 Hanyue Li
Hanyue Li Yini Li
Yini Li Shengyu Luo1
Shengyu Luo1 Zhenhua Feng
Zhenhua Feng Sen Li
Sen Li- 1School of Physical Education, Southwest Medical University, Luzhou, China
- 2Department of Ultrasound, Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 3Luzhou Vocational and Technical College, Luzhou, China
- 4Division of Spine Surgery, Department of Orthopedic Surgery, Affiliated Hospital of Medical School, Nanjing Drum Tower Hospital, Nanjing, Jiangsu, China
Both acute and chronic tendon injuries are the most frequently occurring musculoskeletal diseases in human and veterinary medicine, with a limited repertoire of successful and evidenced-based therapeutic strategies. Inflammation has been suggested as a key driver for the formation of scar and adhesion tissue following tendon acute injury, as well as pathological alternations of degenerative tendinopathy. However, prior efforts to completely block this inflammatory process have yet to be largely successful. Recent investigations have indicated that a more precise targeted approach for modulating inflammation is critical to improve outcomes. The nuclear factor-kappaB (NF-κB) is a typical proinflammatory signal transduction pathway identified as a key factor leading to tendon disorders. Therefore, a comprehensive understanding of the mechanism or regulation of NF-κB in tendon disorders will aid in developing targeted therapeutic strategies for human and veterinary tendon disorders. In this review, we discuss what is currently known about molecular components and structures of basal NF-κB proteins and two activation pathways: the canonical activation pathway and the non-canonical activation pathway. Furthermore, we summarize the underlying mechanisms of the NF-κB signaling pathway in fibrosis and adhesion after acute tendon injury, as well as pathological changes of degenerative tendinopathy in all species and highlight the effect of targeting this signaling pathway in tendon disorders. However, to gain a comprehensive understanding of its mechanisms underlying tendon disorders, further investigations are required. In the future, extensive scientific examinations are warranted to full characterize the NF-κB, the exact mechanisms of action, and translate findings into clinical human and veterinary practice.
1 Introduction
Both acute and chronic tendon injuries represent the most frequent musculoskeletal ailments in human and veterinary medicine (1). In humans, an estimated 30% of musculoskeletal consultations are tendon disorders (2). In veterinary medicine, approximately 50% of tendon disorders influence racehorses, leading to a long shadow of financial losses due to compromised sports performances (3). However, tendon injuries in all species are challenging as their limited regenerative ability makes tendon repair ineffective, resulting in the formation of inferior scar tissue (4, 5). Moreover, scar-mediated healing is particularly problematic in the flexor tendon as excessive scar tissue can form adhesion between the tendon, sheath, and surrounding tissue, further disrupting tendon function (6). On the other hand, the accumulation of microdamage during overloading after unsuccessful tendon regenerative healing is a predisposing factor for the progression of degenerative tendinopathy (7). Degenerative tendinopathy is a broad spectrum of chronic tendon disorders characterized by swelling, pain, ossification, and physical disability (8). Despite the prevalence of these diseases, current therapeutic strategies often fall short of achieving complete functional recovery of the tendon (9). Therefore, to exploit novel therapeutic strategies for enhancing tendon regenerative healing and preventing the progression of degenerative tendinopathy, it is pivotal to understand the basic mechanisms underlying tendon pathologies and scar healing (10).
The significance of inflammation in both the tendon scar healing and the early initiation of degenerative tendinopathy has been highlighted in recent years in patient samples in human and veterinary species (11, 12). To alleviate the inflammation of tendon disorders, anti-inflammatory therapeutic approaches such as non-steroidal anti-inflammatory drugs (NSAIDs) are commonly prescribed to treat tendon disorders (13, 14). However, these therapeutic interventions to completely block the inflammation have been explored with mixed success, possibly because controlled inflammation is beneficial for tendon repair (15). Toward this end, recent investigations have indicated that a more precise targeted approach for modulating inflammation is better than completely blocking it in humans and equines (16).
The NF-κB signaling pathway has been regarded as a typical proinflammatory signal transduction pathway, the activation of which is involved in the regulation of multiple genes, including proinflammatory cytokines, chemokines, adhesion molecules, and enzymes (17, 18). It has been reported that blocking the activation of NF-κB signaling pathway in tendon disorders has been more effective than completely inhibiting all the inflammatory response (19–21). Nevertheless, there is currently insufficient mechanistic understanding of the NF-κB signaling pathway in tendon disorders to manifest this possibility in practice. In this biomedical challenge, rodent models of induced tendon disorders and in vitro cell/tissue culture reports function as preclinical models to obtain critical biological information and as translational models to drive advancements in human and veterinary medicine (22). While the prevalence and importance of tendon disorders vary between species, most of the mechanisms triggering their progression are alike. This suggests that we can learn about the NF-κB signaling pathway in tendon disorders by both using animal data to improve human studies and using human data in animal research. Here, in this review, we provide a summary of our current understanding of the mechanisms of the NF-κB signaling pathway underlying the degenerative tendinopathy and tendon scar healing in both species and highlight the effect of targeting this signaling pathway in tendon disorders, which can give some indications for veterinary clinical application. Nevertheless, future investigations are still warranted to investigate the exact mechanisms of action.
2 NF-κB “identity card”
2.1 Molecular components and structures of the NF-κB family
Transcription factors are proteins responsible for gene expression through binding to gene enhancer and promoter sites (23). NF-κB is a family of transcription factors expressed in almost all cells and participates in the regulation of various cell processes and signaling pathways (24). To date, NF-κB was first discovered as a transcription modulator in 1986, which binds to the intrinsic enhancer of the kappa light chain gene (the κB site) in B lymphocytes (25, 26). Subsequently, NF-κB has been found to be one of the highly conserved protein families, present in many organisms, such as humans, mice, and equines (27).
The mammalian NF-κB represents a family of transcription factors, mainly including RelA (p65), c-Rel, RelB, NF-κB1 (p50), and NF-κB2 (p52; Table 1) (28). These proteins interact with each other to form dimers that can bind to DNA and activate transcription (Figure 1). These members are split into two groups according to their manner of inactivation and structural similarities. The first group contains RelA, c-Rel, and RelB, all of which are characterized by the presence of a C-terminal transcription activation domain (TAD) and an N-terminal rel homology domain (RHD) of the protein structure (29, 30). TAD of the protein structure is required for NF-κB dimer translocation to the nucleus, while RHD within these proteins serves to bind with the inhibitory Kappa B (IκB) proteins to retain NF-κB in the cytoplasm (31). Inconsistent with other members, RelB does not homodimerize, and it is characterized by the presence of a leucine zipper (LZ) motif in its N terminus, which exerts a critical role in gene transcription (28). Nevertheless, it remains to be determined whether the LZ confers other functional specificity to RelB, including heterodimerization with NF-κB2 or another possible coagent. Differently, another group, NF-κB1 and NF-κB2, lack TAD. Thus, NF-κB1 and NF-κB2 homodimers cannot activate gene transcription or act as a transcriptional repressor, but they are synthesized as large inactive precursors p105 and p100 that yield mature NF-κB subunits p50 and p52, respectively (32, 33). The ankyrin repeats (ANK) in the C-terminal region of p105 and p100 participate in their degradation along the ubiquitin-proteasome pathway (34).
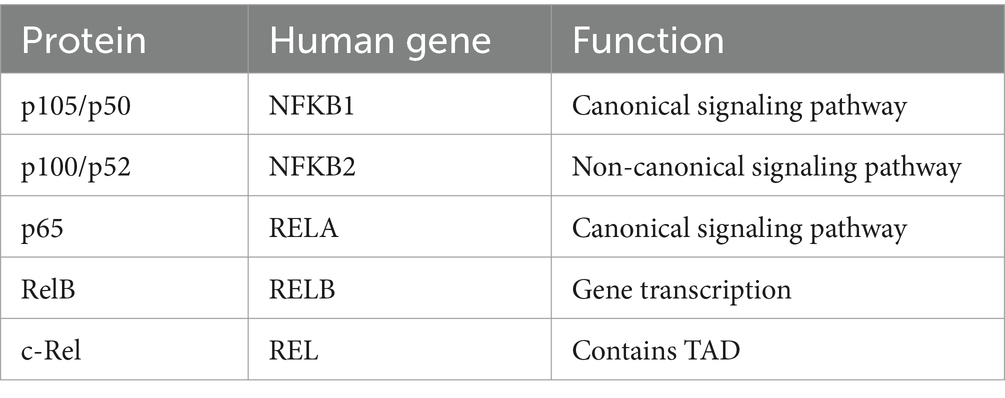
Table 1. The NF-κB transcription factors.
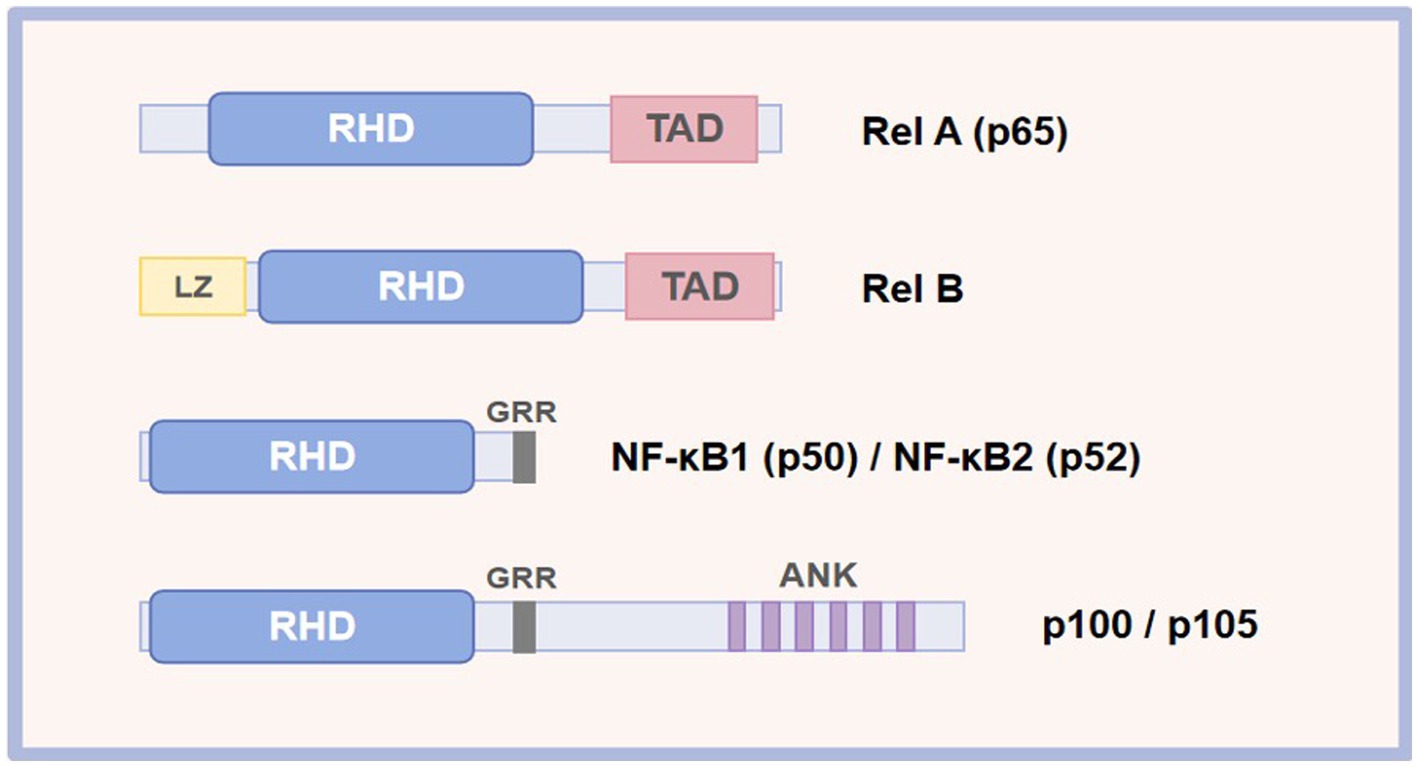
Figure 1. Schematic representation of the NF-κB protein family. RHD, rel homology; TAD, transcriptional activation domain; LZ, leucine zipper; ANK, ankyrin repeat; GRR, glycine-rich repeat.
2.2 General function and regulation of NF-κB
The activation of NF-κB after responding to an external or internal stimulus occurs in two main ways: the canonical and noncanonical (alternative) pathways (35). The canonical pathway will predominately result in the activation of RelA (p65), NF-κB1 (p50), and c-Rel dimers, while the alternative pathway is primarily responsible for the activation of NF-κB2 (p52) and RelB dimers (36). However, it is worth noting that these two signaling pathways are not mutually exclusive. For instance, the canonical p65/p50 dimers have been reported to be involved in the regulation of the alternative pathway (26).
The IκB proteins and IκB kinase (IKK) complex tightly regulate the activation of the NF-κB signaling pathway (37, 38). IκB is a specific inhibitor of NF-κB. The IκB proteins in mammalian contain seven members: IκBα, IκBβ, IκBε, IκBδ, Bcl-3, IκBξ, and IκBNS (Table 2) (39). They block the nucleus localization signal by binding to the RHD region of NF-κB, therefore preventing NF-κB from entering the nucleus to perform its function. The IKK complex consisting of two subunits, IKKα (IKK1) and IKKβ (IKK2), and one regulatory subunit, IKKγ (NEMO), is responsible for the degradation of the IκB proteins, allowing NF-κB proteins free to translocate in the nucleus where it binds to the promoter region of the target genes (40, 41). Moreover, IKKα is primarily responsible for the activation of the noncanonical pathway by phosphorylating and processing of p100. In contrast, IKKβ and IKKγ are required for the ignition of the canonical pathway by phosphorylating of IκB proteins (42).
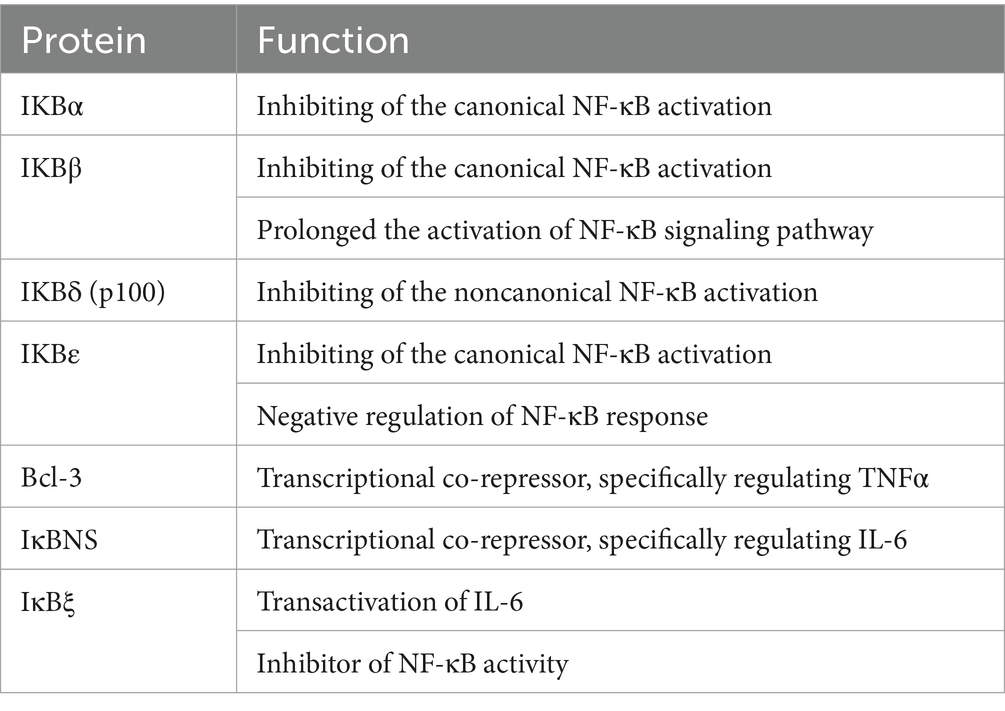
Table 2. The IκΒ family.
The canonical signaling pathway is primarily responsible for sensing tissue damage, infection, and proinflammatory signals (43). After tissue injury, this pathway is activated by various proinflammatory signals (cytokine receptors such as IL-1R and TNF-R), toll-like receptors (TLR), T cell receptors (TCR), and B cell receptors (BCR) (44). When an infection or proinflammatory signals arrive in the cell, the receptors mentioned above can be activated. Activation of these receptors induces the activation of IKK, and then the activated IKK triggers the phosphorylation of IκB, leading to its ubiquitination and subsequent proteasomal degradation. The degradation of IκB proteins unmasks a nuclear localization signal, offering NF-κB dimers entrance ability to the nucleus. This process further accelerates the transcription of various proinflammatory cytokines, proteinases, and prostaglandins, contributing to the activation of inflammatory cells (macrophages and neutrophils) (45). On the other hand, the alternative signaling pathway of NF-κB is primarily involved in lymphoid organogenesis and the activation of the adaptive immune response (46). In contrast to the canonical NF-κB signaling pathway, the alternative signaling pathway is activated by a subset of Tumor Necrosis Factor Receptor (TNFR) superfamily members, including CD40, the lymphotoxin-β receptor (LTβR), receptor activator of NF-κB ligand (RANK), and B lymphocyte stimulating factor receptor (BAFFR) (47). Furthermore, this alternative pathway of NF-κB activation depends on the inducible processing of p100 precursor protein rather than the degradation of IκB proteins of the canonical pathway. The C-terminal ARD domain of p100 functions like IκB proteins to keep NF-κB dimers in an inactive state (48). Its C-terminal part owes its inhibitory effect to the presence of ankyrin repeats-33 amino acid residue motifs, which resemble those present in IκB proteins and are responsible for binding to NF-κB proteins (49, 50). In general, stimulation from these receptors leads to the activation of the NIK protein, which further induces the phosphorylation of the IKKα and promotes the degradation of p100 to generate p52. Following the phosphorylation cascade, the p52 subunit dimerizes RelB, resulting in the expression of NF-κB target genes (51).
Overall, these aspects of NF-κB function and activation are undoubtedly critical to our understanding of this family of NF-κB’s overall behavior. They also provide a foundation for therapeutic targeting in tendon disorders based on NF-κB inhibition (Figure 2).
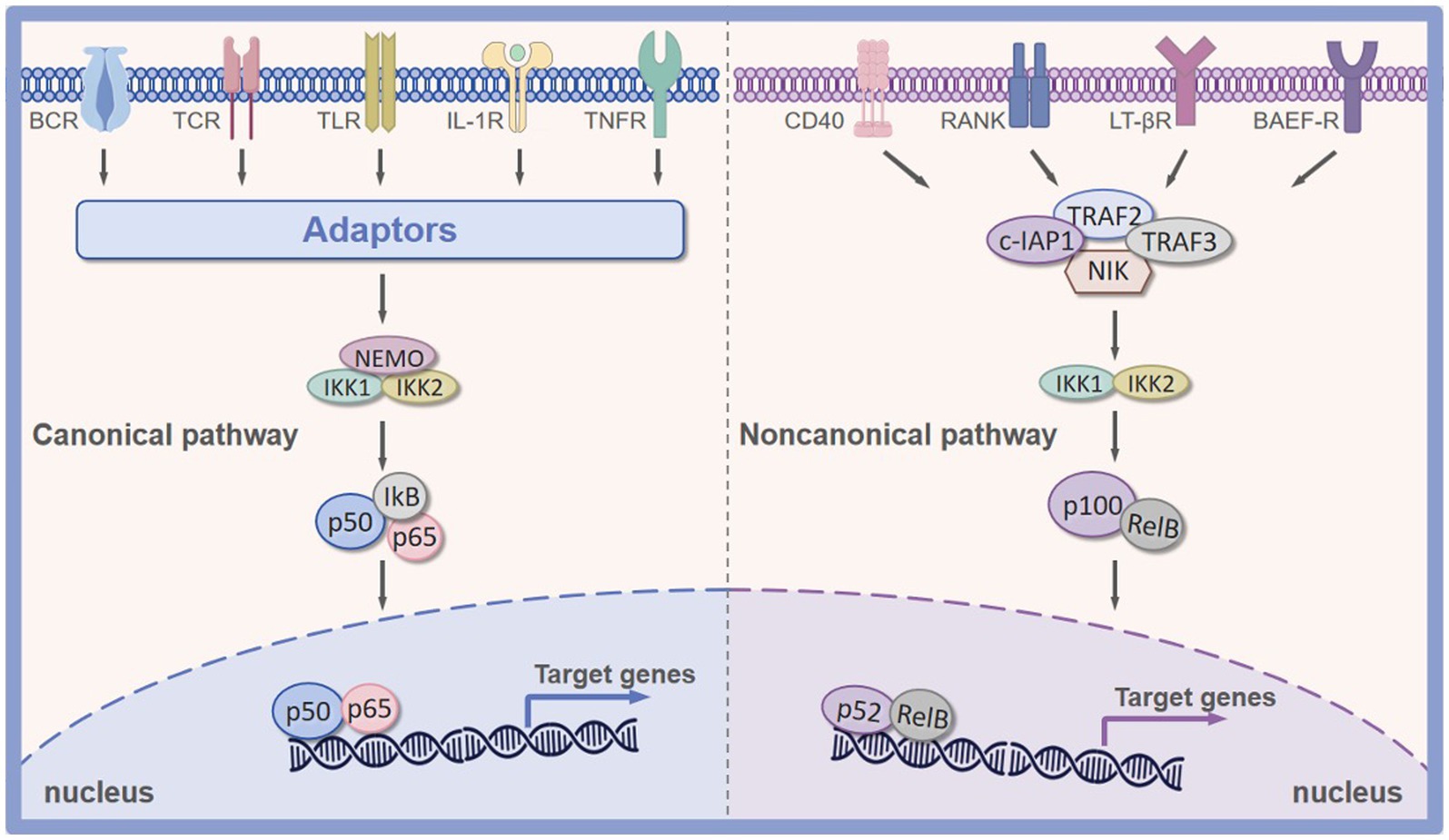
Figure 2. Canonical and noncanonical pathway of NF-κB signaling pathway. Canonical NF-κB signaling pathway is primarily activated by TNFR, IL-1R, TLR, BCR, and TCR. Non-canonical NF-κB signaling pathway is predominated activated by CD40, RANK, LTβR, and BAFFR. BAFFR, B lymphocyte stimulating factor receptor; RANK, receptor activator of nuclear factor kappa-B ligand; TLR, Toll-like receptor; IL-1R, interleukin-1 receptor; TNFR, tumor necrosis factor receptor; LTβR, lymphotoxin β-receptor; BCR, B-cell receptor; TCR, T-cell receptor; CD40, CD40 ligand receptor.
3 Fibrotic-scarring tendon healing, adhesion, and degenerative tendinopathy
In a manner similar to other tissue, tendon healing in veterinary species also consists of three distinct but overlapping phases: (1) the inflammatory phase (a few days), (2) the proliferative phase (several weeks), (3) the remodeling phase (lasting months/years) (52). In the inflammatory phase, the injury site is predominately infiltrated by macrophages. They produce a variety of proinflammatory cytokines, such as TNF-α and IL-1β, that amplify inflammatory response, leading to the activation of other immune cells and activation of tenocytes and tendon stem/progenitor cells (TSPCs) (53). Following this, the proliferative phase is featured by the expansion of ECM, increased cellularity, and depositional of the fibrovascular scar by tenocytes. About 2 weeks after injury, remodeling the injured area during the remodeling phase starts with the reorganization of the newly deposited collagen, with a gradual decrease in cellularity and an increase in the fibrous matrix (54). However, despite efforts to repair, the ensuing healing processes often fail to yield fully regenerative healing of the injured tissue, resulting in biomechanically inferior scar tissue formation (55). Moreover, the proliferation of scar tissue between the tendon and adjacent tissues is also undesirable as this can lead to the adhesion formation, further impeding normal tendon gliding and function (56). If the tendon continues to undergo microdamage after unsuccessful tendon regenerative healing, the local cells in tendon tissue undergo pathological alternations, resulting in the destruction of tendon tissue. This can progress to the degenerative tendinopathy (57).
4 The mechanisms of NF-κB in tendon fibrotic scar healing, adhesion
As mentioned above, scar tissue formation and tendon adhesion are two primary issues encountered after acute tendon injury. The NF-κB signaling pathway is a key player involved in these processes. Mechanically, the NF-κB signaling pathway promotes the tendon scar and adhesion formation by facilitating the activation of M1 macrophages, promoting the production of proinflammatory factors, accelerating the persistent presence of myofibroblasts, and inducing the ECM accumulation (Figure 3), which will be thoroughly described below.
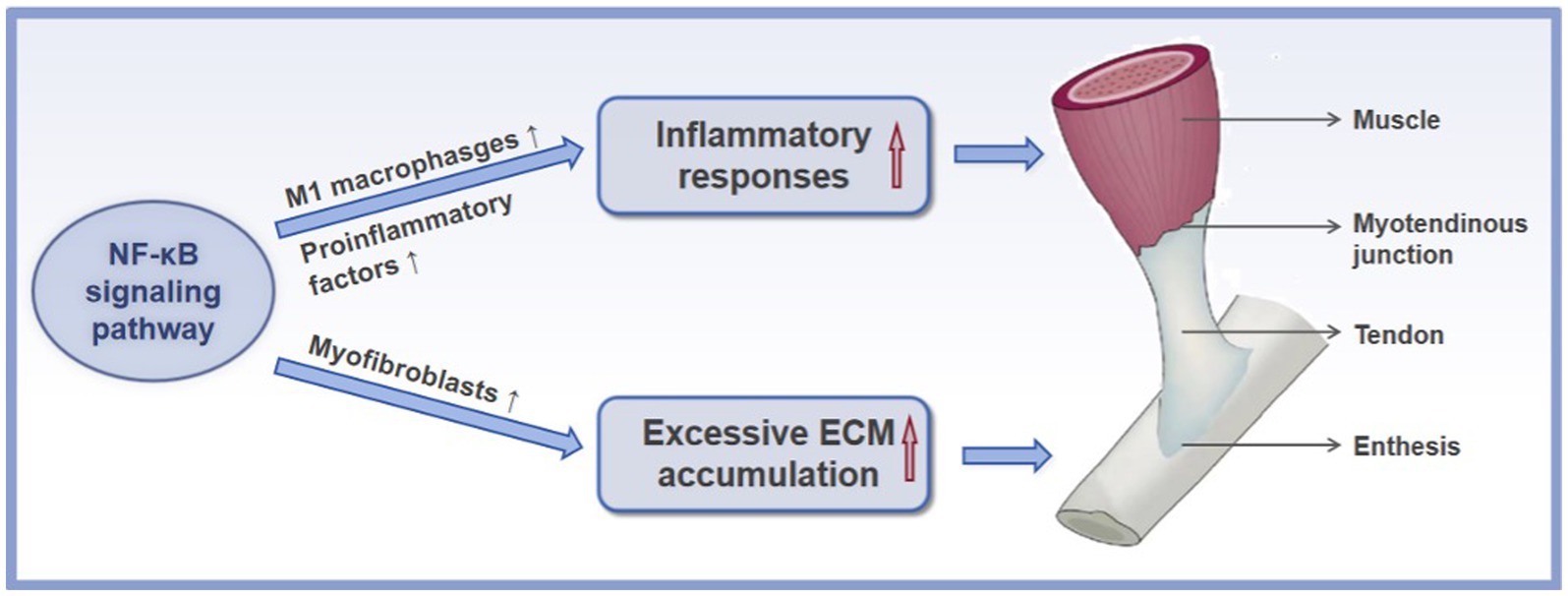
Figure 3. The mechanisms of the NF-κB signaling pathway in the formation of scar and adhesion after acute tendon injury. ↑ refers to upregulation or increase.
4.1 NF-κB amplifies the inflammatory responses
Although inflammation is an essential step for proper tendon regenerative healing, a marked early inflammatory response often leads to the formation of a scar-like tendon and adhesions, which disturb the intrinsic repair process and result in a tendon with poor tissue quality and inferior mechanical properties (58). Controlling the inflammatory environment after tendon injury is, therefore, a potential therapeutic approach for promoting tendon regenerative healing (59). Regulatory mechanisms in inflammation are triggered by inflammatory cell infiltration and cytokine production. Various cellular and gene regulatory mechanisms modulate both inflammatory cell infiltration and cytokine production. One such mechanism is the activation of the NF-κB signaling pathway. This was confirmed in a canine flexor tendon injury model, whereby inhibition of IKK-β activity and thereby decreased the activation of the NF-κB signaling pathway was demonstrated to reduce the early inflammatory response and decrease the formation of scar and adhesion tissue compared to the saline-treated group (60). Some reports describe the underlying mechanisms of NF-κB signaling pathway in the inflammatory response during fibrotic tendon healing.
The excessive inflammation during tendon scar healing has been identified to be mainly driven by macrophage accumulation (61). In fact, macrophages primarily display two different phenotypes, with polarization into proinflammatory phenotype (M1) during the early tendon healing process followed by switching to an anti-inflammatory phenotype (M2) during the latter tendon healing process (62). The M1 macrophage polarization is thought to trigger the formation of scar tissue and adhesion. The M1 macrophage polarization is also revealed to cause the release of harmful proinflammatory factors, which may further cause injury to the tendon tissue. It is well established that the activation of the NF-κB signaling pathway plays a key role in the macrophage polarization toward M1 macrophages during the healing process of the injured tendon tissue. Dates from healing mouse flexor tendons has demonstrated that overactivation of the NF-κB signaling pathway enhanced the presence of proinflammatory macrophages at the injured area, resulting in elevated formation of scar tissue and disrupting the mechanical properties of the healing tendon (63). Additionally, an experimental study in a rat rotator cuff healing model displayed that inhibiting the NF-κB signaling pathway, deceased macrophage polarization toward M1 macrophages and thereby reduced the production of excessive inflammatory factors and finally provoked the regenerative potential of injured tissue (64). Previous reports have indicated that microRNAs (miRNAs) primarily function as a silencer of coding genes and that the expression of miRNAs is thought to play a significant role in injured tissue’s healing process. In a study by Shen et al. (65), the delivery of miR-147-3p mimic to treat the injured Achilles tendon of mice blocked the activation of the NF-κB signaling pathway in a process that is mediated by the activation of TLR receptor, which subsequently led to the reduced M1 macrophage polarization and showed a significant attenuation in proinflammatory factors, and finally resulting in the improved healing process after injury, highlights the critical role of the NF-κB signaling pathway in the inflammatory process during tendon healing. Similarly, blocking the p65 phosphorylation of the NF-κB signaling pathway suppresses M1 macrophage polarization, significantly alleviating inflammation and peritendinous adhesion and scar tissue formation in a rat model of Achilles tendon injury (66). Furthermore, in vivo, both Wang et al. (67) and Li et al. (68) found that mitigating the activation of p65 of the NF-κB signaling pathway inhibited M1 macrophage polarization and thereby effectively reduced proinflammatory factors production during fibrotic tendon healing, resulting in the improved tendon healing process. To sum up, the aforementioned evidence indicates the role of the NF-κB signaling pathway in the formation of scar and adhesion by inducing macrophage polarization toward M1 macrophages. Nevertheless, the interaction between M2 macrophage polarization and the NF-κB signaling pathway is poorly defined. Therefore, further report is required to illuminate the correlation between NF-κB pathway and M2 macrophage polarization in the process of tendon scar healing.
On the other hand, up-regulation of proinflammatory factors occurs as a direct response after acute injury in an independent pathway of M1 macrophages. It has been reported that the NF-κB signaling pathway is directly involved in the synthesis and activity of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α (69). During the inflammatory stage of tendon scar healing, binding these cytokines to their corresponding receptors leads to the activation of the NF-κB signaling pathway (70). Activated NF-κB signaling pathway, in turn, triggers a proinflammatory cytokine storm, resulting in the formation of a positive feedback loop that has the potential to produce chronic and excessive inflammation (71, 72). These findings suggest that the NF-κB signaling pathway is a central medium to enhance the production of proinflammatory cytokines. Indeed, in a rat model of Rotator cuff injury, blocking the activation of the NF-κB signaling pathway through IKKβ small-molecule inhibitor (ACHP) prevented the production of the proinflammatory cytokines, IL-1β, IL-6, and TNF-α, and thereby alleviated the excessive inflammation that occurred in the early stages of rotator cuff tendon-to-bone healing and decreased the formation of scar tissue, finally improved healing results (73). Thus, in light of the critical role of NF-κB in mediating inflammation, the NF-κB signaling pathway could be a key treatment target for attenuating inflammation and inflammation-related processes to enhance tendon regenerative healing. However, there is still limited research conducted for understanding of the NF-κB signaling pathway involved in the inflammatory response of acute tendon injury.
4.2 NF-κB induces excessive ECM accumulation
Generally, adhesion and fibrotic scar formation are thought to be healing responses to a tendon injury, in which the balance of tissue repair and excessive ECM accumulation is perturbed, favoring the latter (74). The contribution of the NF-κB signaling pathway to fibrotic tendon healing has also been reported to induce excessive ECM accumulation.
Myofibroblasts, tenocytes expressing α-smooth muscle actin (αSMA), participate decisively in the remodeling phase during healing process, potentially resulting in scar and adhesion formation (75). They initially appear in the proliferative phase to synthesize and contract the ECM to close gaps within injured tendons (76). However, the persistence of myofibroblasts is recognized as a putative driver of fibrovascular scar healing in the tendon tissue due to exuberant and sustained ECM production (77). In the flexor tendon healing, they also lead to tendon adhesions between the injured site and the surrounding tissue, preventing tendon gliding (78). As such, disrupting myofibroblast persistence has been suggested for the treatment of scar and adhesion formation. The NF-κB signaling pathway is considered to be a therapeutic target for inhibiting myofibroblast persistence during fibrotic tendon healing. Previous investigations have demonstrated that constitutively activated NF-κB signaling pathway in myofibroblasts was observed in human tendon scar tissue, suggesting that the NF-κB signaling pathway may participate in the myofibroblast persistence (79). Moreover, global blocking of p65 of the NF-κB signaling pathway or pharmacological inhibiting of p65 was shown to reduce myofibroblast presence, accompanied by decreased ECM production, leading to the reduced formation of scar tissue in a rat model of flexor tendon injury (80). Above all, the NF-κB signaling pathway plays a potential role in myofibroblast persistence during fibrotic tendon healing. However, the precise mechanisms regulating the promoting effect of the NF-κB signaling pathway on myofibroblast persistence have not yet been fully elucidated and understood. Additionally, in one study, the reverse transcription-polymerase chain reaction analysis revealed that a significantly increased expression of type III collagen and type I collagen, along with the activation of the NF-κB signaling pathway after flexor tendon injury. The activation of the NF-κB signaling pathway was particularly proportional to the increased expression of type III collagen and its promoter genes (81). Therefore, one plausible mechanism for the NF-κB signaling pathway-mediated fibrotic healing process is that the NF-κB signaling pathway can directly trigger the excessive production of type III collagen. Further studies are critical to illuminate the relation between the NF-κB signaling pathway collagen production.
5 The mechanisms of NF-κB in the degenerative tendinopathy
Although the role of inflammation in degenerative tendinopathy has been debated for many years, increasing investigations strongly support that molecular inflammation with elevated inflammatory factors, such as IL-1β, COX2, matrix metalloproteinases (MMPs), and TNF-α, lead to the progression of degenerative tendinopathy (82, 83). The increased expression of these factors initiates a cascade of tendon degeneration, such as ECM destruction, tenocyte apoptosis, TSPCs osteogenesis, senescence, and apoptosis (84). The NF-κB signaling pathway is central to this vicious cycle of degenerative tendinopathy (85). Below, we will thoroughly provide the mechanisms of the NF-κB signaling pathway underlying degenerative tendinopathy (Figure 4).
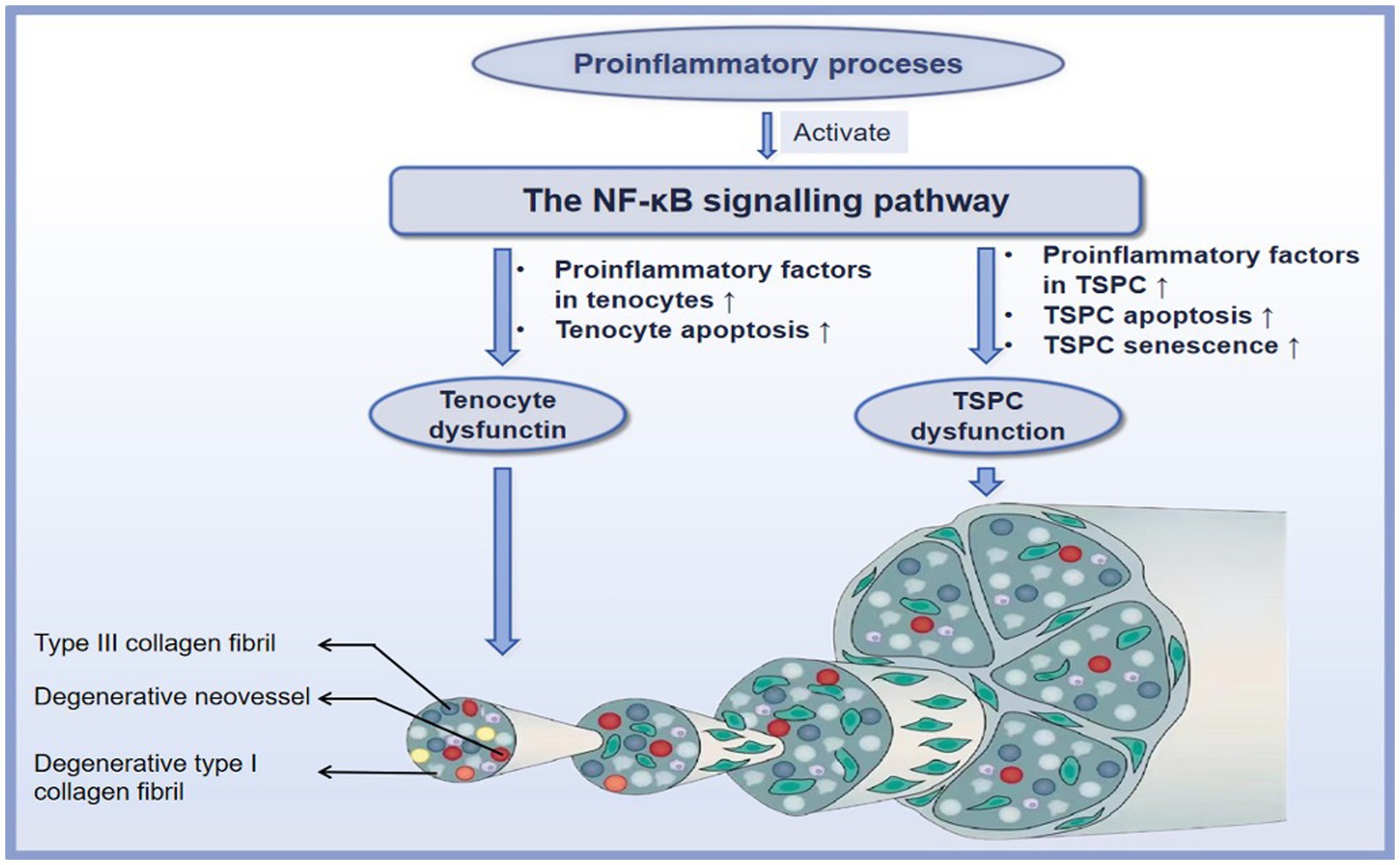
Figure 4. The mechanisms of the NF-κB signaling pathway in the degenerative tendinopathy. ↑ refers to upregulation or increase.
5.1 NF-κB and tenocytes
Tenocytes, the primary local cells of the tendon tissue, exert vital roles in tendon homeostasis. Tenocytes in degenerative tendinopathy have recently been demonstrated to exhibit apoptotic and inflammatory phenotypes. The activation of the NF-κB signaling pathway has been reported to be a critical checkpoint for inflammation-induced degenerative tendinopathy. For example, Buhrmann et al. (86) reported that IL-1β induced the increased expression of COX-2, MMP1, MMP9, and MMP13, and cell apoptosis in human tenocytes and that these mediators resulted in the destruction of tenocytes; subsequently, blocking the activation of the NF-κB signaling pathway can inhibit the detrimental effect of IL-1β. In vitro, knockout of IKK-β in tendon fibroblasts from tendinopathic tissue was potentially unresponsive to IL-1β stimulation: results demonstrated decreases in the transcription of MMP-1a, MMP-3, and MMP-13 and in the translation of proinflammatory cytokines and chemokines (87). Furthermore, signaling investigations with specific inhibitors and western blot analysis reported that the underlying mechanisms of IL-1β participate in the above processes through the NF-κB signaling pathway: IL-1β first binds to its receptor IL-1βR and subsequently induces the phosphorylation of Src, and Src being the upstream molecule of Akt and can enhance phosphorylation of Akt. The phosphorylation of Src/Akt directly phosphorylates IKK1 and IKK2, leading to the activation of p65 and p50 of the NF-κB signaling pathway with the consequences for inducing the expression of COX-2 and enhancing the expression of MMP-1, MMP-9, MMP-13 in human tenocytes (88). Conversely, Mueller et al. (89) reported that Calebin A significantly inhibited inflammation-induced MMP-9, COX-2, and cell apoptosis by promoting the expression of SCX and concomitant blocking the p65 of the NF-κB signaling pathway in tenocytes. Moreover, Sirt-1, a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, has been demonstrated to be associated with the NF-κB signaling pathway (90). Busch et al. (91), in their in vitro investigation, found that Sirt-1 activation inhibited IL-1β-mediated inflammatory factors COX-2, MMP9, and cell apoptosis through inhibiting the activation of the NF-κB signaling pathway in human tenocytes, while down-regulation of Sirt-1 results in similar effects as caused by stimulation with IL-1β. This finding opens the interesting possibility that interfering with NF-κB signaling by activating of Sirt-1 may harbor valuable opportunities for tendon disorders.
5.2 NF-κB and tendon stem/progenitor cell
It has long been considered that the majority of cells in tendons are tenocytes, which is well characterized as a regulator of tendon tissue homeostasis and repair. However, a new type of cell residing in the tendons, termed tendon stem/progenitor cell (TSPC), has been identified in horses (92), humans (93), and several other species (94). TSPCs differ from tenocytes in that they can clonogenicity, self-renewal, and multiple differentiation potential. TSPCs are capable of regenerating tendon tissue through tenogenesis, a process of differentiation into new tenocytes (95). Nevertheless, as degenerative tendinopathy progresses, the pathological environment can induce an erroneous differentiation of TSPCs, resulting in chondrocyte-like cells and ossification sites, which are responsible for heterotopic calcification (HO) (96). Furthermore, TSPCs in degenerative tendinopathy also manifest inflammation, cell apoptosis, and senescence. It has been demonstrated that one of the major contributors to TSPC dysfunction is the NF-κB signaling pathway. For example, in cultured TSPCs, inhibition of the NF-κB signaling pathway restored TSPC tenogenic differentiation and effectively decreased the expression of inflammatory factors, including IL-6, MMP-1, MMP-3, and MMP-13 after exposure to IL-1β (97). Moreover, data revealed that the NF-κB and the MAPK signaling pathways exerted a synergistic role in leading to the HO, senescence, and apoptosis of TSPCs after exposure to IL-1β (98). Along with IL-1β, the proinflammatory cytokine TNF-α can also activate the NF-κB signaling pathway and enhance a series of degenerative cascades (99). For example, TNF-α induced inflammation, apoptosis, and HO of TSPCs by activating the NF-κB and MAPK signaling pathways, leading to dysfunction of TSPCs during degenerative tendinopathy (100). These finding suggests that the interaction between NF-κB and other signaling pathways may reveal many potential therapeutic targets to ameliorate the progression of degenerative tendinopathy.
On the other hand, aging is associated with chronic inflammation. During the aging process, a low-grade inflammation network that contributes to the progression of degenerative tendinopathy is activated, termed (inflamm-aging) (101). It has been demonstrated that one of the key contributors to TSPC senescence is inflamm-aging. Once exposed to inflamm-aging, TSPC will be prone to adopt a senescent phenotype and enhance the proinflammatory cytokine milieu, which accelerates the senescence of other cells (102). The NF-κB signaling pathway has been implicated in this process. For example, it has been reported that the senescence progress of TSPCs was remarkably accelerated by the activation of the NF-κB signaling pathway in TSPCs from the degenerative tendinopathy of patients (103). Wang et al. (104) found that target inhibition of the IKKβ can reduce the TSPC senescence response to the inflamm-aging-triggered rat degenerative rotator cuff tendinopathy by inhibiting the expression of the senescence-associated b-galactosidase (SA-b-gal) and cyclin-dependent kinase inhibitor (p21CIP1A). Overall, these findings suggest opportunities to reverse the aging process of tendon tissues by targeting the activation of the NF-κB signaling pathway. However, there is still limited studies conducted for understanding its complete mechanisms of action as anti-aging.
Taken together, by mediating the interaction between tenocytes, TSPCs, and inflammatory processes, the NF-κB signaling pathway disturbs tendon homeostasis and induces the progression of degenerative tendinopathy. These studies shed new light on the mechanisms underlying tendinopathy, further establishing the potential of the NF-κB signaling pathway as a therapeutic target for chronic tendinopathy.
6 Future prospects of the NF-κB signaling pathway in tendon disorders
Although targeting the activation of the NF-κB signaling pathway has demonstrated promising outcomes in managing tendon disorders, there is still a lot to be explored in this domain.
First, according to investigations, the NF-κB signaling pathway can also interact with other signaling pathways, such as MAPK, to promote the progression of tendinopathy (as also highlighted in Section 5) (105). Also, Fu et al. (106) reported that the NF-κB signaling pathway coordinated with the mTORC1 signaling pathway to induce HO during progression of tendinopathy. On the other hand, Wang et al. (107) found that TGF-β1/Smad3 collaborated with the NF-κB signaling pathway, triggering the tendon scar and adhesion formation following injury. Also, Fu et al. (106) reported that the NF-κB signaling pathway coordinated with the mTORC1 signaling pathway to induce HO during progression of tendinopathy. Considering the interaction of the NF-κB signaling pathway with other signaling pathways that perpetuate the development of tendinopathy, there is a possibility that interfering with NF-κB signaling and these signaling pathways may harbor more promising opportunities for tendon disorders. This possibility remains to be investigated.
Secondly, target blockage of the NF-κB signaling pathway regarding different tendinopathic disease courses is required to be determined. For example, supraspinatus tendon samples from patients with early-stage tendon disorders displayed a mixed inflammation signature with increased expression of genes and proteins mediated by interferon and the NF-κB signaling pathway; intermediate-stage tendon disorders showed an inflammation signature where expression of the NF-κB mediated proinflammatory genes and proteins; advanced-stage tendon disorders showed the increased expression of STAT-6 and glucocorticoid receptor pathway (108). This transition in inflammation activation signatures indicates that therapeutic targeting of the NF-κB signaling pathway may be viable at early and intermediate stages of degenerative tendinopathy. Further investigations to demonstrate the efficacy of targeting the NF-κB signaling pathway at an appropriate time during the progression of tendinopathy are needed.
Lastly, although overwhelming data across the literature indicates that the NF-κB signaling pathway negative affects during tendon healing, another investigation suggests otherwise. For example, blocking IKKβ by gene knockout in the Scx-lineage population was reported to reduced scar tissue and restore tendon failure strength in a mouse model of Rotator Cuff tendon injury (87). In other contradicting studies, it has been reported that deletion of IKKβ in the Scx-lineage population in mice led to enhanced cell apoptosis and ECM accumulation and was not biomechanically beneficial to flexor tendon healing, suggesting that the NF-κB signaling pathway likely plays different roles in a given microenvironment (79). That is, flexor (midsubstance) tendon healing, as used in this study, includes a combination of intrinsic healing and extrinsic healing. Enthesis, as in the supraspinatus tendon, is more complicated by additional cell contributions from the bone, such as osteoblasts and osteoclasts, and the fibrocartilaginous transition region, leading to a different cellular environment. This contradiction likely reflects an incomplete understanding of the action of the NF-κB signaling pathway in mediating tendon healing at different locations. Thus, future studies are warranted to investigate the precise mechanism of the NF-κB signaling pathway in mediated fibrotic tendon healing at different locations.
7 Conclusion
Summing up, activation of the NF-κB signaling pathway is associated with the formation of scar and adhesion tissue after acute tendon injury and pathological alternations of chronic degenerative tendinopathy. In terms of mechanisms, after acute tendon injuries, the NF-κB signaling pathway contributes to tendon scar and adhesion by promoting M1 macrophage polarization, inducing production of the proinflammatory cytokines, and enhancing the persistent presence of myofibroblasts. During the progression of degenerative tendinopathy, the NF-κB signaling pathway is a key checkpoint for inflammation-induced degenerative alternations, including promoting tenocyte to produce inflammatory factors, apoptosis and inducing TSPC to produce inflammatory factors, apoptosis, and HO. However, extensive scientific examinations are still warranted to full characterize the NF-κB, the exact mechanisms of action, and translate findings into clinical human and veterinary practice.
Author contributions
HL: Writing – original draft, Writing – review & editing. SLu: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. ZF: Writing – original draft, Writing – review & editing. SLi: Writing – original draft, Writing – review & editing. YL: Writing – review & editing. YZ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Sichuan Science and Technology Program (2022YFS0609). Supported from the following four projects is also gratefully acknowledged: the Sichuan Provincial Science and Technology Plan Project (2022NSFSC0688), the Luzhou Municipal Government-Southwest Medical University Joint Project (2021LZXNYD-J10), Southwest Medical University Applied Basic Fundamental Research Project (2021ZKMS050), and Hejiang County People’s Hospital-Southwest Medical University Science and Technology Strategic Cooperation Project (2022HJXNYD07).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schnabel, LV, and Koch, DW. Use of mesenchymal stem cells for tendon healing in veterinary and human medicine: getting to the "core" of the problem through a one health approach. J Am Vet Med Assoc. (2023) 261:1435–42. doi: 10.2460/javma.23.07.0388
2. van der Vlist, AC, Breda, SJ, Oei, EHG, Verhaar, JAN, and de Vos, RJ. Clinical risk factors for Achilles tendinopathy: a systematic review. Br J Sports Med. (2019) 53:1352–61. doi: 10.1136/bjsports-2018-099991
3. Pauwels, F, Hartmann, A, Alawneh, J, Wightman, P, and Saunders, J. Contrast enhanced computed tomography findings in 105 horse distal extremities. J Equine Vet. (2021) 104:103704. doi: 10.1016/j.jevs.2021.103704
4. Del Signore, F, De Dominicis, S, Mastromatteo, G, Simeoni, F, Scapolo, PA, Tamburro, R, et al. Sonoelastography of Normal canine common calcaneal tendon: preliminary results. Vet Comp Orthop Traumatol. (2021) 34:200–5. doi: 10.1055/s-0040-1721660
5. Li, H, Luo, S, Wang, H, Chen, Y, Ding, M, Lu, J, et al. The mechanisms and functions of TGF-beta1 in tendon healing. Injury. (2023) 54:111052. doi: 10.1016/j.injury.2023.111052
6. Quam, VG, Altmann, NN, Brokken, MT, and Durgam, SS. Zonal characterization and differential trilineage potentials of equine intrasynovial deep digital flexor tendon-derived cells. BMC Vet Res. (2021) 17:138. doi: 10.1186/s12917-021-02793-1
7. Oshita, T, Tobita, M, Tajima, S, and Mizuno, H. Adipose-derived stem cells improve collagenase-induced tendinopathy in a rat model. Am J Sports Med. (2016) 44:1983–9. doi: 10.1177/0363546516640750
8. Smith, RKW, and McIlwraith, CW. "one health" in tendinopathy research: current concepts. J Orthop Res. (2021) 39:1596–602. doi: 10.1002/jor.25035
9. Bostrom, A, Asplund, K, Bergh, A, and Hyytiainen, H. Systematic review of complementary and alternative veterinary medicine in sport and companion animals: therapeutic ultrasound. Animals. (2022) 12:144. doi: 10.3390/ani12223144
10. Luo, J, Wang, Z, Tang, C, Yin, Z, Huang, J, Ruan, D, et al. Animal model for tendinopathy. J Orthop Translat. (2023) 42:43–56. doi: 10.1016/j.jot.2023.06.005
11. Watts, AE, Millar, NL, Platt, J, Kitson, SM, Akbar, M, Rech, R, et al. MicroRNA29a treatment improves early tendon injury. Mol Ther. (2017) 25:2415–26. doi: 10.1016/j.ymthe.2017.07.015
12. Tamura, N, Nukada, T, Kato, T, Kuroda, T, Kotoyori, Y, Fukuda, K, et al. The use of sonoelastography to assess the recovery of stiffness after equine superficial digital flexor tendon injuries: a preliminary prospective longitudinal study of the healing process. Equine Vet J. (2017) 49:590–5. doi: 10.1111/evj.12665
13. Dakin, SG, Dudhia, J, and Smith, RK. Resolving an inflammatory concept: the importance of inflammation and resolution in tendinopathy. Vet Immunol Immunopathol. (2014) 158:121–7. doi: 10.1016/j.vetimm.2014.01.007
14. Visser, TSS, van Linschoten, R, Vicenzino, B, Weir, A, and de Vos, RJ. Terminating corticosteroid injection in tendinopathy? Hasta la Vista, Baby. J Orthop Sports Phys Ther. (2023) 54:10–3. doi: 10.2519/jospt.2023.11875
15. Dakin, SG, Dudhia, J, and Smith, RK. Science in brief: resolving tendon inflammation. A new perspective. Equine Vet J. (2013) 45:398–400. doi: 10.1111/evj.12030
16. Shen, H, Jayaram, R, Yoneda, S, Linderman, SW, Sakiyama-Elbert, SE, Xia, Y, et al. The effect of adipose-derived stem cell sheets and CTGF on early flexor tendon healing in a canine model. Sci Rep. (2018) 8:11078. doi: 10.1038/s41598-018-29474-8
17. Jasinski, T, Zdrojkowski, L, Kautz, E, Juszczuk-Kubiak, E, Ferreira-Dias, G, and Domino, M. Equine Endometrosis pathological features: are they dependent on NF-kappaB signaling pathway? Animals. (2021) 11:151. doi: 10.3390/ani11113151
18. Lyu, Q, Wawrzyniuk, M, Rutten, V, van Eden, W, Sijts, A, and Broere, F. Hsp70 and NF-kB mediated control of innate inflammatory responses in a canine macrophage cell line. Int J Mol Sci. (2020) 21:6464. doi: 10.3390/ijms21186464
19. Jiang, H, Lin, X, Liang, W, Li, Y, and Yu, X. Friedelin alleviates the pathogenesis of collagenase-induced tendinopathy in mice by promoting the selective Autophagic degradation of p65. Nutrients. (2022) 14:1673. doi: 10.3390/nu14081673
20. Sugioka, K, Nakagawa, K, Murata, R, Ochiai, N, Sasho, T, Arai, M, et al. Radial shock waves effectively introduced NF-kappa B decoy into rat achilles tendon cells in vitro. J Orthop Res. (2010) 28:1078–83. doi: 10.1002/jor.21081
21. Lin, X, Tian, X, Jiang, H, Li, W, Wang, C, Wu, J, et al. Carpaine alleviates tendinopathy in mice by promoting the ubiquitin-proteasomal degradation of p65 via targeting the E3 ubiquitin ligase LRSAM1. Phytomedicine. (2023) 124:155323. doi: 10.1016/j.phymed.2023.155323
22. Faydaver, M, El Khatib, M, Russo, V, Rigamonti, M, Raspa, M, Di Giacinto, O, et al. Unraveling the link: locomotor activity exerts a dual role in predicting Achilles tendon healing and boosting regeneration in mice. Front Vet Sci. (2023) 10:1281040. doi: 10.3389/fvets.2023.1281040
23. Rothenberg, EV. Transcription factors specifically control change. Genes Dev. (2022) 36:1097–9. doi: 10.1101/gad.350308.122
24. Nowak, K, Jablonska, E, and Ratajczak-Wrona, W. NF-kappaB-an important player in Xenoestrogen signaling in immune cells. Cells. (2021) 10:1799. doi: 10.3390/cells10071799
25. Guldenpfennig, C, Teixeiro, E, and Daniels, M. NF-kB's contribution to B cell fate decisions. Front Immunol. (2023) 14:1214095. doi: 10.3389/fimmu.2023.1214095
26. Wibisana, JN, and Okada, M. Encoding and decoding NF-kappaB nuclear dynamics. Curr Opin Cell Biol. (2022) 77:102103. doi: 10.1016/j.ceb.2022.102103
28. Bacher, S, and Schmitz, ML. Open questions in the NF-kappaB field. Biochim Biophys Acta, Mol Cell Res. (2024) 1871:119469. doi: 10.1016/j.bbamcr.2023.119469
29. Kizilirmak, C, Bianchi, ME, and Zambrano, S. Insights on the NF-kappaB system using live cell imaging: recent developments and future perspectives. Front Immunol. (2022) 13:886127. doi: 10.3389/fimmu.2022.886127
30. Wang, AG, Son, M, Kenna, E, Thom, N, and Tay, S. NF-kappaB memory coordinates transcriptional responses to dynamic inflammatory stimuli. Cell Rep. (2022) 40:111159. doi: 10.1016/j.celrep.2022.111159
31. Bacher, S, Meier-Soelch, J, Kracht, M, and Schmitz, ML. Regulation of transcription factor NF-kappaB in its natural habitat: the nucleus. Cells. (2021) 10:753. doi: 10.3390/cells10040753
32. Wibisana, JN, Inaba, T, Shinohara, H, Yumoto, N, Hayashi, T, Umeda, M, et al. Enhanced transcriptional heterogeneity mediated by NF-kappaB super-enhancers. PLoS Genet. (2022) 18:e1010235. doi: 10.1371/journal.pgen.1010235
33. Mucka, P, Lindemann, P, Bosco, B, Willenbrock, M, Radetzki, S, Neuenschwander, M, et al. CLK2 and CLK4 are regulators of DNA damage-induced NF-kappaB targeted by novel small molecule inhibitors, cell. Chem Biol. (2023) 30:1303–1312.e3. doi: 10.1016/j.chembiol.2023.06.027
34. Jimi, E, Fei, H, and Nakatomi, C. NF-kappaB signaling regulates physiological and pathological Chondrogenesis. Int J Mol Sci. (2019) 20:6275. doi: 10.3390/ijms20246275
35. Jaruszewicz-Blonska, J, Kosiuk, I, Prus, W, and Lipniacki, T. A plausible identifiable model of the canonical NF-kappaB signaling pathway. PLoS One. (2023) 18:e0286416. doi: 10.1371/journal.pone.0286416
36. Ziegler, LS, Gerner, MC, Schmidt, RLJ, Trapin, D, Steinberger, P, Pickl, WF, et al. Attenuation of canonical NF-kappaB signaling maintains function and stability of human Treg. FEBS J. (2021) 288:640–62. doi: 10.1111/febs.15361
37. Liu, T, Zhang, L, Joo, D, and Sun, SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
38. Williams, LM, and Gilmore, TD. Looking Down on NF-kappaB. Mol Cell Biol. (2020) 40:20. doi: 10.1128/MCB.00104-20
39. Gautam, P, Maenner, S, Cailotto, F, Reboul, P, Labialle, S, Jouzeau, JY, et al. Emerging role of IkappaBzeta in inflammation: emphasis on psoriasis. Clin Transl Med. (2022) 12:e1032. doi: 10.1002/ctm2.1032
40. Hovelmeyer, N, Schmidt-Supprian, M, and Ohnmacht, C. NF-kappaB in control of regulatory T cell development, identity, and function. J Mol Med. (2022) 100:985–95. doi: 10.1007/s00109-022-02215-1
41. Sehnert, B, Burkhardt, H, Dubel, S, and Voll, RE. Cell-type targeted NF-kappaB inhibition for the treatment of inflammatory diseases. Cells. (2020) 9:1627. doi: 10.3390/cells9071627
42. Jiang, Y, Zhang, J, Shi, C, Li, X, Jiang, Y, and Mao, R. NF-kappaB: a mediator that promotes or inhibits angiogenesis in human diseases? Expert Rev Mol Med. (2023) 25:e25. doi: 10.1017/erm.2023.20
43. Fung, SY, Lu, HY, Sharma, M, Sharma, AA, Saferali, A, Jia, A, et al. MALT1-dependent cleavage of HOIL1 modulates canonical NF-kappaB signaling and inflammatory responsiveness. Front Immunol. (2021) 12:749794. doi: 10.3389/fimmu.2021.749794
44. Lin, TH, Pajarinen, J, Lu, L, Nabeshima, A, Cordova, LA, Yao, Z, et al. NF-kappaB as a therapeutic target in inflammatory-associated bone diseases. Adv Protein Chem Struct Biol. (2017) 107:117–54. doi: 10.1016/bs.apcsb.2016.11.002
45. Tapiero, B, Maari, C, Kokta, V, Fernandez, I, and Touzot, F. Lupus vulgaris revealing an IkappaBalpha gain of function variant. J Clin Immunol. (2023) 43:545–7. doi: 10.1007/s10875-022-01421-3
46. Radzka, J, Lapinska, Z, Szwedowicz, U, Gajewska-Naryniecka, A, Gizak, A, and Kulbacka, J. Alternations of NF-kappaB signaling by natural compounds in muscle-derived cancers. Int J Mol Sci. (2023) 24:11900. doi: 10.3390/ijms241511900
47. Xiao, W. Advances in NF-kappaB signaling transduction and transcription. Cell Mol Immunol. (2004) 1:425–35.
48. Chatterjee, B, Roy, P, Sarkar, UA, Zhao, M, Ratra, Y, Singh, A, et al. Immune differentiation regulator p100 Tunes NF-kappaB responses to TNF. Front Immunol. (2019) 10:997. doi: 10.3389/fimmu.2019.00997
49. Pflug, KM, and Sitcheran, R. Targeting NF-kappaB-inducing kinase (NIK) in immunity, inflammation, and Cancer. Int J Mol Sci. (2020) 21:8470. doi: 10.3390/ijms21228470
50. Kucharzewska, P, Maracle, CX, Jeucken, KCM, van Hamburg, JP, Israelsson, E, Furber, M, et al. NIK-IKK complex interaction controls NF-kappaB-dependent inflammatory activation of endothelium in response to LTbetaR ligation. J Cell Sci. (2019) 132:615. doi: 10.1242/jcs.225615
51. Prescott, JA, Balmanno, K, Mitchell, JP, Okkenhaug, H, and Cook, SJ. IKKalpha plays a major role in canonical NF-kappaB signalling in colorectal cells. Biochem J. (2022) 479:305–25. doi: 10.1042/BCJ20210783
52. Johnson, SA, Valdes-Martinez, A, Turk, PJ, Wayne McIlwraith, C, Barrett, MF, McGilvray, KC, et al. Longitudinal tendon healing assessed with multi-modality advanced imaging and tissue analysis. Equine Vet J. (2021) 54:766–81. doi: 10.1111/evj.13478
53. Chisari, E, Rehak, L, Khan, WS, and Maffulli, N. Tendon healing in presence of chronic low-level inflammation: a systematic review. Br Med Bull. (2019) 132:97–116. doi: 10.1093/bmb/ldz035
54. Leong, NL, Kator, JL, Clemens, TL, James, A, Enamoto-Iwamoto, M, and Jiang, J. Tendon and ligament healing and current approaches to tendon and ligament regeneration. J Orthop Res. (2020) 38:7–12. doi: 10.1002/jor.24475
55. Romero, A, Barrachina, L, Ranera, B, Remacha, AR, Moreno, B, de Blas, I, et al. Comparison of autologous bone marrow and adipose tissue derived mesenchymal stem cells, and platelet rich plasma, for treating surgically induced lesions of the equine superficial digital flexor tendon. Vet J. (2017) 224:76–84. doi: 10.1016/j.tvjl.2017.04.005
56. Spaas, JH, Guest, DJ, and Van de Walle, GR. Tendon regeneration in human and equine athletes: Ubi Sumus-quo Vadimus (where are we and where are we going to)? Sports Med. (2012) 42:871–90. doi: 10.1007/BF03262300
57. Ribitsch, I, Oreff, GL, and Jenner, F. Regenerative medicine for equine musculoskeletal diseases. Animals. (2021) 11:234. doi: 10.3390/ani11010234
58. Ribitsch, I, Bileck, A, Aldoshin, AD, Kandula, MM, Mayer, RL, Egerbacher, M, et al. Molecular mechanisms of fetal tendon regeneration versus adult fibrous repair. Int J Mol Sci. (2021) 22:5619. doi: 10.3390/ijms22115619
59. Ruan, D, Fei, Y, Qian, S, Huang, Z, Chen, W, Tang, C, et al. Early-stage primary anti-inflammatory therapy enhances the regenerative efficacy of platelet-rich plasma in a rabbit Achilles tendinopathy model. Am J Sports Med. (2021) 49:3357–71. doi: 10.1177/03635465211037354
60. Lane, RA, Migotsky, N, Havlioglu, N, Iannucci, LE, Shen, H, Lake, S, et al. The effects of NF-kappaB suppression on the early healing response following intrasynovial tendon repair in a canine model. J Orthop Res. (2023) 41:2295–304. doi: 10.1002/jor.25576
61. Cooper, HE, Bowlby, C, Long, S, and Durgam, SS. Macrophage phenotype impacts in vitro equine intrasynovial deep digital flexor tenocyte matrix metalloproteinase gene expression and secretion. Am J Vet Res. (2023) 84:1–8. doi: 10.2460/ajvr.23.05.0106
62. Dakin, SG, Werling, D, Hibbert, A, Abayasekara, DR, Young, NJ, Smith, RK, et al. Macrophage sub-populations and the lipoxin A4 receptor implicate active inflammation during equine tendon repair. PLoS One. (2012) 7:e32333. doi: 10.1371/journal.pone.0032333
63. Best, KT, Lee, FK, Knapp, E, Awad, HA, and Loiselle, AE. Deletion of NFKB1 enhances canonical NF-kappaB signaling and increases macrophage and myofibroblast content during tendon healing. Sci Rep. (2019) 9:10926. doi: 10.1038/s41598-019-47461-5
64. Song, W, Ma, Z, Wang, X, Wang, Y, Wu, D, Wang, C, et al. Macroporous granular hydrogels functionalized with aligned architecture and small extracellular vesicles stimulate osteoporotic tendon-to-bone healing. Adv Sci. (2023) 10:e2304090. doi: 10.1002/advs.202304090
65. Shen, H, and Lane, RA. Extracellular vesicles from primed adipose-derived stem cells enhance Achilles tendon repair by reducing inflammation and promoting intrinsic healing. Stem Cells. (2023) 41:617–27. doi: 10.1093/stmcls/sxad032
66. Lu, M, Wang, S, Wang, H, Xue, T, Cai, C, Fan, C, et al. Pyrrolidine Dithiocarbamate-loaded electrospun membranes for Peritendinous anti-adhesion through inhibition of the nuclear factor-kappaB pathway. Acta Biomater. (2023) 155:333–46. doi: 10.1016/j.actbio.2022.10.004
67. Wang, H, Song, D, Wei, L, Huang, L, Wei, D, Su, Y, et al. Ethyl caffeate inhibits macrophage polarization via SIRT1/NF-kappaB to attenuate traumatic heterotopic ossification in mice. Biomed Pharmacother. (2023) 161:114508. doi: 10.1016/j.biopha.2023.114508
68. Li, J, Sun, Z, Luo, G, Wang, S, Cui, H, Yao, Z, et al. Quercetin attenuates trauma-induced heterotopic ossification by tuning immune cell infiltration and related inflammatory insult. Front Immunol. (2021) 12:649285. doi: 10.3389/fimmu.2021.649285
69. Xiao, Y, Tao, Z, Ju, Y, Huang, X, Zhang, X, Liu, X, et al. Diamond-like carbon depositing on the surface of Polylactide membrane for prevention of adhesion formation during tendon repair. Nanomicro Lett. (2024) 16:186. doi: 10.1007/s40820-024-01392-7
70. Mimpen, JY, Snelling, SJB, Carr, AJ, and Dakin, SG. Interleukin-17 cytokines and receptors: potential amplifiers of tendon inflammation. Front Bioeng Biotechnol. (2021) 9:795830. doi: 10.3389/fbioe.2021.795830
71. Chen, Q, Lu, H, and Yang, H. Chitosan prevents adhesion during rabbit flexor tendon repair via the sirtuin 1 signaling pathway. Mol Med Rep. (2015) 12:4598–603. doi: 10.3892/mmr.2015.4007
72. Almeida Mdos, S, Guerra Fda, R, de Oliveira, LP, Vieira, CP, and Pimentel, ER. A hypothesis for the anti-inflammatory and mechanotransduction molecular mechanisms underlying acupuncture tendon healing. Acupunct Med. (2014) 32:178–82. doi: 10.1136/acupmed-2013-010455
73. Golman, M, Li, X, Skouteris, D, Abraham, AA, Song, L, Abu-Amer, Y, et al. Enhanced tendon-to-bone healing via IKKbeta inhibition in a rat rotator cuff model. Am J Sports Med. (2021) 49:780–9. doi: 10.1177/0363546520985203
74. Paredes, J, Pekmezian, A, and Andarawis-Puri, N. MRL/MpJ tendon matrix-derived therapeutic promotes improved healing outcomes in scar-mediated canonical tendon healing. J Orthop Res. (2021) 39:1548–60. doi: 10.1002/jor.24754
75. Ackerman, JE, Muscat, SN, Adjei-Sowah, E, Korcari, A, Nichols, AEC, Buckley, MR, et al. Identification of Periostin as a critical niche for myofibroblast dynamics and fibrosis during tendon healing. Matrix Biol. (2024) 125:59–72. doi: 10.1016/j.matbio.2023.12.004
76. Abdelhakiem, MAH, Hussein, A, Seleim, SM, Abdelbaset, AE, and Abd-Elkareem, M. Silver nanoparticles and platelet-rich fibrin accelerate tendon healing in donkey. Sci Rep. (2023) 13:3421. doi: 10.1038/s41598-023-30543-w
77. Alenchery, RG, Ajalik, RE, Jerreld, K, Midekksa, F, Zhong, S, Alkatib, B, et al. PAI-1 mediates TGF-beta1-induced myofibroblast activation in tenocytes via mTOR signaling. J Orthop Res. (2023) 41:2163–74. doi: 10.1002/jor.25594
78. Pownder, SL, Hayashi, K, Lin, BQ, Meyers, KN, Caserto, BG, Breighner, RE, et al. Differences in the magnetic resonance imaging parameter T2* may be identified during the course of canine patellar tendon healing: a pilot study. Quant Imaging Med Surg. (2021) 11:1234–46. doi: 10.21037/qims-20-684
79. Best, KT, Nichols, AEC, Knapp, E, Hammert, WC, Ketonis, C, Jonason, JH, et al. NF-kappaB activation persists into the remodeling phase of tendon healing and promotes myofibroblast survival. Sci Signal. (2020) 13:7209. doi: 10.1126/scisignal.abb7209
80. Chen, S, Jiang, S, Zheng, W, Tu, B, Liu, S, Ruan, H, et al. RelA/p65 inhibition prevents tendon adhesion by modulating inflammation, cell proliferation, and apoptosis. Cell Death Dis. (2017) 8:e2710. doi: 10.1038/cddis.2017.135
81. Tang, JB, Xu, Y, Ding, F, and Wang, XT. Expression of genes for collagen production and NF-kappaB gene activation of in vivo healing flexor tendons. J Hand Surg Am. (2004) 29:564–70. doi: 10.1016/j.jhsa.2003.12.019
82. Ge, Z, Tang, H, Lyu, J, Zhou, B, Yang, M, Tang, K, et al. Conjoint analysis of lncRNA and mRNA expression in rotator cuff tendinopathy. Ann Transl Med. (2020) 8:335. doi: 10.21037/atm.2020.02.149
83. Millar, NL, Murrell, GA, and McInnes, IB. Inflammatory mechanisms in tendinopathy—towards translation. Nat Rev Rheumatol. (2017) 13:110–22. doi: 10.1038/nrrheum.2016.213
84. Chen, Y, Xie, Y, Liu, M, Hu, J, Tang, C, Huang, J, et al. Controlled-release curcumin attenuates progression of tendon ectopic calcification by regulating the differentiation of tendon stem/progenitor cells. Mater Sci Eng C Mater Biol Appl. (2019) 103:109711. doi: 10.1016/j.msec.2019.04.090
85. Semis, HS, Gur, C, Ileriturk, M, Kandemir, FM, and Kaynar, O. Evaluation of therapeutic effects of quercetin against Achilles tendinopathy in rats via oxidative stress, Inflammation, Apoptosis, Autophagy, and Metalloproteinases. Am J Sports Med. (2022) 50:486–98. doi: 10.1177/03635465211059821
86. Buhrmann, C, Mobasheri, A, Busch, F, Aldinger, C, Stahlmann, R, Montaseri, A, et al. Curcumin modulates nuclear factor kappaB (NF-kappaB)-mediated inflammation in human tenocytes in vitro: role of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem. (2011) 286:28556–66. doi: 10.1074/jbc.M111.256180
87. Abraham, AC, Shah, SA, Golman, M, Song, L, Li, X, Kurtaliaj, I, et al. Targeting the NF-kappaB signaling pathway in chronic tendon disease. Sci Transl Med. (2019) 11:4319. doi: 10.1126/scitranslmed.aav4319
88. Busch, F, Mobasheri, A, Shayan, P, Lueders, C, Stahlmann, R, and Shakibaei, M. Resveratrol modulates interleukin-1beta-induced phosphatidylinositol 3-kinase and nuclear factor kappaB signaling pathways in human tenocytes. J Biol Chem. (2012) 287:38050–63. doi: 10.1074/jbc.M112.377028
89. Mueller, AL, Brockmueller, A, Kunnumakkara, AB, and Shakibaei, M. Calebin a, a compound of turmeric, Down-regulates inflammation in tenocytes by NF-kappaB/Scleraxis signaling. Int J Mol Sci. (2022) 23:1695. doi: 10.3390/ijms23031695
90. Sarmah, D, Datta, A, Kaur, H, Kalia, K, Borah, A, Rodriguez, AM, et al. Sirtuin-1—mediated NF-kappaB pathway modulation to mitigate Inflammasome signaling and cellular apoptosis is one of the neuroprotective effects of intra-arterial mesenchymal stem cell therapy following ischemic stroke. Stem Cell Rev Rep. (2022) 18:821–38. doi: 10.1007/s12015-021-10315-7
91. Busch, F, Mobasheri, A, Shayan, P, Stahlmann, R, and Shakibaei, M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J Biol Chem. (2012) 287:25770–81. doi: 10.1074/jbc.M112.355420
92. Rajpar, I, and Barrett, JG. Multi-differentiation potential is necessary for optimal tenogenesis of tendon stem cells. Stem Cell Res Ther. (2020) 11:152. doi: 10.1186/s13287-020-01640-8
93. Perucca Orfei, C, Bowles, AC, Kouroupis, D, Willman, MA, Ragni, E, Kaplan, LD, et al. Human tendon stem/progenitor cell features and functionality are highly influenced by in vitro culture conditions. Front Bioeng Biotechnol. (2021) 9:711964. doi: 10.3389/fbioe.2021.711964
94. Stauber, T, Wolleb, M, Duss, A, Jaeger, PK, Heggli, I, Hussien, AA, et al. Extrinsic macrophages protect while tendon progenitors degrade: insights from a tissue engineered model of tendon compartmental crosstalk. Adv Healthc Mater. (2021) 10:e2100741. doi: 10.1002/adhm.202100741
95. Walia, B, and Huang, AH. Tendon stem progenitor cells: understanding the biology to inform therapeutic strategies for tendon repair. J Orthop Res. (2019) 37:1270–80. doi: 10.1002/jor.24156
96. Tachibana, N, Chijimatsu, R, Okada, H, Oichi, T, Taniguchi, Y, Maenohara, Y, et al. RSPO2 defines a distinct undifferentiated progenitor in the tendon/ligament and suppresses ectopic ossification. Sci Adv. (2022) 8:eabn2138. doi: 10.1126/sciadv.abn2138
97. Ge, Z, Yang, M, Wei, D, Wang, D, Zhao, R, Deng, X, et al. Inhibition of IKKbeta via a DNA-based in situ delivery system improves Achilles tendinopathy healing in a rat model. Am J Sports Med. (2023) 51:3533–45. doi: 10.1177/03635465231198501
98. Xu, K, Lin, C, Ma, D, Chen, M, Zhou, X, He, Y, et al. Spironolactone ameliorates senescence and calcification by modulating autophagy in rat tendon-derived stem cells via the NF-kappaB/MAPK pathway. Oxidative Med Cell Longev. (2021) 2021:5519587. doi: 10.1155/2021/5519587
99. Avey, AM, Devos, F, Roberts, AG, Essawy, ESE, and Baar, K. Inhibiting JAK1, not NF-kappaB, reverses the effect of pro-inflammatory cytokines on engineered human ligament function. Matrix Biol. (2024) 125:100–12. doi: 10.1016/j.matbio.2023.12.007
100. Moqbel, SAA, Xu, K, Chen, Z, Xu, L, He, Y, Wu, Z, et al. Tectorigenin alleviates inflammation, apoptosis, and ossification in rat tendon-derived stem cells via modulating NF-kappa B and MAPK pathways, front cell. Dev Biol. (2020) 8:568894. doi: 10.3389/fcell.2020.568894
101. Salminen, A, and Kaarniranta, K. Genetics vs. entropy: longevity factors suppress the NF-kappaB-driven entropic aging process. Ageing Res Rev. (2010) 9:298–314. doi: 10.1016/j.arr.2009.11.001
102. Dakin, SG, Dudhia, J, Werling, NJ, Werling, D, Abayasekara, DR, and Smith, RK. Inflamm-aging and arachadonic acid metabolite differences with stage of tendon disease. PLoS One. (2012) 7:e48978. doi: 10.1371/journal.pone.0048978
103. Cai, Z, Zhang, Y, Liu, S, and Liu, X. Celecoxib, beyond anti-inflammation, alleviates tendon-derived stem cell senescence in degenerative rotator cuff tendinopathy. Am J Sports Med. (2022) 50:2488–96. doi: 10.1177/03635465221098133
104. Wang, C, Zhou, Z, Song, W, Cai, Z, Ding, Z, Chen, D, et al. Inhibition of IKKbeta/NF-kappaB signaling facilitates tendinopathy healing by rejuvenating inflamm-aging induced tendon-derived stem/progenitor cell senescence. Mol Ther Nucleic Acids. (2022) 27:562–76. doi: 10.1016/j.omtn.2021.12.026
105. Jiao, X, Zhang, Y, Li, W, Zhou, X, Chu, W, Li, Y, et al. HIF-1alpha inhibition attenuates severity of Achilles tendinopathy by blocking NF-kappaB and MAPK pathways. Int Immunopharmacol. (2022) 106:108543. doi: 10.1016/j.intimp.2022.108543
106. Fu, J, Zhang, J, Jiang, T, Ao, X, Li, P, Lian, Z, et al. mTORC1 coordinates NF-kappaB signaling pathway to promote chondrogenic differentiation of tendon cells in heterotopic ossification. Bone. (2022) 163:116507. doi: 10.1016/j.bone.2022.116507
107. Wang, K, Chen, D, Wang, Z, Yang, J, and Liu, W. An injectable and antifouling supramolecular polymer hydrogel with microenvironment-regulatory function to prevent Peritendinous adhesion and promote tendon repair. Macromol Biosci. (2023) 23:e2300142. doi: 10.1002/mabi.202300142
Keywords: NF-κB signaling pathway, tendon injury, tendon scar healing, tendon adhesion tendinopathy, tendon repair
Citation: Li H, Li Y, Luo S, Zhang Y, Feng Z and Li S (2024) The roles and mechanisms of the NF-κB signaling pathway in tendon disorders. Front. Vet. Sci. 11:1382239. doi: 10.3389/fvets.2024.1382239
Edited by:
Louis Charles Penning, Utrecht University, NetherlandsReviewed by:
Iris Maria Gerner, University of Veterinary Medicine Vienna, AustriaDebbie Guest, Royal Veterinary College (RVC), United Kingdom
Copyright © 2024 Li, Li, Luo, Zhang, Feng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Feng, ZmVuZ3poZW5odWExMjIyQDE2My5jb20=; Sen Li, amh0MTg3QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship