 Brian P. Bourke1,2,3*
Brian P. Bourke1,2,3* Robert J. Dusek4
Robert J. Dusek4 Koray Ergunay1,2,3,5
Koray Ergunay1,2,3,5 Yvonne-Marie Linton1,2,3Sergei V. Drovetski6
Yvonne-Marie Linton1,2,3Sergei V. Drovetski6- 1Walter Reed Biosystematics Unit, Museum Support Center MRC-534, Smithsonian Institution, Suitland, MD, United States
- 2One Health Branch, Walter Reed Army Institute of Research, Silver Spring, MD, United States
- 3Department of Entomology, Smithsonian Institution—National Museum of Natural History, Washington, DC, United States
- 4U.S. Geological Survey, National Wildlife Health Center, Madison, WI, United States
- 5Hacettepe University, Department of Medical Microbiology, Ankara, Türkiye
- 6U.S. Geological Survey, Eastern Ecological Science Center at the Patuxent Research Refuge, Laurel, MD, United States
The threat posed by emerging infectious diseases is a major concern for global public health, animal health and food security, and the role of birds in transmission is increasingly under scrutiny. Each year, millions of mass-reared game-farm birds are released into the wild, presenting a unique and a poorly understood risk to wild and susceptible bird populations, and to human health. In particular, the shedding of enteric pathogens through excrement into bodies of water at shared migratory stop-over sites, and breeding and wintering grounds, could facilitate multi-species long-distance pathogen dispersal and infection of high numbers of naive endemic birds annually. The Mallard (Anas platyrhynchos) is the most abundant of all duck species, migratory across much of its range, and an important game species for pen-rearing and release. Major recent population declines along the US Atlantic coast has been attributed to game-farm and wild mallard interbreeding and the introduction maladaptive traits into wild populations. However, pathogen transmission and zoonosis among game-farms Mallard may also impact these populations, as well as wildlife and human health. Here, we screened 16 game-farm Mallard from Wisconsin, United States, for enteric viral pathogens using metatranscriptomic data. Four families of viral pathogens were identified – Picobirnaviridae (Genogroup I), Caliciviridae (Duck Nacovirus), Picornaviridae (Duck Aalivirus) and Sedoreoviridae (Duck Rotavirus G). To our knowledge, this is the first report of Aalivirus in the Americas, and the first report of Calicivirus outside domestic chicken and turkey flocks in the United States. Our findings highlight the risk of viral pathogen spillover from peri-domestically reared game birds to naive wild bird populations.
Introduction
Emerging infectious diseases are a major threat to global public and animal health (1). Since the beginning of the 21st century, there have been a series of severe global disease outbreaks primarily caused by zoonotic viral pathogens (2–5). Investigations into the zoonotic origins of recent outbreaks have flagged the risks posed by birds, most notably in the transmission of West Nile Virus (WNV) (6, 7) and Highly Pathogenic Avian Influenza (HPAI) (8, 9).
Each year, billions of birds undertake seasonal migration, with some moving between the high latitudes of the northern and southern hemispheres and travelling tens of thousands of kilometers annually. Such vagility provides bird pathogens a unique opportunity to be rapidly dispersed across the globe (10). Migratory birds often congregate at stopover sites, which offer shelter to rest and feed, and the high bird densities and multi-species clustering increases opportunities for pathogen transmission (11, 12). The migratory routes of birds have played a key role in the global transmission of HPAI (13). The arrival of HPAI in North America in 2014, and 2022, is attributed to birds using migratory flyways on their way from summer breeding grounds in Siberia (14) and northwest Europe (15), respectively. The transmission of this virus occurs via the fecal-oral route and typically through inhalation of infectious aerosols, or contact with contaminated water (16, 17). Predatory and scavenging birds can also become infected by consumption of infected prey and/or carcasses (18, 19). In addition to birds serving as highly mobile pathogen reservoirs, they can transport ectoparasites, which may in turn serve as vectors of pathogens. Birds can thus also be vehicles for the transport of vector-borne pathogens along migratory flyways. Studies from Europe and North America indicate migratory birds carry exotic ectoparasites, including ticks (20, 21). Studies screening these hitch-hiking ticks have confirmed Crimean-Congo hemorrhagic fever (CCHF) virus (22–25), and Borrelia (26) and Babesia (27) species that can cause disease in many vertebrates, including humans.
In Europe and North America, game birds (e.g., Common pheasant [Phasianus colchicus], Quail [Colinus virginianus], Chukar partridge [Alectoris chukar], red-legged partridges [Alectoris rufa], Mallard [Anas platyrhynchos]) are reared en mass and released into the wild to bolster game populations for hunting (28–30). The scale of this industry is vast, involving many tens of millions of birds annually. In Europe, game-farm Mallard releases are estimated at almost 3 million annually and are comparable in size to the continent’s wild mallard breeding population (30–33). In North America more than 300 licensed hunting preserves, located primarily along the Atlantic and Mississippi flyways, release almost 300,000 game-farm Mallard annually (34). Interbreeding between game-farm and wild Mallard populations is believed to have formed a Mallard hybrid swarm in North America, resulting in population decline due to the introduction of artificially created and maladaptive genes (35, 36). However, interactions among game-farm and wild Mallard can also introduce emerging and novel pathogens into naive wild bird populations, which may also explain population declines. While pathogen transmission risks associated with wild and commercial poultry flocks has been well documented (37–39), the transmission risk from game-farm birds to wild birds, and other vertebrate hosts, including humans, is poorly understood (40).
The global scale and magnitude of avian viral outbreaks has the potential not only to threaten vulnerable species with extinction (41–43) and decimate wild and domestic bird populations (44–46) across the globe, but is also known to spill over into terrestrial and marine mammal species, offering opportunities for adaptation, new transmission cycles, and zoonosis (47). Characterizing viral pathogen diversity particularly at avian, human, and environment interfaces is therefore a key element to safeguarding global health security. Although the risks and impacts of HPAI and WNV have been widely reported, a variety of other, less well-characterized viral pathogens also pose a threat to bird populations, the food industry, and human health. These include Newcastle Disease Virus (NDV), which causes disease in domestic and wild birds (48), and Rotavirus (ROV) and Eastern equine encephalitis virus (EEEV), which cause disease in both birds (49, 50) and humans (51, 52).
Advances in whole genome sequencing technologies allow new opportunities to screen, identify, and characterize viral pathogens. Metagenomics and metatranscriptomics allows the full nucleotide content of a sample to be sequenced concurrently, thus providing an agnostic approach to characterizing the viral community. Here we use a metatranscriptomics approach to analyze the viromes of game-farm Mallard to expose known and novel pathogens that may pose a risk to wild mallard and susceptible wild bird populations and offer a potential route for reverse zoonosis in wildlife.
Materials and methods
Sampling and nucleic acid extraction
We sampled the cecal content of 16 fully grown 8-week-old (juvenile) female Mallard that were raised in an open-air pond in Wisconsin, United States. None of the birds showed signs of ill health. Approximately 100 mg of cecal content was sampled from each bird immediately after euthanasia by CO2 according to the protocol approved by the ACUC of the U.S. Geological Survey, National Wildlife Health Center (tracking number EP210811). The cecum wall was punctured with a sterile scalpel blade and cecal content (two pin-head size drops) was pushed directly into a 2 mL screw cap tube pre-filled with 1 mL of DNA/RNA Shield and a mix of 0.5 mm and 0.1 mm ultra-high density BashingBeads (Zymo Research, Irvine, CA, United States). Samples were stored at 4°C until nucleic acid extraction within 30 days of collection. Total RNA from the cecal samples was extracted using the ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research), according to the manufacturer’s instructions. The RNA concentration of each sample was then measured on a Qubit 4.0 (ThermoFisher Scientific, Waltham, MA, United States) using the High-Sensitivity RNA Assay Kit (ThermoFisher Scientific).
Library preparation and sequencing
Samples were prepared for sequencing by depleting ribosomal RNA using QIAseq FastSelect -rRNA HMR Kit (QIAGEN, Germantown MD) and RNA libraries were then prepared using the NEB Ultra II RNA kit (New England Biolabs, Inc., Ipswich, MA, United States). All libraries were sequenced simultaneously on an Illumina HiSeq 4,000 platform (2 × 150 bp; Illumina, Inc., San Diego, CA, United States). Library preparation and sequencing was performed by GeneWiz/Azenta Life Sciences (South Plainfield, NJ, United States). The number of pair-ended reads obtained from individual samples ranged from 49,218,452 to 74,399,926 [median 60,564,664].
Data analysis
Demultiplexed raw Illumina data were first adaptor trimmed and quality filtered using fastp v0.23.3 (−-qualified_quality_phred = 15; −-unqualified_percent_limit = 40) (53, 54). Following this step, host genome reads were removed from the dataset using KneadData v0.12.01 and the Mallard reference assembly.2 The bowtie alignment option within KneadData was set to “--very-sensitive-local” (i.e., -D 20; -R 3; -N 0; -L 20; -i S,1,0.50).
The cleaned data was then de-novo assembled using MEGAHIT v1.2.9 and its “basic usage” setting for paired end libraries (55). De novo assembled contigs were then aligned to the National Center for Biotechnology Information (NCBI) protein non-redundant (nr) database (http://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz; accessed April 5 2023) using Diamond (−-long-reads; −-evalue 1e-9) (56, 57) and taxonomically binned using Megan v6.24.20 (−-minSupport 1; −-minPercentIdentity 70; −-maxExpected 1.0E-9; −-lcaAlgorithm longReads; −-lcaCoveragePercent 51; −-longReads) (58, 59). Contigs classified as viruses of potential pathogenic importance (vertebrate pathogens) based on a review of the literature were then exported from Megan. Putative Open Reading Frames (ORFs) and amino acid sequences were predicted using ORF finder.3 ORFs were then aligned with close and/or congeneric relatives, as described in the International Committee on Taxonomy of Viruses (ICTV) report chapters,4 using Muscle implemented in SeaView v5.0.4 (60). The removal of spurious and/or poorly aligned regions from the multiple sequence alignment was achieved using trimAl v1.4 (61) and its automated heuristic approach (option-automated1).
Maximum likelihood phylogenetic analysis was performed in IQ-TREE 2 v2.2.0 (62) with 1,000 replicates. Optimal model selection was performed using the -m MFP option, and the models were restricted to those designed for viruses using the -msub viral option. The resulting consensus trees contain UFBoot support values, which are more unbiased than standard nonparametric bootstrap values, and support for a clade considered at ≥95%.
Screening for recombination among virus genomes was performed through algorithms implemented in the RDP4 software using the seven primary algorithms available (RDP, GENECONV, BOOTSCAN/RECSCAN, MAXCHI, Chimaera, 3SEQ and SISCAN) (63).
Results
A variety of viruses were detected in the metatranscriptomic data from these Mallard gut samples (Table 1). None of the viruses detected displayed a signal of recombination in a majority of the seven default RDP4 tests.
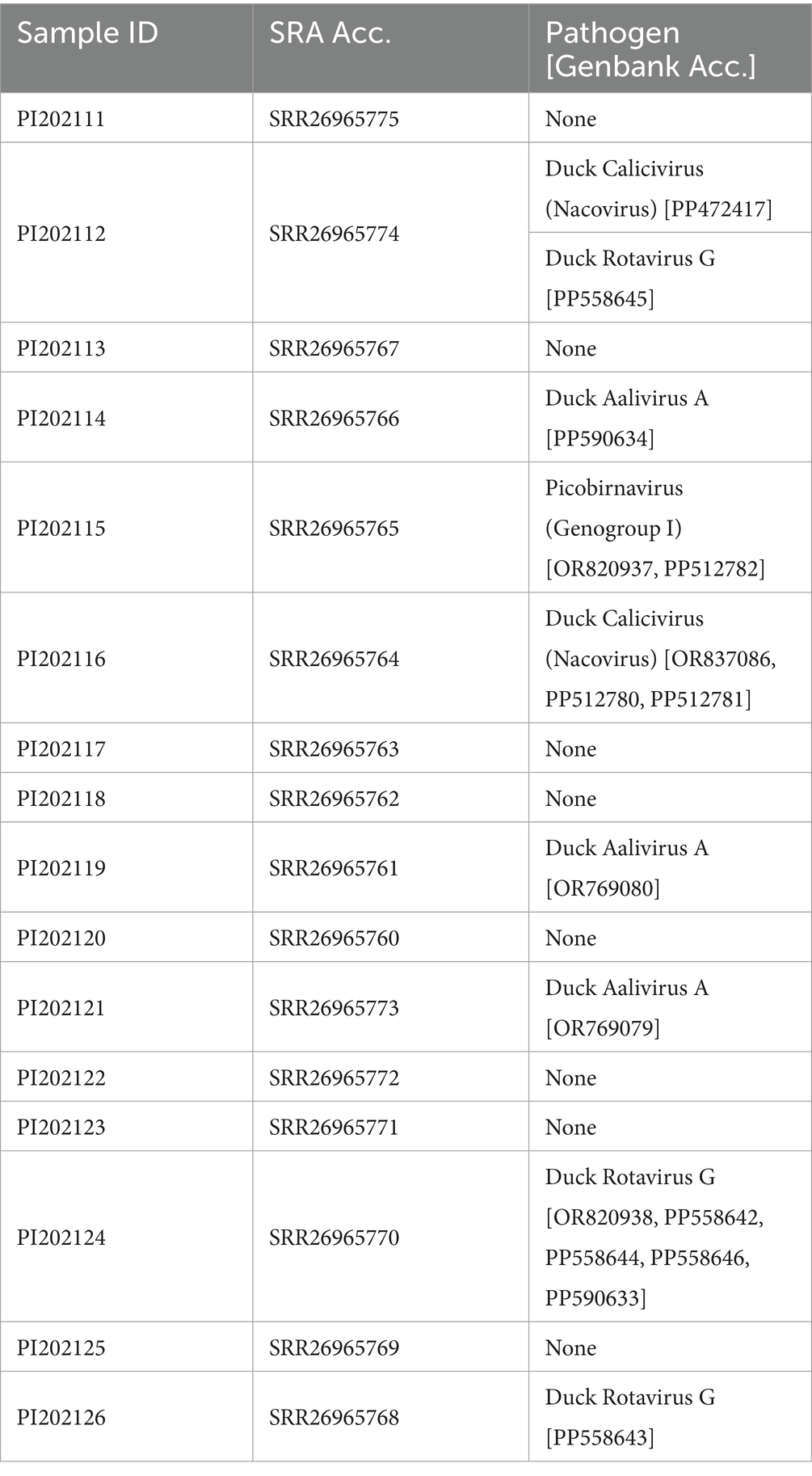
Table 1. Summary of metatranscriptomic data and viral pathogen detection from the cecum of 16 captive Mallard from Wisconsin, United States (Bioproject: PRJNA1045981).
Picobirnaviruses
Two de novo assembled contigs from sample PI202115 were classified as Picobirnavirus in Diamond-Megan analysis. They were from the two linear dsRNA Picobirnavirus segments (dsRNA1 and dsRNA2). The contig from the dsRNA1 segment (1,375 base pairs) represented the viral capsid protein (ORF3), which was 457 amino acids in length. Although the phylogenetic signal from the viral capsid protein was poor, based on the lack of branch support (Supplementary Figure S1), the PI202115 sequence clustered (UFBoot support: 100%) with chicken picobirnavirus from Brazil (Genbank Acc. AXL64599), at 91.98% identity.
The contig from the dsRNA2 segment (1,669 base pairs; Genbank Acc. OR820937) represented the viral RNA-dependent RNA polymerase (RdRp) and was 529 amino acids in length. The sequence contains the seven conserved core polymerase domain motifs of picobirnaviruses (64). Phylogenetic analysis of the RdRP region showed that the PI202115 sequence clustered (UFBoot support: 95%) with a pig picobirnavirus (UAW00636), within the Genogroup I clade (UFBoot support: 100%), which can be resolved from the Genogroup II (UFBoot support: 100%) and Genogroup III (UFBoot support: 100%) found in the Picobirnaviridae tree (Figure 1), as described in the ICTV Report on Picobirnaviridae (65). The picobinavirus RdRp region from PI202115 shared 80.64% identity (based on amino acid sequence) with the pig picobirnavirus (UAW00636).
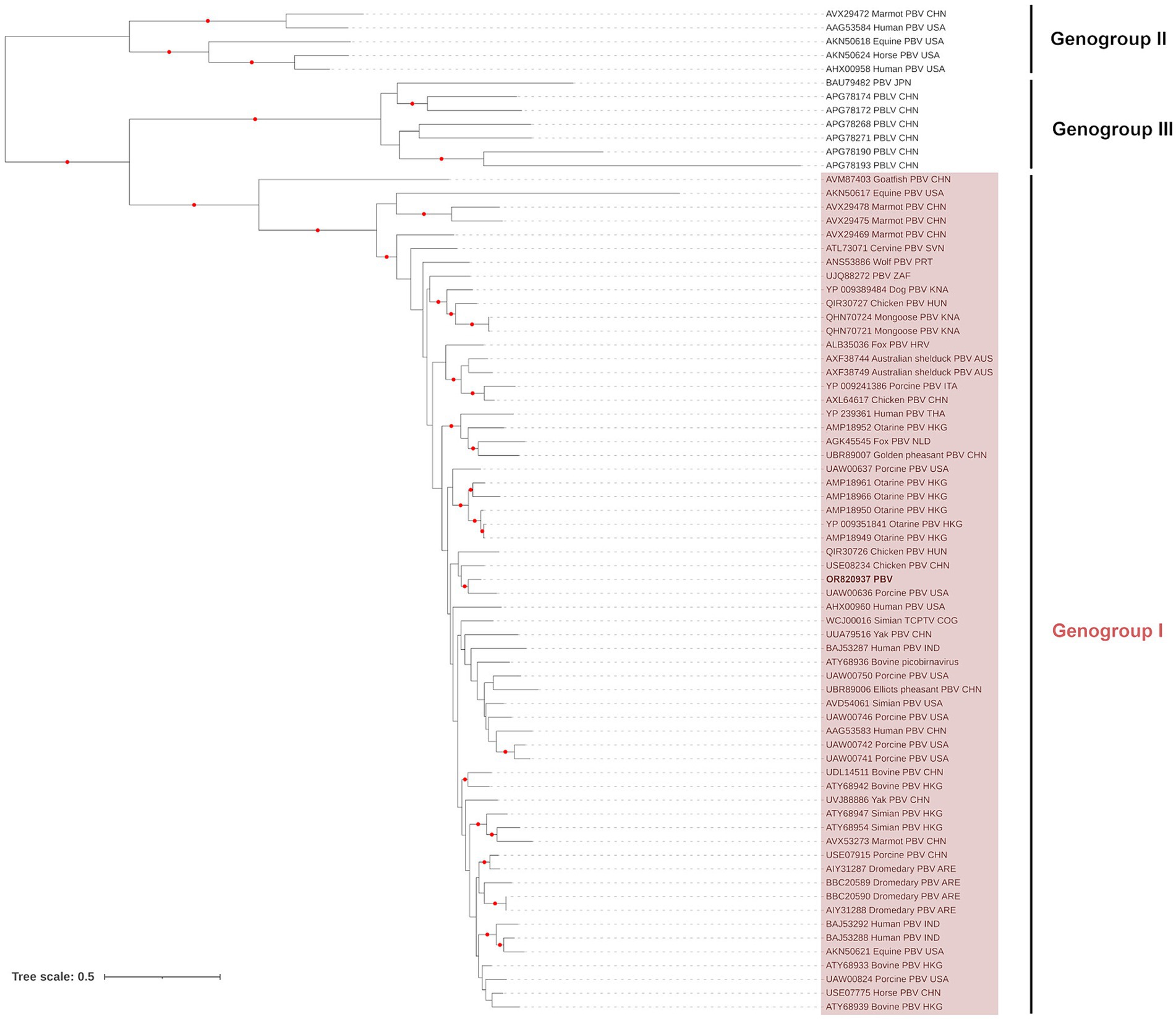
Figure 1. Maximum likelihood (extended majority-rule consensus) tree of the family Picobirnaviridae using the RdRP region. Red circles indicate branches with UFBoot support greater than 95%. Picobirnavirus from sample PI202115 (Genbank Acc. OR820937) is highlighted in boldface.
Caliciviruses
Taxonomic binning of the de novo assembled contigs found one (2,560 bp) and six (1,020–5,850 bp) contigs with Calicivirus (CV) and descendant taxa classifications from samples PI202112 and PI202116, respectively. Although the contigs did not represent the complete genome, the genome’s two open reading frames (ORF1 and ORF2) were detected in sample PI202116, whereas only ORF1 was identified from sample PI202112. Although coverage was complete for ORF2, individual contig coverage did not exceed 70% for ORF1.
Only a single contig, from PI202116 (Genbank Acc. OR837086), overlapped with the complete major capsid protein (VP1) amino acid sequence of ORF1, which was the region recommended for CV genus demarcation and species resolution by the ICTV. Phylogenetic analysis of VP1 showed the PI202116 contig belonged to the Nacovirus group of CVs, which are typically associated with avian species (Figure 2). The contig was a sister (UFBoot support: 100%) to the nacovirus detected in a Mallard from Canada (Genbank Acc. MN175552), with which it shared 95.58% identity (based on amino acid sequence). This sister pair formed a clade (UFBoot support: 100%) with an additional nacovirus detected in an American black duck, Anas rubripes, from Canada (Genbank Acc. MN175556).
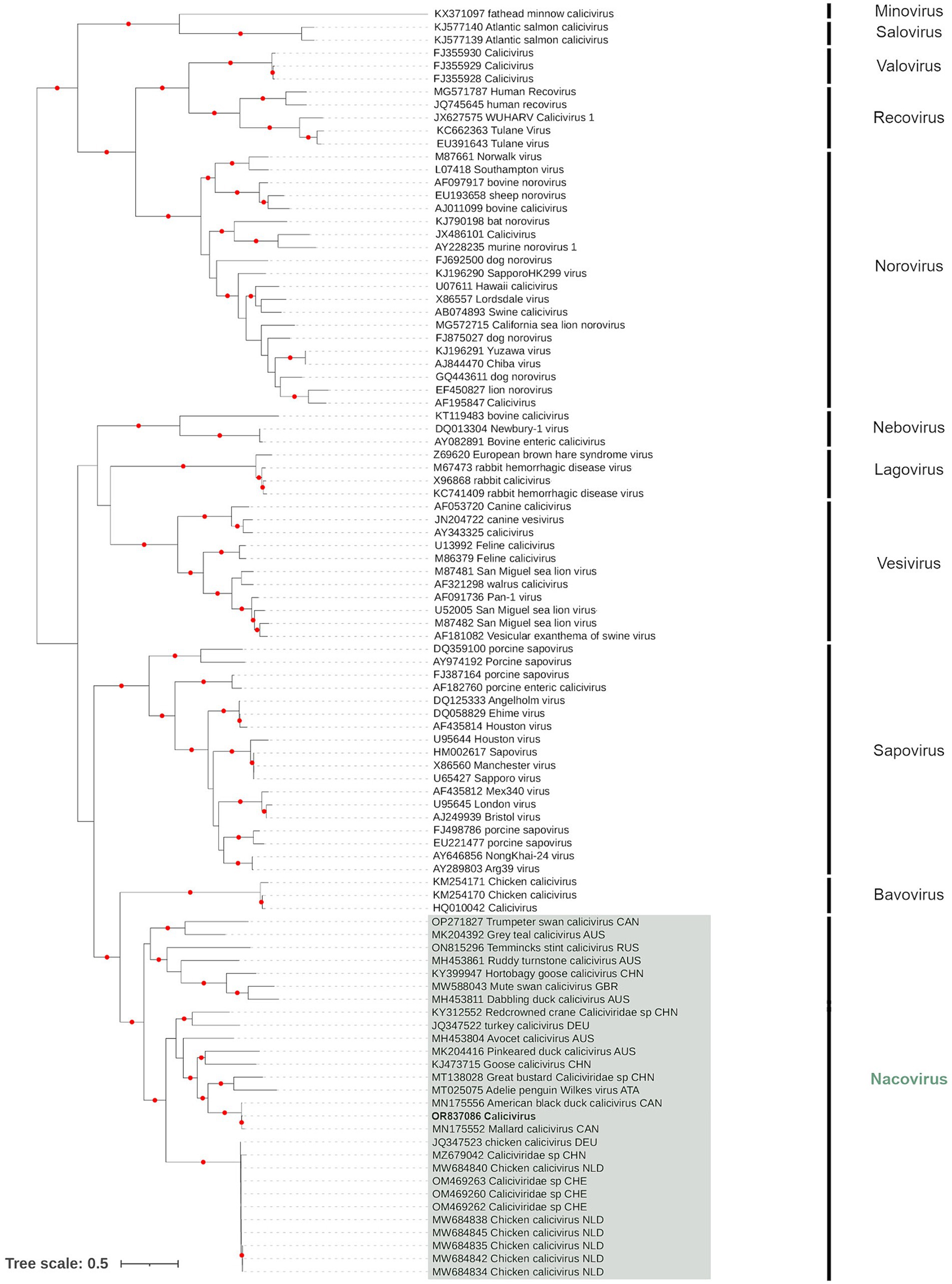
Figure 2. Maximum likelihood (extended majority-rule consensus) tree of the family Caliciviridae using the VP1 region. Red circles indicated branches are supported at greater than 95% (UFBoot support). Calicivirus from sample PI202116 (Genbank Acc. OR837086) is highlighted in boldface.
When the full ORF1 region was analyzed, the contigs from PI202112 and PI202116 overlapping this region were found within the nacovirus group, but within distinct clades. Although nacovirus from PI202116 remained within a clade formed by the nacovirus from Mallard and American black duck, the nacovirus from PI202112 is most closely associated with one identified in a “Dabbling Duck” from Australia (Genbank Acc. AXF38657), at 87.37% amino acid base identity (Supplementary Figure S2). These two sequences formed a sister relationship with a Hortobagy goose calicivirus (Genbank Acc. ARM65436), which has previously been proposed as a new CV genus, denoted “Sanovirus” (66). Analysis of the minor structural protein (VP2 located in ORF2) found little phylogenetic signal with a poorly supported tree.
Aaliviruses
Taxonomic binning of the de-novo assemblies resulted in one (1,073 bp), five (1,195–3,105 bp) and three (1,229–3,198 bp) contigs with Aalivirus (Pacific black duck [Anas superciliosa] Aalivirus A/Aalivirus A) consensus classifications for samples PI202114, PI202119 and PI202121, respectively. The genome of Aalivirus is organized into viral structural proteins (P1) and non-structural proteins (P2 and P3). Contigs from all three proteins were detected in the data, with P1 found in sample PI202114 and PI202121, and P2 and P3 found in PI202119 and PI202121. All four canonical cleavage sites (DxExNPG|P) described for the P2 protein (2A region) in Aalivirus (67) were detected in contigs. Characteristic motifs conserved in picornaviruses were detected in helicase, from the P2 protein (GEPGSGKS and DDLGQ), and in cysteine protease (GSCG) and RdRp (KDELR, DFKKFD, GGMCSGSPCTTVLNNT and FLKR) from the P3 protein.
Phylogenetic analysis of the RdRp region in P3 showed that contigs from samples PI202119 (Genbank Acc. OR769080) and PI202121 (Genbank Acc. OR769079) clustered together (Figure 3), and these formed a sister relationship with the Duck aalivirus A found in a domestic Mallard from China (Genbank Acc. YP_009026377). These PI202119 and PI202121 contigs had percent identities (based on amino acid sequence) of 98.99 and 98.78% with Duck aalivirus A (Genbank Acc. YP_009026377), respectively. Phylogenetic analysis of the Aalivirus from sample PI202114 using a P1 region sequence alignment (not shown) placed it as a sister to Duck aalivirus A from Australia (Genbank Acc. QMI57977). Analysis of helicase and peptidase C3, regions previously used for the phylogenetic analysis of the aalivirus group (67), found little phylogenetic signal, with poorly supported trees.
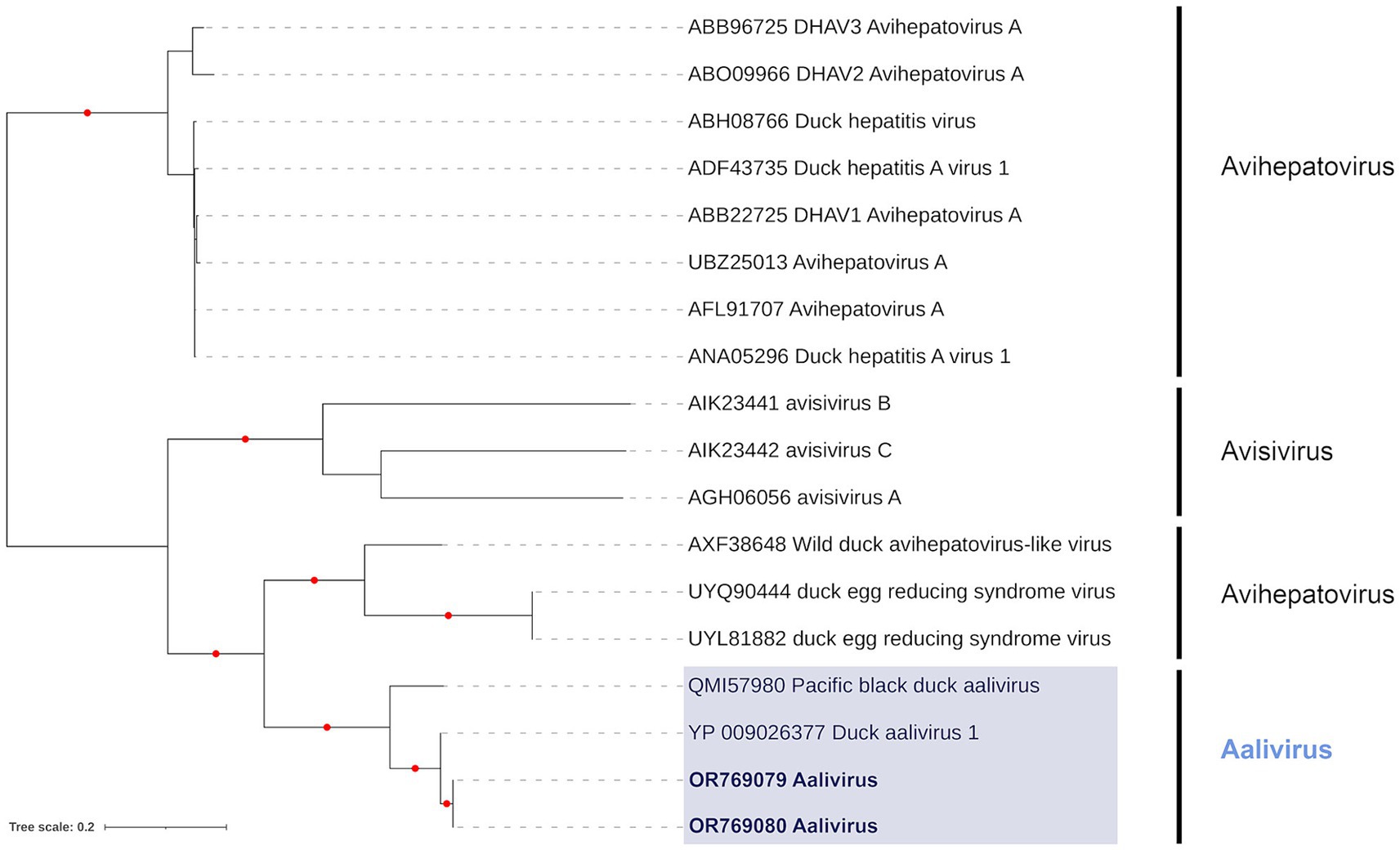
Figure 3. Maximum likelihood (extended majority-rule consensus) tree of the Genus Aalivirus and its closest relatives using the RdRp region. Red circles indicated branches are supported at greater than 95% (UFBoot support). Aaliviruses from samples PI202119 (Genbank Acc. OR769080) and PI202121 (Genbank Acc. OR769079) are highlighted in bold.
Rotavirus G
Taxonomic binning of contigs resulted in two (1,266–1,589 bp), seven (1,022–3,507 bp) and one (1,345 bp) contigs with Rotavirus G (Rotavirus G and Pacific black duck Rotavirus G) consensus classifications for samples PI202112, PI202124, and PI202126, respectively. The Rotavirus G genome is organized into 11 linear dsRNA segments and contigs from seven of these segments (Segments 1 to 7) were detected among the samples analyzed. Three conserved VP1 protein motifs characteristic of rotaviruses (ANKIILYTDVSQWDAS, LKIRYHGVASGEKTTKIGNSYANVALI, and LRVDGDDNVIT) (68, 69) were identified from segment 1 in sample PI202124. An additional conserved rotavirus motif from the VP3 protein in segment 3 (ALYSISN) was also found in sample PI202124.
In accordance with the criteria for genus demarcation and species demarcation by the ICTV, we aligned our data with all known species of Rotavirus (Rotavirus A–D, F–J) at the inner capsid protein (VP6) region of segment 6. Only sample PI202124 had Rotavirus G contigs overlapping this region. The Rotavirus G from PI202124 (Genbank Acc. OR820938) was most closely related to Pacific black duck Rotavirus G (Genbank Acc. QQD36994) and Mallard Rotavirus G (Genbank Acc. UAJ21473) identified from Australia (Figure 4) with 100 and 99.75% identity (based on amino acid sequence), respectively.
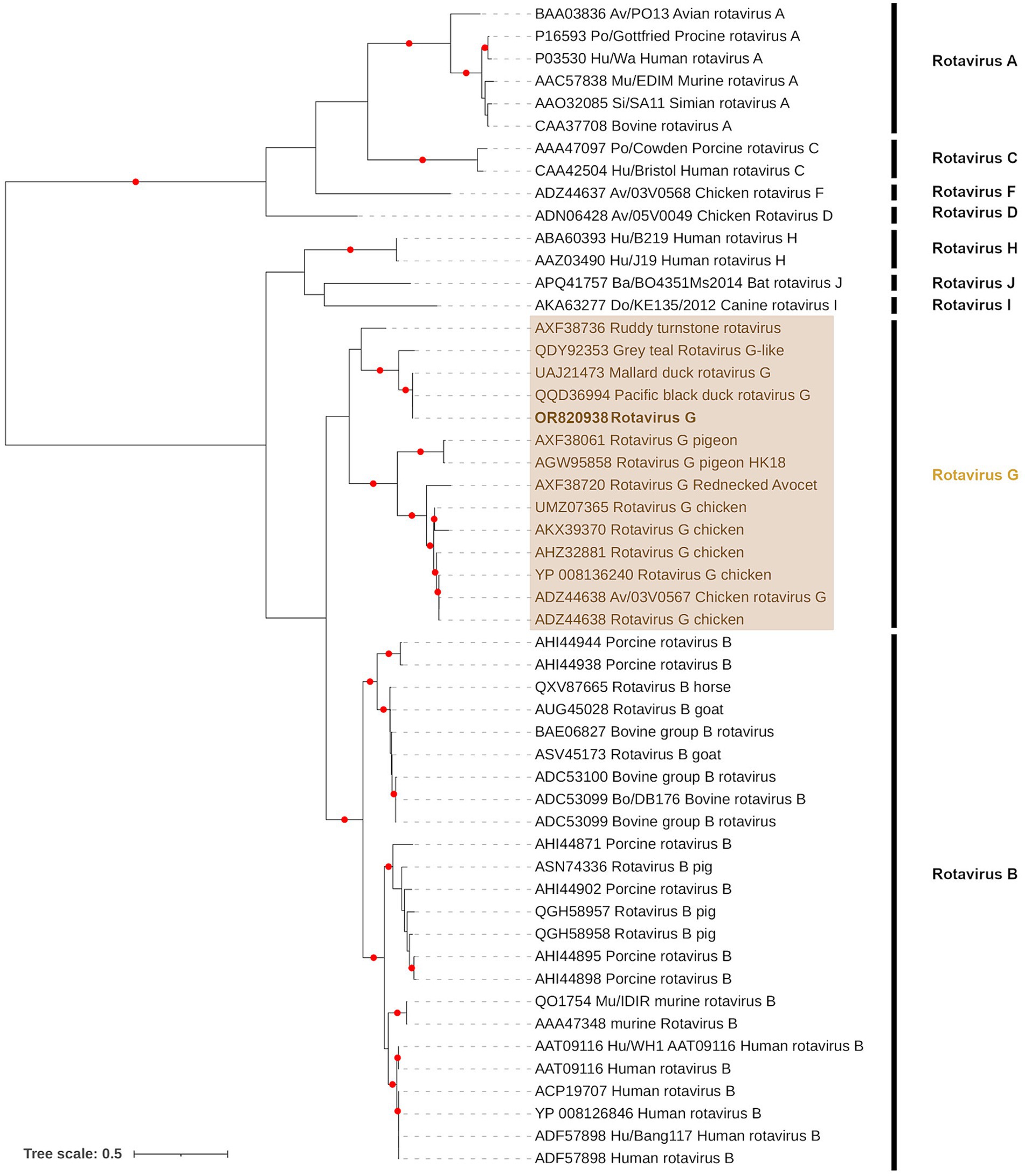
Figure 4. Maximum likelihood (extended majority-rule consensus) tree of the Genus Rotavirus using the VP6 region. Red circles indicated branches are supported at greater than 95% (UFBoot support). Calicivirus from sample PI202124 (Genbank Acc. OR820938) is highlighted in bold.
Although phylogenetic branch support was poor in Segments 5 (Sample PI202124) and 7 (Samples PI202112 and PI202124) contigs, the remaining Segments 1 to 4 contigs from samples PI202112, PI202124, and PI202126 were placed in sister positions with either Pacific black duck Rotavirus G or Mallard Rotavirus G from Australia (Supplementary Figure S3).
Discussion
Birds are among the most vagile of vertebrates and have the capacity to transport a range of viral pathogens that pose a risk to other wild birds, commercial poultry, wildlife, domestic animals, and human health. Viral pathogens that can be transmitted through enteric routes are of considerable risk to commercial poultry flocks and global food supply chains, and the potential risks of zoonosis, most notably in relation to HPAI in recent years, are of major public health concern (70). Characterizing the viral diversity present in migratory birds is thus an essential component of a viral pathogen surveillance, diagnostic, and early warning system (10). Here we have characterized viral pathogens present in the enteric system (cecum) of a group of Mallard captive-raised for release during hunting season. Mallard is the most abundant of all duck species and one that is highly migratory across much of its range with a potential to move between continents (71). We detected four families of viral pathogens in our dataset - Picobirnaviridae (Genogroup I), Caliciviridae (duck Nacovirus), Picornaviridae (duck Aalivirus) and Sedoreoviridae (duck Rotavirus G).
The first group of viruses detected from our study group belong to the genus Aalivirus from the family Picornaviridae. To our knowledge, this is the first record of aalivirus in the Americas, with records from only Australia (72), China (67), and Iran (73). This genus comprises linear, positive sense, unsegmented ssRNA genomes (~9,000 bp), which are made up of a polyprotein divided into three functional regions, P1, P2, and P3. The first aalivirus was detected in diseased domestic Mallard from China in 2014 (67). These were found to be most closely related to duck hepatitis A virus from the genus Avihepatovirus, which causes high mortality among young ducks and is of considerable importance to the poultry industry (74). Work on Aalivirus since its discovery has failed to establish effective culturing methods, and the epidemiology of aalivirus remains largely unknown. In the current study, the aaliviruses detected were most closely related to the original duck aalivirus first described in China (67) and one subsequently identified from Australia (75). Further agnostic sequencing of the enteric systems of potential host species across broad geographical ranges could substantially contribute to novel Aalivirus discovery, characterizing phylogenomic relationships, and better understanding this viruses phylodynamics.
The second group detected is from the family Caliciviridae. This is a family of viruses comprising linear, positive sense, unsegmented ssRNA genomes (6,400–8,500 bp), and consist of a major capsid protein, VP1, a minor structural protein, VP2, and VPg. Caliciviruses have been detected in a wide variety of animals, where they are associated with veterinary diseases, such as gastroenteritis, respiratory illnesses, and hemorrhagic fever (76–79). They are believed to be spread primarily by the fecal-oral route (80). In humans, they are among the most important causes of non-bacterial gastroenteritis outbreaks and epidemics (81–83). They are routinely detected in birds but, without reliable virus isolation, their epidemiology remains poorly known. The caliciviruses detected in our study group belonged to the genus Nacovirus, which is one of the 11 recognized genera within the family Caliciviridae, and whose genome is comprised of two open reading frames (ORF1 And ORF2). The nacovirus that was detected in PI202116 and could be characterized according to ICTV criteria (the VP1 amino acid sequence of ORF1) was found to be closely related to a duck nacovirus detected in a Mallard from Canada (84). Although calicivirus has been detected in a variety of wild waterfowl in Canada (84, 85) and from domestic poultry in the United States (86), to our knowledge this is the first time this virus has been reported outside domestic chicken and turkey flocks in the United States. Further characterization of calicivirus diversity extant in wild bird populations from North America could enhance our understanding of the potential for transmission between wild, captive reared and domestic birds.
The third group of viruses detected belong to the family Picobirnaviridae. This family of viruses comprises bi-segmented dsRNA genomes (4,100–4,600 bp total length). The longer of the two segments, dsRNA1, comprises three open reading frames, whereas the smaller segment, dsRNA2, encodes a viral RNA-dependent RNA polymerase (RdRp). Picobirnaviruses (PBVs) are widespread in animal gastrointestinal tracts and feces but the potential link between PBV and gastroenteritis in mammals remains controversial (87). Although information is scant, there is some evidence for inter-species transmission and zoonoses (88–90). Currently, only three species have been delimited by the ICTV – Genogroups I, II and III - based on host specificity and the strong sequence divergence of their capsid proteins. The PBV detected in the current study (found in sample PI202115) and characterized by ICTV criteria belonged to the Genogroup I. It was found to be most closely related to a Genogroup I metagenome assembled genome (MAG) detected in a pig from North Carolina, United States (91). Given the enteric nature of PBVs, there is elevated risk of transmission among birds and other animals at water bodies utilized by resident and migratory birds.
The final group of viruses detected belong to the genus Rotavirus (RV) from the family Sedoreoviridae. This genus comprises double-stranded RNA genomes with 11 segments (700 bp to 3,200 bp) contained within a non-enveloped, triple-layered capsid. These segments encompass structural proteins, including VP1, VP2, VP3, VP4, VP6, and VP7, and non-structural proteins, including NSP1, NSP2, NSP3, NSP4, NSP5, and NSP6. The genus is currently divided into nine species - rotavirus A–D and F–J – which are major causes of gastroenteritis in a wide variety of animals. In humans, RV infection is the most common cause of acute gastroenteritis worldwide, with the species responsible primarily identified as rotavirus A–C (92). The species Rotavirus D (93), F, and G (94) have only ever been detected in avian species. Avian rotavirus (AvRV) infection is most often encountered among young birds (1–2 weeks (50)) and prevalence of up to 85% have been found among some poultry flocks (95). Despite such high prevalence, AvRV vaccines remain unavailable. The mode of RV transmission is primarily via the fecal-oral route, and an important source of infection is contaminated water (96, 97). Although RV zoonotic risk is considered low, the frequency of reassortment among RVs may facilitate viral novelty and opportunities for major evolutionary changes that are required for successful zoonotic transmission. Such zoonotic transmission may have a variety of animal origins (98, 99). In the current study, taxonomic classification of the de novo assemblies revealed the presence of the bird-specific rotavirus G species. Characterization according to ICTV species demarcation found the rotavirus G species present was the same species as that found in Mallard and Pacific black duck from Australia (72). These hosts are closely related migratory species and have the potential to transport rotavirus along migratory routes and may be an important source of transmission, as has been proposed in other migratory waterfowl (100).
Each of the enteric viral pathogens detected in this study may be shed at high concentrations in feces, leading to the contamination of large water bodies utilized by waterfowl and a great variety of birds and other animals, including humans, sharing these water resources. Surface water has been shown to be a highly important medium for the transmission of viruses, including HPAI (101). The viral pathogens detected in this study are therefore potentially important candidates for water-borne disease outbreaks in wildlife, domestic animals, and humans. Although pathogen transmission between wildlife and commercial poultry is well documented (37–39), the transmission risk of pathogens from captive-bred game birds and wildlife remains poorly understood. Commercial farms each typically produce between 20,000 to 300,000 game birds annually, where individuals are initially housed in indoor brooder houses before acclimation to the outdoors after 1–2 months (28). Such high host density present can facilitate pathogen transmission and increase disease susceptibility among the captive-bred flock. The release of these flocks onto private and public lands where they mix with wild conspecifics and other wildlife may therefore represent an important source for viral transmission between wildlife, domestic animals, and humans (102, 103). It is proposed that the pervasive interbreeding of game-farm and wild Mallard in North America east (35, 104) has led to the introduction of maladaptive traits, which has been an important factor in recent population decline (35, 36). Findings from the current study propose pathogen spillover from game-farm Mallard as another possible contributing factor in recent population declines. Additionally, the Mallard is the most closely related extant species to, and possibly the common ancestor of, domestic ducks (105), thus making the interface between wildlife and domestic birds highly permeable and increasing the risks of viral spillover and spillback.
Understanding the epidemiology and etiology of game-farm pathogens first requires establishing systems of surveillance to quantify pathogen prevalence, characterize diversity, elucidate evolutionary relationships and phylodynamics and, in the absence of effective culturing approaches, proposing etiological hypotheses by investigating relationships between pathology and infection prevalence. Increasing efforts to screen game-farm flocks prior to release and redoubling efforts to characterize game-farm and wild bird viromes using agnostic sequencing approaches could help elucidate the pathogen transmission dynamics between game-farm and wild birds and reveal spillover events and may provide a means to control the release of infectious game-farm birds. In our study, despite sampling Mallard from only a single age cohort, season, and game-farm, we find a rich and important diversity of viruses. This preliminary data therefore provides a strong impetus for us to further sample game-farm and wild Mallard to test our hypothesis of spillover between game-farm and wild populations and better characterize viral diversity within these populations. Enhancing and increasing monitoring of enteric viral pathogens in water bodies near release sites, and at important stopover sites, and breeding/wintering grounds, could strengthen our ability to track emerging pathogens, identify potential hotspots and better understand their epidemiology. Agnostic sequencing approaches offer a valuable opportunity surveil a rich diversity of viruses in these water bodies, but further testing and optimization could aid in understanding the sensitivity of these sequencing approaches to presumably lower viral concentrations present in this medium. These approaches possess the means to establish a baseline for viral pathogen prevalence, diversity, and dynamics at important interfaces and among globally important bird populations and could be used to develop an early warning system for outbreaks of water-borne disease of avian origin. Such information could be utilized by One Health networks, which focus on the relationships between animal, human, and environmental health where outbreaks of emerging and neglected diseases can occur, to better develop strategies to detect, characterize and mitigate potentially pandemic pathogens.
Data availability statement
Metatranscriptomic data generated and analyzed in this study is available from BioProject PRJNA1045981 (https://www.ncbi.nlm.nih.gov/bioproject). GenBank accessions for the viral sequences used in phylogenetic analyses are indicated in Table 1, and also available in a U.S. Geological Survey data release (106).
Ethics statement
The animal study was approved by ACUC, U.S. Geological Survey, National Wildlife Health Center (tracking number EP210811). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
BB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. RD: Conceptualization, Resources, Writing – review & editing. KE: Formal analysis, Writing – review & editing. Y-ML: Resources, Writing – review & editing, Supervision. SD: Conceptualization, Data curation, Funding acquisition, Investigation, Resources, Writing – review & editing, Project administration, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This manuscript was prepared whilst BB and KE held a National Research Council (NRC) Research Associateship Awards at the Walter Reed Biosystematics Unit (WRBU), through the Walter Reed Army Institute of Research (WRAIR), Silver Spring, MD. Y-ML and BB were financially supported by the Armed Forces Health Surveillance Division—Global Emerging Infections Surveillance (AFHSD-GEIS) award P0055_24_WR. This research was supported by U.S. Geological Survey Ecosystems Mission Area Environmental Health Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
All or portions of the laboratory and/or data analysis were conducted in and with the support of the Laboratories of Analytical Biology (https://ror.org/05b8c0r92) of the National Museum of Natural History. The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official, or as reflecting true views of the Department of the Army, Navy, or the Department of Defense. Material contained within this publication has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1396552/full#supplementary-material
Footnotes
1. ^https://github.com/biobakery/kneaddata
2. ^https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_015476345.1/
References
1. Allen, T, Murray, KA, Zambrana-Torrelio, C, Morse, SS, Rondinini, C, Di Marco, M, et al. Global hotspots and correlates of emerging zoonotic diseases. Nat Commun. (2017) 8:1124. doi: 10.1038/s41467-017-00923-8
2. Sullivan, SJ, Jacobson, RM, Dowdle, WR, and Poland, GA. 2009 H1N1 Influenza. Mayo Clin Proc. (2010) 85:64–76. doi: 10.4065/mcp.2009.0588
3. Coltart, CEM, Lindsey, B, Ghinai, I, Johnson, AM, and Heymann, DL. The Ebola outbreak, 2013–2016: old lessons for new epidemics. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160297. doi: 10.1098/rstb.2016.0297
4. Baud, D, Gubler, DJ, Schaub, B, Lanteri, MC, and Musso, D. An update on Zika virus infection. Lancet. (2017) 390:2099–109. doi: 10.1016/S0140-6736(17)31450-2
5. Al-Awwal, N, Dweik, F, Mahdi, S, El-Dweik, M, and Anderson, SH. A review of SARS-CoV-2 disease (COVID-19): pandemic in our time. Pathogens. (2022) 11:368. doi: 10.3390/pathogens11030368
6. Garmendia, AE, Van Kruiningen, HJ, and French, RA. The West Nile virus: its recent emergence in North America. Microbes Infect. (2001) 3:223–9. doi: 10.1016/S1286-4579(01)01374-0
7. Ronca, SE, Ruff, JC, and Murray, KO. A 20-year historical review of West Nile virus since its initial emergence in North America: has West Nile virus become a neglected tropical disease? PLoS Negl Trop Dis. (2021) 15:e0009190. doi: 10.1371/journal.pntd.0009190
8. de Jong, JC, Claas, EC, Osterhaus, AD, Webster, RG, and Lim, WL. A pandemic warning? Nature. (1997) 389:554. doi: 10.1038/39218
9. Sellwood, C, Asgari-Jirhandeh, N, and Salimee, S. Bird flu: if or when? Planning for the next pandemic. Postgrad Med J. (2007) 83:445–50. doi: 10.1136/pgmj.2007.059253
10. Georgopoulou, I, and Tsiouris, V. The potential role of migratory birds in the transmission of Zoonoses. Vet Ital. (2008) 44:671–7.
11. de Angeli Dutra, D, Fecchio, A, Martins Braga, É, and Poulin, R. Migratory birds have higher prevalence and richness of avian Haemosporidian parasites than residents. Int J Parasitol. (2021) 51:877–82. doi: 10.1016/j.ijpara.2021.03.001
12. de Angeli Dutra, D, Filion, A, Fecchio, A, Braga, ÉM, and Poulin, R. Migrant birds disperse Haemosporidian parasites and affect their transmission in avian communities. Oikos. (2021) 130:979–88. doi: 10.1111/oik.08199
13. Kilpatrick, AM, Chmura, AA, Gibbons, DW, Fleischer, RC, Marra, PP, and Daszak, P. Predicting the global spread of H5N1 avian influenza. Proc Natl Acad Sci. (2006) 103:19368–73. doi: 10.1073/pnas.0609227103
14. Lee, D-H, Torchetti, MK, Winker, K, Ip, HS, Song, C-S, and Swayne, DE. Intercontinental spread of Asian-origin H5N8 to North America through Beringia by migratory birds. J Virol. (2015) 89:6521–4. doi: 10.1128/JVI.00728-15
15. Günther, A, Krone, O, Svansson, V, Pohlmann, A, King, J, Hallgrimsson, GT, et al. Iceland as stepping stone for spread of highly pathogenic avian influenza virus between Europe and North America. Emerg Infect Dis. (2022) 28:2383–8. doi: 10.3201/eid2812.221086
16. Webster, RG, Bean, WJ, Gorman, OT, Chambers, TM, and Kawaoka, Y. Evolution and ecology of influenza a viruses. Microbiol Rev. (1992) 56:152–79. doi: 10.1128/mr.56.1.152-179.1992
17. Roche, B, Lebarbenchon, C, Gauthier-Clerc, M, Chang, C-M, Thomas, F, Renaud, F, et al. Water-borne transmission drives avian influenza dynamics in wild birds: the case of the 2005–2006 epidemics in the Camargue area. Infect Genet Evol. (2009) 9:800–5. doi: 10.1016/j.meegid.2009.04.009
18. van den Brand, JM, Krone, O, Wolf, PU, van de Bildt, MW, van Amerongen, G, Osterhaus, AD, et al. Host-specific exposure and fatal neurologic disease in wild raptors from highly pathogenic avian influenza virus H5N1 during the 2006 outbreak in Germany. Vet Res. (2015) 46:24. doi: 10.1186/s13567-015-0148-5
19. Nemeth, NM, Ruder, MG, Poulson, RL, Sargent, R, Breeding, S, Evans, MN, et al. Bald eagle mortality and Nest failure due to clade 2.3.4.4 highly pathogenic H5N1 influenza a virus. Sci Rep. (2023) 13:191. doi: 10.1038/s41598-023-27446-1
20. Chitimia-Dobler, L, Schaper, S, Rieß, R, Bitterwolf, K, Frangoulidis, D, Bestehorn, M, et al. Imported Hyalomma ticks in Germany in 2018. Parasit Vectors. (2019) 12:134. doi: 10.1186/s13071-019-3380-4
21. Cohen, EB, Auckland, LD, Marra, PP, and Hamer, SA. Avian migrants facilitate invasions of Neotropical ticks and tick-borne pathogens into the United States. Appl Environ Microbiol. (2015) 81:8366–78. doi: 10.1128/AEM.02656-15
22. Mancuso, E, Toma, L, Pascucci, I, d’Alessio, SG, Marini, V, Quaglia, M, et al. Direct and indirect role of migratory birds in spreading CCHFV and WNV: a multidisciplinary study on three stop-over islands in Italy. Pathogens. (2022) 11:1056. doi: 10.3390/pathogens11091056
23. Palomar, AM, Portillo, A, Santibáñez, P, Mazuelas, D, Arizaga, J, Crespo, A, et al. Crimean-Congo hemorrhagic fever virus in ticks from migratory birds, Morocco. Emerg Infect Dis. (2013) 19:260–3. doi: 10.3201/eid1902.121193
24. Leblebicioglu, H, Eroglu, C, Erciyas-Yavuz, K, Hokelek, M, Acici, M, and Yilmaz, H. Role of migratory birds in spreading Crimean-Congo hemorrhagic fever. Turkey Emerg Infect Dis. (2014) 20:1331–4. doi: 10.3201/eid2008.131547
25. Lindeborg, M, Barboutis, C, Ehrenborg, C, Fransson, T, Jaenson, TGT, Lindgren, P-E, et al. Migratory birds, ticks, and Crimean-Congo hemorrhagic fever virus. Emerg Infect Dis. (2012) 18:2095–7. doi: 10.3201/eid1812.120718
26. Wilhelmsson, P, Jaenson, TGT, Olsen, B, Waldenström, J, and Lindgren, P-E. Migratory birds as disseminators of ticks and the tick-borne pathogens Borrelia Bacteria and tick-borne encephalitis (TBE) virus: a seasonal study at Ottenby bird Observatory in South-Eastern Sweden. Parasit Vectors. (2020) 13:607. doi: 10.1186/s13071-020-04493-5
27. Wilhelmsson, P, Pawełczyk, O, Jaenson, TGT, Waldenström, J, Olsen, B, Forsberg, P, et al. Three Babesia species in Ixodes ricinus ticks from migratory birds in Sweden. Parasit Vectors. (2021) 14:183. doi: 10.1186/s13071-021-04684-8
28. Wallner-Pendleton, E., and Hulet, M. “Game Bird Industry,” in Foreign Animal Disease Preparedness and Response Plan (FAD PReP), editor. Poultry Industry Manual. Center for Food Security and Public Health; Iowa State University of Science and Technology: Ames, IA, USA (2013).
29. Arroyo, B, and Beja, P. Impact of hunting management practices on biodiversity. Reconciling gamebird hunting and biodiversity (REGHAB) report. Brussels: European Commission Brussels (2002).
30. Madden, JR. How many gamebirds are released in the UK each year? Eur J Wildl Res. (2021) 67:72. doi: 10.1007/s10344-021-01508-z
31. Aebischer, NJ. Fifty-year trends in UK hunting bags of birds and mammals, and calibrated estimation of National bag Size, using GWCT’s National Gamebag Census. Eur J Wildl Res. (2019) 65:64. doi: 10.1007/s10344-019-1299-x
32. Champagnon, J, Gauthier-Clerc, M, Lebreton, J-D, Mouronval, J-B, and Guillemain, M. Les Canards Colverts Lâchés Pour La Chasse Interagissent-Ils Avec Les Populations Sauvage. Faune Sauvage. (2013) 298:4–9.
33. BirdLife International. Birds in Europe: Populations estimates, Trends and Conservation Status. Cambridge: BirdLife International (2004).
34. U.S. Fish and Wildlife Service. Review of captive-reared mallard regulations on shooting preserves - final report. Washington DC: U.S. Fish and Wildlife Service (2013).
35. Schummer, ML, Simpson, J, Shirkey, B, Kucia, SR, Lavretsky, P, and Tozer, DC. Population genetics and geographic origins of mallards harvested in northwestern Ohio. PLoS One. (2023) 18:e0282874. doi: 10.1371/journal.pone.0282874
36. Lavretsky, P, McInerney, NR, Mohl, JE, Brown, JI, James, HF, McCracken, KG, et al. Assessing changes in genomic divergence following a century of human-mediated secondary contact among wild and captive-bred ducks. Mol Ecol. (2020) 29:578–95. doi: 10.1111/mec.15343
37. Miller, RS, Farnsworth, ML, and Malmberg, JL. Diseases at the livestock-wildlife interface: status, challenges, and opportunities in the United States. Prev Vet Med. (2013) 110:119–32. doi: 10.1016/j.prevetmed.2012.11.021
38. Ayala, AJ, Yabsley, MJ, and Hernandez, SM. A review of pathogen transmission at the backyard chicken-wild bird Interface. Front Vet Sci. (2020) 7:7. doi: 10.3389/fvets.2020.539925
39. Si, Y, de Boer, WF, and Gong, P. Different environmental drivers of highly pathogenic avian influenza H5N1 outbreaks in poultry and wild birds. PLoS One. (2013) 8:e53362. doi: 10.1371/journal.pone.0053362
40. Vandegrift, KJ, Sokolow, SH, Daszak, P, and Kilpatrick, AM. Ecology of avian influenza viruses in a changing world. Ann N Y Acad Sci. (2010) 1195:113–28. doi: 10.1111/j.1749-6632.2010.05451.x
41. Banyard, AC, Lean, FZX, Robinson, C, Howie, F, Tyler, G, Nisbet, C, et al. Detection of highly pathogenic avian influenza virus H5N1 clade 2.3.4.4b in great Skuas: a species of conservation concern in Great Britain. Viruses. (2022) 14:212. doi: 10.3390/v14020212
42. Alexandrou, O, Malakou, M, and Catsadorakis, G. The impact of avian influenza 2022 on Dalmatian pelicans was the worst ever wildlife disaster in Greece. Oryx. (2022) 56:813–3. doi: 10.1017/S0030605322001041
43. Gamarra-Toledo, V, Plaza, PI, Gutiérrez, R, Luyo, P, Hernani, L, Angulo, F, et al. Avian flu threatens Neotropical birds. Science. (1979) 379:246–6. doi: 10.1126/science.adg2271
44. Klaassen, M, and Wille, M. The plight and role of wild birds in the current bird flu Panzootic. Nat Ecol Evol. (2023) 7:1541–2. doi: 10.1038/s41559-023-02182-x
45. Stokstad, E. Deadly flu spreads through north American birds. Science. (1979) 376:441–2. doi: 10.1126/science.abq7228
46. Hunter, P. Europe’s worst ever bird flu outbreak. EMBO Rep. (2022) 23:e56048. doi: 10.15252/embr.202256048
47. Abbasi, J. Bird flu has begun to spread in mammals—Here’s What’s important to know. JAMA. (2023) 329:619–21. doi: 10.1001/jama.2023.1317
48. Ganar, K, Das, M, Sinha, S, and Kumar, S. Newcastle disease virus: current status and our understanding. Virus Res. (2014) 184:71–81. doi: 10.1016/j.virusres.2014.02.016
49. Molaei, G, Thomas, MC, Muller, T, Medlock, J, Shepard, JJ, Armstrong, PM, et al. Dynamics of vector-host interactions in avian communities in four eastern equine encephalitis virus foci in the northeastern U.S. PLoS Negl Trop Dis. (2016) 10:e0004347. doi: 10.1371/journal.pntd.0004347
50. Dhama, K, Saminathan, M, Karthik, K, Tiwari, R, Shabbir, MZ, Kumar, N, et al. Avian rotavirus enteritis – an updated review. Vet Q. (2015) 35:142–58. doi: 10.1080/01652176.2015.1046014
51. Armstrong, PM, and Andreadis, TG. Eastern equine encephalitis virus — old enemy, new threat. N Engl J Med. (2013) 368:1670–3. doi: 10.1056/NEJMp1213696
52. Parashar, UD, Burton, A, Lanata, C, Boschi-Pinto, C, Shibuya, K, Steele, D, et al. Global mortality associated with rotavirus disease among children in 2004. J Infect Dis. (2009) 200:S9–S15. doi: 10.1086/605025
53. Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using Fastp. iMeta. (2023) 2:e107. doi: 10.1002/imt2.107
54. Chen, S, Zhou, Y, Chen, Y, and Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
55. Li, D, Liu, C-M, Luo, R, Sadakane, K, and Lam, T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
56. Buchfink, B, Reuter, K, and Drost, H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. (2021) 18:366–8. doi: 10.1038/s41592-021-01101-x
57. Buchfink, B, Xie, C, and Huson, DH. Fast and Sensitive Protein Alignment Using DIAMOND. Nat Methods. (2015) 12:59–60. doi: 10.1038/nmeth.3176
58. Bağcı, C, Patz, S, and Huson, DH. DIAMOND+MEGAN: fast and easy taxonomic and functional analysis of short and long microbiome sequences. Curr Protoc. (2021) 1:e59. doi: 10.1002/cpz1.59
59. Huson, DH, Albrecht, B, Bağcı, C, Bessarab, I, Górska, A, Jolic, D, et al. MEGAN-LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and Contigs. Biol Direct. (2018) 13:6. doi: 10.1186/s13062-018-0208-7
60. Gouy, M, Guindon, S, and Gascuel, O. SeaView version 4: a multiplatform graphical user Interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. (2010) 27:221–4. doi: 10.1093/molbev/msp259
61. Capella-Gutiérrez, S, Silla-Martínez, JM, and Gabaldón, T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. (2009) 25:1972–3. doi: 10.1093/bioinformatics/btp348
62. Minh, BQ, Schmidt, HA, Chernomor, O, Schrempf, D, Woodhams, MD, von Haeseler, A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
63. Martin, DP, Murrell, B, Khoosal, A, and Muhire, B. Detecting and analyzing genetic recombination using RDP4 (2017) 1525:433–60. doi: 10.1007/978-1-4939-6622-6_17,
64. Collier, AM, Lyytinen, OL, Guo, YR, Toh, Y, Poranen, MM, and Tao, YJ. Initiation of RNA polymerization and polymerase Encapsidation by a small DsRNA virus. PLoS Pathog. (2016) 12:e1005523. doi: 10.1371/journal.ppat.1005523
65. Delmas, B, Attoui, H, Ghosh, S, Malik, YS, Mundt, E, and Vakharia, VN. ICTV Virus Taxonomy Profile: Picobirnaviridae. J Gen Virol. (2019) 100:133–4. doi: 10.1099/jgv.0.001186
66. Wang, F, Wang, M, Dong, Y, Zhang, B, and Zhang, D. Genetic characterization of a novel Calicivirus from a goose. Arch Virol. (2017) 162:2115–8. doi: 10.1007/s00705-017-3302-8
67. Wang, X, Liu, N, Wang, F, Ning, K, Li, Y, and Zhang, D. Genetic characterization of a novel duck-origin picornavirus with six 2A proteins. J Gen Virol. (2014) 95:1289–96. doi: 10.1099/vir.0.063313-0
68. Mitchell, DB, and Both, GW. Conservation of a potential metal binding motif despite extensive sequence diversity in the rotavirus nonstructural protein NS53. Virology. (1990) 174:618–21. doi: 10.1016/0042-6822(90)90117-a
69. Chen, F, Knutson, TP, Porter, RE, Ciarlet, M, Mor, SK, and Marthaler, DG. Genome characterization of Turkey rotavirus G strains from the United States identifies potential recombination events with human rotavirus B strains. J Gen Virol. (2017) 98:2931–6. doi: 10.1099/jgv.0.000963
70. Huang, P, Sun, L, Li, J, Wu, Q, Rezaei, N, Jiang, S, et al. Potential cross-species transmission of highly pathogenic avian influenza H5 subtype (HPAI H5) viruses to humans calls for the development of H5-specific and universal influenza vaccines. Cell Discov. (2023) 9:58. doi: 10.1038/s41421-023-00571-x
71. Drilling, N, Titman, RD, and McKinney, F. Mallard (Anas Platyrhynchos) In: SM Billerman, editor. Birds of the world. Ithaca, NY: Cornell Lab of Ornithology (2020)
72. Vibin, J, Chamings, A, Klaassen, M, and Alexandersen, S. Metagenomic characterisation of additional and novel avian viruses from Australian wild ducks. Sci Rep. (2020) 10:22284. doi: 10.1038/s41598-020-79413-9
73. Ebnalnassir, M, Ghalyanchi Langeroudi, A, Najafi, H, Hosseini, H, Ziafati Kafi, Z, Sadri, N, et al. Molecular identification and phylogenetic analysis of a novel strain of duck picornavirus (Aalivirus) in Iran: the first report. Iran J Virol. (2022) 16:54–8.
74. Yugo, DM, Hauck, R, Shivaprasad, HL, and Meng, X-J. Hepatitis virus infections in poultry. Avian Dis. (2016) 60:576–88. doi: 10.1637/11229-070515-Review.1
75. Vibin, J, Chamings, A, Klaassen, M, Bhatta, TR, and Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci Rep. (2020) 10:12800. doi: 10.1038/s41598-020-69557-z
76. Abrantes, J, van der Loo, W, Le Pendu, J, and Esteves, PJ. Rabbit Haemorrhagic disease (RHD) and rabbit Haemorrhagic disease virus (RHDV): a review. Vet Res. (2012) 43:12. doi: 10.1186/1297-9716-43-12
77. Hofmann-Lehmann, R, Hosie, MJ, Hartmann, K, Egberink, H, Truyen, U, Tasker, S, et al. Calicivirus infection in cats. Viruses. (2022) 14:937. doi: 10.3390/v14050937
78. Guo, M, Evermann, JF, and Saif, LJ. Detection and molecular characterization of cultivable Caliciviruses from clinically Normal mink and enteric Caliciviruses associated with diarrhea in mink. Arch Virol. (2001) 146:479–93. doi: 10.1007/s007050170157
79. Ike, AC, Roth, BN, Böhm, R, Pfitzner, AJP, and Marschang, RE. Identification of bovine enteric Caliciviruses (BEC) from cattle in Baden-Württemberg. Dtsch Tierarztl Wochenschr. (2007) 114:12–5.
80. Katpally, U, and Smith, TJ. The Caliciviruses. Curr Top Microbiol Immunol. (2010) 343:23–41. doi: 10.1007/82_2010_36
81. Goodgame, RW. Viral causes of diarrhea. Gastroenterol Clin N Am. (2001) 30:779–95. doi: 10.1016/s0889-8553(05)70210-7
82. Hall, AJ, Lopman, BA, Payne, DC, Patel, MM, Gastañaduy, PA, Vinjé, J, et al. Norovirus disease in the United States. Emerg Infect Dis. (2013) 19:1198–205. doi: 10.3201/eid1908.130465
83. Karst, SM. Pathogenesis of noroviruses, emerging RNA viruses. Viruses. (2010) 2:748–81. doi: 10.3390/v2030748
84. Canuti, M, Kroyer, ANK, Ojkic, D, Whitney, HG, Robertson, GJ, and Lang, AS. Discovery and characterization of novel RNA viruses in aquatic north American wild birds. Viruses. (2019) 11:768. doi: 10.3390/v11090768
85. Canuti, M, Wilson, L, Bowes, V, Redford, T, Dufour, SC, Lang, AS, et al. A novel Calicivirus discovered in trumpeter swans (Cygnus Buccinator) expands the richness of known avian Caliciviruses. Curr Res Microb Sci. (2022) 3:100169. doi: 10.1016/j.crmicr.2022.100169
86. Day, JM, Ballard, LL, Duke, MV, Scheffler, BE, and Zsak, L. Metagenomic analysis of the Turkey gut RNA virus community. Virol J. (2010) 7:313. doi: 10.1186/1743-422X-7-313
87. Reddy, MV, Gupta, V, Nayak, A, and Tiwari, SP. Picobirnaviruses in animals: a review. Mol Biol Rep. (2023) 50:1785–97. doi: 10.1007/s11033-022-08133-2
88. Wang, Y, Bányai, K, Tu, X, and Jiang, B. Simian Genogroup I Picobirnaviruses: prevalence, genetic diversity, and zoonotic potential. J Clin Microbiol. (2012) 50:2779–82. doi: 10.1128/JCM.00634-12
89. Bányai, K, Martella, V, Bogdán, Á, Forgách, P, Jakab, F, Meleg, E, et al. Genogroup I Picobirnaviruses in pigs: evidence for genetic diversity and relatedness to human strains. J Gen Virol. (2008) 89:534–9. doi: 10.1099/vir.0.83134-0
90. Li, W, Qiang, X, Qin, S, Huang, Y, Hu, Y, Bai, B, et al. Virome diversity analysis reveals novel enteroviruses and a human Picobirnavirus in stool samples from African green monkeys with diarrhea. Infect Genet Evol. (2020) 82:104279. doi: 10.1016/j.meegid.2020.104279
91. Ramesh, A, Bailey, ES, Ahyong, V, Langelier, C, Phelps, M, Neff, N, et al. Metagenomic characterization of swine slurry in a north American swine farm operation. Sci Rep. (2021) 11:16994. doi: 10.1038/s41598-021-95804-y
92. Parashar, UD, Bresee, JS, Gentsch, JR, and Glass, RI. Rotavirus. Emerg Infect Dis. (1998) 4:561–70. doi: 10.3201/eid0404.980406
93. Trojnar, E, Otto, P, Roth, B, Reetz, J, and Johne, R. The genome segments of a group D rotavirus possess group A-like conserved termini but encode group-specific proteins. J Virol. (2010) 84:10254–65. doi: 10.1128/JVI.00332-10
94. Johne, R, Otto, P, Roth, B, Löhren, U, Belnap, D, Reetz, J, et al. Sequence analysis of the VP6-encoding genome segment of avian group F and G rotaviruses. Virology. (2011) 412:384–91. doi: 10.1016/j.virol.2011.01.031
95. Otto, PH, Ahmed, MU, Hotzel, H, Machnowska, P, Reetz, J, Roth, B, et al. Detection of avian rotaviruses of groups a, D, F and G in diseased chickens and turkeys from Europe and Bangladesh. Vet Microbiol. (2012) 156:8–15. doi: 10.1016/j.vetmic.2011.10.001
96. Gerba, CP, Rose, JB, Haas, CN, and Crabtree, KD. Waterborne rotavirus: a risk assessment. Water Res. (1996) 30:2929–40. doi: 10.1016/S0043-1354(96)00187-X
97. Cook, N, Bridger, J, Kendall, K, Gomara, MI, El-Attar, L, and Gray, J. The zoonotic potential of rotavirus. J Infect. (2004) 48:289–302. doi: 10.1016/j.jinf.2004.01.018
98. Geletu, US, Usmael, MA, and Bari, FD. Rotavirus in calves and its zoonotic importance. Vet Med Int. (2021) 2021:6639701–18. doi: 10.1155/2021/6639701
99. McDonald, SM, Nelson, MI, Turner, PE, and Patton, JT. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol. (2016) 14:448–60. doi: 10.1038/nrmicro.2016.46
100. Fujii, Y, Hirayama, M, Nishiyama, S, Takahashi, T, Okajima, M, Izumi, F, et al. Characterization of an avian rotavirus a strain isolated from a velvet scoter (Melanitta Fusca): implication for the role of migratory birds in global spread of avian rotaviruses. J Gen Virol. (2022) 103:103 (2). doi: 10.1099/jgv.0.001722
101. Pepin, KM, Leach, CB, Barrett, NL, Ellis, JW, VanDalen, KK, Webb, CT, et al. Environmental transmission of influenza a virus in mallards. MBio. (2023) 14:e0086223. doi: 10.1128/mbio.00862-23
102. Hofer, U. Multitude of viruses in game animals. Nat Rev Microbiol. (2022) 20:253–3. doi: 10.1038/s41579-022-00716-x
103. Dwight, IA, Coates, PS, Stoute, ST, and Pitesky, ME. Health surveillance of a potential bridge host: pathogen exposure risks posed to avian populations augmented with captive-bred pheasants. Transbound Emerg Dis. (2022) 69:1095–107. doi: 10.1111/tbed.14068
104. Davis, JB, Outlaw, DC, Ringelman, KM, Kaminski, RM, and Lavretsky, P. Low levels of hybridization between domestic and wild mallards wintering in the lower Mississippi flyway. Ornithology. (2022) 139:139 (4). doi: 10.1093/ornithology/ukac034
105. Hou, Z-C, Yang, F-X, Qu, L-J, Zheng, J-X, Brun, J-M, Basso, B, et al. Genetic structure of Eurasian and north American mallard ducks based on MtDNA data. Anim Genet. (2012) 43:352–5. doi: 10.1111/j.1365-2052.2011.02248.x
Keywords: Mallard, pathogen, virus, game-farm, spillover
Citation: Bourke BP, Dusek RJ, Ergunay K, Linton Y-M and Drovetski SV (2024) Viral pathogen detection in U.S. game-farm mallard (Anas platyrhynchos) flags spillover risk to wild birds. Front. Vet. Sci. 11:1396552. doi: 10.3389/fvets.2024.1396552
Edited by:
Zorica D. Dakić, University of Belgrade, SerbiaReviewed by:
Kurt Vandegrift, The Pennsylvania State University (PSU), United StatesBranislav Kureljušić, University of Belgrade, Serbia
Copyright © 2024 Bourke, Dusek, Ergunay, Linton and Drovetski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian P. Bourke, Ym91cmtlYkBzaS5lZHU=