center, College of Veterinary Medicine, University of Minnesota, Saint Paul, MN, United States
Background: Several epidemiological studies have been carried out using Multiple-Locus Variable-number tandem repeat Analysis (MLVA) for M. hyopneumoniae typing. However, a global perspective on the implications of the genetic diversity of this pathogen is lacking.
Objective: This study aimed to determine and to analyze the genetic structure of M. hyopneumoniae based on the p97R1-p146R3 MLVA typing scheme and to characterize, analyze and compare MLVA types among countries where the information was publicly available.
Methods: A set of 797 publicly available data of M. hyopneumoniae p97R1-p146R3 MLVA genetic types from six different countries were analyzed using Genalex 6.41 software to characterize loci polymorphism and using Structure 2.3.4 software in order to identify the genetic structure.
Results: A total of 185 MLVA types were identified among the analyzed data. For the p97R1 and p146R3 loci, most of the molecular variation in M. hyopneumoniae populations was identified within countries. Three genetic clusters and their recombinations were identified globally.
Conclusion: M. hyopneumoniae is a genetically diverse pathogen with limited clonality and three well-defined clusters and their combinations were identified in this investigation. The greatest genetic variation of M. hyopneumoniae was observed within countries.
1 Introduction
Mycoplasma hyopneumoniae is the primary agent involved in porcine enzootic pneumonia and the Porcine Respiratory Disease Complex. Infections with this bacterium are highly prevalent in almost all swine-producing areas, causing significant economic losses to the pig industry worldwide (1). Information on the genetic diversity of M. hyopneumoniae can have implications in the design, identification, and application of pig health intervention strategies, such as emergence of new and drug-resistant strains, development of precision diagnostics, therapeutics and vaccines, as well as epidemiological studies.
To identify genetic variability in M. hyopneumoniae, different typing schemes have been reported, such as Random Amplification of Polymorphic DNA [RAPD-PCR (2)], Amplified Fragment Length Polymorphism [AFLP (2, 3)], Arbitrarily Primed-PCR [AP-PCR (4)] and Pulsed Field Gel Electrophoresis [PFGE (5)]. Although some of the above mentioned techniques have demonstrated high discriminatory power, currently, they are not widely used. In addition, Multiple-Locus Sequence Typing (MLST) and Multiple-Locus Variable-Number Tandem Repeat Analysis (MLVA) have been or are used for genetic typing of this microorganism worldwide (6–9, 43, 45, 46). While MLST can be laborious, costly, and has mainly been employed for research purposes (10–14), MLVA can be applied with ease, is less expensive, and has been increasingly used for M. hyopneumoniae typing (7, 9, 15, 16), even in field based investigations (17, 43). From a proposed repertoire of twenty-two Variable Number Tandem Repeats (VNTR) regions, four have proven to be highly polymorphic (6), namely p97R1, p146R3, H4 and H5. Although analyzing the four loci of the MLVA scheme has shown a high discriminatory power (6, 18), the detection of different M. hyopneumoniae genetic types has been evidenced using three (15, 19), two (7) or even one VNTR (20, 21).
Numerous epidemiological studies have been carried out using different MLVA schemes in various countries, at farm, regional and/or country level (7, 15–17). However, a global perspective on the implications of the genetic diversity of this etiological agent is lacking. To this extent, M. hyopneumoniae has been pointed out as a microorganism with limited clonality (13). Notwithstanding, information is needed on the overall allele frequency, distribution of MLVA types, and genetic structure of this pathogen. Therefore, the objectives of this study were to determine and to analyze the genetic structure of M. hyopneumoniae based on the p97R1-p146R3 MLVA typing scheme and to characterize, analyze and compare MLVA types among countries where the information was publicly available.
2 Materials and methods
2.1 Study inclusion criteria and data extraction
Considering that the majority of published studies using MLVA as a typing scheme for M. hyopneumoniae included the p97R1 and p146R3 loci, and that both genes encode well-known adhesins (22–24), only studies in which at least these two loci were typed were included in this investigation. Moreover, this criterion was applied as it has been previously suggested that two loci are to be the minimum in M. hyopneumoniae typing schemes (25). Additionally, a small number of reports using three or four loci were available in the literature. The data set used in this investigation also included unpublished data from our research group. From the selected studies, only tandem repeats from the p97R1 and p146R3 loci were included. Table 1 shows the number of VNTR types according to country. Thus, 797 VNTR type data sets (VNTR types, from hereon) were recovered and analyzed.
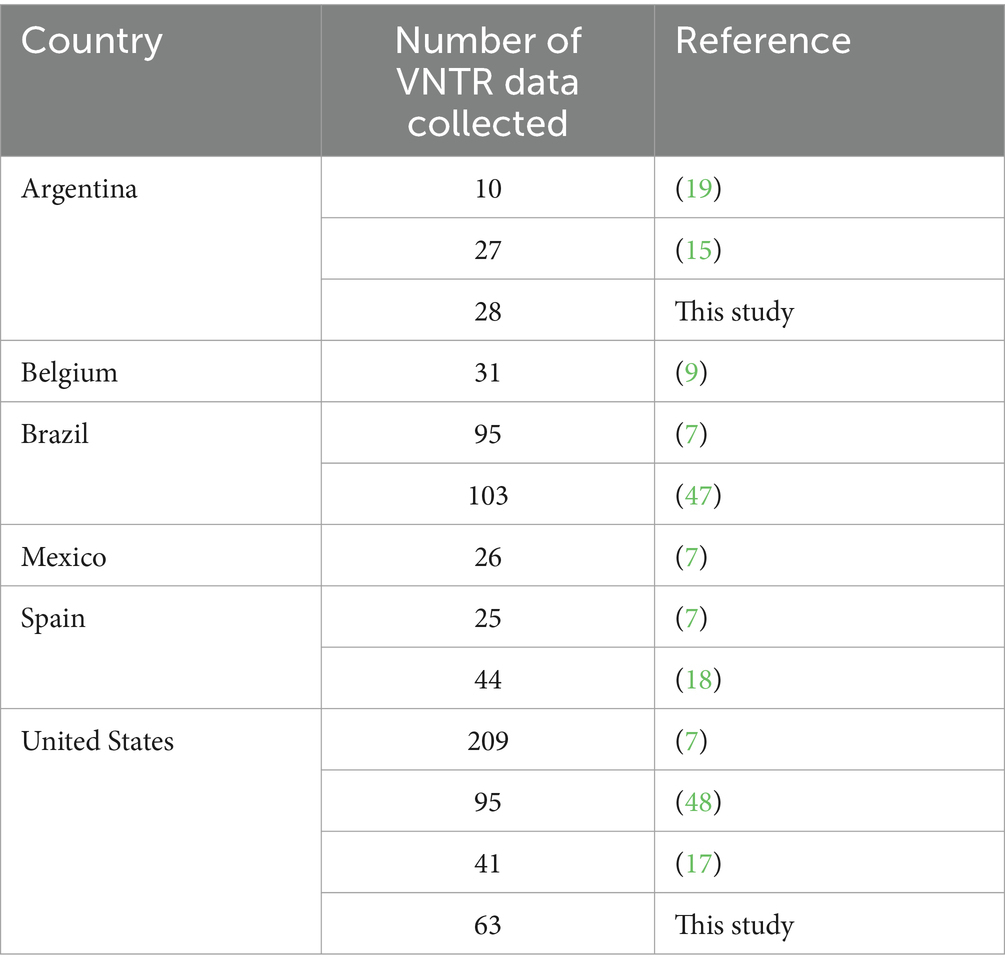
Table 1. Variable number tandem repeat data collected by country, according to the reference.
2.2 Data analysis
The number of alleles (Na), number of effective alleles (Ne), Shannon’s Information Index (I) and values of diversity (h) and unbiased diversity (uh) were calculated using the program Genalex 6.41 (26) as a way to characterize the polymorphism of M. hyopneumoniae loci in each country for which data was available. The pairwise differentiation among countries using the Nei unbiased genetic distance was also calculated using the above-mentioned program and the allele frequencies by population and by locus was plotted. Analysis of molecular variance (AMOVA) among countries, regions (America vs. Europe), and within populations (countries), was performed for both loci, separately and together.
The genetic structure of M. hyopneumoniae was examined using the Bayesian algorithm implemented in Structure 2.3.4 (27) assuming a model of population admixture and correlated allele frequencies among groups. The analysis was performed without the inclusion of any geographic information. Thus, the software could identify the most likely number of genetic clusters without a priori information. Analyses for a number of K ranging from 1 to 7, for 2,000,000 iterations, including a burn-in of 500,000 iterations was performed. The parameter Alphapropsd = 0.125 to improve mixing was used. The procedure was repeated five times for each K to check for consistency. The optimal value of K was calculated according to the ΔK method described by Evanno et al. (28) using the online Structure Harvester 0.6.7 software.1 Replicate runs of Structure were analyzed using Clumpp 1.1.2 (29) to summarize each type’s membership across runs using a greedy algorithm. Results were averaged to build summary bar plots for each VNTR type using the program Distruct 1.1 (30). For a VNTR type to be considered part of a cluster, the membership to one cluster should had been more than 0.90, otherwise, admixed ancestry was assumed (31).
3 Results
Locus p97R1 revealed 20 alleles, while locus p146R3 revealed 38 alleles, with tandem repeats (VNTRs) ranging from 10 to 48 (Supplementary Table S1; Figures 1, 2). For p97R1, the VNTR types with 9, 10 and 11 tandem repeats were identified in all countries, while unique alleles were reported in the United States (VNTRs 4, 16, 18 and 19), Belgium (VNTR 1), and Mexico (VNTR 20). For p146R3, the only type shared in all the countries was VNTR 17, while unique types were observed in the United States (VNTRs 10 and 11), Spain (VNTRs 30, 36, 46 and 47), and Brazil (VNTRs 33 and 48) (Supplementary Table S1). For p97R1, the country with the highest number of alleles (Na), effective alleles (Ne) and most diversity (h) and unbiased diversity (uh) by population was the United States (Table 2). For p146R3, the country with the highest number of alleles (Na) was Brazil. The highest number of effective alleles (Ne) and the most diversity (h) and unbiased (uh) diversity by population was Belgium (Table 2).
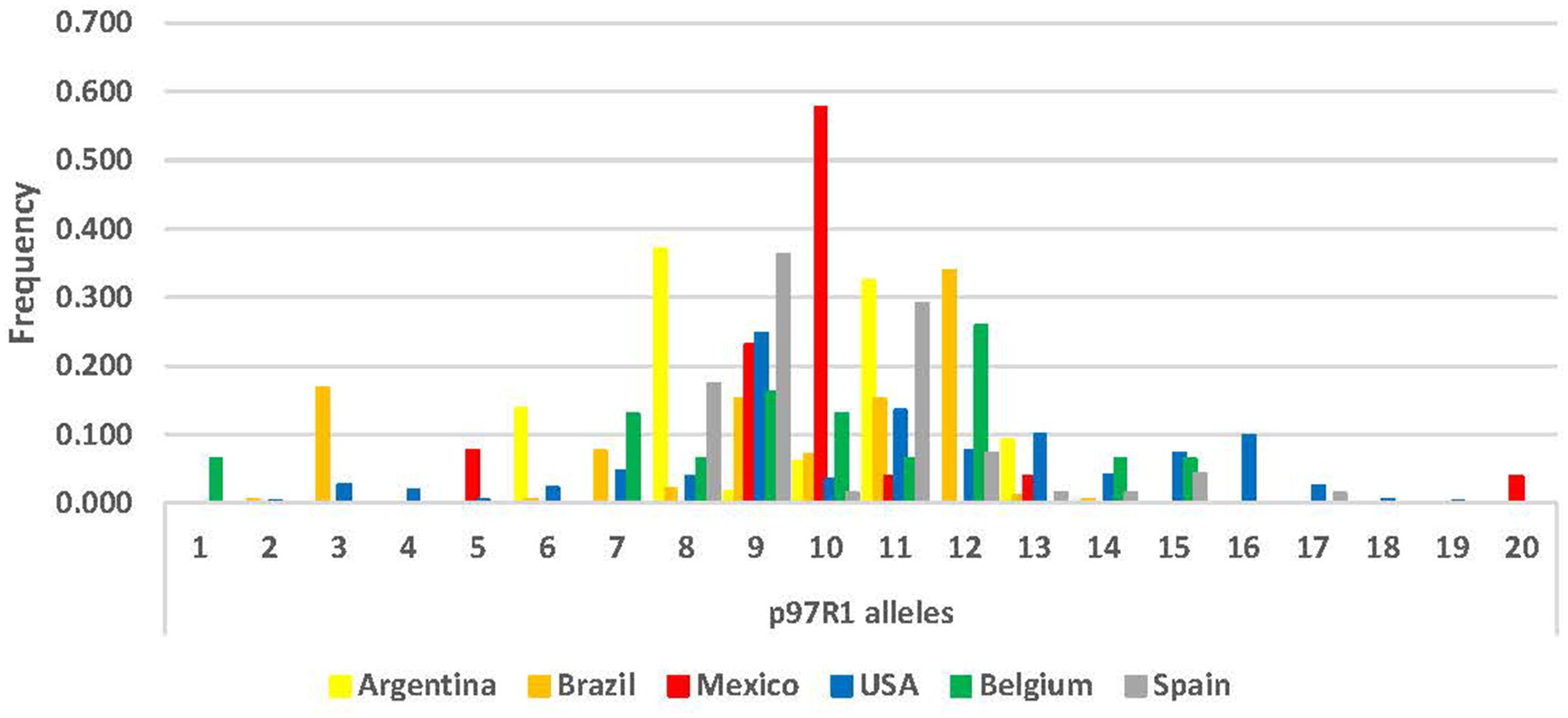
Figure 1. Allele frequency distribution for p97R1 locus, according to number of Tandem Repeats (TR), by country.
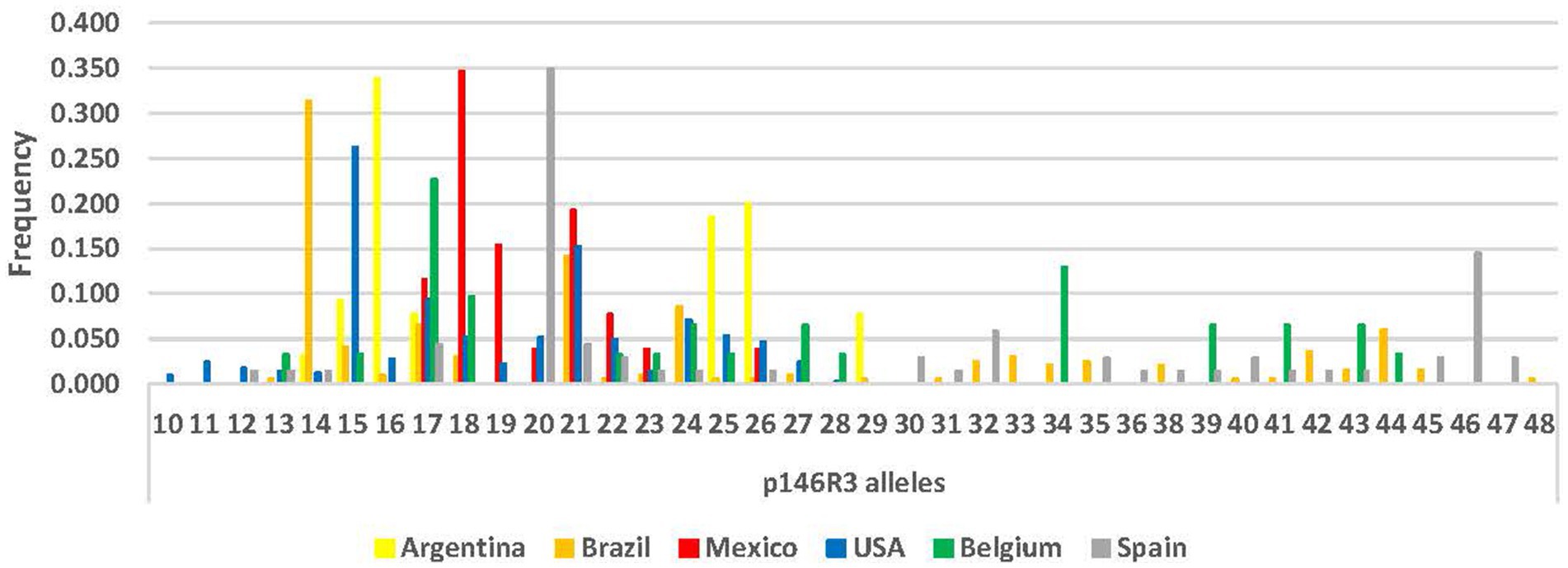
Figure 2. Allele frequency distribution for p146R3 locus, according to number of Tandem Repeats (TR), by country.
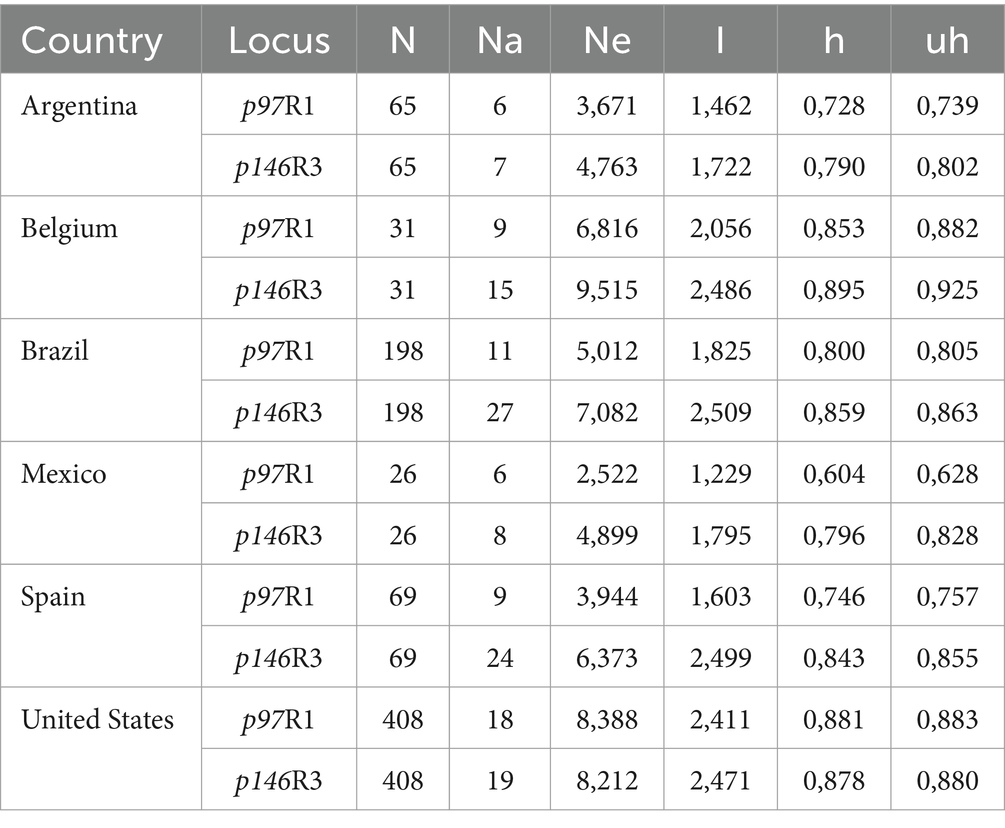
Table 2. Sample size (N), Number of alleles (Na), Number of effective alleles (Ne), information index (I), Diversity (h) and Unbiased Diversity (uh) by country (population) for the p97R1 and p146R3 loci.
A total of 185 M. hyopneumoniae typologies were identified among the 797 VNTR types analyzed. The most frequent VNTR types (p97R1-p146R3, respectively) were: 9–15 (8.4%), 11–21 (7.4%), 14–14 (6.5%), 13–17 (3.4%), 9–20 (3.3%), 16–24 (2.9%) and 8–16 (2.4%), which were present in all countries included in the study, except Belgium (Supplementary material S2). The remaining VNTR types were identified in less than 2.1% of the analyzed data. The 44.3% (82/185) of the identified types were deemed unique and were distributed in all countries (Supplementary material S2). In Argentina there were 13 VNTR types, being the most frequent 8–16 (27.7%) and 11–26 (15.4%). In Belgium, 20 types were recognized, with 12–17 (19.4%) and 7–34 (9.7%) being the most prevalent. In Brazil 52 typologies were identified, the most frequent: 12–14 (24.2%) and 11–21 (13.6%). In Mexico 12 types were observed, with 10–18, 10–19 and 10–17 in 30.8, 15.4 and 11.5%, respectively. In Spain 32 types were identified, including 9–20 (34.8%) and 11–46 (14.5%). In the United States a total of 101 types were observed, the most prominent being 9–15 (16.2%) and 11–21 (7.6%) (Supplementary material S2). All MLVA types identified in this investigation were not present in all countries, with no type present in more than three countries.
Similarity coefficients of Nei’s unbiased genetic distance between M. hyopneumoniae populations ranged from 0.471 to 1.943 (Table 3). The highest distance was observed between Mexico and Argentina (1.943%), while the highest Nei unbiased genetic identity was observed between Brazil and Belgium (0.6%; Table 4).
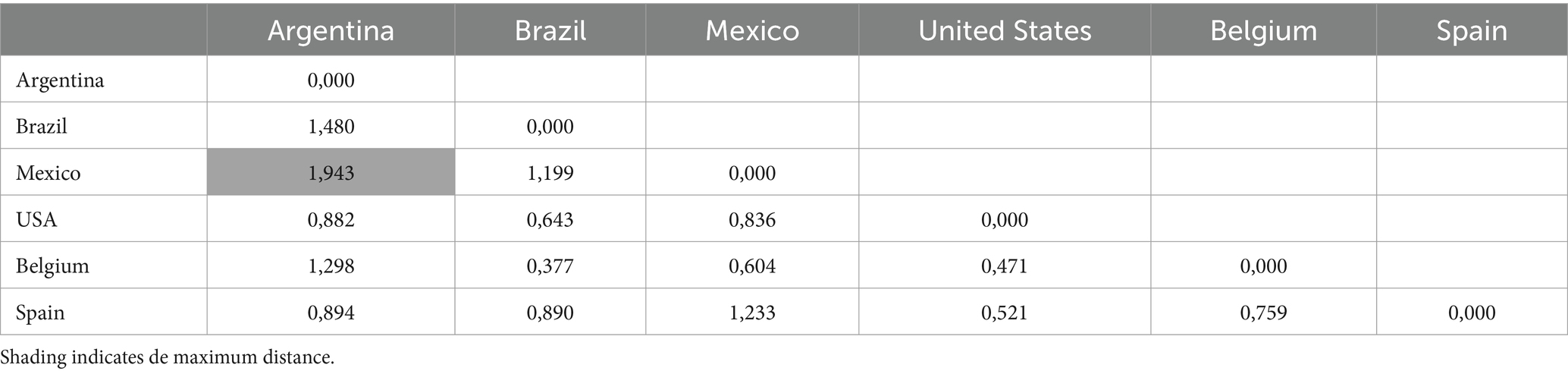
Table 3. Pairwise population matrix of nei unbiased genetic distance.
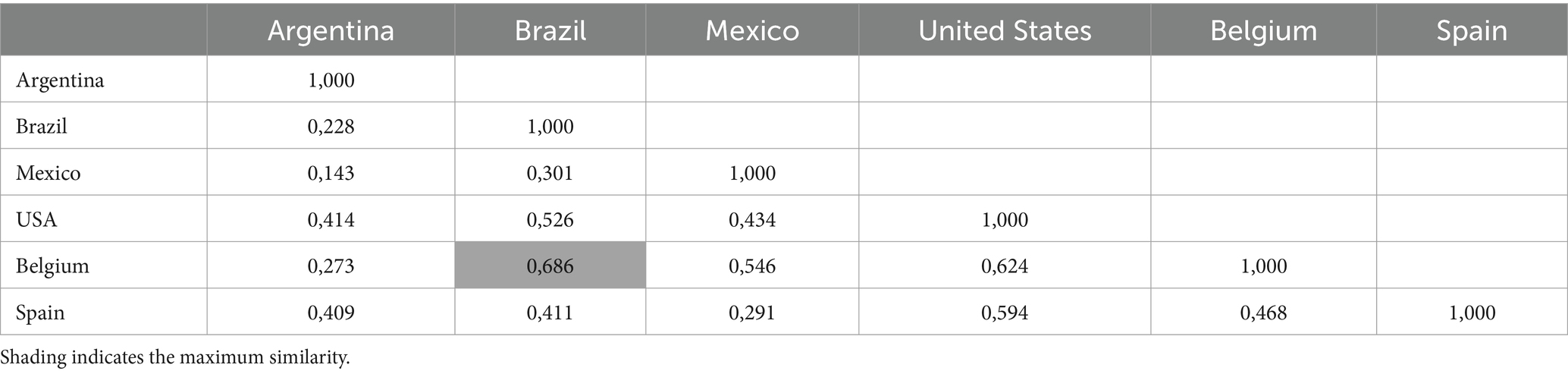
Table 4. Pairwise population matrix of nei unbiased genetic identity.
Results of the AMOVA indicated that, for both loci, most of the molecular variation in M. hyopneumoniae populations (65%), existed within populations (countries), with less variation between regions (28%) and among populations (7%). For the p97R1 locus, most of the variation existed within populations (90%) and 10% among populations, with no difference between regions. For the p146R3 locus, most of the variation existed within populations (61%), with less variation among regions (32%) and populations (7).
Analyses revealed three genetic clusters, with log posterior probabilities of −3611.72 ± 2.83 for the 797 types (Figure 3). Different countries in the study were marked in Figure 3 for ease of visualization, although a geographical differentiation was not set as part of the priors of the analysis. The frequency of each of the three clusters varied considerably among countries (Table 5). VNTR types whose genome belonged to a single cluster were deemed “pure” and typologies with admixed ancestry were identified in all clusters. At a general level, there were more VNTR types with admixed ancestry (43.3%) than pure types (Table 5). The largest proportion of VNTR types belonging to cluster I were reported in Brazil (41.9%), and in Spain for clusters II and III (34.8 and 20.3%, respectively). Mexico showed the largest proportion of admixed ancestry types (80.8%).
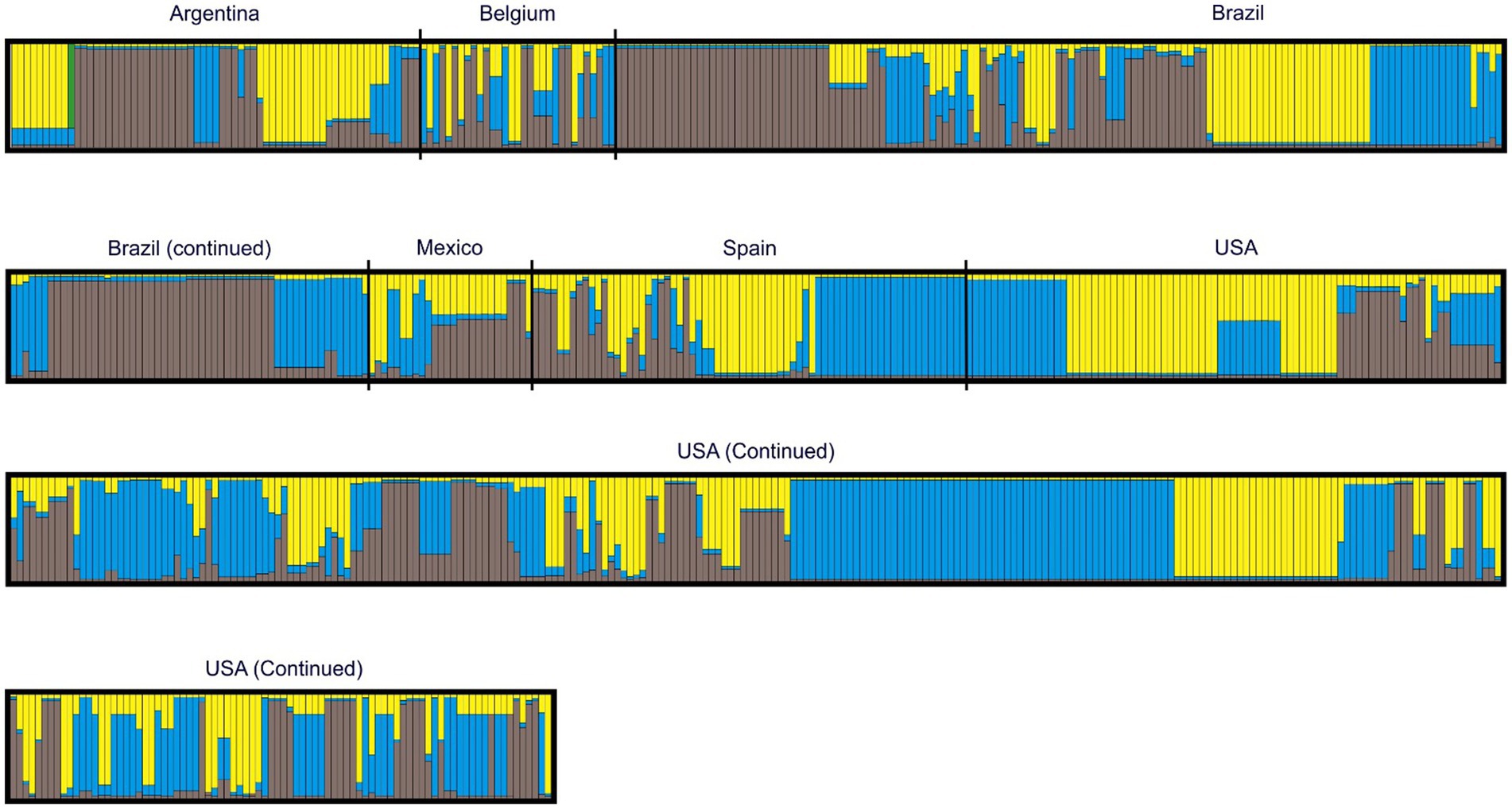
Figure 3. Mycoplasma hyopneumoniae individual Bayesian assignment probabilities of p97R1-p146R3 MLVA using the program STRUCTURE for k clusters. Each vertical line represents the probability of an individual belonging to one of three clusters (represented by different colors) or a combination if ancestry is mixed.
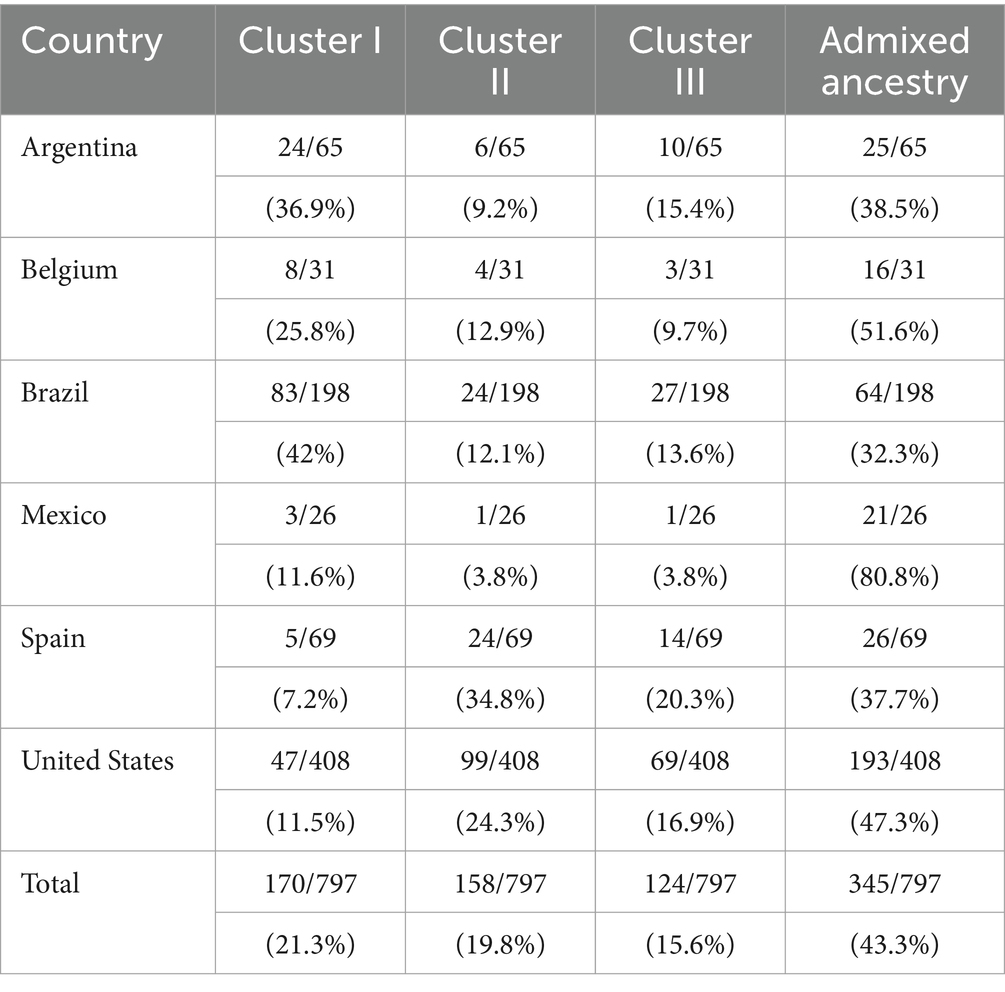
Table 5. Proportion and percentage of specimens belonging to clusters I-III and admixed ancestry of M. hyopneumoniae populations by country, with a 90% membership rate.
4 Discussion
Bacterial population structures range from panmictic or non-clonal, to strictly clonal, but intermediate structures have also been recognized, such as epidemic (32). The different population genetic structures may be due to events like genetic recombination, geographical or ecological isolation, genetic drift, horizontal gene transfer, among others (32–35). While Salmonella enterica is a model of a clonal population, Neisseria gonorrhoeae and Helicobacter pylori are representative of non-clonal models (36). In the former, the same or very similar types of individuals can cause disease and/or outbreaks, and in the latter genetically unrelated individuals of a given type lead to malaise.
In the present study, the fact that more than 40% of the analyzed VNTR types belonged to admixed ancestry, three defined genetic clusters were identified, differences in diversity among countries was minimal, and no p97R1-p146R3 MLVA type was reported in all countries suggests that the genetic structure of M. hyopneumoniae is predominantly, but not exclusively, panmictic or non-clonal. On the other hand, the abundance of VNTR types, in addition to unique alleles identified in different countries strongly suggests that M. hyopneumoniae is a highly diverse microorganism.
Our findings indicate that three M. hyopneumoniae MLVA genetic clusters, which possibly evolved separately, are now seen combined globally, or at least in the analyzed countries. A similar pattern has been described for M. bovis, where three genetic clusters have been reported, with a preponderance of two (37). In the above mentioned study, the authors used MLST typing for 137 M. bovis isolates from 12 countries and observed the first cluster including isolates from multiple geographical origins (United Kingdom, Lithuania, Hungary, Germany, Italy, Israel, and Australia) and European origin only (Spain, Germany, France, and Lithuania). The second cluster including also isolates from several origins and all the strains from China and most of those of Israeli and Australian origin, and the third, including only two Israeli isolates (37). Moreover, similar patterns of geographical distribution of M. bovis isolates has been recognized using the MLVA typing method (38).
Numerous sequence-based typing approaches for M. hyopneumoniae have been developed, being MLST and MLVA the most commonly used currently. However, the use of one or the other techniques should not be arbitrary. While the MLST scheme, which like Multiple Locus Enzyme Electrophoresis (MLEE) detects DNA changes in conserved genome areas, MLVA was developed for high discrimination between closely related strains (39). This information, combined with the intermediate genetic structure of the agent might explain some incongruities reported to date. For instance, while some studies using MLST associated enzootic pneumonia (EP) with identical or very similar M. hyopneumoniae genotypes (12, 13, 40), others using MLVA typing reported a higher prevalence and severity of EP-like lung lesions with higher diversity of the pathogen (41). Particularly for M. hyopneumoniae, the use of the MLVA assay has been recommended for differentiation of strains with identical MLST sequence types (10). Considering that the MLST scheme is useful for phylogenetic analysis, but often not discriminative enough for outbreak detection (39), and that MLVA outcomes can be highly polymorphic, the use of both typing approaches in combination for EP outbreak studies could yield insightful information.
In spite of the fact that VNTR types with mixed ancestry were identified in the six analyzed countries, in Mexico, it reached almost 81%. The source of this type of VNTR could be due, for instance, to genetic drive or horizontal gene transfer. Future studies are needed for a better understanding of this phenomenon. In addition to this, Mexico is the only country among those analyzed in which a live attenuated M. hyopneumoniae vaccine has recently been licensed (42). Further longitudinal studies to evaluate the stability of VNTR types from M. hyopneumoniae hosted in pigs vaccinated with live vaccines should be conducted. Interestingly, the VNTR type of the J strain (9–18), the most used strain for inactivated vaccines against EP worldwide (18), did not match exactly any of the analyzed VNTR types. Similar types, namely 9–17, 9–19 and 8–17, were also identified in Argentina, Belgium, Spain and the United States. Contrary to what was observed in Mexico, in Brazil more VNTR types belonging to cluster I than with admixed ancestry were identified, suggesting that gene flow may be restricted between the Brazilian M. hyopneumoniae populations and those of other countries. In addition, if the Brazilian populations originated from a single source of infection and if not many types with a different genetic composition have entered the country, it could be expected for the M. hyopneumoniae populations to have a predominance of a single cluster with very little presence of the other genetic combinations, which has been observed in this study.
Results from this investigation suggest that most variation in M. hyopneumoniae types (61–90%) was observed within populations (countries). Although it has been suggested that a certain M. hyopneumoniae MLVA type predominates and persists within each herd (17, 19), circulating M. hyopneumoniae genotypes are abundant, at herd and even at pig level (15, 43, 44). The molecular variation among populations (7–10%) was probably due to the existence of unique alleles in some countries and to that a small number of p97R1 and p146R3 alleles were present in all countries. Results also indicated that for both loci in combination, and for the p146R3 locus alone, approximately 30% of variation was observed between regions (America vs. Europe), which was probably due to the high polymorphism of the p146R3 locus (6). A practical implication of this finding would be to use p146R3 together with other polymorphic loci when making comparisons between the two continents.
In conclusion, this study reinforces the concept that M. hyopneumoniae is a genetically diverse pathogen with limited clonality. When typed by MLVA, three well-defined populations or clusters and their combinations were identified. The major cause of genetic variation of M. hyopneumoniae might be within populations, recognizing the bias in the analyzed data, as not all countries were analyzed using the same number of farms, nor randomly. This study constitutes a starting point for future investigations to provide greater detail of the molecular epidemiological behavior of this pathogen. The difficulty in culturing and isolating M. hyopneumoniae strains and the scarcity of genomes available, continues to be a limitation for the application of whole genome sequencing (WGS). Analysis of WGS of this pathogen could offer greater insight into genetic variability than that of the comparison of a small number of loci.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
PT: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Funding acquisition, Project administration, Visualization. VG: Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. RG-I: Conceptualization, Data curation, Formal analysis, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. MP: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was financially supported in part by FONCyT-ANPCyT-MinCyT, República Argentina [PICT 02148/2018] and SeCyT-UNRC, República Argentina [PPI 2020(PPI -2022)].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1510825/full#supplementary-material
Footnotes
References
1. Pieters, M, and Maes, D. Mycoplasmosis In: J Zimmerman, editor. Diseases of swine. New Jersey, United States: John Wiley and Sons, Inc. (2019). 863–83.
2. Stakenborg, T, Vicca, J, Maes, D, Peeters, J, de Kruif, A, Haesebrouck, F, et al. Comparison of molecular techniques for the typing of Mycoplasma hyopneumoniae isolates. J Microbiol Methods. (2006) 66:263–75. doi: 10.1016/j.mimet.2005.12.002
3. Kokotovic, B, Friis, NF, Jensen, JS, and Ahrens, P. Amplified-fragment length polymorphism fingerprinting of Mycoplasma species. J Clin Microbiol. (1999) 37:3300–7. doi: 10.1128/JCM.37.10.3300-3307.1999
4. Artiushin, S, and Minion, FC. Arbitrarily primed PCR analysis of Mycoplasma hyopneumoniae field isolates demonstrates genetic heterogeneity. Int J Syst Bacteriol. (1996) 46:324–8. doi: 10.1099/00207713-46-1-324
5. Stakenborg, T, Vicca, J, Butaye, P, Maes, D, Peeters, J, de Kruif, A, et al. The diversity of Mycoplasma hyopneumoniae within and between herds using pulsed-field gel electrophoresis. Vet Microbiol. (2005) 109:29–36. doi: 10.1016/j.vetmic.2005.05.005
6. de Castro, LA, Rodrigues Pedroso, T, Kuchiishi, SS, Ramenzoni, M, Kich, JD, Zaha, A, et al. Variable number of tandem aminoacid repeats in adhesion-related CDS products in Mycoplasma hyopneumoniae strains. Vet Microbiol. (2006) 116:258–69. doi: 10.1016/j.vetmic.2006.04.022
7. Dos Santos, LF, Sreevatsan, S, Torremorell, M, Moreira, MA, Sibila, M, and Pieters, M. Genotype distribution of Mycoplasma hyopneumoniae in swine herds from different geographical regions. Vet Microbiol. (2015) 175:374–81. doi: 10.1016/j.vetmic.2014.11.018
8. Mayor, D, Zeeh, F, Frey, J, and Kuhnert, P. Diversity of Mycoplasma hyopneumoniae in pig farms revealed by direct molecular typing of clinical material. Vet Res. (2007) 38:391–8. doi: 10.1051/vetres:2007006
9. Vranckx, K, Maes, D, Calus, D, Villarreal, I, Pasmans, F, and Haesebrouck, F. Multiple-locus variable-number tandem-repeat analysis is a suitable tool for differentiation of Mycoplasma hyopneumoniae strains without cultivation. J Clin Microbiol. (2011) 49:2020–3. doi: 10.1128/JCM.00125-11
10. Felde, O, Kreizinger, Z, Sulyok, KM, Marton, S, Bányai, K, Korbuly, K, et al. Genotyping Mycoplasma hyopneumoniae isolates based on multi-locus sequence typing, multiple-locus variable-number tandem repeat analysis and analysing gene p146. Vet Microbiol. (2018) 222:85–90. doi: 10.1016/j.vetmic.2018.07.004
11. Kuhnert, P, Overesch, G, and Belloy, L. Genotyping of Mycoplasma hyopneumoniae in wild boar lung samples. Vet Microbiol. (2011) 152:191–5. doi: 10.1016/j.vetmic.2011.04.026
12. Kuhnert, P, and Overesch, G. Molecular epidemiology of Mycoplasma hyopneumoniae from outbreaks of enzootic pneumonia in domestic pig and the role of wild boar. Vet Microbiol. (2014) 174:261–6. doi: 10.1016/j.vetmic.2014.08.022
13. Mayor, D, Jores, J, Korczak, BM, and Kuhnert, P. Multilocus sequence typing (MLST) of Mycoplasma hyopneumoniae: a diverse pathogen with limited clonality. Vet Microbiol. (2008) 127:63–72. doi: 10.1016/j.vetmic.2007.08.010
14. Overesch, G, and Kuhnert, P. Persistence of Mycoplasma hyopneumoniae sequence types in spite of a control program for enzootic pneumonia in pigs. Prev Vet Med. (2017) 145:67–72. doi: 10.1016/j.prevetmed.2017.06.007
15. Sosa, C, Blois, A, Ibáñez, F, and Tamiozzo, P. Genetic diversity of Mycoplasma hyopneumoniae in Mendoza province. Rev Argent Microbiol. (2019) 51:229–33. doi: 10.1016/j.ram.2018.07.004
16. Tamiozzo, P, Zamora, R, Lucchesi, PM, Estanguet, A, Parada, J, Carranza, A, et al. MLVA typing of Mycoplasma hyopneumoniae bacterins and field strains. Vet Rec Open. (2015) 2:e000117. doi: 10.1136/vetreco-2015-000117
17. Betlach, AM, Fano, E, Sponheim, A, Valeris-Chacin, R, Dalquist, L, Singer, RS, et al. Mycoplasma hyopneumoniae genetic variability within swine production flows. Can J Vet Res. (2020) 84:310–3.
18. Garza-Moreno, L, Segalés, J, Aragón, V, Correa-Fiz, F, Pieters, M, Carmona, M, et al. Characterization of Mycoplasma hyopneumoniae strains in vaccinated and non-vaccinated pigs from Spanish slaughterhouses. Vet Microbiol. (2019) 231:18–23. doi: 10.1016/j.vetmic.2019.02.023
19. Rebaque, F, Camacho, P, Parada, J, Lucchesi, P, Ambrogi, A, and Tamiozzo, P. Persistence of the same genetic type of Mycoplasma hyopneumoniae in a closed herd for at least two years. Rev Argent Microbiol. (2018) 50:147–50. doi: 10.1016/j.ram.2017.05.002
20. Savic, B, Ivetic, V, Milicevic, V, Pavlovic, I, Zutic, M, and Gagrcin, M. Genetic diversity of Mycoplasma hyopneumoniae isolates from conventional farrow-to-finish pig farms in Serbia. Acta Vet Hung. (2010) 58:297–308. doi: 10.1556/AVet.58.2010.3.3
21. Tamiozzo, PJ, Lucchesi, PMA, and Ambrogi, A. Monitoring for Mycoplasma hyopneumoniae before and after a partial depopulation program using a typing scheme based on the polyserine repeat motif of p146. J Swine Health Prod. (2013) 21:309–12. doi: 10.54846/jshap/782
22. Bogema, DR, Deutscher, AT, Woolley, LK, Seymour, LM, Raymond, BB, Tacchi, JL, et al. Characterization of cleavage events in the multifunctional cilium adhesin Mhp684 (P146) reveals a mechanism by which Mycoplasma hyopneumoniae regulates surface topography. MBio. (2012) 3:282. doi: 10.1128/mBio.00282-11
23. Hsu, T, Artiushin, S, and Minion, FC. Cloning and functional analysis of the P97 swine cilium adhesin gene of Mycoplasma hyopneumoniae. J Bacteriol. (1997) 179:1317–23. doi: 10.1128/jb.179.4.1317-1323.1997
24. Tacchi, JL, Raymond, BB, Haynes, PA, Berry, IJ, Widjaja, M, Bogema, DR, et al. Post-translational processing targets functionally diverse proteins in Mycoplasma hyopneumoniae. Open Biol. (2016) 6:150210. doi: 10.1098/rsob.150210
25. Betlach, AM, Maes, D, Garza-Moreno, L, Tamiozzo, P, Sibila, M, Haesebrouck, F, et al. Mycoplasma hyopneumoniae variability: current trends and proposed terminology for genomic classification. Transbound Emerg Dis. (2019) 66:1840–54. doi: 10.1111/tbed.13233
26. Peakall, R, and Smouse, PE. GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research--an update. Bioinformatics. (2012) 28:2537–9. doi: 10.1093/bioinformatics/bts460
27. Falush, D, Stephens, M, and Pritchard, JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. (2007) 7:574–8. doi: 10.1111/j.1471-8286.2007.01758.x
28. Evanno, G, Regnaut, S, and Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. (2005) 14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x
29. Jakobsson, M, and Rosenberg, NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. (2007) 23:1801–6. doi: 10.1093/bioinformatics/btm233
30. Rosenberg, NA. Distruct: a program for the graphical display of population structure. Molec Ecol Notes. (2004) 4:137–8. doi: 10.1046/j.1471-8286.2003.00566.x
31. Vähä, JP, and Primmer, CR. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol. (2006) 15:63–72. doi: 10.1111/j.1365-294X.2005.02773.x
32. Maynard Smith, J, Smith, NH, O'Rourke, M, and Spratt, BG. How clonal are bacteria? Proc Natl Acad Sci U S A. (1993) 90:4384–8. doi: 10.1073/pnas.90.10.4384
33. Citti, C, Baranowski, E, Dordet-Frisoni, E, Faucher, M, and Nouvel, LX. Genomic Islands in mycoplasmas. Genes (Basel). (2020) 11:836. doi: 10.3390/genes11080836
34. Citti, C, Dordet-Frisoni, E, Nouvel, LX, Kuo, CH, and Baranowski, E. Horizontal gene transfers in mycoplasmas (Mollicutes). Curr Issues Mol Biol. (2018) 29:3–22. doi: 10.21775/cimb.029.003
35. Spratt, BG. Exploring the concept of clonality in bacteria. Met Mol Biol. (2004) 266:323–52. doi: 10.1385/1-59259-763-7:323
36. Spratt, BG, and Maiden, MC. Bacterial population genetics, evolution and epidemiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. (1999) 354:701–10. doi: 10.1098/rstb.1999.0423
37. Rosales, RS, Churchward, CP, Schnee, C, Sachse, K, Lysnyansky, I, Catania, S, et al. Global multilocus sequence typing analysis of Mycoplasma bovis isolates reveals two main population clusters. J Clin Microbiol. (2015) 53:789–94. doi: 10.1128/JCM.01910-14
38. Amram, E, Freed, M, Khateb, N, Mikula, I, Blum, S, Spergser, J, et al. Multiple locus variable number tandem repeat analysis of Mycoplasma bovis isolated from local and imported cattle. Vet J. (2013) 197:286–90. doi: 10.1016/j.tvjl.2013.03.023
39. Lindstedt, BA. Multiple-locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis. (2005) 26:2567–82. doi: 10.1002/elps.200500096
40. Balestrin, E, Kuhnert, P, Wolf, JM, Wolf, LM, Fonseca, ASK, Ikuta, N, et al. Clonality of Mycoplasma hyopneumoniae in swine farms from Brazil. Vet Microbiol. (2019) 238:108434. doi: 10.1016/j.vetmic.2019.108434
41. Michiels, A, Vranckx, K, Piepers, S, Del Pozo Sacristán, R, Arsenakis, I, Boyen, F, et al. Impact of diversity of Mycoplasma hyopneumoniae strains on lung lesions in slaughter pigs. Vet Res. (2017) 48:2. doi: 10.1186/s13567-016-0408-z
42. Maes, D, Boyen, F, Dellagostin, O, Shao, G, and Haesebrouck, F. Vaccines and vaccination against Mycoplasma hyopneumoniae In: D Maes, M Sibila, and M Pieters, editors. Mycoplasmas in swine. Leuven, Belgium: Acco Publishers (2020). 207–28.
43. Tonni, M, Boniotti, MB, Gasparrini, S, Guarneri, F, Formenti, N, Pieters, M, et al. Genomic variability of Mycoplasma hyopneumoniae within pig lung lobes. Porc Health Managem. (2021) 7:14. doi: 10.1186/s40813-021-00195-1
44. Vranckx, K, Maes, D, Sacristán, RP, Pasmans, F, and Haesebrouck, F. A longitudinal study of the diversity and dynamics of Mycoplasma hyopneumoniae infections in pig herds. Vet Microbiol. (2012) 156:315–21. doi: 10.1016/j.vetmic.2011.11.007
45. Panneitz, AK, Braga, ER, Petri, FAM, Menegatt, JCO, Driemeier, D, Maes, D, et al. Exploring the genetic diversity of Mycoplasma hyopneumoniae in pigs with pneumonia and pleurisy at slaughter. Microorganisms. (2024) 12:1988. doi: 10.3390/microorganisms12101988
46. Zhang, H, Wang, Y, Gao, L, Wang, Y, and Wei, R. Genotype diversity of Mycoplasma hyopneumoniae in Chinese swine herds based on multilocus sequence typing. BMC Vet Res. (2021) 17:347. doi: 10.1186/s12917-021-03059-6
47. Takeuti, KL, de Barcellos, DESN, de Andrade, CP, de Almeida, LL, and Pieters, M. Infection dynamics and genetic variability of Mycoplasma hyopneumoniae in self-replacement gilts. Vet Micro. (2017) 208:18–24. doi: 10.1016/j.vetmic.2017.07.007
48. Pantoja, LG, Pettit, K, Dos Santos, LF, Tubbs, R, and Pieters, M. Mycoplasma hyopneumoniae genetic variability within a swine operation. Journal of veterinary diagnostic investigation: official publication of the American Association of Veterinary Laboratory Diagnosticians, Inc. (2016) 28:175–79. doi: 10.1177/1040638716630767
Keywords: Mycoplasma hyopneumoniae , genetic structure, panmictic-clonality, MLVA, porcine enzootic pneumonia, admixed ancestry
Citation: Tamiozzo P, García V, González-Ittig RE and Pieters M (2025) Genetic structure and diversity of Mycoplasma hyopneumoniae based on a MLVA typing scheme. Front. Vet. Sci. 11:1510825. doi: 10.3389/fvets.2024.1510825
Edited by:
Amro Hashish, Iowa State University, United StatesReviewed by:
Rachel Jean Derscheid, Iowa State University, United StatesGaurav Rawal, Clinvet South Dakota, United States
Copyright © 2025 Tamiozzo, García, González-Ittig and Pieters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Pieters, cGlldDAwOTRAdW1uLmVkdQ==