
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Vet. Sci., 04 November 2024
Sec. Veterinary Neurology and Neurosurgery
Volume 11 - 2024 | https://doi.org/10.3389/fvets.2024.1485454
This article is part of the Research TopicCase Reports in Veterinary Neurology and NeurosurgeryView all 16 articles
 Gabriel Utida Eguchi1
Gabriel Utida Eguchi1 Mariana Isa Poci Palumbo1*
Mariana Isa Poci Palumbo1* Fabrício Moreira Cerri2
Fabrício Moreira Cerri2 Roberta Martins Basso2
Roberta Martins Basso2 José Paes de Oliveira-Filho2
José Paes de Oliveira-Filho2 Silvana Marques Caramalac1
Silvana Marques Caramalac1 Alexandre Secorun Borges2
Alexandre Secorun Borges2At 4 months of age, a male dog was presented with a complaint of a stiff gait following a startle response. Neurological examination revealed no deficits, but clinical myotonia was easily induced upon requesting the patient to jump. Additionally, myotonia of the upper lip muscles was observed upon manipulation. Hereditary myotonia was suspected, and electromyography confirmed the presence of myotonic potentials. Genetic testing of the myotonic patient identified a complex of mutations, including c.[1636_1639 delins AACGGG] and c.[1644 A>T], both located in exon 15 of the CLCN1 gene leading to the formation of a premature stop codon. Genetic investigations of the mother and four littermates revealed that, except for one littermate who was wild type, all others carried a copy of the mutated gene. To the best of the authors' knowledge, these mutations have not been previously reported.
Myotonia, defined as a delayed relaxation of muscles after contraction, is the prominent sign of hereditary myotonia (1). In animals, non-dystrophic hereditary myotonia associated with abnormal chloride channel 1 has been previously described in many species, including goats (2), dogs (3–15), cats (16–18), pigs (19), and buffaloes (20). These species have been confirmed to be affected. In dogs, it was previously associated with purebred dogs (3–11). Only one study illustrates the genetic mutation associated with hereditary myotonia in a family of mixed-breed dogs (12).
The knowledge of the mutations involved in the CLCN1 gene could prevent the propagation of the defect, consequently reducing the incidence of dogs suffering from the condition. CLCN1 gene mutations exhibit complex heterogeneity with different mutations found across various dog lineages (5–8, 10–12). As more mutations are discovered, more comprehensive genetic screening for suspected dogs can be developed.
Here, we describe a recessive form of hereditary myotonia in a mixed-breed dog including the clinical presentation, electrophysiological abnormalities, and genetic characterization of a novel complex exon 15 mutation of the CLCN1 gene, along with the genetic investigation of its relatives.
A 4-month-old male dog was referred with the main complaint of a stiff gait when startled or during any sudden movement initiation. This transient motor difficulty had been present since the first months of life. Otherwise, there were no other disclosed abnormalities, and the patient was reported as a normal puppy regarding growth and behavior. During the clinical evaluation, general physical parameters and neurological examination were all within normal limits. The dog had generalized muscular hypertrophy. Myotonia was easily induced, most prominently when the patient was requested to jump high steps. Additionally, due to the dog's defensive behavior, picking him up caused him to growl, and upper lip myotonia was also observed. The “warm-up phenomenon” (improvement in muscle stiffness and the ease of movement that occurs after repeated use of the affected muscles) was also present (Supplementary Video 1). The owners did not perceive the myotonic episodes as having a significant impact on the animal's overall quality of life. They noted that during the episodes, the patient might be in discomfort, but no signs of vocalization or pain behavior were described.
Considering the clinical myotonia, warm-up phenomenon, muscle hypertrophy, and absence of weakness, non-dystrophic hereditary myotonia associated with abnormal chloride channel 1 was suspected. Therefore, electromyography (EMG) was performed, and whole blood was sent for genetic investigation. Additional blood tests, including a complete blood count, creatinine, urea, alanine aminotransferase, alkaline phosphatase, total protein, and creatine kinase enzyme, were also conducted.
Blood samples were also collected from the mother and four littermates for genetic analysis. Unfortunately, no neurological examination was possible for these animals. However, no similar clinical signs were reported, and based on videos, photos, and a general physical examination performed by a generalist veterinarian at the time of blood collection, no abnormalities were found.
EMG was performed (Neurotec Neuromap® EQPE041) without any anesthetic medications. The band-pass filter was set between 30 and 10,000 Hz, and display settings were configured to 100 μV/division sensitivity and 10 ms/division sweep speed. The electrode was a concentric needle (Neurotecnologia® D039035408 40 mm, 28 G), and the grounding was a surface electrode with an alligator clip placed on the inguinal skin.
Myotonic potentials were present and myotonia was also characterized by its “wax and wane” sound in the tibialis cranialis, gastrocnemius, biceps femoris, and extensor carpi radialis muscles (Figure 1; Supplementary Video 2). Ancillary blood analysis showed no abnormalities.
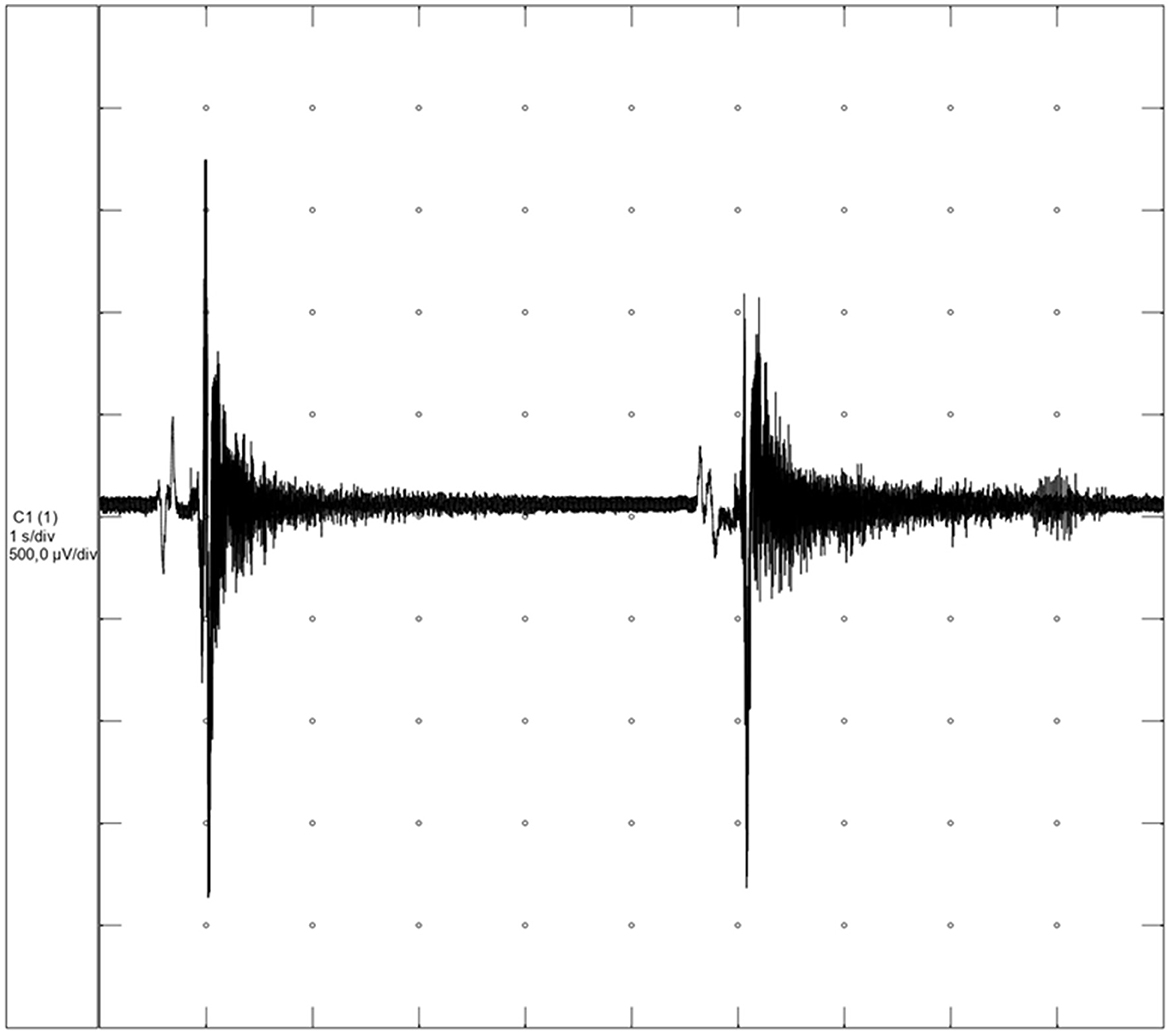
Figure 1. Myotonic discharges from the dog with hereditary myotonia (Neurotec Neuromap® EQPE041; 500 μV/division sensitivity and 1 s/division).
Blood samples obtained from the myotonic dog (dog 1), its mother (dog 2), littermates (dogs 3–6), and an unrelated wild-type dog (dog 7, sourced from the DNA bank of the Veterinary Molecular Biology Laboratory of the Department of Veterinary Clinical Science of the FMVZ-Unesp) underwent genomic DNA extraction and purification using the DNeasy Blood & Tissue kit (Qiagen) following the manufacturer's instructions. DNA concentration was then measured using spectrophotometry (Nanodrop® 2000—Thermo Scientific™).
The primers used to amplify the 23 exons (coding sequence) of the canine CLCN1 gene (Supplementary Table 1), along with the PCR conditions and thermocycling, were previously described (12). The amplicons were analyzed by 1.5% agarose gel electrophoresis, purified with magnetic beads, and sequenced via Sanger sequencing. The electropherograms were examined using Geneious Prime® 2019.1.3 software. The obtained sequences were compared to the reference of the CLCN1 gene from Canis lupus familiaris (GenBank NP_001003124.1).
The alignment of sequences from the myotonic dog (dog 1), its mother (dog 2), littermates (dogs 3–6), and a wild-type dog (dog 7), along with the GenBank reference sequence (NP_001003124.1), revealed a complex of mutations c.[1636_1639delinsAACGGG] and c.[1644A>T], both in exon 15. This second mutation, c.[1644A>T], resulted in the modification of the amino acid sequence p.Glu548Asp. The chloride channel in the mutated animal has the initial 545 amino acids preserved, followed by the generation of four different amino acids (546–549). The variant complex c.[1636_1639delinsAACGGG] caused the formation of a premature stop codon in exon 15 (positions c.1646-1648). This results in a truncated protein with ~400 fewer amino acids compared to the normal CLC protein (NP_001003124.1), which has 976 amino acids in the dog (Figure 2).
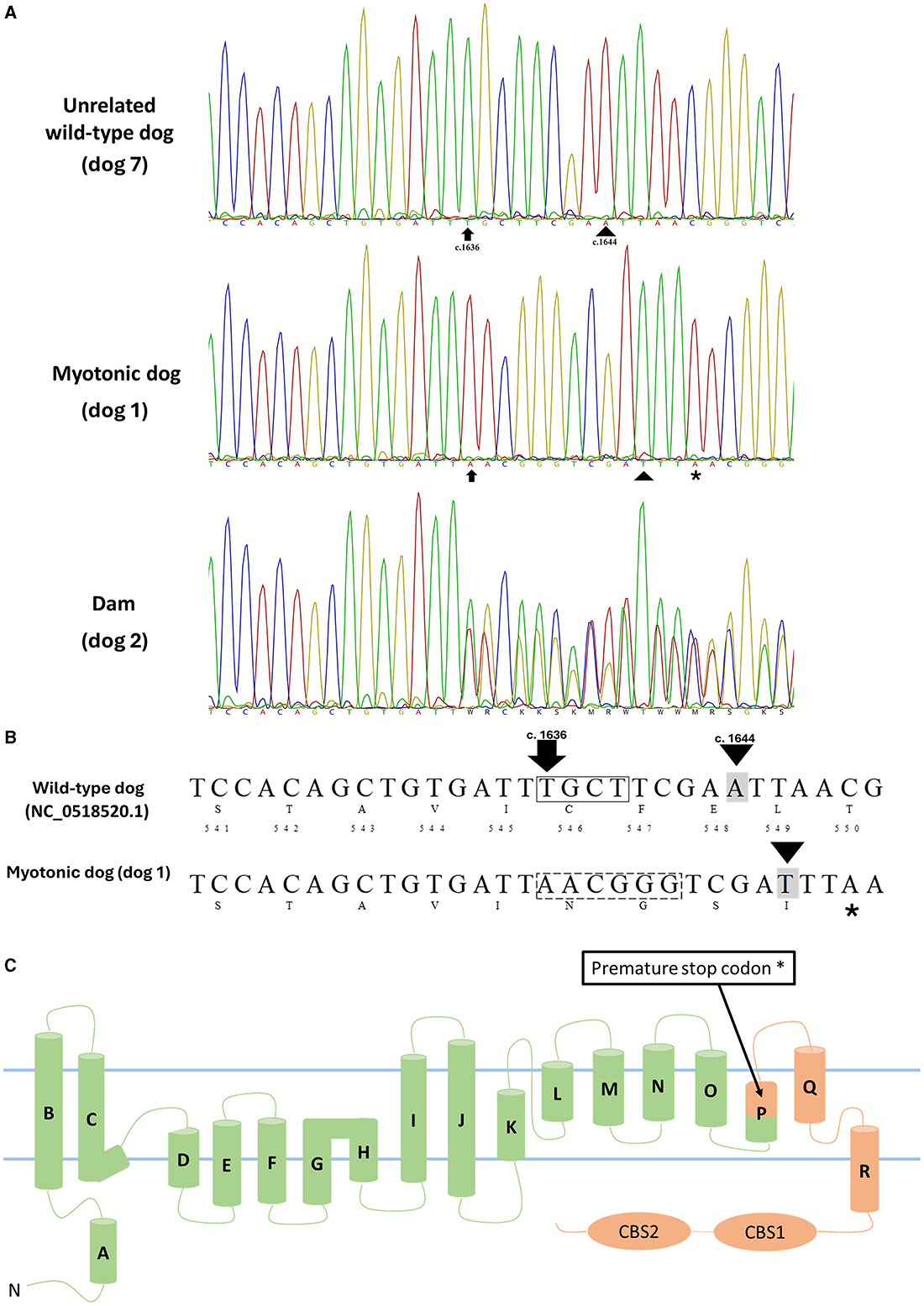
Figure 2. (A) Partial capillary sequencing chromatogram for exon 15 of CLCN1 in an unrelated wild-type dog (dog 7), a myotonic dog (dog 1), and the dam (dog 2). The arrow indicates the start of the mutation (c.1636-1639delinsAACGGG), and the arrowhead indicates the second observed mutation (c.1644 A>T). (B) Schematic representation of nucleotide and amino acid sequences of the wild-type dog (top, NC_0518520.1) and the myotonic dog (bottom) with the corresponding nucleotide numbers and amino acids. The box representing four nucleotides in the wild-type sequence indicates the amino acids deleted in the myotonic animal (c.1636-1639del TGCT), while the hatched (- - -) box in the myotonic dog sequence (AACGGG) results from the insertion of 6 nucleotides (c.1636_1639delins AACGGG). The described complex mutation results in a frameshift and the formation of a premature stop codon. Nucleotide highlighted in gray indicates the observed substitution (c.1644 A>T). *Indicates a premature stop codon (TAA - c.1646-1648). (C) Membrane topology model of the human skeletal muscle chloride channel monomer, ClC-1 [modified from Brenes et al. (25)], representing the premature stop codon formation and compromising the structure of α-helices P (encompassing amino acids 541-551), Q and R as well as the CBS1 and 2 domains in addition to the P-Q and Q-R loops and the C-terminal, all of which play key roles in the proper functioning of this channel. In green are the structures that are preserved, and in orange are the structures that are absent in the dog with myotonia.
Dog 4 had no alterations in the region of the observed mutation (wild type). Dogs 3, 5, and 6 each presented a copy of the mutated gene. The mother (dog 2) also showed the presence of a copy of the mutated sequence. Dogs 2, 3, 5, and 6 were considered heterozygous for the described mutations.
No treatment was instituted as the condition didn't seem to impact the patient's quality of life. Nonetheless, owner education was emphasized as a crucial aspect of managing this condition. Educating the owners about the nature of clinical myotonia is important because the episodes can cause psychological distress for some people who may mistakenly believe the dog is in active pain during these events. Additionally, owners were advised to remove all affected relatives from breeding programs to prevent the propagation of the genetic mutation.
Two years after the initial evaluation, the patient showed no worsening of the condition in terms of frequency and severity. The dog exhibited normal growth and behavior, and despite some visible generalized muscle hypertrophy (Supplementary Video 1), no other clinical abnormalities were found.
Delayed relaxation of skeletal muscles after onset of movements or startle, muscle hypertrophy, and the “warm-up” phenomenon are highly indicative of hereditary myotonia related to chloride channel 1 abnormalities. The “warm-up” phenomenon serves as a valuable clinical tool in diagnostic criteria for hereditary myotonia (HC), distinguishing myotonic events from paradoxical myotonia, where muscle rigidity typically worsens with exercise (12, 21). Myotonia in domestic animals can also occur with sodium channel abnormalities, but these are usually associated with weakness, which was not observed in this dog.
While myotonia caused by mutations in CLCN1 has been well-documented in purebred dogs (3–11), reports and genetic investigations in mixed-breed dogs remain limited but have been described (12). To the best of our investigation, no pure-breed ancestors could be identified in our patient.
Clinical myotonia was confirmed through EMG evaluation, which detected myotonic discharges [frequencies may range from 20 to 150 Hz, potential amplitudes range from 10 to 1,000 mV and last 500 ms or longer (22, 23)] resulting in a characteristic sound and distinguishes them from cases of pseudomyotonia, where clinical myotonia is present without myotonic potentials (1, 21). A limitation of this study is the lack of histological analysis of the musculature, as the biopsy was not authorized by the owner. This analysis is important to rule out cases of dystrophic myotonia, which was not the initial suspicion for this dog. In cases of hereditary myotonia, the muscle is usually only characterized by variation in the diameter of the fibers (15, 16).
Mutations in the CLCN1 gene responsible for hereditary myotonia have been documented in various dog breeds, including Miniature Schnauzers (6), Australian Cattle Dogs (8), Labrador Retrievers (10), American Bulldogs (11). However, in the dog with myotonia from the present study, none of the previously reported mutations in the CLCN1 gene associated with myotonia in dogs or other domestic animals were identified. Only one study has reported a mutation in the CLCN1 gene in a family of mixed-breed dogs, and that mutation was a complex variant in exon 6, different from the variant found in our study, which is located in exon 15 (12). Several mutations have been described in humans in exon 15, but not at the same position as the complex variant found in our study (3, 14, 24). The complex mutation observed in this patient resulting in a premature stop codon affected the structure of α-helices P (encompassing amino acids 541–551), Q (555–571), and R (576–585), as well as the 2 intracellular C-terminal cystathionin-β-synthase (CBS) segments (609–876), in addition to the P-Q and Q-R and the C-terminal loops, all of which play key roles in the proper functioning of CLCN1. This importance is underscored by ~110 mutations located in this region that have been reported in humans, where amino acid alterations have led to congenital myotonia (25). Further supporting the significance of these α-helices (P, Q, and R) and CBS domains, congenital myotonia has also been described in animals, with point mutations in this region reported in a horse (26), cat (16), dogs (8–10), and goats (27). To highlight the importance of this region of the CLCN1 gene in cases of myotonia in animals, we can mention goats, which were the first domesticated species described with myotonia and the responsible mutation is a single nucleotide change that results in the substitution of proline for a conserved alanine residue in the CBS (27). Therefore, it is reasonable to consider that, even in the absence of patch-clamp studies, the truncated CLCN1 (~400 aa shorter than the normal chloride channel 1) observed in the dog from the present study is responsible for the clinical myotonia. Considering the distribution of findings in the studied family and the sequencing results, it is concluded that this is an autosomal recessive form of the disease, similar to all other cases of hereditary myotonia associated with chloride channel mutations described in domestic animals to date.
Despite being a disease that typically does not severely compromise the quality of life of affected dogs, myotonia can still cause some limitations and predispose patients to accidental falls. In some cases, myotonic episodes may be frequent enough to hinder normal daily activities and require treatment attempts (10). Hereditary myotonia does not seem to reduce the life expectancy of affected individuals. Patients are often diagnosed as adults (7, 8, 10, 12) and typically experience no severe decline in quality of life or developmental issues. The patient described in our study had a follow-up period of 2 years, during which no deterioration in condition was observed. Nevertheless, from an owner's perspective, these episodes may resemble completely different physiological conditions, such as cramping syndromes, and thus owners may extrapolate the discomfort from any muscle cramp they have experienced themselves. Therefore, owner education is crucial regarding the nature of this condition and any available treatment options. When treatment is chosen, details such as type of medication, dosage, and expected outcomes have been previously reported.
Moreover, it is essential not to overlook owner education regarding the hereditary nature of this disease, and preventing mating and breeding of affected individuals should be strongly advised.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The animal described in this study were received as clinical patient in the Veterinary Hospital of Universidade Federal de Mato Grosso do Sul—UFMS (Animal Ethics Committee protocol number 1.281/2023). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners of the animals for the publication of this case report.
GE: Data curation, Investigation, Writing – original draft, Writing – review & editing. MP: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. FC: Data curation, Writing – original draft, Writing – review & editing. RB: Data curation, Writing – original draft, Writing – review & editing. JO-F: Writing – original draft, Writing – review & editing. SC: Writing – review & editing. AB: Data curation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was financed in part by the Universidade Federal de Mato Grosso do Sul—UFMS/MEC - Brazil.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1485454/full#supplementary-material
Supplementary Video 1 | Dog with hereditary myotonia at first presentation with 4 months old and at 2-years follow-up; clinical myotonia episodes and “warm-up” phenomenon are observed. Note the muscle hypertrophy and upper lip myotonia (2-years).
Supplementary Video 2 | EMG evaluation of the dog with hereditary myotonia (Neurotec Neuromap® EQPE041; band pass filter, 30 to 10,000 Hz; display settings, 100 μV/division sensitivity and 10 ms/division sweep speed; sixty Hz filter “on”). Note myotonic discharges that wax and wane and characteristic sound.
Supplementary Table 1 | Primers for PCR amplification.
1. Vite CH. Myotonia and disorders of altered muscle cell membrane excitability. Vet Clin North Am Small Anim Pract. (2002) 32:169–87. doi: 10.1016/S0195-5616(03)00084-6
2. Bryant SH. Myotonia in the goat. Ann N Y Acad Sci. (1979) 317:314–25. doi: 10.1111/j.1749-6632.1979.tb37355.x
3. Farrow BRH, Malik R. Hereditary myotonia in the Chow Chow. J Small Anim Practc. (1981) 22:451–65. doi: 10.1111/j.1748-5827.1981.tb00629.x
4. Hill SL, Shelton GD, Lenehan TM. Myotonia in a cocker spaniel. JAAHA. (1995) 31:506–9. doi: 10.5326/15473317-31-6-506
5. Rhodes TH, Vite CH, Giger U, Patterson DF, Fahlke C, George AL Jr, et al. missense mutation in canine ClC-1 causes recessive myotonia congenita in the dog. FEBS Lett. (1999) 456:54–8. doi: 10.1016/S0014-5793(99)00926-6
6. Vite CH, Melniczek J, Patterson D, Giger U. Congenital myotonic myopathy in the Miniature Schnauzer: an autosomal recessive trait. J Hered. (1999) 90:578–80. doi: 10.1093/jhered/90.5.578
7. Bhalerao DP, Rajpurohit Y, Vite CH, Giger U. Detection of a genetic mutation for myotonia congenita among Miniature Schnauzers and identification of a common carrier ancestor. Am J Vet Res. (2002) 63:1443–7. doi: 10.2460/ajvr.2002.63.1443
8. Finnigan DF, Hanna WJB, Poma R, Bendall AJ. A novel mutation of the CLCN1 gene associated with myotonia hereditaria in an Australian cattle dog. J Vet Intern Med. (2007) 21:458–63. doi: 10.1111/j.1939-1676.2007.tb02990.x
9. Lobetti RG. Myotonia congenita in a Jack Russell terrier. J S Afr Vet Assoc. (2009) 80:106–7. doi: 10.4102/jsava.v80i2.181
10. Quitt PR, Hytönen MK, Matiasek K, Rosati M, Fischer A, Lohi H. Myotonia congenita in a Labrador Retriever with truncated CLCN1. Neuromuscul Disord. (2018) 28:597–605. doi: 10.1016/j.nmd.2018.05.002
11. Rodrigues DDJ, Damasceno AD, Araújo CETD, Torelli SR, Fonseca LGH, Delfiol DJZ, et al. Hereditary myotonia in American Bulldog associated with a novel frameshift mutation in the CLCN1 gene. Neuromuscul Disord. (2020) 30:991–8. doi: 10.1016/j.nmd.2020.10.007
12. Chimenes ND, Caramalac SM, Caramalac SM, Fernandes TD, Basso RM, Cerri FM, et al. A complex CLCN1 variant associated with hereditary myotonia in a mixed-breed dog. J Vet Diagn Invest. (2023) 35:414–7. doi: 10.1177/10406387231176736
13. Mazón MJ, Barros F De la Pena P, Quesada JF, Escudero A, Cobo AM, Pascual-Pascual SI, et al. Screening for mutations in Spanish families with myotonia functional analysis of novel mutations in CLCN1 gene. Neuromuscul Disord. (2012) 22:231–43. doi: 10.1016/j.nmd.2011.10.013
14. Palma Milla C, Prior De Castro C, Gómez-González C, Martínez-Montero PI, Pascual Pascual S, Molano Mateos J. Myotonia congenita: mutation spectrum of CLCN1 in Spanish patients. J Genet. (2019) 98:1–10. doi: 10.1007/s12041-019-1115-0
15. Shelton GD, Mickelson JR, Friedenberg SG, Cullen JN, Graham K, Carpentier MC, et al. Variants in CLCN1 and PDE4C associated with muscle hypertrophy, dysphagia, and gait abnormalities in young French bulldogs. Animals. (2024) 14:722. doi: 10.3390/ani14050722
16. Gandolfi B, Daniel RJ, O'Brien DP, Guo LT, Youngs MD, Leach SB, et al. A novel mutation in CLCN1 associated with feline myotonia congenita. PLoS ONE. (2014) 9:e109926. doi: 10.1371/journal.pone.0109926
17. Corrêa S, Basso RMB, Cerri FM, Oliveira-Filho JP, Araújo JP Jr, Torelli SR, et al. Hereditary myotonia in cats associated with a new homozygous missense variant pAla331Pro in the muscle chloride channel ClC-1. J Vet Intern Med. (2023) 37:2498–503. doi: 10.1111/jvim.16837
18. Woelfel C, Meurs K, Friedenberg S, DeBruyne N, Olby NJ, A. novel mutation of the CLCN1 gene in a cat with myotonia congenita: diagnosis and treatment. J Vet Int Med. (2022) 36:1454. doi: 10.1111/jvim.16471
19. Araújo CET, Oliveira CMC, Barbosa JD, Oliveira-Filho JP, Resende LAL, Badial PR, et al. A large intragenic deletion in the CLCN1 gene causes hereditary myotonia in pigs. Sci Rep. (2019) 9:15632. doi: 10.1038/s41598-019-51286-7
20. Borges AS, Barbosa JD, Resende LAL, Mota LSLS, Amorim RM, Carvalho TL, et al. Clinical and molecular study of a new form of hereditary myotonia in Murrah water buffalo. Neuromuscul Disord. (2013) 23:206–13. doi: 10.1016/j.nmd.2012.11.008
21. Stee K, Van Poucke M, Peelman L, Lowrie M. Paradoxical pseudomyotonia in English Springer and Cocker Spaniels. J Vet Intern Med. (2020) 34:253–7. doi: 10.1111/jvim.15660
22. Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practice. 3rd ed. New York, NY: Oxford University Press (2001). p. 991.
23. Heatwole CR, Moxley RT. The nondystrophic myotonias. Neurotherapeutics. (2007) 4:238–51. doi: 10.1016/j.nurt.2007.01.012
24. Ivanova EA, Dadali EL, Fedotov VP, Kurbatov SA, Rudenskaya GE, Proskokova TN, et al. The spectrum of CLCN1 gene mutations in patients with nondystrophic Thomsen's and Becker's myotonias. Genetika. (2012) 48:952–61. doi: 10.1134/S1022795412090049
25. Brenes O, Pusch M, Morales F. ClC-1 chloride channel: inputs on the structure-function relationship of myotonia congenita-causing mutations. Biomedicines. (2023) 11:2622. doi: 10.3390/biomedicines11102622
26. Wijnberg ID, Owczarek-Lipska M, Sacchetto R, Mascarello F, Pascoli F, Grünberg W, et al. A missense mutation in the skeletal muscle chloride channel 1 (CLCN1) as candidate causal mutation for congenital myotonia in a New Forest pony. Neuromusc Disord. (2012) 22:361–7. doi: 10.1016/j.nmd.2011.10.001
Keywords: chloride channel, neuromuscular disorder, hereditary disease, electroneuromyography, congenital myotonia
Citation: Eguchi GU, Palumbo MIP, Cerri FM, Basso RM, Oliveira-Filho JPd, Caramalac SM and Borges AS (2024) Case report: A CLCN1 complex variant mutation in exon 15 in a mixed-breed dog with hereditary myotonia. Front. Vet. Sci. 11:1485454. doi: 10.3389/fvets.2024.1485454
Received: 23 August 2024; Accepted: 24 September 2024;
Published: 04 November 2024.
Edited by:
Edward E. Patterson, University of Minnesota Twin Cities, United StatesReviewed by:
Anita Shea, Massey University, New ZealandCopyright © 2024 Eguchi, Palumbo, Cerri, Basso, Oliveira-Filho, Caramalac and Borges. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Isa Poci Palumbo, bWFyaWFuYS5wYWx1bWJvQHVmbXMuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.