
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 23 May 2024
Sec. Animal Behavior and Welfare
Volume 11 - 2024 | https://doi.org/10.3389/fvets.2024.1394290
This article is part of the Research Topic Animal Health and Production: Identifying Challenges and Finding a Way Forward View all 40 articles
 Zhiyu He1,2†Xuejian Li1,2†Xi Zhang1,2
Zhiyu He1,2†Xuejian Li1,2†Xi Zhang1,2 Qingyuan Ouyang1,2Jiwei Hu1,2
Qingyuan Ouyang1,2Jiwei Hu1,2 Shenqiang Hu1,2Hua He1,2
Shenqiang Hu1,2Hua He1,2 Liang Li1,2
Liang Li1,2 Hehe Liu1,2
Hehe Liu1,2 Jiwen Wang1,2*
Jiwen Wang1,2*There is a gradual transition from water to dryland rearing of geese. In this study, we performed 16S rRNA sequencing (16S rRNA-seq) and transcriptome sequencing (RNA-seq) to reveal the effects of cage rearing (CR) and floor rearing (FR) systems on the microbial composition and transcriptome of the goose ileum. Through 16S rRNA-seq, Linear Discriminant Analysis Effect Size (LEfSe) analysis identified 2 (hgcI_clade and Faecalibacterium) and 14 (Bacteroides, Proteiniphilum, Proteiniclasticum, etc.) differential microbiota in CR and FR, respectively. The rearing system influenced 4 pathways including biosynthesis of amino acids in ileal microbiota. Moreover, we identified 1,198 differentially expressed genes (DEGs) in the ileum mucosa, with 957 genes up-regulated in CR and 241 genes up-regulated in FR. In CR, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed the significant enrichment (p < 0.05) of 28 KEGG pathways, most of which were associated with amino acid metabolism. In FR, up-regulated DEGs were mainly enriched in KEGG pathways associated with cellular processes, including apoptosis, necroptosis, and cellular senescence. Spearman correlation analysis of differential microbiota and amino acid metabolism-related DEGs in CR showed a significant positive correlation. Additionally, differential microbiota of FR, Phascolarctobacterium and Sutterella, were positively correlated with FGF10 (p < 0.05) and PIK3R1 (p < 0.01), respectively. In conclusion, there might be differences in ileal amino acid metabolism levels between CR and FR geese, and the observed increase in harmful bacterial species in FR might impact the activity of ileal cells.
Rearing systems constitute pivotal non-genetic factors that significantly impact productivity and individual health in goose farming (1). Cage rearing (CR) and floor rearing (FR) systems, as prevalent dryland rearing systems, can reduce the incidence of intestinal disease outbreaks caused by waterborne pathogens (2). However, knowledge on the effects of different dryland rearing systems on the intestines of geese is limited.
Most intestinal microbiota studies have focused on the cecum (3, 4) or the more accessible excreta (5, 6). However, the small intestine microbiota also plays an important role in host metabolic homeostasis (7, 8). The ileum, as a part of the small intestine, undertakes the final process of food digestion and absorption (9), and its microbial community is important for normal avian growth, including Lactobacillus and bacteria with butyrate-producing activity, like Clostridium, Streptococcus and Enterococcus (10). Moreover, compared to other intestinal segments, there is an abundance of immune cells in the ileum (11). Thus, the ileal microbiota may acts in the maintenance of intestinal health by constituting a well-developed immune system. Numerous studies have demonstrated that rearing systems exert significant influence on ileal development and microbial composition. In chickens, the rearing system was shown to alter the relative weight, microbial composition, and expression levels of immune factors (IL-1β, TNF-α, and IFN-γ) in the ileum (12). Ground litter broilers exhibited higher ileal microbiota α diversity (13), meanwhile, ground rearing increased the abundance of litter breeding bacteria (Facklamia, Globicatella, and Jeotgalicoccus) and potentially pathogenic bacteria (Streptococcus and Staphylococcus) in the ileum (14). In ducks, diverse floor rearing environments altered the dominant bacterial phyla in the ileum of Shaoxing ducks, with ducks reared on plastic mesh floor showing significantly higher ileal villus height and villus height/crypt depth ratio compared to those reared on litter floor (15). Importantly, dryland rearing on netting floors has been noted to enhance the intestinal immunity and reduce the mortality rate (2). Studies on Nonghua ducks demonstrated that floor rearing individuals had significantly higher relative weight, relative length and relative weight/relative length ratios of the ileum compared to net rearing individuals (16). In geese, previous research has indicated that cage rearing geese exhibited a higher villus height/crypt depth ratio, suggesting the potential benefits of cage rearing in enhancing geese resistance against diseases and toxins (17). The impact of rearing systems on avian intestinal microbial composition has been extensively studied, while there is a paucity of research focused on elucidating the effects of alterations in avian intestinal microbial composition induced by rearing systems on the intestinal transcriptome.
In this study, we compared the ileal microbial composition and ileal mucosal transcriptome of cage rearing (CR) and floor rearing (FR) geese to deepen our understanding of how rearing systems affect intestines, which provides a theoretical basis for the management of intestinal health in geese.
All animal handling procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Sichuan Agricultural University (Chengdu campus, Sichuan, China, Permit No. DKY20170913).
The same batch of male goslings, from the Sichuan Agricultural University Waterfowls Breeding Farm (Ya’an, Sichuan, China), were reared under the same rearing environment with free access to feed and water until 120 days. Afterwards, they were randomly divided into 2 groups: CR and FR. At 270 days old, 8 geese were randomly selected for slaughter from each group. The experimental geese were euthanised by carbon dioxide inhalation and cervical dislocation after approximately 12 h of fasting. The intestinal digesta and mid-ileum mucosa were quickly collected and rapidly frozen in liquid nitrogen. Digesta was used for 16S rRNA-seq and mucosa was used for RNA-seq, and both were stored at −80°C prior to sequencing.
In this study, microbial DNA extraction was conducted using the E.Z.N.A. Stool DNA Kit (Omega Bio-Tek, Norcross, GA). DNA concentration and purity were characterized by NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and electrophoresis on 1% agarose gels. DNA with (OD260/OD280) range from 1.8 to 2.0 and (OD260/OD230) range from 2.0 to 2.5 could be used for subsequent experiments. Based on the concentration, DNA was diluted to 1ug/μL with sterile water. And, the V3−V4 hypervariable region was targeted for amplification, utilizing primers 338-F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806-R (5′-GGACTACNNGGGTATCTAAT-3′). The PCR reaction was performed on a thermocycling PCR system (Bio-Rad T100, Germany) using high-fidelity polymerase according to the following procedure: 98°C for 60 s; 30 cycles of 98°C for 10 s, 50°C for 30 s, and 72°C for 30 s; 72°C for 5 min. The same volume of IX loading buffer (contained SYB green) was mixed with the PCR products and detected by electrophoresis on 2% agarose gels. The PCR products were mixed at an equidensity ratio. Then, the mixture PCR products were purified with Qiagen Gel Extraction Kit (Qiagen, Germany). Following the manufacturer’s recommendations, sequencing libraries were generated using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, United States) and index codes were added. Library quality was assessed by Qubit@2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, United States). Purified amplicons were sequenced on Illumina NovaSeq 6000 platform (2 × 250 paired ends) by Novogene Co., Ltd. (Beijing, China).
The assembly of paired-end reads, characterized by overlaps exceeding 10 bp, was accomplished using FLASH software (version 1.2.11) (18). Subsequently, reads of low quality, containing ambiguous characters and sequences shorter than 400 bp, were excluded. QIIME2 software (version 2023.5) (19) was employed for processing and assignment of these assembly readings. The denoise-paired method in DADA2 was applied to identify amplicon sequence variants (ASVs). Annotation of results utilized the SILVA 138 database (20), providing classifications at the kingdom, phylum, class, order, family and genus. QIIME2 (version 2023.5) facilitated the calculation of α and β diversity, and unweighted UniFrac distance metrics were used to generate principal coordinate analysis (PCoA). Differences in microbial composition between the 2 groups were assessed using Linear Discriminant Analysis Effect Size (LEfSe) (21), employing screening criteria of LDA score > 3 and p < 0.05. Additionally, we then predicted ileal microbial metabolic pathways using the PICRUSt2 (22) and used STAMP software (version 2.1.3) (23) to compare differences in function. Welch t-test (two-sided) was used for intergroup comparison, and the Welch’s inverted confidence interval (CI) method was used for CI calculation. p < 0.05 indicated a significant difference.
Total RNA extraction from the ileum was accomplished using Trizol (Invitrogen, Carlsbad, CA, United States), following the manufacturer’s instructions. RNA integrity was assessed using the Fragment Analyzer 5400 (Agilent Technologies, CA, United States). RNA integrity values range from 6.8 to 8.9. The library construction utilizing the obtained RNA was undertaken by Novogene Co., Ltd. (Beijing, China). All Illumina PE libraries were constructed and 2 × 150 bp RNA-seq was completed using the Illumina sequencing platform (NovaSeq 6000). The datasets presented in this study can be found in the National Center for Biotechnology Information (NCBI) under BioProject ID PRJNA1054312.
Standard quality control measures were implemented using Fastp software (version 0.23.1) (24) to filter out low-quality reads, ensuring the retention of clean reads for subsequent analyses. The obtained clean reads were aligned to the goose reference genome (BioProject ID PRJNA801885, data not released) using HISAT2 software (version 2.2.1) (25). The resulting SAM (sequencing alignment/mapping) files were subsequently converted to BAM (binary alignment/mapping) files and sorted using SAMtools (version 1.15.1) (26). The expression levels of each transcript were computed using featureCounts (version 2.0.3) (27), while gene expression was quantified using the transcripts per million (TPM) method.
DEseq2 (28) was employed to identify differentially expressed genes (DEGs) between groups. Genes exhibiting |Log2(FC)| ≥ 1 and p < 0.01 were designated as DEGs. And the online tool KOBAS (version 3.0) (29) was utilized for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional analysis, accessible at http://kobas.cbi.pku.edu.cn/kobas3/?t=1. The functional gene analysis was conducted using G. gallus as a reference.
Statistical analysis was performed using SPSS 27.0 software. Spearman’s correlation coefficients were calculated to analyze the correlation. Differences were considered statistically significant at p < 0.05.
Following quality control and filtering procedures, a total of 957,019 effective reads were generated from 16 samples, averaging 59,814 reads per sample (Supplementary Table S1). A total of 745 ASVs were identified through QIIME2 analysis. Of these, 321 ASVs were found to be common to both rearing systems, while 221 ASVs were exclusive to the CR and 203 ASVs were unique to the FR (Figure 1). Subsequent taxonomic classification categorized these ASVs into 17 phyla, 34 classes, 88 orders, 167 families, 215 genera.
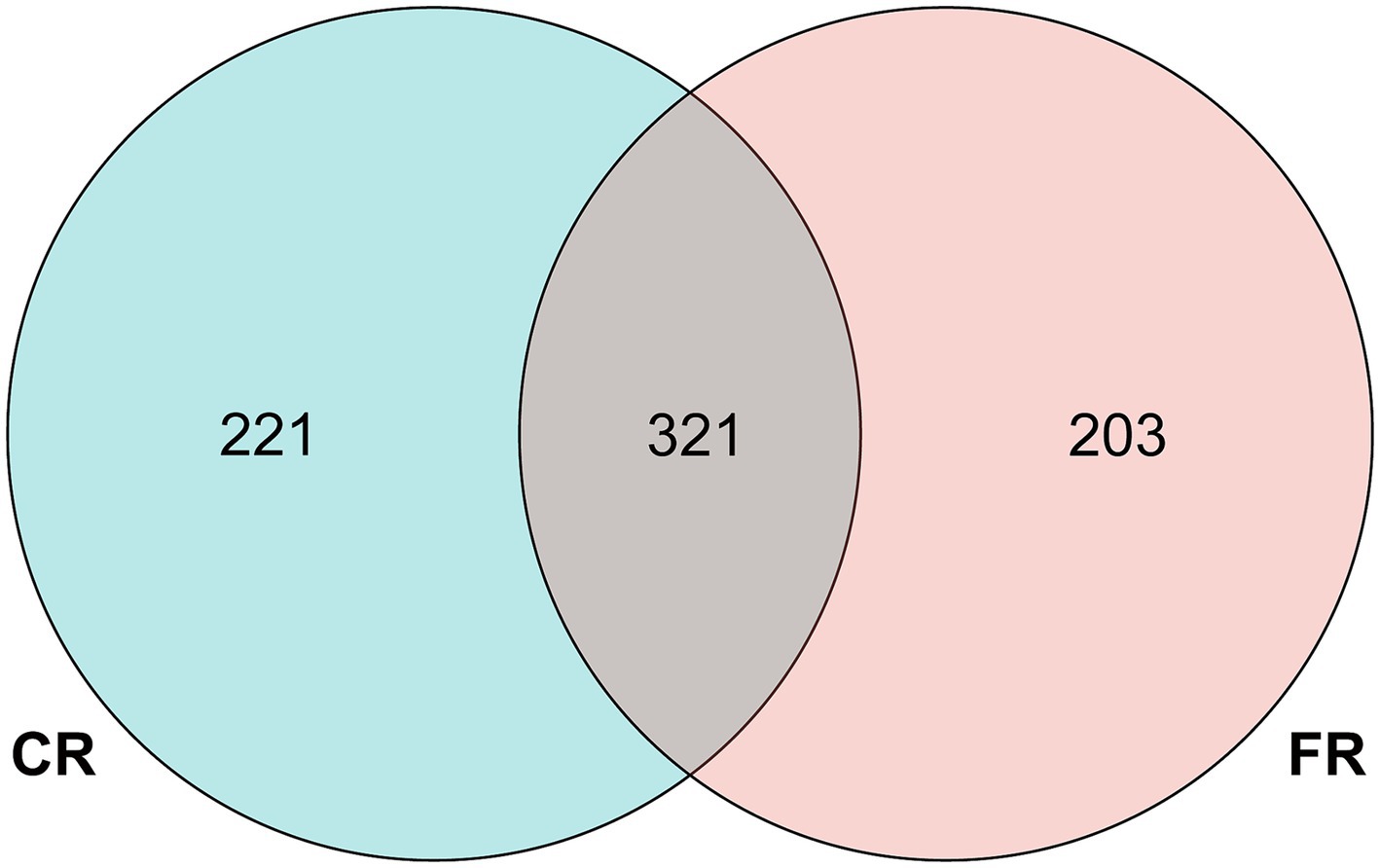
Figure 1. Venn diagram of the number of ASVs.
The complexity of the ileal microbiota was estimated on the basis of α-diversity indices (Observed_features, Shannon, Chao1, Faith’s PD, Evenness). The analysis revealed that the Faith’s PD α-diversity of CR was significantly lower than that of FR (Figure 2A). PCoA analyses demonstrated that the first principal coordinate (PCo1) explained 30.02% of the variations among samples and the second principal coordinate (PCo2) explained 18.34% of the variations (Figure 2B). The ANOSIM test further confirmed significant differences in ileal microbial communities between CR and FR (R = 0.22, p = 0.04). Comparison analysis of ileal microbiota at the phylum level and genus levels revealed broad similarities in dominant microbiota between the 2 rearing systems. At the phylum level, the dominant phyla in CR were Firmicutes (72.11%), Proteobacteria (10.39%) and Fusobacteriota (6.27%); in FR the dominant phyla were Firmicutes (54.64%), Fusobacteriota (19.76%) and Bacteroidetes (10.62%) (Figure 2C). At the genus level, in CR the top 3 genera were Romboutsia (45.03%), Clostridium_sensu_stricto_1 (12.26%) and Fusobacterium (6.78%); in FR, the top 3 genera were also Romboutsia (23.38%), Fusobacterium (21.13%) and Clostridium_sensu_stricto_1 (8.85%) (Figure 2D).
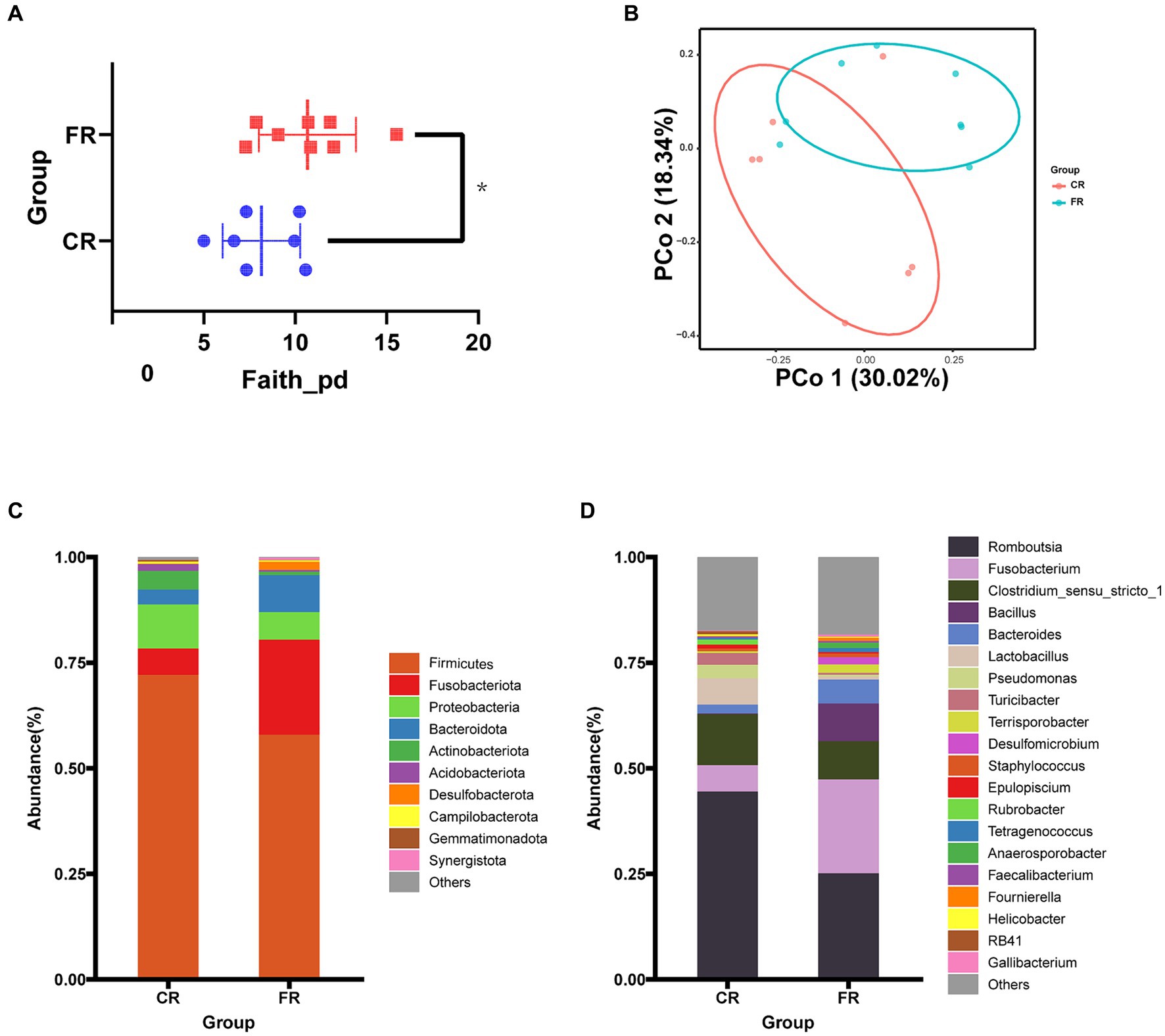
Figure 2. Ileal microbiological composition comparison between CR and FR. (A) Faith’s PD α-diversity. (B) The PCoA based on the unweighted UniFrac distance. (C) Phylum-level microbial composition. (D) Genus-level microbial composition.
At the genus level, LEfSe analyses identified 2 and 14 differential microbiota from CR and FR, respectively (Figures 3A,B). In CR, the abundance of hgcI_clade and Faecalibacterium was significantly higher than that of FR. Meanwhile, Bacteroides, Proteiniphilum, Proteiniclasticum, Syner_01, Phascolarctobacterium, Sutterella, Thermovirga, Colidextribacter, Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium, Paraclostridium, Petrimonas, Succinivibrio, Desulfovibrio, and Campylobacter had higher abundance in FR. In addition, PICRUSt2 predictive function analyses indicated that 4 metabolic pathways differed between the 2 groups, including Alzheimer disease, biosynthesis of amino acids, nicotinate and nicotinamide metabolism and pantothenate and CoA biosynthesis (Figure 3C).
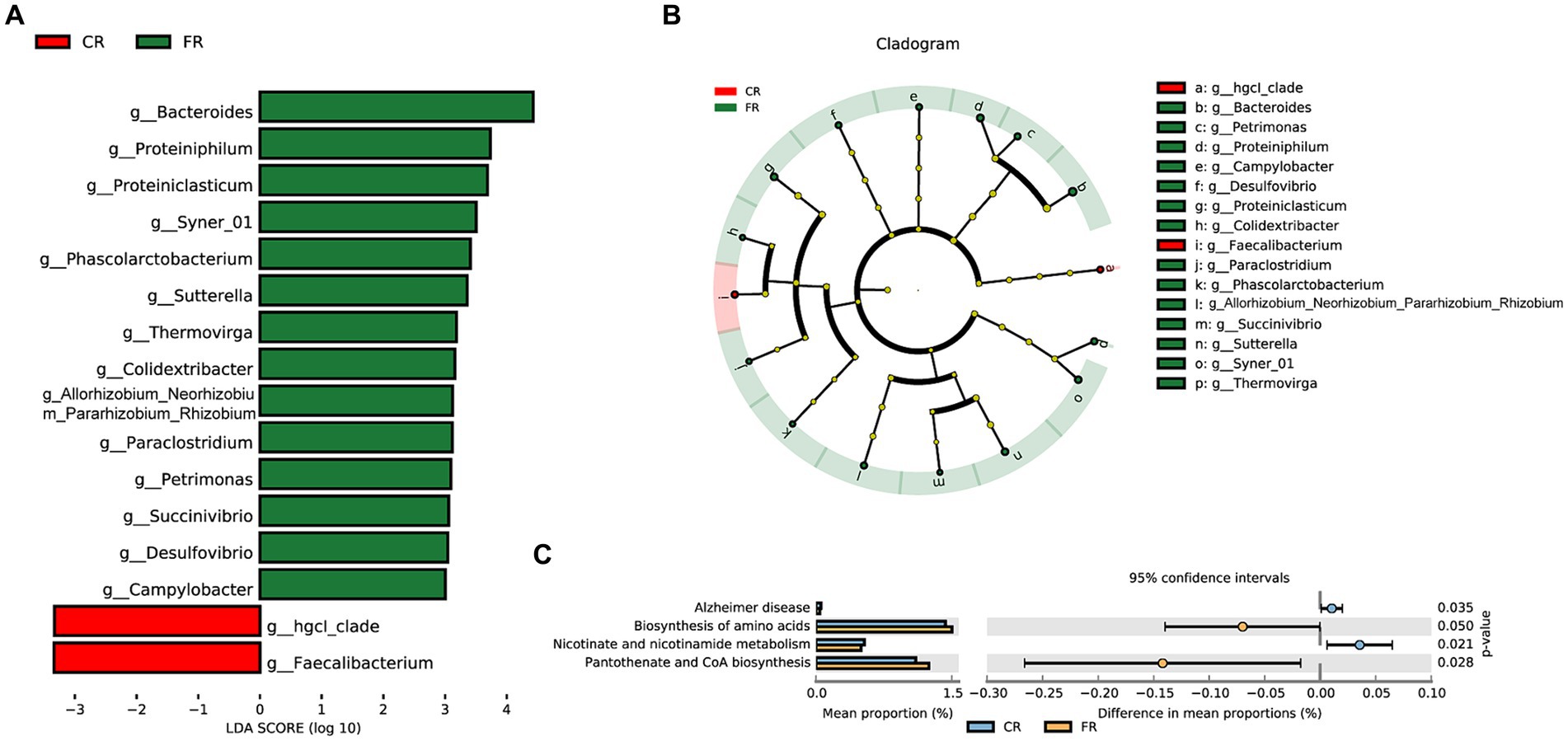
Figure 3. Differential microbiota identification and function prediction. (A) Differential microbiota in the CR and FR ileum. (B) Cladogram of differential microbiota. (C) Differential metabolic pathways predicted.
A comprehensive set of 292,451,214 raw reads were generated across the 7 samples, and subsequent stringent filtering yielded an average of 40,961,820 clean reads for each sample. The quality metrics, including Q20, Q30, GC content, and mapping rates, exhibited favorable ranges of 97.15–97.81%, 92.82–94.08%, 44.72–52.03%, and 86.86–93.52%, respectively (Supplementary Table S2). These results attested to the robust sequencing quality essential for subsequent analyses. We identified a total of 1,198 DEGs (Figure 4A), of which 241 were up-regulated and 957 were down-regulated (Figure 4B). The clustering heatmap of TPM illustrated the expression profiles of DEGs in the ileum of CR and FR geese in a visually informative manner (Figure 4C).
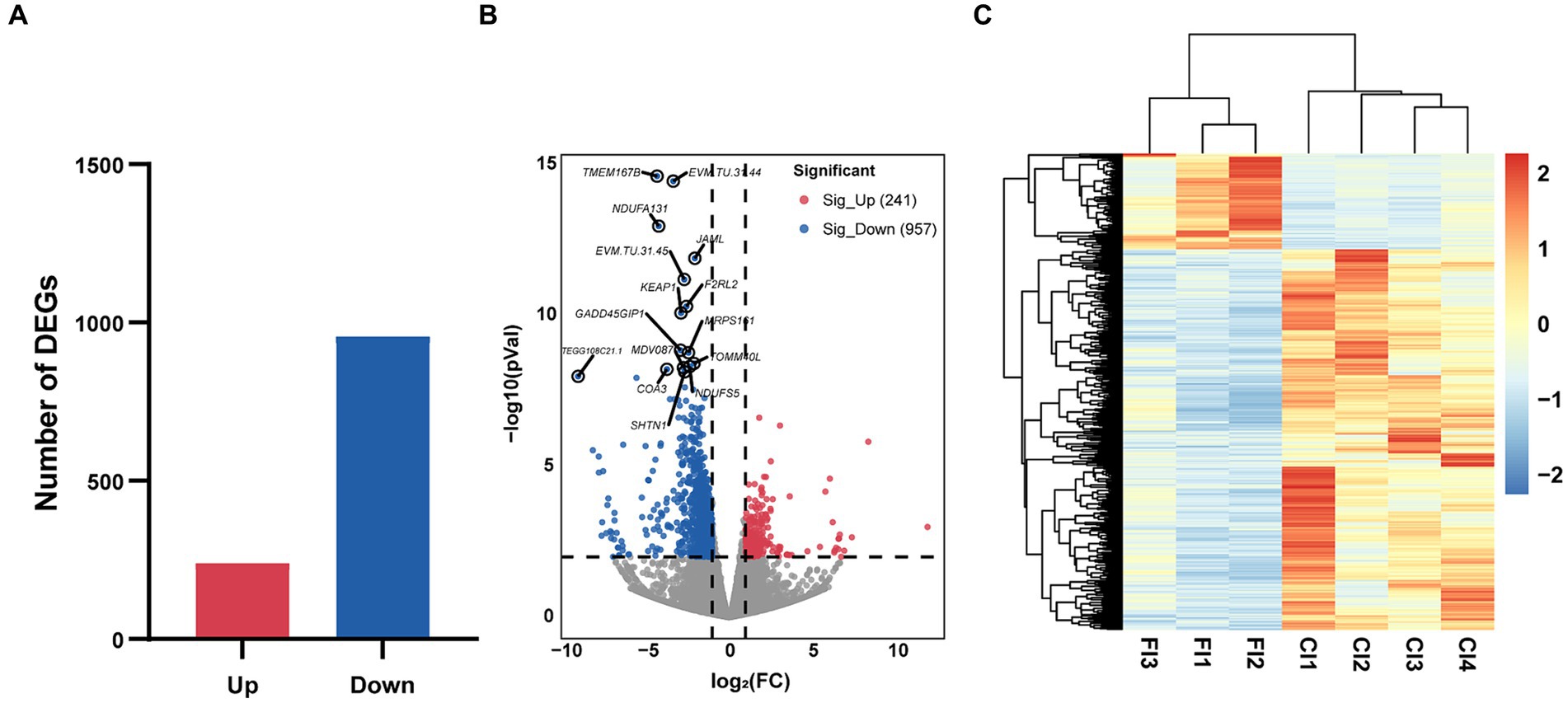
Figure 4. Characterizations of ileal transcriptome variation between CR and FR. (A) The number of DEGs. (B) Volcano map. The red dots represent up-regulated genes and blue dots represent down-regulated genes. (C) Hierarchical clustering of DEGs. DEG, differentially expressed gene; CI, ileum of cage rearing geese; FI, ileum of floor rearing geese.
Compared to FR, 957 genes were up-regulated in CR. These genes were enriched to 220 GO terms, including 129 biological processes (BP), 44 cellular components (CC), and 47 molecular functions (MF) (p < 0.05) (Figure 5A). The top 3 GO terms were cytosol, protein homodimerization activity, and lysosome. Based on KEGG pathway enrichment analysis, 28 pathways were identified significantly (p < 0.05) (Figure 5B). Metabolic pathways, lysosome, and glycosaminoglycan degradation were included. Further analysis revealed that 15 of the 28 pathways were related to metabolism, 5 to genetic information processing, 5 to cellular processes, 2 to environmental information processing, and 1 to organismal systems. Metabolism was the most up-regulated function, with 46.67% of the pathways associated with amino acid metabolism. These pathways included glycosaminoglycan degradation, biosynthesis of amino acids, glycosphingolipid biosynthesis—ganglio series, glutathione metabolism, taurine and hypotaurine metabolism, selenocompound metabolism, and glycine, serine and threonine metabolism.
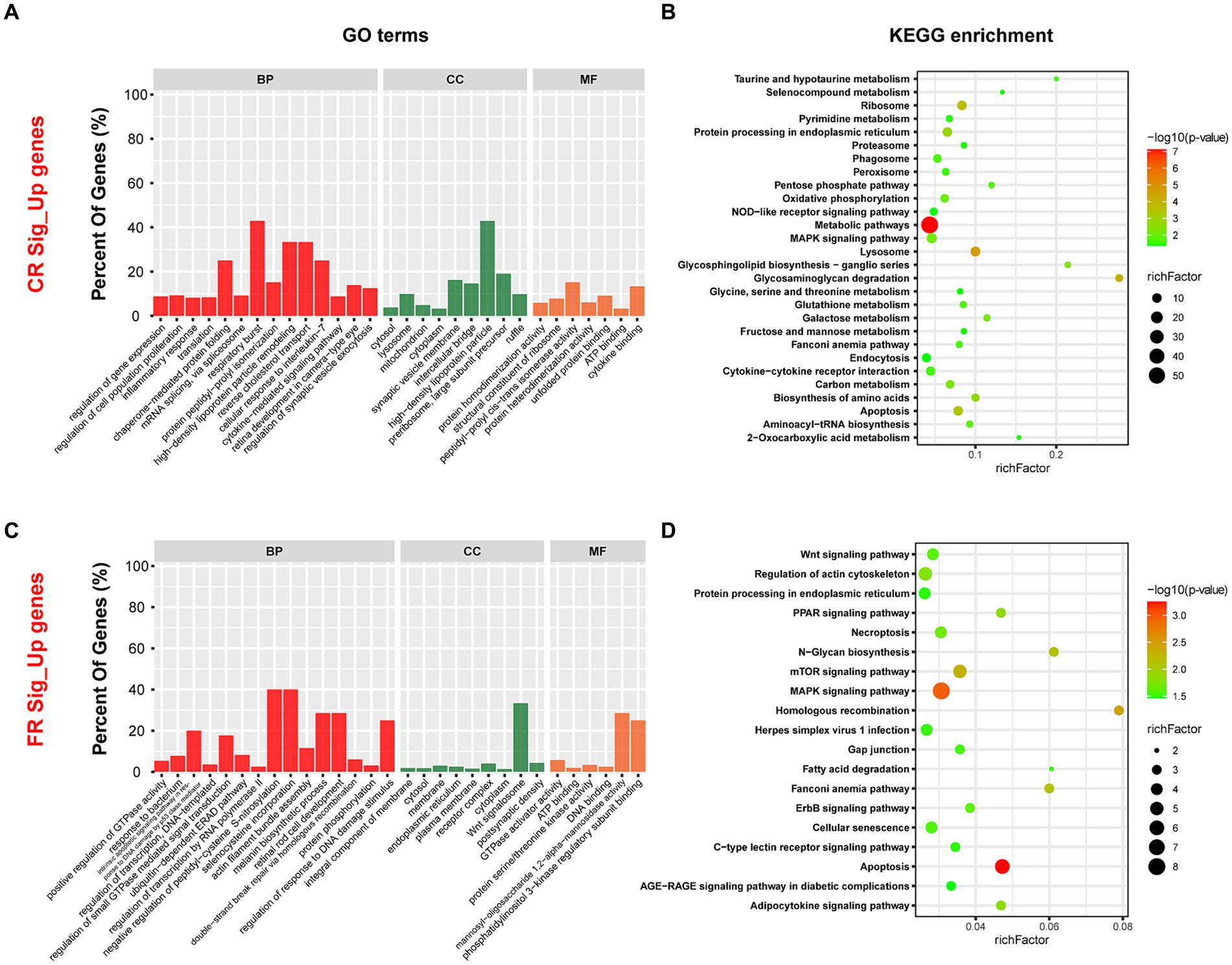
Figure 5. GO terms and KEGG pathways enriched by DEGs. (A) Top 30 GO terms enriched by DEGs up-regulated in CR. (B) The KEGG pathways significantly enriched by DEGs up-regulated in CR. (C) Top 30 GO terms enriched by DEGs up-regulated in FR. (D) The KEGG pathways significantly enriched by DEGs up-regulated in FR. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, biological processes; CC, cellular components; MF, molecular functions.
Compared to CR, 241 genes were up-regulated in the FR and they included 119 BP, 39 CC, and 41 MF (p < 0.05) (Figure 5C). Integral component of membrane, cytosol, and GTPase activator activity were the GO terms with the highest significance. Through KEGG enrichment analysis, we identified 19 signaling pathways (p < 0.05), with the highest percentage of pathways associated with cellular processes (Figure 5D). Pathways associated with cellular processes included apoptosis, regulation of actin cytoskeleton, necroptosis, cellular senescence, and gap junction were up-regulated in FR.
Combining these findings, we generated heat maps based on the results of Spearman correlation analysis. Illustrated in Figure 6A, we performed Spearman correlation analysis between up-regulated DEGs involved in amino acid metabolism and differential microbiota in CR. The abundance of hgcI_clade was significant positively correlated with the expression of MARS, NAGLU, ACY1, IDH2, ST6GALNAC6, HYAL1, SGSH, SCLY, GGT7, and GRHPR, while demonstrating a significant negative correlation with the expression of GLB1 (p < 0.05). Simultaneously, we analyzed the correlation between up-regulated DEGs involved in cellular processes and differential microbiota in FR, and the results showed that FGF10 was significantly and positively correlated with Phascolarctobacterium (p < 0.05), and, PIK3R1 was significantly and positively correlated with Sutterella (p < 0.01) (Figure 6B).
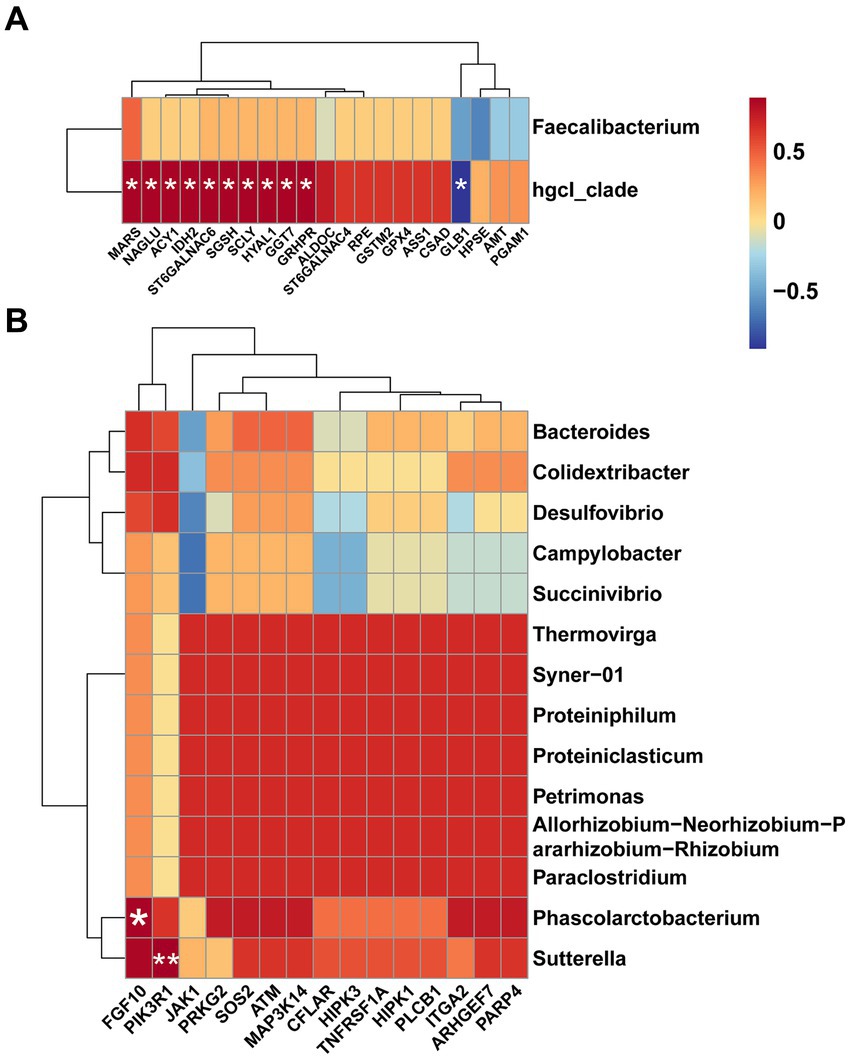
Figure 6. Heat maps for Spearman correlation analysis. (A) Spearman correlation analysis of differential microbiota and amino acid metabolism-related DEGs in CR. (B) Spearman correlation analysis of differential microbiota and cellular processes-related DEGs in FR.
The intestinal microbiota of animals represents a complex and dynamic entity susceptible to environmental influences (8). Several studies have highlighted the profound impact of rearing systems on avian intestinal development (30) and microbial composition (4, 31, 32), thereby influencing intestinal functions, digestion, and nutrient absorption (33). However, the consequences of altered microbial composition on intestinal transcriptome have received limited attention.
Consistent with previous reports (34), the rearing system was found to impact ileal microbial composition. Comparison of the microbial composition in CR and FR revealed dominant phyla such as Firmicutes, Proteobacteria, Fusobacteriota, and Bacteroidetes, consistent with previous goose studies (35). Firmicutes, Bacteroidetes, and Proteobacteria have also been identified as the major phyla in the intestinal tracts of chickens and turkeys, collectively constituting over 90% of all sequences (36). Additionally, Romboutsia emerged as the primary genus in both rearing systems, a finding somewhat divergent from certain investigations into ileal microbial composition in chickens (37, 38). Notably, Romboutsia has been identified as the predominant genus in the ileum of cage rearing chickens, potentially linked to body weight maintenance (39), lipid metabolism (40), and intestinal water metabolism (41). Further analysis revealed 2 and 14 differential microbiota in CR and FR, respectively.
Differential microbiota identified in CR, hgcI_clade, belongs to Actinobacteria (42), which has been repeatedly reported to be present in water bodies (43, 44). Some reports considered it as a potential probiotic (45). The results of Spearman’s correlation analysis showed, hgcI_clade was significant correlated with MARS, NAGLU, ACY1, IDH2, ST6GALNAC6, HYAL1, SGSH, SCLY, GGT7, GRHPR, and GLB1. This suggested that hgcI_clade may influence amino acid metabolism by modulating enzyme levels. MARS, encoding the methionyl-tRNA synthetase, may play a role in regulating the cell cycle by linking methionine and cyclin-dependent kinase 4 (46). The NAGLU encodes an enzyme involved in the catabolism of glycosaminoglycans through hydrolysis of the terminal N-acetyl-D-glucosamine residue in N-acetyl-alpha-D-glucosaminides (47). A Study on rats demonstrated that the expression of ACY1 (aminoacylase 1) was associated with the development of the jejunal crypt-villus axis and could be used as a marker for the metabolism of intestinal N-α-acetylated protein metabolism (48). Isocitrate dehydrogenase-2 (IDH2) is a marker of mitochondrial function (49), and its main function is catalyzing the oxidative decarboxylation of isocitrate to producing α-ketoglutarate (50). The ST6GALNAC6 gene encodes ST6 N-acetylgalactosaminide α-2,6-sialyltransferase 6, an enzyme belonging to the family of sialyltransferase that may catalyze the addition of sialic acid to N-acetylgalactosamine via an α-2,6 linkage (51), and its expression is associated with colon health (52). Degradation of intracellular hyaluronan acid is the main function of hyaluronidase 1 (HYAL1), which affects cell proliferation, migration and differentiation (53) and participates in neuroimmunomodulators in the microbiota-gut axis (54). SGSH, SCLY, GGT7, GRHPR, and GLB1, encoding N-sulfoglucosamine sulfohydrolase (55), selenocysteine lyase (56), gamma-glutamyltransferase 7 (57), glyoxylate and hydroxypyruvate reductase (58), and galactosidase beta 1 (59), are involved in various processes of amino acid metabolism. Another differential microbiota in CR, Faecalibacterium is known to exert vital effects in immune system regulation, intestinal barrier protection, and microbiota regulation (60). Recent findings have shown that spermidine produced by Faecalibacterium could improve intestinal function in geese (61) and chickens (62). Integrating the insights from our previous studies (17) and the current study, Faecalibacterium may be involved in enhancing ileal development in CR geese.
In our study, the FR system appeared to increase the number of harmful genera in the goose ileum, such as Campylobacter, Sutterella, Paraclostridium, Succinivibrio, and Desulfovibrio. Campylobacter is a common cause of gastroenteritis in humans worldwide (63) and poultry is the primary host (64, 65). Our findings suggested that cage rearing is essential for reducing Campylobacter colonization in the ileum, thus preventing contamination of goose products (66). Sutterella, a gram-negative microaerophilic bacterium, has been implicated in various human diseases such as autism (67, 68), Down syndrome (69) and inflammatory bowel disease (70). And it has been found in the liver and breast of chickens, which could be a potential source of contamination for humans (71). Some species of Paraclostridium have been linked to fatal infections in humans and animals (72), but the mechanism is unknown (73). It was certain that Paraclostridium was associated with poultry meat spoilage and was difficult to eradicate (74). Succinivibrio, belonging to the family Succinivibrionaceae, ferments glucose and other carbohydrates to produce large amounts of acetic acid and succinic acid (75), which might have pro-inflammatory effects (76). Studies in pigs have implicated Desulfovibrio as a major contributor in the utilization of feces for H2S production (77). And, H2S has been hypothesized to contribute to intestinal diseases such as inflammatory bowel disease (78), particularly ulcerative colitis (79). The RNA-seq results seemed to reflect the effect of harmful genera on ileal function. Because, the increase of these bacteria up-regulated pathways associated with apoptosis, necroptosis, and cellular senescence, suggesting adverse effects on ileal cells (80, 81).
In conclusion, there might be differences in ileal amino acid metabolism levels between CR and FR geese. Besides, the increase in harmful bacterial species in FR might affect the activity of ileal cells. This study provides new insights into the selection of appropriate dryland rearing systems to maintain intestinal health in geese. And more comprehensive molecular regulatory networks remain to be further investigated.
The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) repository, accession number PRJNA1054312.
The animal study was approved by the Institutional Animal Care and Use Committee (IACUC) of Sichuan Agricultural University (Chengdu campus, Sichuan, China, Permit No. DKY20170913). The study was conducted in accordance with the local legislation and institutional requirements.
ZH: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. XL: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing. XZ: Data curation, Investigation, Methodology, Writing – review & editing. QO: Data curation, Investigation, Methodology, Writing – review & editing. JH: Data curation, Investigation, Methodology, Writing – review & editing. SH: Resources, Writing – review & editing. HH: Data curation, Investigation, Methodology, Writing – review & editing. LL: Investigation, Writing – review & editing. HL: Investigation, Writing – review & editing. JW: Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by funding from the China Agriculture Research System of MOF and MARA (CARS-42-4), the School Cooperation Project of Ya’an (21SXHZ0028), and the Key Technology Support Program of Sichuan Province (2021YFYZ0014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1394290/full#supplementary-material
1. Boz, MA, Sarica, M, and Yamak, US. Production traits of artificially and naturally hatched geese in intensive and free-range systems: I. Growth traits. Br Poult Sci. (2017) 58:132–8. doi: 10.1080/00071668.2016.1261997
2. Zhao, Y, Li, X, Sun, S, Chen, L, Jin, J, Liu, S, et al. Protective role of dryland rearing on netting floors against mortality through gut microbiota-associated immune performance in Shaoxing ducks. Poult Sci. (2019) 98:4530–8. doi: 10.3382/ps/pez268
3. Tian, C, Wang, L, Liu, M, Liu, J, Qiu, M, and Chen, Y. Isolation and identification of chicken-derived lactic acid bacteria: in vitro probiotic properties and antagonistic effects against Salmonella pullorum, Staphylococcus aureus, and Escherichia Coli. Microorganisms. (2024) 12:795. doi: 10.3390/microorganisms12040795
4. Lin, YY, Chang, PE, Shen, SY, and Wang, SD. Effects of indoor and outdoor rearing system on geese biochemical parameters and cecal microbial composition. Poult Sci. (2023) 102:102731. doi: 10.1016/j.psj.2023.102731
5. Keshri, J, Smith, KM, Svendsen, MK, Keillor, HR, Moss, ML, Jordan, HJ, et al. Phenotypic characterization and draft genome sequence analyses of two novel endospore-forming Sporosarcina spp. isolated from Canada goose (Branta canadensis) feces. Microorganisms. (2024) 12:70. doi: 10.3390/microorganisms12010070
6. Moriarty, EM, Weaver, L, Sinton, LW, and Gilpin, B. Survival of Escherichia Coli, enterococci and Campylobacter Jejuni in Canada goose Faeces on pasture. Zoonoses Public Health. (2012) 59:490–7. doi: 10.1111/zph.12014
7. Zhang, X, Akhtar, M, Chen, Y, Ma, Z, Liang, Y, Shi, D, et al. Chicken Jejunal microbiota improves growth performance by mitigating intestinal inflammation. Microbiome. (2022) 10:107. doi: 10.1186/s40168-022-01299-8
8. Jha, R, and Mishra, P. Dietary Fiber in poultry nutrition and their effects on nutrient utilization, performance, gut health, and on the environment: a review. J Anim Sci Biotechnol. (2021) 12:51. doi: 10.1186/s40104-021-00576-0
9. Hiżewska, L, Osiak-Wicha, C, Tomaszewska, E, Muszyński, S, Dobrowolski, P, Andres, K, et al. Morphometric analysis of developmental alterations in the small intestine of goose. Animals (Basel). (2023) 13:3292. doi: 10.3390/ani13203292
10. Borda-Molina, D, Seifert, J, and Camarinha-Silva, A. Current perspectives of the chicken gastrointestinal tract and its microbiome. Comput Struct Biotechnol J. (2018) 16:131–9. doi: 10.1016/j.csbj.2018.03.002
11. Liu, Y, Feng, Y, Yang, X, Lv, Z, Li, P, Zhang, M, et al. Mining chicken ileal microbiota for immunomodulatory microorganisms. ISME J. (2023) 17:758–74. doi: 10.1038/s41396-023-01387-z
12. Yan, L, Lv, ZZ, An, S, Xing, K, Wang, ZG, Lv, MB, et al. Effects of rearing system and Narasin on growth performance, gastrointestinal development, and gut microbiota of broilers. Poult Sci. (2021) 100:100840. doi: 10.1016/j.psj.2020.10.073
13. Song, B, Li, P, Xu, H, Wang, Z, Yuan, J, Zhang, B, et al. Effects of rearing system and antibiotic treatment on immune function, gut microbiota and metabolites of broiler chickens. J Anim Sci Biotechnol. (2022) 13:144. doi: 10.1186/s40104-022-00788-y
14. Song, B, Yan, S, Li, P, Li, G, Gao, M, Yan, L, et al. Comparison and correlation analysis of immune function and gut microbiota of broiler chickens raised in double-layer cages and litter floor pens. Microbiol Spectr. (2022) 10:e0004522. doi: 10.1128/spectrum.00045-22
15. Wang, S, Chen, L, He, M, Shen, J, Li, G, Tao, Z, et al. Different rearing conditions Alter gut microbiota composition and host physiology in Shaoxing ducks. Sci Rep. (2018) 8:7387. doi: 10.1038/s41598-018-25760-7
16. Chen, X, Huang, L, Cheng, L, Hu, B, Liu, H, Hu, J, et al. Effects of floor-and net-rearing systems on intestinal growth and microbial diversity in the ceca of ducks. BMC Microbiol. (2022) 22:76. doi: 10.1186/s12866-022-02478-1
17. Li, X, Hu, S, Wang, W, Tang, B, Zheng, C, Hu, J, et al. Effects of cage versus floor rearing system on goose intestinal histomorphology and cecal microbial composition. Poult Sci. (2022) 101:101931. doi: 10.1016/j.psj.2022.101931
18. Magoč, T, and Salzberg, SL. Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
19. Bolyen, E, Rideout, JR, Dillon, MR, Bokulich, NA, Abnet, CC, Al-Ghalith, GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
20. Quast, C, Pruesse, E, Yilmaz, P, Gerken, J, Schweer, T, Yarza, P, et al. The Silva ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. (2013) 41 (Database issue): D590-6) 41:D590–6. doi: 10.1093/nar/gks1219
21. Segata, N, Izard, J, Waldron, L, Gevers, D, Miropolsky, L, Garrett, WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
22. Langille, MG, Zaneveld, J, Caporaso, JG, McDonald, D, Knights, D, Reyes, JA, et al. Predictive functional profiling of microbial communities using 16s Rrna marker gene sequences. Nat Biotechnol. (2013) 31:814–21. doi: 10.1038/nbt.2676
23. Parks, DH, and Beiko, RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. (2010) 26:715–21. doi: 10.1093/bioinformatics/btq041
24. Chen, S, Zhou, Y, Chen, Y, and Gu, J. Fastp: An ultra-fast all-in-one Fastq preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
25. Kim, D, Langmead, B, and Salzberg, SL. Hisat: a fast spliced aligner with low memory requirements. Nat Methods. (2015) 12:357–60. doi: 10.1038/nmeth.3317
26. Li, H, Handsaker, B, Wysoker, A, Fennell, T, Ruan, J, Homer, N, et al. The sequence alignment/map format and Samtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
27. Liao, Y, Smyth, GK, and Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
28. Love, MI, Huber, W, and Anders, S. Moderated estimation of fold change and dispersion for Rna-Seq data with Deseq 2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
29. Bu, D, Luo, H, Huo, P, Wang, Z, Zhang, S, He, Z, et al. Kobas-I: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. (2021) 49:W317–25. doi: 10.1093/nar/gkab447
30. Li, J, Miao, Z, Tian, W, Yang, Y, Wang, J, and Yang, Y. Effects of different rearing systems on growth, small intestinal morphology and selected indices of fermentation status in broilers. Anim Sci J. (2017) 88:900–8. doi: 10.1111/asj.12697
31. Wang, L-d, Zhang, Y, Kong, L-l, Wang, Z-x, Bai, H, Jiang, Y, et al. Effects of rearing system (floor vs. cage) and sex on performance, meat quality and enteric microorganism of yellow feather broilers. J Integr Agric. (2021) 20:1907–20. doi: 10.1016/S2095-3119(20)63420-7
32. Shi, S, Qi, Z, Gu, B, Cheng, B, Tu, J, Song, X, et al. Analysis of high-throughput sequencing for Cecal microbiota diversity and function in hens under different rearing systems. 3 Biotech. (2019) 9:438. doi: 10.1007/s13205-019-1970-7
33. Gong, J, Forster, RJ, Yu, H, Chambers, JR, Wheatcroft, R, Sabour, PM, et al. Molecular analysis of bacterial populations in the ileum of broiler chickens and comparison with Bacteria in the cecum. FEMS Microbiol Ecol. (2002) 41:171–9. doi: 10.1111/j.1574-6941.2002.tb00978.x
34. Kers, JG, Velkers, FC, Fischer, EAJ, Hermes, GDA, Stegeman, JA, and Smidt, H. Host and environmental factors affecting the intestinal microbiota in chickens. Front Microbiol. (2018) 9:235. doi: 10.3389/fmicb.2018.00235
35. Yang, H, Xiao, Y, Gui, G, Li, J, Wang, J, and Li, D. Microbial community and short-chain fatty acid profile in gastrointestinal tract of goose. Poult Sci. (2018) 97:1420–8. doi: 10.3382/ps/pex438
36. Wei, S, Morrison, M, and Yu, Z. Bacterial census of poultry intestinal microbiome. Poult Sci. (2013) 92:671–83. doi: 10.3382/ps.2012-02822
37. Bjerrum, L, Engberg, RM, Leser, TD, Jensen, BB, Finster, K, and Pedersen, K. Microbial community composition of the ileum and cecum of broiler chickens as revealed by molecular and culture-based techniques. Poult Sci. (2006) 85:1151–64. doi: 10.1093/ps/85.7.1151
38. Maki, JJ, Klima, CL, Sylte, MJ, and Looft, T. The microbial pecking order: utilization of intestinal microbiota for poultry health. Microorganisms. (2019) 7:376. doi: 10.3390/microorganisms7100376
39. Wiersema, ML, Koester, LR, Schmitz-Esser, S, and Koltes, DA. Comparison of intestinal permeability, morphology, and ileal microbial communities of commercial hens housed in conventional cages and cage-free housing systems. Poult Sci. (2021) 100:1178–91. doi: 10.1016/j.psj.2020.10.052
40. Yin, H, Huang, J, Guo, X, Xia, J, and Hu, M. Romboutsia Lituseburensis Jcm1404 supplementation ameliorated endothelial function via gut microbiota modulation and lipid metabolisms alterations in obese rats. FEMS Microbiol Lett. (2023) 370:fnad016. doi: 10.1093/femsle/fnad016
41. Li, J, Sun, Y, Wang, R, Ma, S, Shi, L, Wang, K, et al. Seasonal differences in intestinal Flora are related to Rats' intestinal water metabolism. Front Microbiol. (2023) 14:1109696. doi: 10.3389/fmicb.2023.1109696
42. Ali, S, Xie, J, Zada, S, Hu, Z, Zhang, Y, Cai, R, et al. Bacterial community structure and bacterial isolates having antimicrobial potential in shrimp pond aquaculture. AMB Express. (2022) 12:82. doi: 10.1186/s13568-022-01423-9
43. Zhang, M, Zeng, G, Liang, D, Xu, Y, Li, Y, Huang, X, et al. An analysis of the Colony structure of prokaryotes in the Jialing River waters in Chongqing. Int J Environ Res Public Health. (2022) 19:5525. doi: 10.3390/ijerph19095525
44. Yue, Y, Cai, L, Tang, Y, Zhang, Y, Yang, M, and Wang, F. Vertical distribution of bacterial community in water columns of reservoirs with different trophic conditions during thermal stratification. Front Environ Sci. (2021) 9:9. doi: 10.3389/fenvs.2021.632089
45. Das, S, Ward, LR, and Burke, C. Prospects of using marine Actinobacteria as probiotics in aquaculture. Appl Microbiol Biotechnol. (2008) 81:419–29. doi: 10.1007/s00253-008-1731-8
46. Yu, YC, Han, JM, and Kim, S. Aminoacyl-Trna Synthetases and amino acid signaling. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118889. doi: 10.1016/j.bbamcr.2020.118889
47. Jokela, H, Hakkarainen, J, Kätkänaho, L, Pakarinen, P, Ruohonen, ST, Tena-Sempere, M, et al. Deleting the mouse Hsd17b1 gene results in a Hypomorphic Naglu allele and a phenotype mimicking a lysosomal storage disease. Sci Rep. (2017) 7:16406. doi: 10.1038/s41598-017-16618-5
48. Cigna, N, Nicoletti, C, Durand, A, Chaix, JC, Giardina, T, and Perrier, J. Acylase 1 expression in rat intestinal crypt--villus Axis. Cell Biol Int. (2007) 31:966–73. doi: 10.1016/j.cellbi.2007.03.006
49. Anderson, G, and Mazzoccoli, G. Left ventricular hypertrophy: roles of mitochondria Cyp1b1 and melatonergic pathways in co-ordinating wider pathophysiology. Int J Mol Sci. (2019) 20:4068. doi: 10.3390/ijms20164068
50. Rhee, YY, Kim, KJ, and Kang, GH. Cpg Island Methylator phenotype-high colorectal cancers and their prognostic implications and relationships with the serrated neoplasia pathway. Gut Liver. (2017) 11:38–46. doi: 10.5009/gnl15535
51. Poulsen, NA, Robinson, RC, Barile, D, Larsen, LB, and Buitenhuis, B. A genome-wide association study reveals specific transferases as candidate loci for bovine Milk oligosaccharides synthesis. BMC Genomics. (2019) 20:404. doi: 10.1186/s12864-019-5786-y
52. Huang, H-C, Chao, C-C, Wu, P-H, Chung, H-Y, Lee, H-Y, Suen, C-S, et al. Epigenetic silencing of the synthesis of immunosuppressive Siglec ligand Glycans by Nf-Κb/Ezh 2/Yy1 axis in early-stage colon cancers. Biochim Biophys Acta Gene Regul Mech. (2019) 1862:173–83. doi: 10.1016/j.bbagrm.2019.01.002
53. Šimek, M, Turková, K, Schwarzer, M, Nešporová, K, Kubala, L, Hermannová, M, et al. Molecular weight and gut microbiota determine the bioavailability of orally administered hyaluronic acid. Carbohydr Polym. (2023) 313:120880. doi: 10.1016/j.carbpol.2023.120880
54. Bosi, A, Banfi, D, Bistoletti, M, Moretto, P, Moro, E, Crema, F, et al. Hyaluronan: a neuroimmune modulator in the microbiota-gut axis. Cells. (2021) 11:126. doi: 10.3390/cells11010126
55. Costanzi, E, Beccari, T, Stinchi, S, Bibi, L, Hopwood, JJ, and Orlacchio, A. Gene encoding the mouse Sulphamidase: cDNA cloning, structure, and chromosomal mapping. Mamm Genome. (2000) 11:436–9. doi: 10.1007/s003350010083
56. Seale, LA. Selenocysteine Β-Lyase: biochemistry, regulation and physiological role of the Selenocysteine decomposition enzyme. Antioxidants (Basel). (2019) 8:357. doi: 10.3390/antiox8090357
57. Heisterkamp, N, Groffen, J, Warburton, D, and Sneddon, TP. The human gamma-Glutamyltransferase gene family. Hum Genet. (2008) 123:321–32. doi: 10.1007/s00439-008-0487-7
58. Katane, M, Matsuda, S, Saitoh, Y, Miyamoto, T, Sekine, M, Sakai-Kato, K, et al. Glyoxylate reductase/hydroxypyruvate reductase regulates the free D-aspartate level in mammalian cells. J Cell Biochem. (2021) 122:1639–52. doi: 10.1002/jcb.30110
59. Kwak, JE, Son, M-Y, Son, YS, Son, MJ, and Cho, YS. Biochemical and molecular characterization of novel mutations in Glb1 and Neu1 in patient cells with lysosomal storage disorders. Biochem Biophys Res Commun. (2015) 457:554–60. doi: 10.1016/j.bbrc.2015.01.023
60. Martín, R, Rios-Covian, D, Huillet, E, Auger, S, Khazaal, S, Bermúdez-Humarán, LG, et al. Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol Rev. (2023) 47:fuad 039. doi: 10.1093/femsre/fuad039
61. Wang, Z, Jiang, D, Wang, X, Jiang, Y, Sun, Q, Ling, W, et al. Spermidine improves the antioxidant capacity and morphology of intestinal tissues and regulates intestinal microorganisms in Sichuan white geese. Front Microbiol. (2023) 14:1292984. doi: 10.3389/fmicb.2023.1292984
62. Zhu La, AT, Li, D, Cheng, Z, Wen, Q, Hu, D, Jin, X, et al. Enzymatically prepared Neoagarooligosaccharides improve gut health and function through promoting the production of spermidine by Faecalibacterium in chickens. Sci Total Environ. (2023) 912:169057. doi: 10.1016/j.scitotenv.2023.169057
63. Costa, D, and Iraola, G. Pathogenomics of emerging campylobacter species. Clin Microbiol Rev. (2019) 32:e00072-18. doi: 10.1128/cmr.00072-18
64. Dearlove, BL, Cody, AJ, Pascoe, B, Méric, G, Wilson, DJ, and Sheppard, SK. Rapid host switching in generalist Campylobacter strains erodes the signal for tracing human infections. ISME J. (2016) 10:721–9. doi: 10.1038/ismej.2015.149
65. Skarp, CPA, Hänninen, ML, and Rautelin, HIK. Campylobacteriosis: the role of poultry meat. Clin Microbiol Infect. (2016) 22:103–9. doi: 10.1016/j.cmi.2015.11.019
66. Keener, KM, Bashor, MP, Curtis, PA, Sheldon, BW, and Kathariou, S. Comprehensive review of Campylobacter and poultry processing. Compr Rev Food Sci Food Saf. (2004) 3:105–16. doi: 10.1111/j.1541-4337.2004.tb00060.x
67. Williams, BL, Hornig, M, Parekh, T, and Lipkin, WI. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. MBio. (2012) 3:e00261-11. doi: 10.1128/mBio.00261-11
68. Wang, L, Christophersen, CT, Sorich, MJ, Gerber, JP, Angley, MT, and Conlon, MA. Increased abundance of Sutterella Spp. and Ruminococcus Torques in feces of children with autism Spectrum disorder. Mol Autism. (2013) 4:42. doi: 10.1186/2040-2392-4-42
69. Biagi, E, Candela, M, Centanni, M, Consolandi, C, Rampelli, S, Turroni, S, et al. Gut microbiome in down syndrome. PLoS One. (2014) 9:e112023. doi: 10.1371/journal.pone.0112023
70. Mangin, I, Bonnet, R, Seksik, P, Rigottier-Gois, L, Sutren, M, Bouhnik, Y, et al. Molecular inventory of faecal microflora in patients with Crohn's disease. FEMS Microbiol Ecol. (2004) 50:25–36. doi: 10.1016/j.femsec.2004.05.005
71. Derqaoui, S, Oukessou, M, Attrassi, K, Elftouhy, FZ, and Nassik, S. Detection of Sutterella spp. in broiler liver and breast. Front Vet Sci. (2022) 9:859902. doi: 10.3389/fvets.2022.859902
72. Gohil, P, Patel, K, Patel, S, Pandit, R, Suthar, V, Duggirala, S, et al. In-depth analysis of an obligate anaerobe Paraclostridium bifermentans isolated from uterus of Bubalus Bubalis. Animals (Basel). (2022) 12:1765. doi: 10.3390/ani12141765
73. Magome, TG, Ramatla, T, Mokgokong, P, Thekisoe, O, and Lekota, KE. The draft genome and Pan-genome structure of Paraclostridium bifermentans strain T2 isolated from sheep Faeces. Data Brief. (2023) 51:109660. doi: 10.1016/j.dib.2023.109660
74. Song, M, Lei, Y, Ali, A, Xu, Y, Sheng, K, Huang, T, et al. Inhibitory effect of licorice extract on the germination and outgrowth of Paraclostridium bifermentans spores. Front Microbiol. (2022) 13:13. doi: 10.3389/fmicb.2022.1076144
75. Li, Z, Shen, J, Xu, Y, and Zhu, W. Metagenomic analysis reveals significant differences in microbiome and metabolic profiles in the rumen of sheep fed low N diet with increased urea supplementation. FEMS Microbiol Ecol. (2020) 96:fiaa117. doi: 10.1093/femsec/fiaa117
76. Marquez-Ortiz, RA, Leon, M, Abril, D, Escobar-Perez, J, Florez-Sarmiento, C, Parra-Izquierdo, V, et al. Colonoscopy aspiration lavages for mucosal metataxonomic profiling of Spondylarthritis-associated gastrointestinal tract alterations. Sci Rep. (2023) 13:7015. doi: 10.1038/s41598-023-33597-y
77. Karnachuk, OV, Rusanov, II, Panova, IA, Grigoriev, MA, Zyusman, VS, Latygolets, EA, et al. Microbial sulfate reduction by Desulfovibrio is an important source of hydrogen sulfide from a large swine finishing facility. Sci Rep. (2021) 11:10720. doi: 10.1038/s41598-021-90256-w
78. Stummer, N, Feichtinger, RG, Weghuber, D, Kofler, B, and Schneider, AM. Role of hydrogen sulfide in inflammatory bowel disease. Antioxidants (Basel). (2023) 12:1570. doi: 10.3390/antiox12081570
79. Pitcher, MC, and Cummings, JH. Hydrogen Sulphide: a bacterial toxin in ulcerative colitis? Gut. (1996) 39:1–4. doi: 10.1136/gut.39.1.1
80. D'Arcy, MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. (2019) 43:582–92. doi: 10.1002/cbin.11137
Keywords: goose, ileum, 16S rRNA sequencing, transcriptome, rearing system
Citation: He Z, Li X, Zhang X, Ouyang Q, Hu J, Hu S, He H, Li L, Liu H and Wang J (2024) Effects of rearing systems (cage versus floor) on the microbial composition and transcriptome of goose ileum. Front. Vet. Sci. 11:1394290. doi: 10.3389/fvets.2024.1394290
Edited by:
Izhar Hyder Qazi, Shaheed Benazir Bhutto University of Veterinary and Animal Sciences, PakistanReviewed by:
Monika Proszkowiec-Weglarz, Agricultural Research Service, United StatesCopyright © 2024 He, Li, Zhang, Ouyang, Hu, Hu, He, Li, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiwen Wang, d2p3Mjg4NjE2NkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.