 Jin-Tao Chen
Jin-Tao Chen Kang-Jing Chen2
Kang-Jing Chen2 Jian-Wei Shao
Jian-Wei Shao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 02 May 2024
Sec. Veterinary Infectious Diseases
Volume 11 - 2024 | https://doi.org/10.3389/fvets.2024.1389264
The genus Hepacivirus comprises a diverse range of genetically distinct viruses that infect both mammalian and non-mammalian hosts, with some posing significant risks to human and animal health. Members of the genus Hepacivirus are typically classified into fourteen species (Hepacivirus A–N), with ongoing discoveries of novel hepaciviruses like Hepacivirus P and Hepacivirus Q. In this study, a novel Hepacivirus was identified in duck liver samples collected from live poultry markets in Hunan province, China, using unbiased high-throughput sequencing and meta-transcriptomic analysis. Through sequence comparison and phylogenetic analysis, it was determined that this newly discovered Hepacivirus belongs to a new subspecies of Hepacivirus Q. Moreover, molecular screening revealed the widespread circulation of this novel virus among duck populations in various regions of Hunan province, with an overall prevalence of 13.3%. These findings significantly enhence our understanding of the genetic diversity and evolution of hepaciviruses, emphasizing the presence of genetically diverse hepaciviruses duck populations in China. Given the broad geographical distribution and relatively high positive rate, further investigations are essential to explore any potential associations between Hepacivirus Q and duck-related diseases.
The genus of Hepacivirus, along with the genera Flavivirus, Pegivirus and Pestivirus, is currently classified within the family Flaviviridae, which includes a genetically diverse group of human and animal pathogens (1, 2). Members of the genus Hepacivirus are enveloped viruses with unsegmented, single-stranded, positive-sense RNA genomes that are approximately 10 kb in length (1). These genomes contain the 5′ untranslated regions (UTR) and 3′ UTR, as well as a single large open reading frame (ORF) that encodes a polyprotein (3). The polyprotein is cleaved by signal peptidase, NS2/NS3 protease, and NS3 protease enzymes, resulting in the production of three structural proteins (Core, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (4).
Hepacivirus was initially identified in 1989 as a human pathogen, with humans believed to be its sole natural host for a long time (5). However, since 2011, novel and genetically diverse hepaciviruses have been found in a wide range of mammalian and non-mammalian hosts (6). Moreover, recently discovered hepaciviruses named Hepacivirus P and Hepacivirus Q were detected in long-tailed ground squirrels and domestic ducks in China, respectively (7–9). These distinct hepaciviruses found in different host species are currently mainly classified as Hepacivirus A–N based on their phylogenetic relationships and host range (2).
The first identification of Hepacivirus in avian species was documented in 2019, specifically in domestic ducks across various areas in China (9). This virus has been associated with significant declines in egg production, with viral RNA detection rates ranging from 38.5 to 88% in different areas (9). Subsequently, additional novel hepaciviruses were also discovered in avian species (10, 11). More recently, another novel Hepacivirus, named Hepacivirus Q, was identified in domestic ducks in China (8), highlighting the genetic diversity of hepaciviruses in avian species. In this study, a new subspecies of Hepacivirus Q was discovered in domestic ducks from Hunan province, China using unbiased high-throughput sequencing and meta-transcriptomic analysis. Furthermore, the prevalence of this novel virus in duck populations in specific regions of Hunan province, China was examined. This study not only enhances our understanding of the genetic diversity and evolution of hepaciviruses but also underscores the importance of investigating whether Hepacivirus Q is associated with any duck disease.
In a poultry viral agent discovery project, 40 duck liver samples were collected from live poultry market located in Hengyang city, Hunan province, China, between August and September 2023 (Figure 1). Approximately 50 mg of liver tissue was homogenized with 500 μL sterile phosphate-buffered saline (PBS). Total RNA was extracted from 200 μL homogenates by using RNAiso Plus reagent (TaKaRa, Dalian, China) and subsequently purified using the RNeasy Plus Mini Kit (Qiagen, Germany). The quantity and quality of the extracted RNA were assessed using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, United States). Subsequently, the individual RNA solutions were pooled in equal quantities, and the quality of the pooled RNA was evaluated using an Agilent 2,100 Bioanalyzer (Agilent Technologies) before library construction and sequencing.
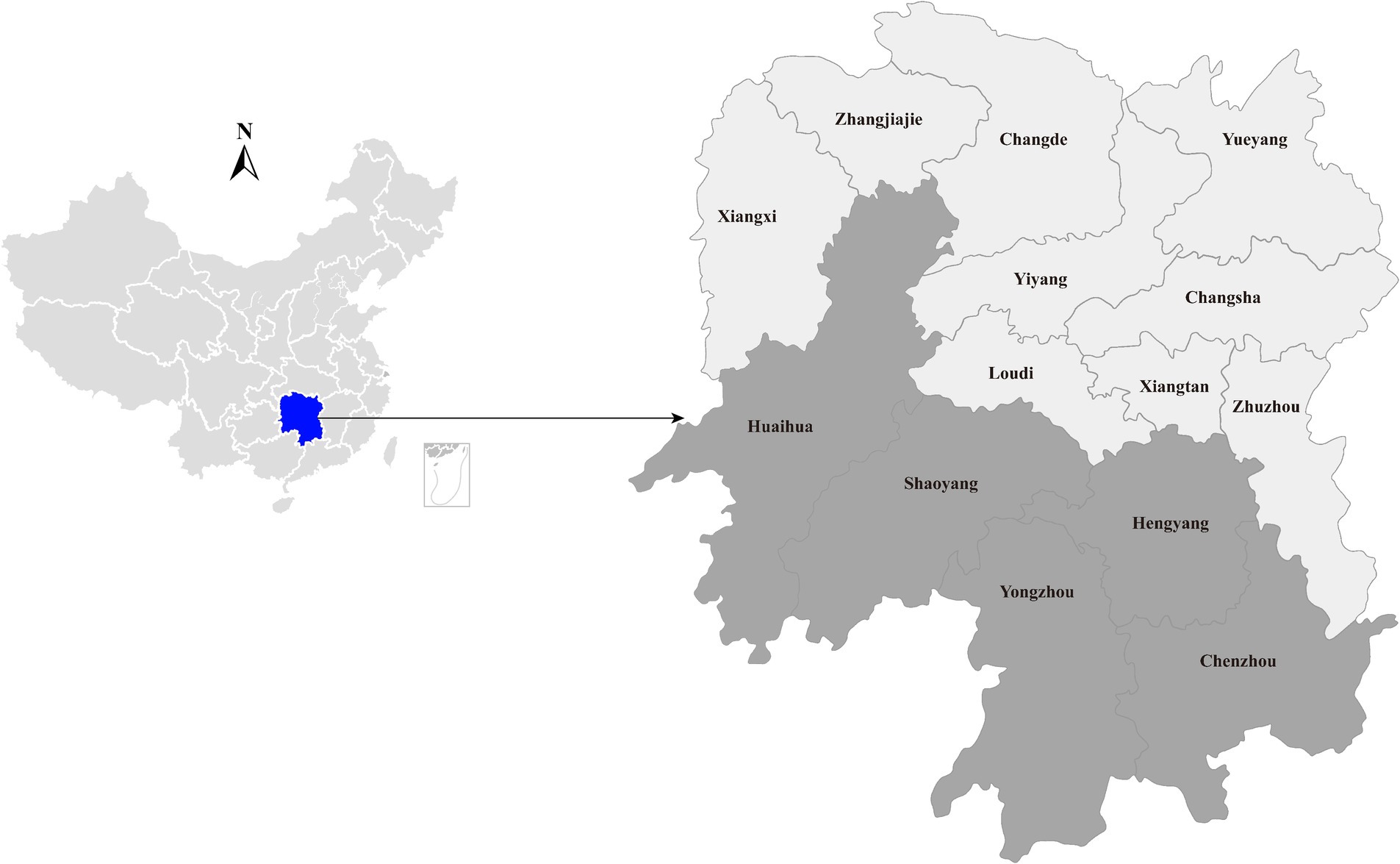
Figure 1. Geographic maps showing the location of sampling sites where the duck liver samples collected in this study. This map was plotted by combination of Surfer software version-4 (Golden Software, United States) and Adobe illustrator version CC2017 (Adobe, United States).
The RNA library preparation followed the methodology as previously described (12, 13). Briefly, ribosomal RNA (rRNA) was depleted using a Ribo-Zero-Gold (Epidemiology) kit (Illumina Inc., United States) following the manufacturer’s instructions. The remaining RNA was fragmented, reverse-transcribed, adapted, and purified using the TruSeq total RNA library preparation kit (Illumina Inc., United States). The quality of the library quality was assessed using the Qubit high-sensitivity RNA/DNA assays (Thermo Fisher Scientific) and the Agilent 2,100 Bioanalyzer (Agilent Technologies). Paired-end sequencing with 150-bp read length was conducted on the Illumina Hiseq2500 platform. All library preparation and sequencing procedures were carried out by Novogene (Tianjin, China).
The sequencing reads were demultiplexed, trimmed for the adaptor, and subjected to quality control using the fastp program (14). Subsequently, de novo assembly was performed using the Megahit program (15) with default parameters. The resulting contigs were compared against the entire non-redundant protein (nr) database downloaded from GenBank using the diamond blastx program (16) with an e-value threshold of 1e–3. Viral contigs with unassembled overlaps or originating from the same scaffold were merged using the SeqMan program implemented in the Lasergene software package (version 7.1, DNAstar).
To confirm the assembly results, reads were mapped back to the target contigs with Bowtie2 (17), and assembly errors were inspected using Integrated Genomics Viewer (IGV). Any gaps between these contigs were filled by reverse transcription PCR (RT-PCR) and Sanger sequencing (Table 1). The virus genome termini were determined using 5′/3′ RACE kits (TaKaRa, Dalian, China) following the producer’s instructions. The final virus genome sequence was obtained by consensus mapping assembly and confirmed by Sanger sequencing using overlapping primers that covered the entire sequence.
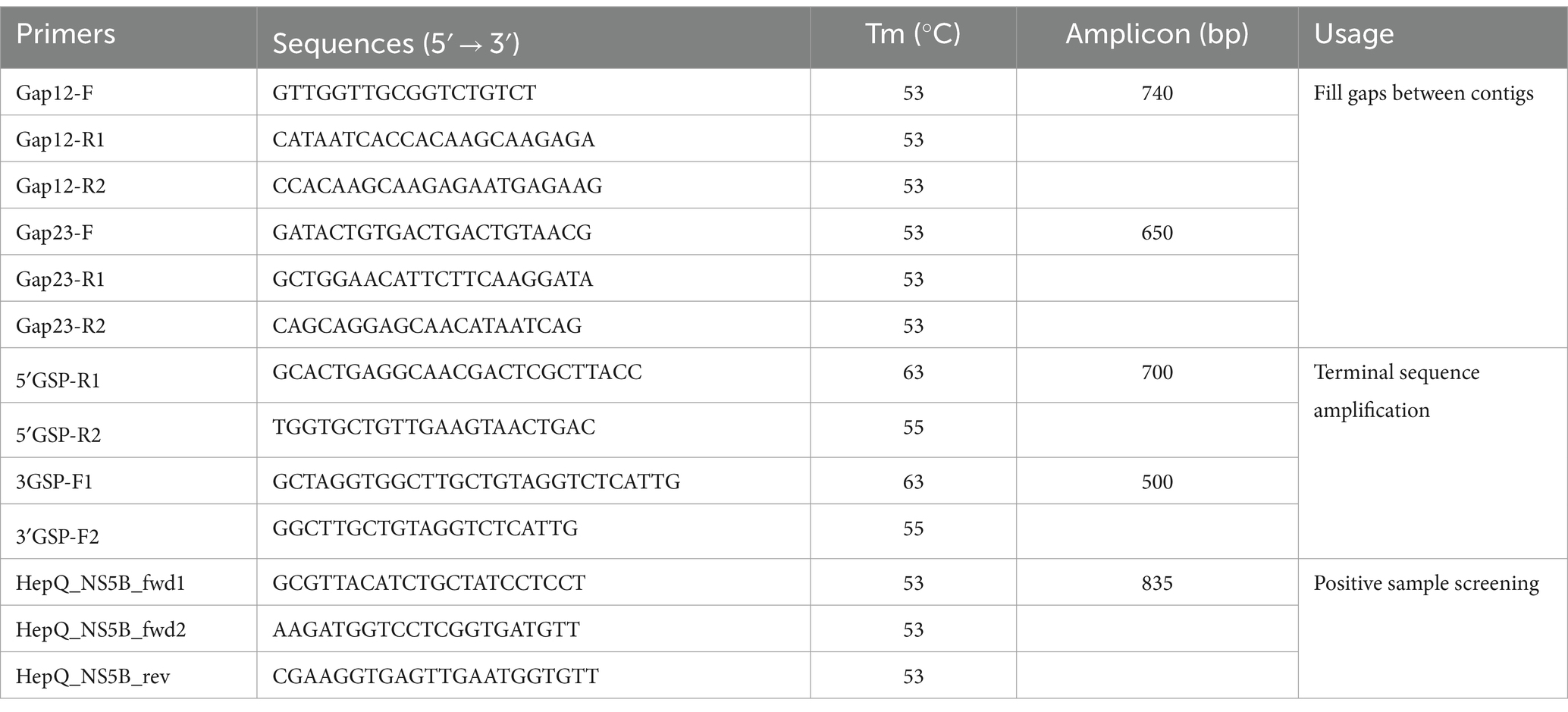
Table 1. Primers used in this study.
The identification of potential open reading frames (ORFs) was conducted using ORFfinder1 and annotated by comparing against the non-redundant protein database. The nucleotide sequence similarity between this newly identified Hepacivirus and other hepaciviruses was calculated using the MegAlign program, available in the Lasergene software package (version 7.1, DNAstar).
To determine potential recombination events that occurred in the evolutionary history of this newly identified Hepacivirus, seven methods (RDP, GENECONV, bootscan, maximum chi square, Chimera, SISCAN, and distance plot) within RDP4 program (18) were employed. Analyzes were conducted using complete genome sequences with default settings for the different test methods and a Bonferroni corrected p-value cutoff of 0.05. Only sequences with significant evidence (p < 0.05) of recombination detected by at least two methods and confirmed by phylogenetic analysis were considered to have strong evidence for recombination. Furthermore, similarity plot analyzes were performed to further characterize potential recombination events, including the location of possible breakpoints, using Simplot version 3.5.1 (19).
To determine the phylogenetic relationship between the newly identified Hepacivirus and other known hepaciviruses, amino acid sequences of the complete polyprotein, NS3 (peptidase and helicase), and NS5 (RNA-dependent RNA polymerase) proteins were aligned using the E-INS-i algorithm implemented in MAFFT program (20). Phylogenetic trees were reconstructed using the maximum-likelihood method (ML) implemented in PhyML version 3.0 (21). The LG amino acid substitution model with a gamma (Γ)-distribution model (i.e., LG + Γ) determined using Prot-Test 3 (22), was employed. Bootstrap support values were calculated from 1,000 replicate trees using a Subtree Pruning and Regrafting (SPR) branch-swapping algorithm. For clarity purposes, all phylogenetic trees were rooted at the mid-point.
To gain insight into the prevalence of the newly identified Hepacivirus circulating in domestic ducks, liver tissues were collected from live poultry markets located in five cities in Hunan (Figure 1). All individual samples, including those that subjected to meta-transcriptome sequencing, were screened for the presence of the virus using PCR with primers targeting the NS5B region (Table 1). The PCR product with the expected size was confirmed by Sanger sequencing.
Statistical analyzes were conducted using Statistical Package for Social Sciences (SPSS) Version 21.0 software. Descriptive statistics were used to calculate frequency and percentage, while Fisher exact test was utilized to determine the p-value and assess the differences in positive rates between sampling sites. A p value <0.05 was considered as statistically significant.
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received. The procedures for sampling and sample processing were approved by the ethics committee of Foshan University. All animals were treated in strict accordance with the Rules for the Implementation of Laboratory Animal Medicine (1998) from the Ministry of Health, China.
In a viral agent discovery project involving domestic ducks, RNA solutions extracted from 40 individual liver tissues were pooled as one sample. This sample was screened for both known and putative novel viruses through meta-transcriptome sequencing. After de novo assembly and comparison against the nr database, 20 contigs ranging from 257 to 995 nt in length were annotated as Hepacivirus Q, with 95.4 to 100% amino acid identities. After filling the gaps between these contigs through RT-PCR, and determining the terminal sequences using 5′/3′ RACE, the complete genome sequence of this virus was obtained. Using this sequence as the reference sequence, 393 reads were remapped, providing a genome coverage of 92.8% (9,194 nt/9,904 nt) and a pairwise identity of 96.2% at a mean depth of 9.9× (Figure 2).
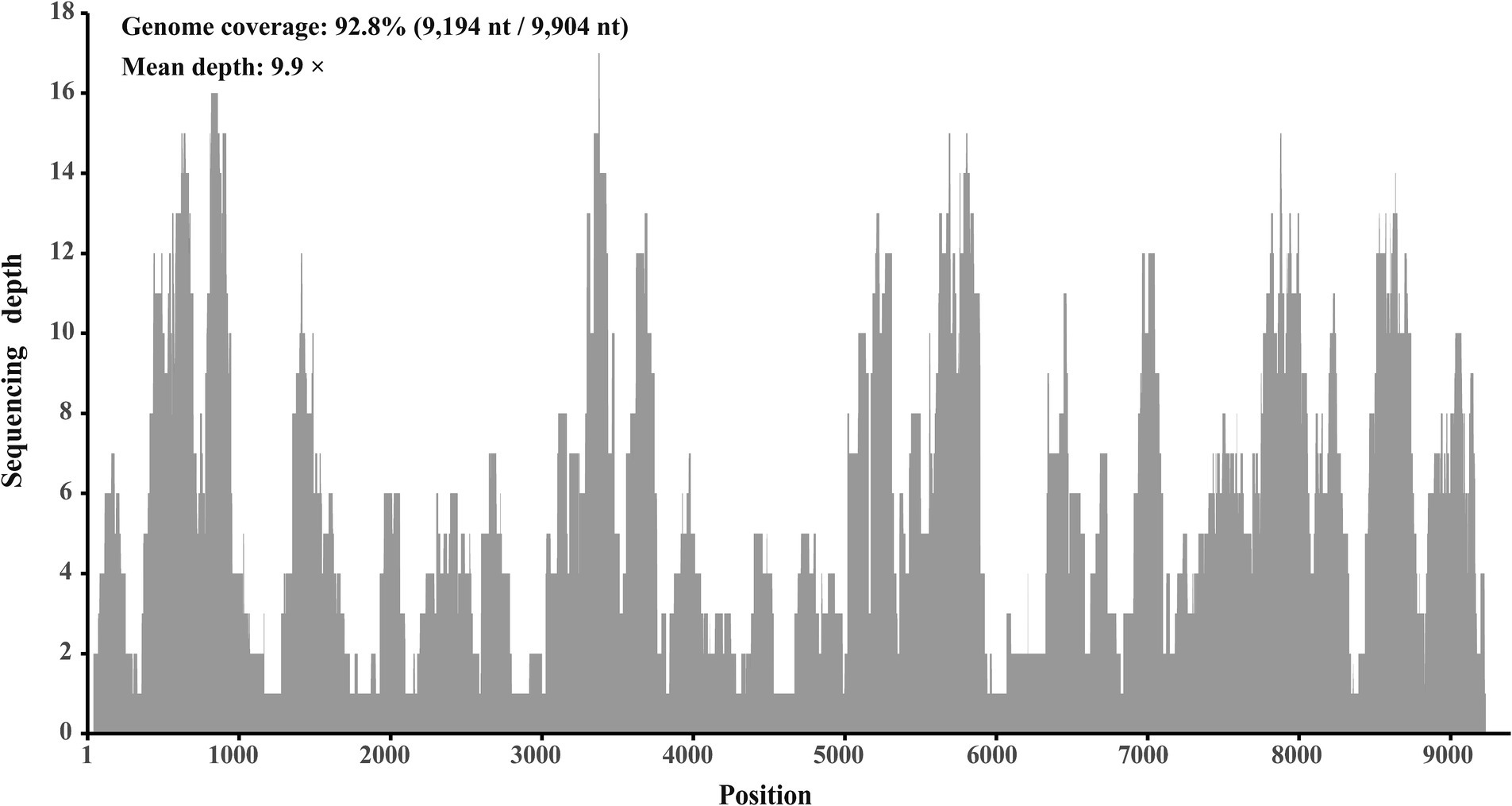
Figure 2. Mapped read count plot of the genome of Hepacivirus Q strain HN. The histograms show the coverage depth per base of genome of Hepacivirus Q strain HN and the mean sequencing depth of was 9.9 ×.
This newly identified Hepacivirus has a complete genome sequence comprising 9,893 nucleotides and contains a single large ORF that encodes a polyprotein of 2,999 amino acids. Comparative analysis of the sequence revealed that this novel virus shares a genome-wide nucleotide identity of 94.95 to 96.94% with the available sequences of Hepacivirus Q in GenBank. Additionally, the putative complete polyprotein of this novel Hepacivirus exhibits an amino acid identity of 98.13 to 98.93% with the polyprotein of Hepacivirus Q. Consequently, this newly identified Hepacivirus can be classified as a new subspecies of Hepacivirus Q.
No statistically supported recombination event was detected within this novel Hepacivirus after systematic analyzes. Phylogenetic trees reconstructed using the amino acid sequence of the complete polyprotein, NS3, and NS5B proteins consistently revealed that this newly identified Hepacivirus grouped together with the known Hepacivirus Q strains and formed a sister lineage to a Hepacivirus that was identified from Bald eale in the USA (Figure 3). Moreover, the Hepacivirus Q group, which includes this newly identified hepacivirus, exhibits significant genetic divergence from other duck Hepacivirus identified in China, indicating a high level of genetic diversity within hepaciviruses. Notably, the NS3 tree shows a closer phylogenetic relationship among the Hepacivirus Q group and both Bald eagle Hepacivirus and Jogalong virus, while the NS5B tree reveals a closer relationship with Jogalong virus (Figure 3). This phylogenetic incongruence suggests that recombination events may have occurred in the evolutionary history of Hepacivirus Q, although no statistically supported recombination event was detected within Hepacivirus Q strains or other hepaciviruses during systematic analysis.
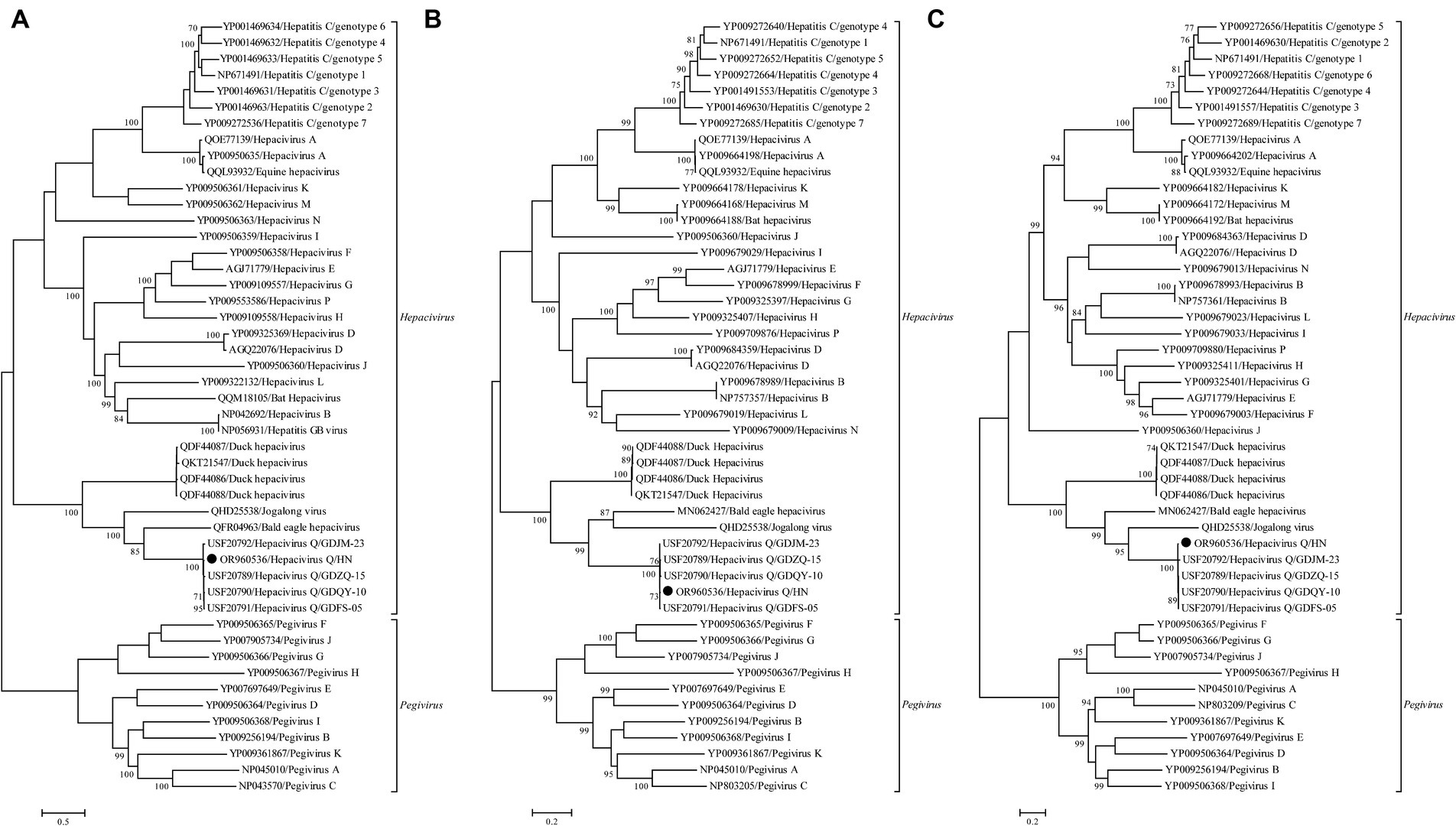
Figure 3. Phylogenetic analysis based on the complete polyprotein (A), NS3 (B), and NS5B (C) protein of hepaciviruses. The trees were constructed using the maximum likelihood method implemented in PhyML v3.0 and were mid-point rooted for clarity. Bootstrap values were calculated based on 100 replicates of the alignment, and only bootstrap values >70% are shown at relevant nodes. The sequence determined in this study is denoted by a black dot.
A total of 480 domestic duck liver tissues, including those previously subjected to meta-transcriptomic sequencing, were screened for the presence of Hepacivirus Q using nested RT-PCR. Following PCR screening, sequencing, and BLAST analysis, 64 individual RNA samples tested positive for Hepacivirus Q, resulting in an overall prevalence rate of 13.3% (95% CI: 10.3–16.3%) (Table 2). All NS5B sequences determined herein shared 98.9 to 99.8%. Additionally, the positive rates of Hepacivirus Q in Hengyang, Chenzhou, Yongzhou, Shaoyang, and Huaihua were 19.2, 11.4, 10.0, 14.1, and 10.0%, respectively. No significant difference was observed among the positive rates across the different sampling sites (χ2 = 5.610, p = 0.230).
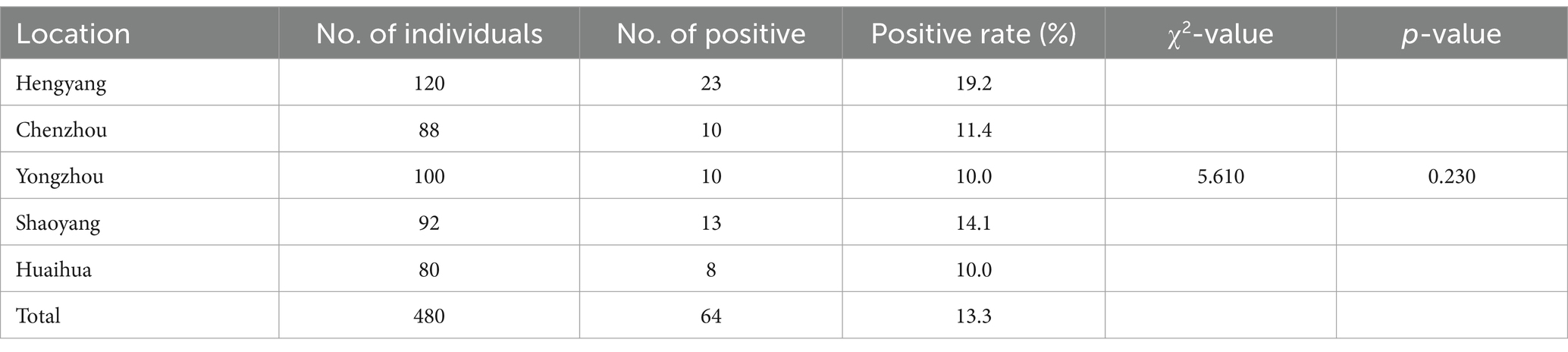
Table 2. Prevalence of Hepacivirus Q in ducks in specific regions of Hunan province.
Hepacivirus Q was recently identified in domestic ducks from Guangdong province, China, utilizing meta-transcriptomic sequencing in our prior research (8). The widespread application of high-throughput sequencing has significantly aided in the unearthing of numerous new viruses, broadening our comprehension of genetic diversity and viral evolution (23). In this context, various novel hepaciviruses have been identified in a diverse array of host species (8–10, 24–30). Within this study, a novel Hepacivirus was uncovered in duck liver samples via meta-transcriptomic sequencing. Through sequence comparison and phylogenetic analysis, it was verified that this novel Hepacivirus is part of a new subspecies of Hepacivirus Q, highlighting the extensive genetic diversity of hepaciviruses worldwide.
In our prior investigation, Hepacivirus Q was observed to be present in duck populations across several cities in Guangdong province, China, exhibiting an overall prevalence of 15.9% (8). The current study identified Hepacivirus Q in ducks from various cities in Hunan province, China, with corresponding positive rates of 19.2, 11.4, 10.0, 14.1, and 10.0%. These results collectively suggest the broad geographical dissemination of Hepacivirus Q within China. Additionally, the overall positive rate of Hepacivirus Q in this study (13.3%) is lower than that documented in our previous study conducted in Guangdong province, China. Nevertheless, the relatively high infection rate of Hepacivirus Q detected in duck populations from both Guangdong and Hunan provinces highlights the necessity to investigate potential associations between Hepacivirus Q and duck-related ailments, although further investigations are needed for definitive.
The Hepatitis C virus, belonging to the genus Hepacivirus, is widely recognized as a significant causative agent of hepatitis, liver cirrhosis, and hepatocellular carcinoma in humans, presenting a substantial global public health concern (6). Despite the identification of diverse hepaciviruses in various animal species, few instances have linked animal hepaciviruses to clinical diseases (31, 32). For instance, duck Hepacivirus was discovered during an investigation into the etiology of severely ill ducks, with uncertain pathogenicity observed as the virus was detected in both diseased and healthy ducks (14). Similarly, Hepacivirus Q was identified in randomly sampled liver tissues from duck populations without any associated clinical disease manifestations in this and our previous study. However, the high prevalence of Hepacivirus Q in duck populations throughout southern China, across a broad geographic range, implies potential nonpathogenicity in ducks and the ability to establish persistent infections. Further study is warranted to delve into the possible pathogenicity of this novel virus in ducks.
In conclusion, a new subspecies of Hepacivirus Q was identified in duck specimens collected from multiple cities in Hunan province, China, exhibiting a notably high positive rate. These results enhance insights into the genetic variability and evolutionary traits of hepaciviruses, underscoring the necessity to explore potential associations between Hepacivirus Q and diseases affecting ducks.
The original contributions presented in the study are publicly available. This data can be found at: https://www.ncbi.nlm.nih.gov/nuccore/; OR960536.
The animal studies were approved by the ethics committee of Foshan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was not obtained from the owners for the participation of their animals in this study because all samples were collected from live poultry market.
J-TC: Formal analysis, Investigation, Visualization, Writing – original draft. K-JC: Resources, Writing – original draft. K-WW: Resources, Writing – original draft. S-HY: Investigation, Writing – original draft. J-WS: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Guangdong Basic and Applied Basic Research Foundation, grant numbers 2021A1515110450 and 2022A1515012194. The funding institution has not contributed to experimental research design, sample collection, data analysis, and writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Smith, DB, Becher, P, Bukh, J, Gould, EA, Meyers, G, Monath, T, et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J Gen Virol. (2016) 97:2894–907. doi: 10.1099/jgv.0.000612
2. Simmonds, P, Becher, P, Bukh, J, Gould, EA, Meyers, G, Monath, T, et al. ICTV virus taxonomy profile: Flaviviridae. J Gen Virol. (2017) 98:2–3. doi: 10.1099/jgv.0.000672
3. Scheel, TK, Simmonds, P, and Kapoor, A. Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antivir Res. (2015) 115:83–93. doi: 10.1016/j.antiviral.2014.12.014
4. Penin, F, Dubuisson, J, Rey, FA, Moradpour, D, and Pawlotsky, JM. Structural biology of hepatitis C virus. Hepatology. (2004) 39:5–19. doi: 10.1002/hep.20032
5. Boonstra, A, van der Laan, LJ, Vanwolleghem, T, and Janssen, HL. Experimental models for hepatitis C viral infection. Hepatology. (2009) 50:1646–55. doi: 10.1002/hep.23138
6. Hartlage, AS, Cullen, JM, and Kapoor, A. The strange, expanding world of animal hepaciviruses. Annu Rev Virol. (2016) 3:53–75. doi: 10.1146/annurev-virology-100114-055104
7. Li, LL, Liu, MM, Shen, S, Zhang, YJ, Xu, YL, Deng, HY, et al. Detection and characterization of a novel hepacivirus in long-tailed ground squirrels (Spermophilus undulatus) in China. Arch Virol. (2019) 164:2401–10. doi: 10.1007/s00705-019-04303-z
8. Zhang, XL, Yao, XY, Zhang, YQ, Lv, ZH, Liu, H, Sun, J, et al. A highly divergent hepacivirus identified in domestic ducks further reveals the genetic diversity of hepaciviruses. Viruses. (2022) 14:371. doi: 10.3390/v14020371
9. Chu, L, Jin, M, Feng, C, Wang, X, and Zhang, D. A highly divergent hepacivirus-like flavivirus in domestic ducks. J Gen Virol. (2019) 100:1234–40. doi: 10.1099/jgv.0.001298
10. Lu, G, Zhao, J, Ou, J, and Li, S. Novel HCV-like virus detected in avian livers in southern China and its implications for natural recombination events. Virol Sin. (2021) 36:149–51. doi: 10.1007/s12250-020-00256-9
11. Goldberg, TL, Sibley, SD, Pinkerton, ME, Dunn, CD, Long, LJ, White, LC, et al. Multidecade mortality and a homolog of hepatitis C virus in bald eagles (Haliaeetus leucocephalus), the national bird of the USA. Sci Rep. (2019) 9:14953. doi: 10.1038/s41598-019-50580-8
12. Shi, M, Lin, XD, Chen, X, Tian, JH, Chen, LJ, Li, K, et al. The evolutionary history of vertebrate RNA viruses. Nature. (2018) 556:197–202. doi: 10.1038/s41586-018-0012-7
13. Zhang, XL, Li, WF, Yuan, S, Guo, JY, Li, ZL, Chi, SH, et al. Meta-transcriptomic analysis reveals a new subtype of genotype 3 avian hepatitis E virus in chicken flocks with high mortality in Guangdong, China. BMC Vet Res. (2019) 15:131. doi: 10.1186/s12917-019-1884-y
14. Chen, S, Zhou, Y, Chen, Y, and Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
15. Li, D, Liu, CM, Luo, R, Sadakane, K, and Lam, TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
16. Buchfink, B, Xie, C, and Huson, DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. (2015) 12:59–60. doi: 10.1038/nmeth.3176
17. Langmead, B, and Salzberg, SL. Fast gapped-read alignment with bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
18. Martin, DP, Murrell, B, Golden, M, Khoosal, A, and Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. (2015) 1:vev003. doi: 10.1093/ve/vev003
19. Lole, KS, Bollinger, RC, Paranjape, RS, Gadkari, D, Kulkarni, SS, Novak, NG, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. (1999) 73:152–60. doi: 10.1128/JVI.73.1.152-160.1999
20. Katoh, K, and Standley, DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
21. Guindon, S, and Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. (2003) 52:696–704. doi: 10.1080/10635150390235520
22. Darriba, D, Taboada, GL, Doallo, R, and Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. (2011) 27:1164–5. doi: 10.1093/bioinformatics/btr088
23. Zhang, YZ, Chen, YM, Wang, W, Qin, XC, and Holmes, EC. Expanding the RNA virosphere by unbiased metagenomics. Annu Rev Virol. (2019) 6:119–39. doi: 10.1146/annurev-virology-092818-015851
24. Lu, G, Jia, K, Ping, X, Huang, J, Luo, A, Wu, P, et al. Novel bovine hepacivirus in dairy cattle, China. Emerg. Microbes Infect. (2018) 7:1–3. doi: 10.1038/s41426-018-0055-8
25. Canal, CW, Weber, MN, Cibulski, SP, Silva, MS, Puhl, DE, Stalder, H, et al. A novel genetic group of bovine hepacivirus in archival serum samples from Brazilian cattle. Biomed Res Int. (2017) 2017:1–4. doi: 10.1155/2017/4732520
26. Porter, AF, Pettersson, JH, Chang, WS, Harvey, E, Rose, K, Shi, M, et al. Novel hepaci-and pegi-like viruses in native Australian wildlife and non-human primates. Virus Evol. (2020) 6:veaa064. doi: 10.1093/ve/veaa064
27. Guo, H, Cai, C, Wang, B, Zhuo, F, Jiang, R, Wang, N, et al. Novel hepacivirus in Asian house shrew, China. Sci China Life Sci. (2019) 62:701–4. doi: 10.1007/s11427-018-9435-7
28. de Souza, WM, Fumagalli, MJ, Sabino-Santos, G Jr, Motta Maia, FG, Modha, S, Teixeira Nunes, MR, et al. A novel hepacivirus in wild rodents from South America. Viruses. (2019) 11:297. doi: 10.3390/v11030297
29. Shao, JW, Guo, LY, Yuan, YX, Ma, J, Chen, JM, and Liu, Q. A novel subtype of bovine hepacivirus identified in ticks reveals the genetic diversity and evolution of bovine hepacivirus. Viruses. (2021) 13:2206. doi: 10.3390/v13112206
30. Breitfeld, J, Fischer, N, Tsachev, I, Marutsov, P, Baymakova, M, Plhal, R, et al. Expanded diversity and host range of bovine hepacivirus-genomic and serological evidence in domestic and wild ruminant species. Viruses. (2022) 14:1457. doi: 10.3390/v14071457
31. Baechlein, C, Fischer, N, Grundhoff, A, Alawi, M, Indenbirken, D, Postel, A, et al. Identification of a novel hepacivirus in domestic cattle from Germany. J Virol. (2015) 89:7007–15. doi: 10.1128/JVI.00534-15
Keywords: Hepacivirus, duck, genetic diversity, epidemiology, China
Citation: Chen J-T, Chen K-J, Wu K-W, Yi S-H and Shao J-W (2024) Identification and epidemiology of a novel Hepacivirus in domestic ducks in Hunan province, China. Front. Vet. Sci. 11:1389264. doi: 10.3389/fvets.2024.1389264
Edited by:
Shixing Yang, Jiangsu University, ChinaReviewed by:
Gang Lu, South China Agricultural University, ChinaCopyright © 2024 Chen, Chen, Wu, Yi and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Wei Shao, andzaGFvMTk4OEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.