 Jiajin Huang1,2,3
Jiajin Huang1,2,3 Yong Ruan1,2,3
Yong Ruan1,2,3 Meimei Xiao1,2,3Lingang Dai1,2,3Chuanmei Jiang1,2,3Jifeng Li1,2,3Jiali Xu1,2,3
Meimei Xiao1,2,3Lingang Dai1,2,3Chuanmei Jiang1,2,3Jifeng Li1,2,3Jiali Xu1,2,3 Xiang Chen1,2,3Houqiang Xu1,2,3*
Xiang Chen1,2,3Houqiang Xu1,2,3*- 1Key Laboratory of Animal Genetics, Breeding and Reproduction in the Plateau Mountainous Region, Ministry of Education, Guizhou University, Guiyang, China
- 2Guizhou Provincial Key Laboratory of Animal Genetics, Breeding and Reproduction, Guizhou University, Guiyang, China
- 3College of Animal Science, Guizhou University, Guiyang, China
The newborn ovary homeobox gene (NOBOX) regulates ovarian and early oocyte development, and thus plays an essential role in reproduction. In this study, the mRNA expression level and single nucleotide polymorphism (SNP) of NOBOX in various tissues of Xiangsu pigs were studied to explore the relationship between its polymorphism and litter size traits. Also, bioinformatics was used to evaluate the effects of missense substitutions on protein structure and function. The results revealed that NOBOX is preferentially expressed in the ovary. Six mutations were detected in the NOBOX sequence, including g.1624 T>C, g.1858 G>A, g.2770 G>A, g.2821 A>G, g.5659 A>G, and g.6025 T>A, of which g.1858 G>A was a missense mutation. However, only g.1858 G>A, g.5659 A>G, and g.6025 T>A were significantly associated with litter size traits (p < 0.05). Further prediction of the effect of the missense mutation g.1858 G>A on protein function revealed that p.V82M is a non-conservative mutation that significantly reduces protein stability and thus alters protein function. Overall, these findings suggest that NOBOX polymorphism is closely related to the litter size of Xiangsu pigs, which may provide new insights into pig breeding.
1 Introduction
Litter size is one of the most important reproductive traits in sows that directly affect economic efficiency (1). It is a low heritability trait that is affected by many factors, such as genetics, environment, management, and nutrition (2). To improve economic efficiency, priority should be given to improving the reproductive quality of sows. However, relying on traditional crossbreeding selection techniques for improvement is significantly limiting. Given that SNPs in genes and trait association analysis methods are not affected by the environment, they are more efficient and accurate for the seed selection and expansion of high propagation populations (3, 4).
The newborn ovary homeobox (NOBOX) gene, an oocyte-specific homeobox gene, transcriptionally regulates oocyte-specific genes that play key roles in ovarian development, early oogenesis, and fertility (5–7). Research on the NOBOX gene has focused on its role in ovarian development and oogenesis. It is one of the most highly mutated genes in women with premature ovarian failure (8). Besides, NOBOX hypohydroxymethylation leads to ovarian dysfunction in offspring adult rats (9). At the same time, compound heterozygous truncating mutations in NOBOX characterized by double allele deletion mutations cause severe primary premature ovarian insufficiency (POI) with primary amenorrhea in patients in consanguineous marriages (10).
Pure heterozygous NOBOX truncation variants induce defective transcriptional activation, leading to primary ovarian insufficiency (11). Of note, immunolocalization, protein imprinting, and transcriptional assay have validated NOBOX mutations as the plausible causes of POI pathogenesis in HEK293T and CHO cells (12). NOBOX is also essential for signaling between somatic and germ cells during mouse embryonic development (13). In livestock, NOBOX is an essential maternal transcription factor during early bovine embryogenesis, thereby regulating embryonic genome activation, pluripotency gene expression, and blastocyst cell allocation (14). In zebrafish, NOBOX deletion does not affect testicular development and spermatogenesis; however, it plays an important role in ovarian differentiation and early follicular development. This suggests that NOBOX is closely related to reproduction (15).
The Xiangsu pig is a new pig line developed via crossbreeding after obtaining desirable production traits by backcrossing multiple Guizhou Congjiang Xiang pig (female parent) and Sutai pig (sire) generations with the Congjiang Xiang pig (sire) (16). This new line is characterized by early sexual maturity and strong disease resistance. In addition, it bears the high adaptability of the Congjiang Xiang pig and the high litter size and fast growth rate of the Sutai pig. In this study, we report for the first time six SNPs in the NOBOX gene of the Xiangsu pig. NOBOX function was predicted using bioinformatic techniques, and the correlation between the six SNPs and litter size was investigated. These results may guide the screening of candidate genes for sow reproductive performance, thereby benefiting the Xiangsu pig breeding program.
2 Materials and methods
2.1 Experimental animals
The animal experiments used in this study strictly complied with the guidelines of the Animal Welfare Committee of Guizhou University (EAE-GZU-2023-E015). A total of 142 healthy Xiangsu breeding sows under uniform feeding conditions were selected from the Guizhou University pig farm. The litter size traits, including the total number born (TNB), number born alive (NBA), and weaned piglet number were recorded per sow for the first and second litters (Supplementary material). Subsequently, blood samples were collected from the anterior vena cava of 142 pigs using a 5 mL EDTA anticoagulant tube and stored at −20°C. After the data collection of farrowing traits, three sows were randomly selected for slaughter, and their heart, liver, spleen, lung, kidney, longissimus dorsi muscle, and ovary tissue samples were collected and stored at −80°C in a refrigerator.
2.2 Extraction of whole blood DNA and tissue RNA
Genomic DNA was extracted from the anterior vena cava blood samples using a DNA extraction kit (DP348; Tiangen, Beijing, China) following the manufacturer’s guidelines. In addition, the total RNA was extracted from the heart, liver, spleen, lung, kidney, longissimus dorsi muscle, and ovary tissue samples using the TRIzol extraction kit (9109, TaKaRa, Dalian, China). The first strand of cDNA was synthesized via reverse transcription using a reverse transcription kit (A230; Kangrun, Beijing, China) according to the manufacturer’s interactions.
2.3 Primer design
The primers for DNA and cDNA amplification were designed according to the pig NOBOX DNA (accession number: NC_010451.4) and RNA (accession number: NM_001195116.1) sequences published on NCBI using Primer Premier 5.0 software (PREMIER Biosoft International, Palo Alto, CA, United States). Primers were synthesized by Qingdao Biotechnology Co., Ltd. (Chongqing, China), and the primer information is shown in Supplementary Table S1.
2.4 PCR amplification and real-time fluorescent quantitative PCR analysis
The PCR amplification of the genomic DNA was performed in a total volume of 20 μL, including 10 μL of 2× Taq PCR Master Mix (GeneStar, Beijing, China), 1 μL of forward primer, 1 μL of reverse primer, 1 μL of genomic DNA, and 7 μL of double-distilled water (ddH2O). The amplification conditions consisted of 35 cycles of 3 min of pre-denaturation at 94°C, 30 s of denaturation at 94°C, 30 s of denaturation at 63°C, 72°C annealing for 1 min, 72°C extension for 5 min, and preservation at 4°C. Next, 5 μL of amplification product was aspirated and subjected to 1% agarose gel electrophoresis for 25 min. The target bands were verified using a gel imaging system, and the amplicons were sent to Qingdao Biotech (Chongqing, China) for sequencing. The cDNA was amplified in a 20 μL q-PCR reaction system consisting of 1 μL of cDNA template, 0.5 μL of forward primer, 0.5 μL of reverse primer, 10 μL of 2× RealStar Fast SYBR qPCR Mix (GeneStar, Beijing, China), and 8 μL of ddH2O. The cDNA amplification was replicated four times. The reaction conditions were pre-denaturation at 95°C for 2 min, denaturation at 95°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s for 40 cycles. GAPDH was the fluorescence quantitative reference gene.
2.5 Bioinformatics analysis
NOBOX amino acid sequences from eight species, including Homo sapiens (XP_016867231.1), Mus musculus (NP_570939.1), Equus caballus (XP_ 023494578.1), Sus scrofa (NP_001182045.1), Ovis aries (XP_042104903.1), Bos taurus (XP_024846980.1), Cervus elaphus (XP_043727747.1), and Gallus gallus (XP_040516952.1) were obtained from NCBI,1 and a phylogenetic tree was constructed using MEGA72 (17). Subsequently, the common motifs in the super secondary structure were predicted using the MEME tool3 to reveal the structural characteristics and functions of NOBOX proteins in the selected eight species (18). Besides, the multiple sequence comparison of the amino acid sequences of NOBOX was performed using Clustal Omega4 (19), and the results were uploaded to the online server Con Surf5 for sequence conservation evaluation. For the functional and stability studies of NOBOX proteins, PhD-SNP,6 SNAP2,7 I-Mutant2.0,8 and MuPro9 were used for prediction analysis (20–23). Additionally, Sopma10 and AlphaFold2 were used to predict the secondary structure and construct the 3D model of the tertiary structure of the protein (24, 25).
2.6 Statistical analysis
The presence of SNPs in NOBOX sequence was determined via peak plotting against the PCR sequencing reads using the SeqMan software (26). Wild-type and mutant sequences were aligned and compared using MegAlign and ClustalW software in the DNA Star package. Genotype and gene frequencies at each mutation locus were calculated directly. Hardy–Weinberg equilibrium (HWE) was evaluated using the chi-squared (χ2) test, and the gene polymorphism parameters included homozygosity (Ho), heterozygosity (He), number of effective alleles (Ne), and polypeptide information content (PIC) (27). Linkage disequilibrium (LD) and haplotype analyses among SNPs were performed using the SHEsis Main11 software (28), and the degree of chain imbalance was evaluated using the r2 value, where r2 > 0.33 indicated a strong chain imbalance state (29). On the other hand, diplotypes were evaluated based on haplotypes.
The difference in the number of litters between the different genotype groups was compared using one-way analysis of variance in the average drop-down option in IBM SPSS Statistics 25 software. The following general linear model formula was used: Yijk = μ + Gi + Sk + Aj + eijk, where Yijk is the litter size and number of pigs weaned, μ is the mean, Gi is the fixed effect of genotype, Sk is the random effect of sire, Aj is fixed effect of age, and eijk is the residual effect. The results are presented as the mean ± standard error (30).
NOBOX expression levels at different mutation sites were calculated using the 2−ΔΔCtd method (31), and its expression patterns were mapped using the GraphPad Prism 8 software. Data are expressed as mean ± standard deviation at two decimal places.
3 Results
3.1 Expression profile of NOBOX in different tissues
The tissue expression profiles of NOBOX are shown in Figure 1A NOBOX was expressed in the heart, liver, kidney, and ovary, with the highest expression level in the ovarian tissues, which was significantly higher than that in the heart, liver, and kidney (p < 0.01). On the contrary, there were no significant differences in NOBOX expression levels among the heart, liver, and kidney (p > 0.05). In addition, NOBOX was not expressed in the spleen, lung, and longest dorsal muscle.
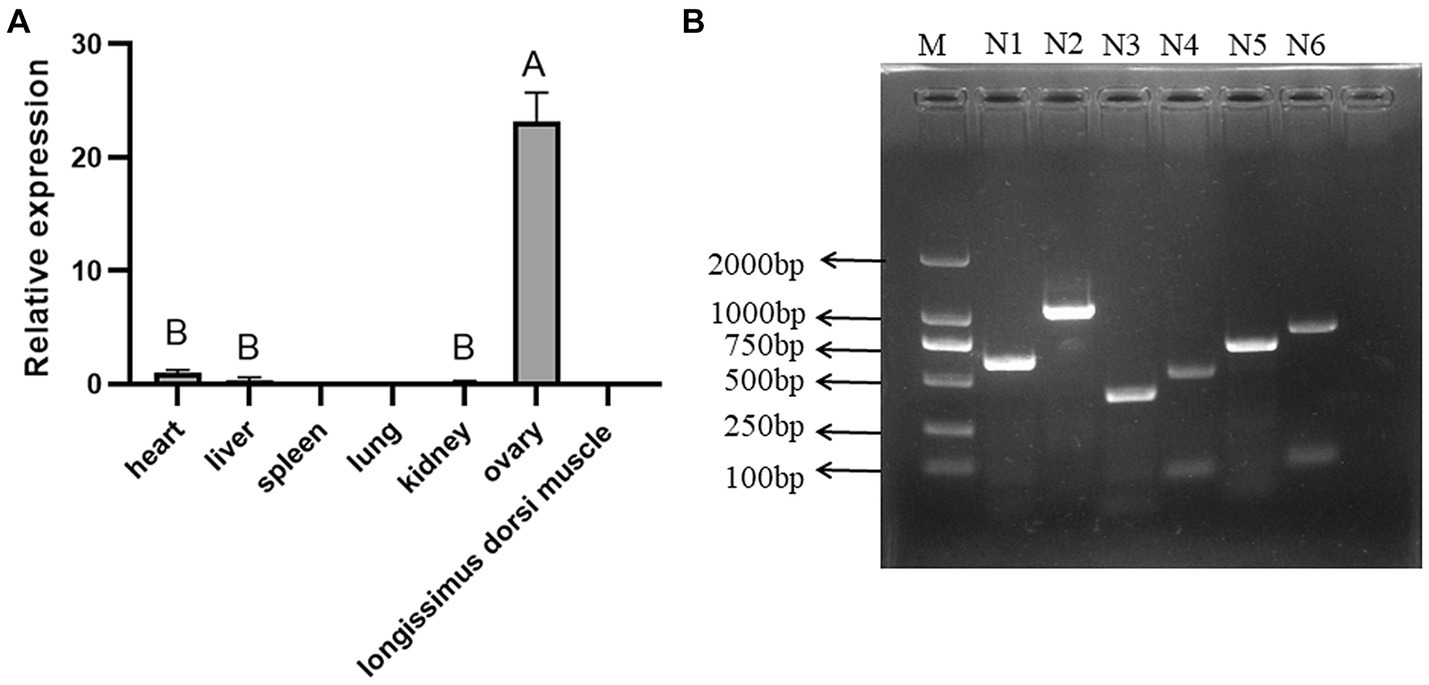
Figure 1. (A) Tissue expression of NOBOX in Xiangsu pig. Different capital letters indicated that the difference between different tissues was extremely significant (p < 0.01). (B) The results of gel electrophoresis imaging of PCR amplification products. M: DL2000 marker, N1–N6: NOBOX gene Exon1–Exon6.
3.2 PCR gel electrophoresis imaging analysis
The gel electrophoresis imaging is shown in Figure 1B. The PCR amplification products were consistent with the target fragment size, with clear and single bands, non-specific amplification, and no obvious trailing phenomenon, implying the primers had good specificity.
3.3 Identification of NOBOX polymorphic loci
Sequence alignment between the sequencing results and the reference sequence of porcine NOBOX (Accession number: NC_010451.4) revealed six SNPs in NOBOX, which were labelled g.1624 T>C, g.1858 G>A, g.2770 G>A, g.2821 A>G, g.5659 A>G. Notably, two alleles and three genotypes were present in all the six SNPs (Figure 2). The wild-type and mutant sequence alignment revealed that the base G at the g.1858 G>A locus was mutated to A, altering the codon-GUG- to -AUG-. Consequently, methionine (M) replaced valine (V), and g.1858 G>A was a missense SNP. The mutation at g.5659 A>G changed the codon-CCA- to -CCG-, resulting in a synonymous mutation because-CCA- and-CCG- are simple codons and proline (P) was not replaced (Supplementary Figure S1).
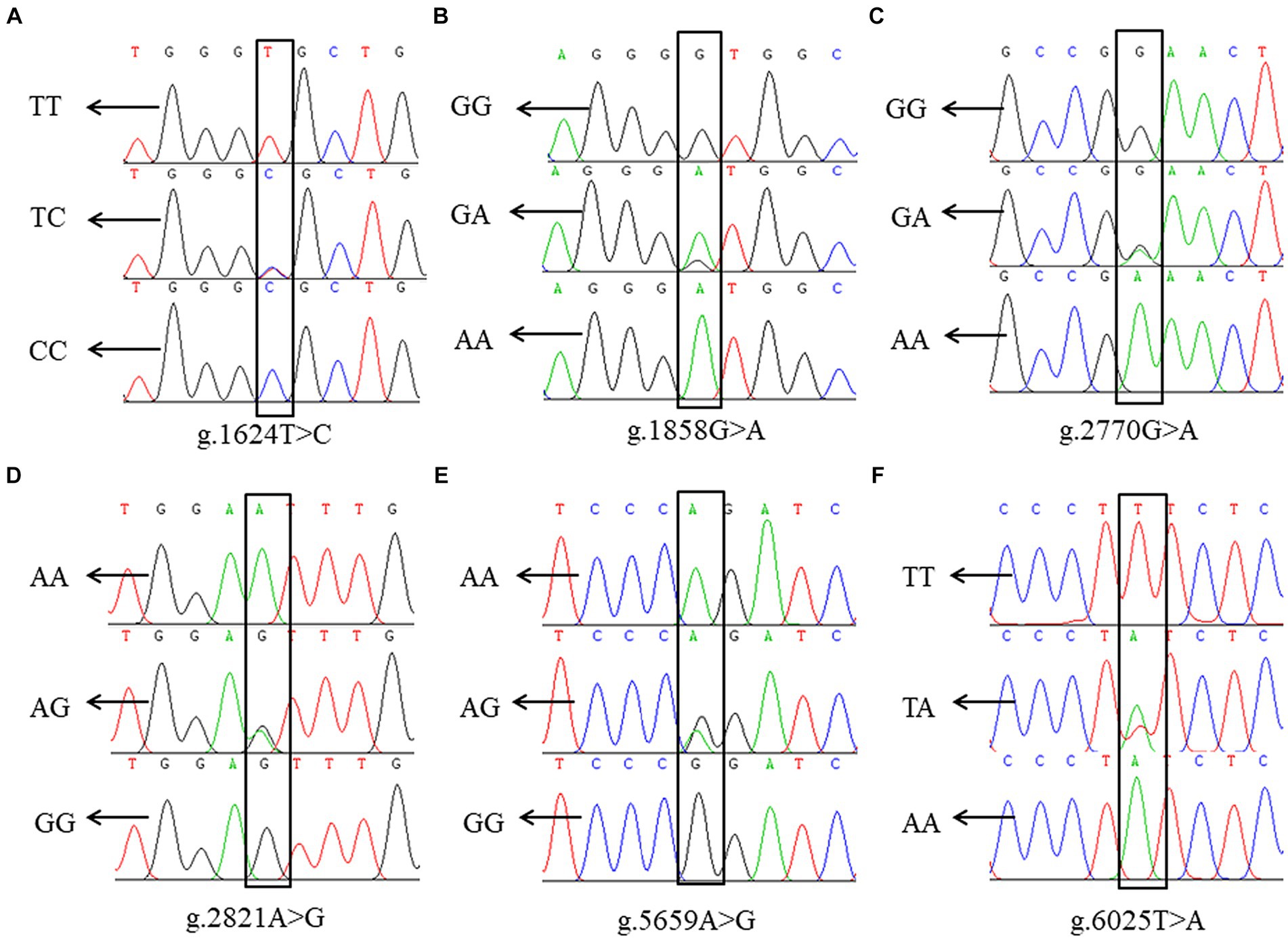
Figure 2. SNP locus of NOBOX in Xiangsu pig. (A) g.1624 T>C, (B) g.1858 G>A, (C) g.2770 G>A, (D) g.2821 A>G, (E) g.5659 A>G, (F) g.6025 T>A.
3.4 Biological evolution and protection
The phylogenetic tree of NOBOX sequences from the eight species is presented in Figure 3. Among the NOBOX species affinities, pig (S. scrofa) was most closely related to human (H. sapiens), followed by house mouse (M. musculus) and horse (E. caballus), and was furthest removed from the chicken (G. gallus). Fifteen significant amino acid sequences were detected in the eight species, indicating functional similarity at the super-secondary structure (Supplementary Figure S2). In addition, NOBOX was poorly conserved across species, with p.V82M as the nonconserved mutation (Figure 4).

Figure 3. Phylogenetic tree (left) and motif structural analysis (right) for eight species. Fifteen significant motifs were identified. The length of the color block shows the position, strength and significance of a particular motif site. The length of the color block shows the position, strength and significance of a particular motif site. The length of the motif is proportional to the negative logarithm of the p-value of the motif site, truncated at the height for a p-value of 1 × 10−10. The colors were generated through motif analysis performed via the MEME suit system.
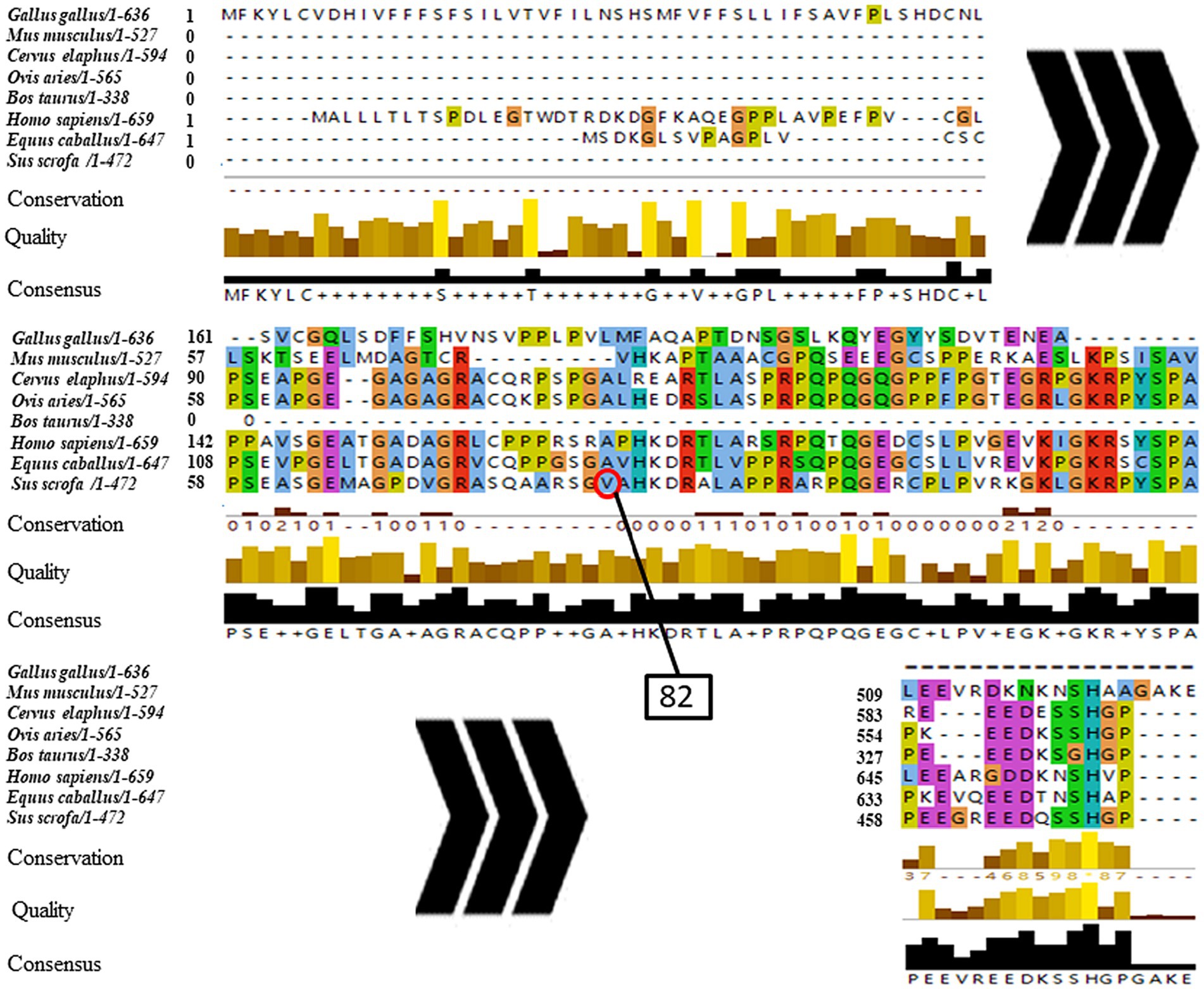
Figure 4. Conservative analysis of NOBOX SNP amino acid mutation sites. The red circle corresponds to p.V82M.
3.5 Missense SNPs affect protein structure and function prediction
The predicted effect of missense SNPs on protein function based on the online prediction tool PhD-SNP yielded a score of 8, indicating a neutral effect. However, the prediction tool SNAP2 yielded a score of 37, suggesting altered protein function. Additionally, the prediction of protein stability using I-Mutant 2.0 and MuPro revealed the free energy changes of −1.21 and −0.53, respectively, with the P.V82M mutation reducing protein stability. A comparison of the secondary structure prediction results between the wild type and mutant revealed that p.V82M mutation increased the α helix and β turn and decreased the random coil (Supplementary Tables S2–S4).
The 3D model of the NOBOX protein at p.V82M constructed using AlphaFold2 is presented in Figure 5. The p.V82M mutation replaced nonpolar, uncharged valine with a large, nonpolar, uncharged methionine, which altered the polar interactions with surrounding amino acids and affected the protein structure and function after the mutation.
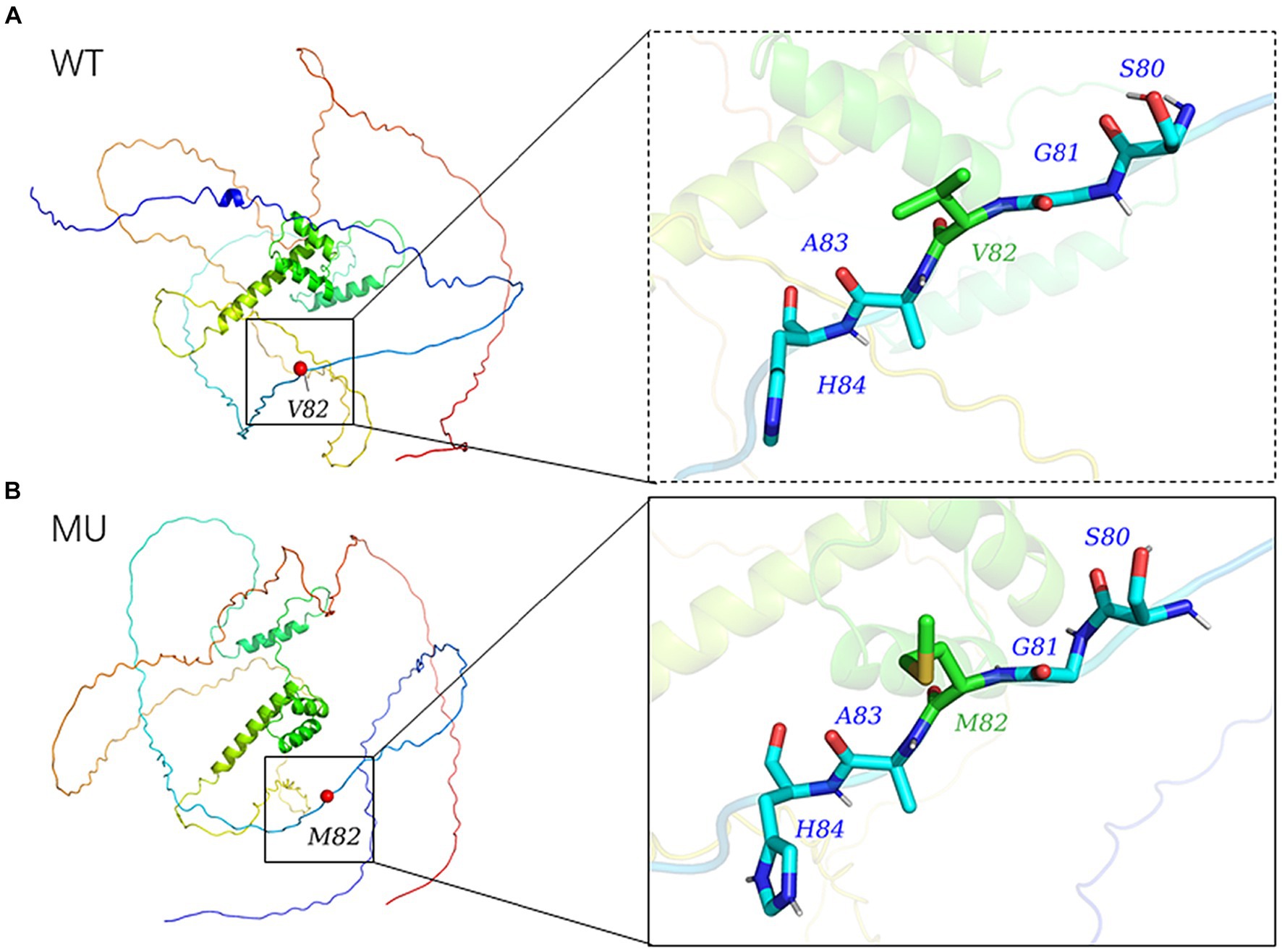
Figure 5. Modelled tertiary structure of the protein encoded by NOBOX. Different colors in the figure represent different secondary structures, (A) wild-type, (B) mutant type.
3.6 NOBOX polymorphism in Xiangsu pig
According to NOBOX population genetic analyses (Table 1), the purity of each SNP locus was greater than the heterozygosity and the effective allele number ranged from 0.12 to 0.37. Besides, the polypeptide of SNP loci g.1624 T>C, g.1858 G>A, g.2821 A>G, g.5659 A>G, and g.6025 T>A was at the moderate polymorphism level (0.25 < PIC < 0.50). These five loci were in the Shangsu hybridization annotation, with strong selection potential and rich genetic diversity. The χ2 test revealed that all five SNP loci were in HWE (p > 0.05). However, the polypeptide content at locus g.2770 G>A was 0.12, a lower polymorphism level (PIC <0.25), contrary to the HWE based on the χ2 test (p < 0.05).
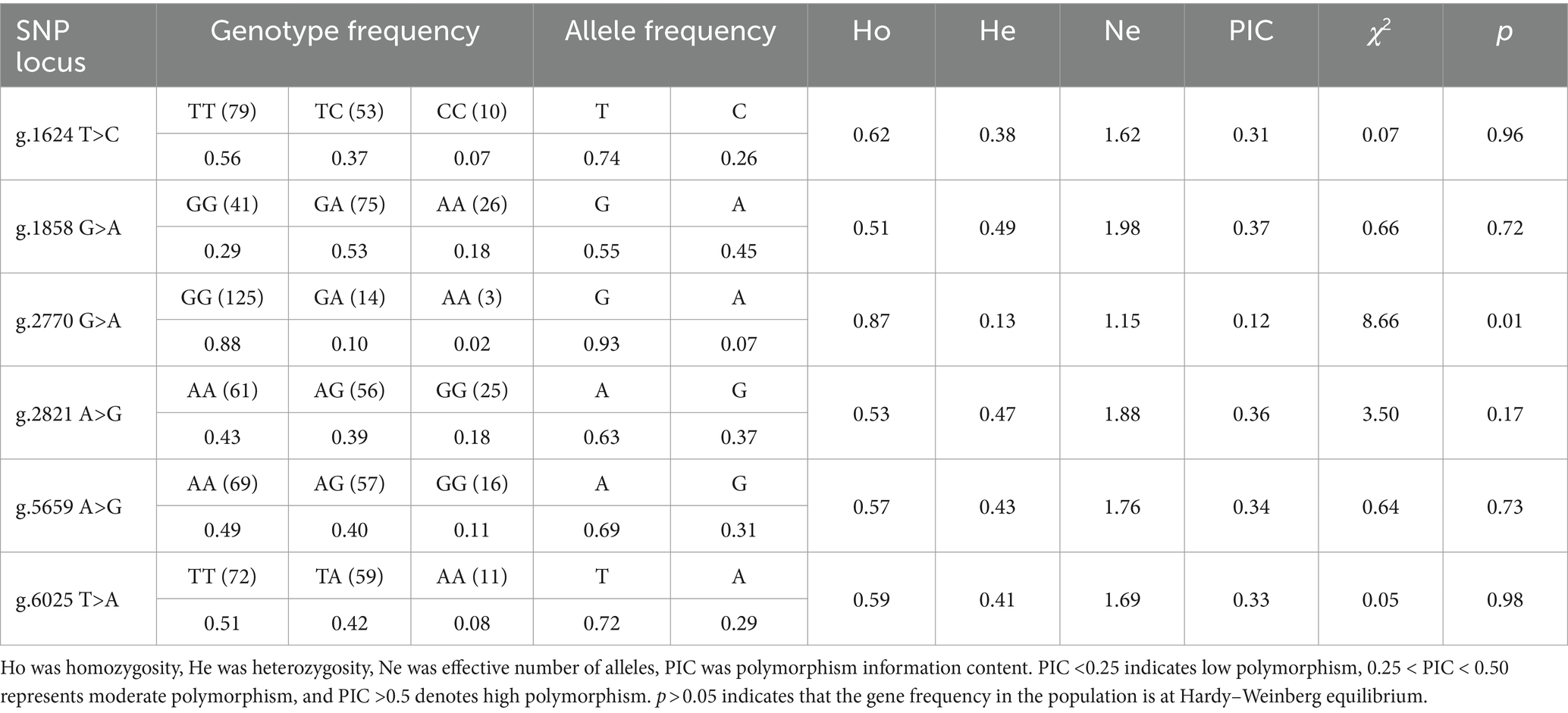
Table 1. Population genetic information of NOBOX SNPs.
3.7 NOBOX LD and haplotype analyses
The LD analysis of NOBOX SNPs using D′ and r2 tests is shown in Figure 6 (32). LD analysis revealed that the D′ values ranged from 0.08 to 1.00 and r2 values from 0 to 0.874. SNP loci between g.1624 T>C and g.5659 A>G, g.1624 T>C and g.6025 T>A, g.2821 A>G and g.5659 A>G, and g.5659 A>G, and g.6025 T>A, with the r2 of 0.44, 0.52, 0.37, and 0.87, respectively, were n strong chain imbalance, with the strongest degree of chain imbalance between g.5659 A>G and g.6025 T>A (Table 2).
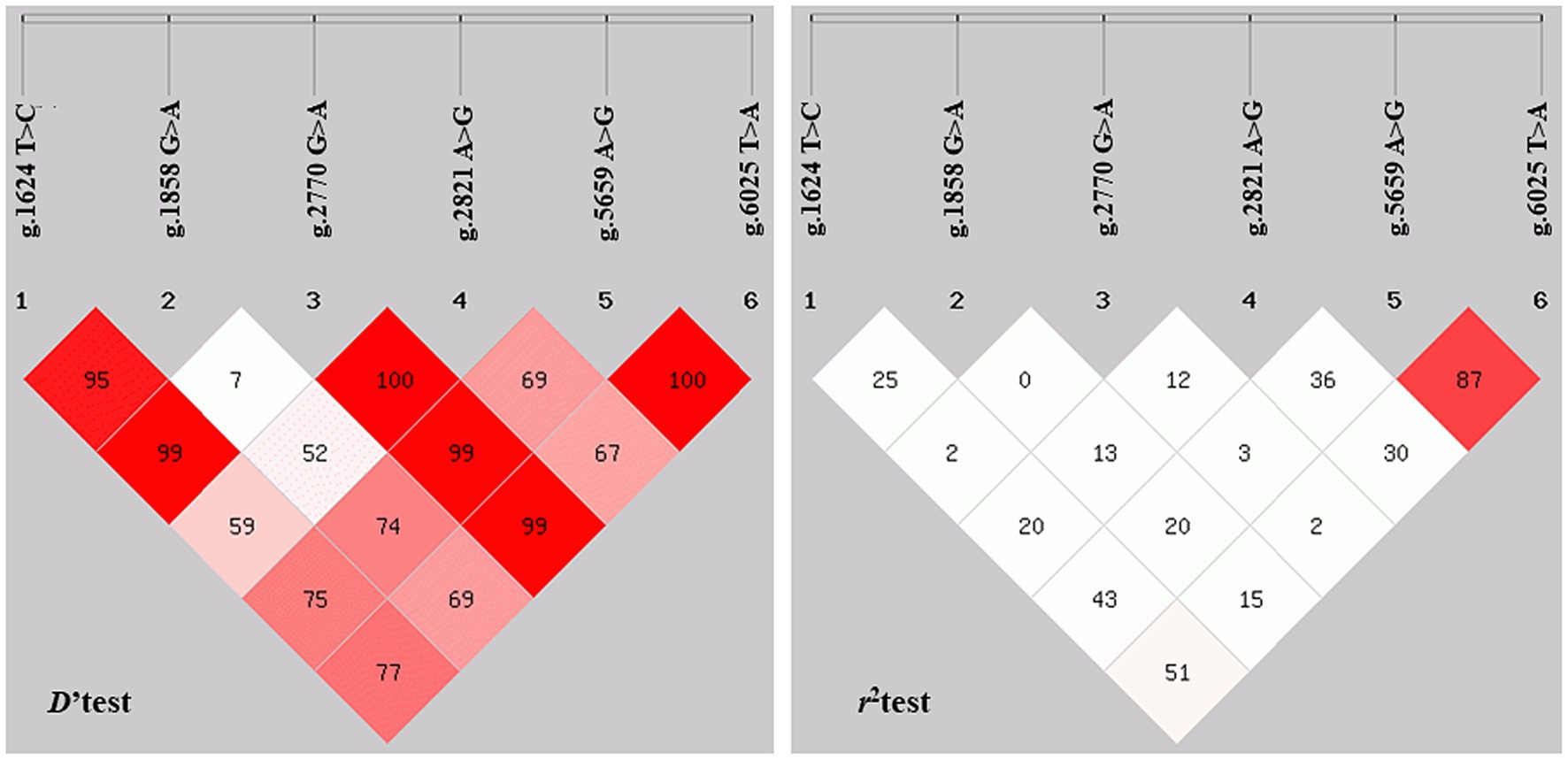
Figure 6. Analysis of linkage disequilibrium. r2 represents the correlation between a pair of loci, and D′ denotes the difference between the observed and the expected frequency of a given haplotype.
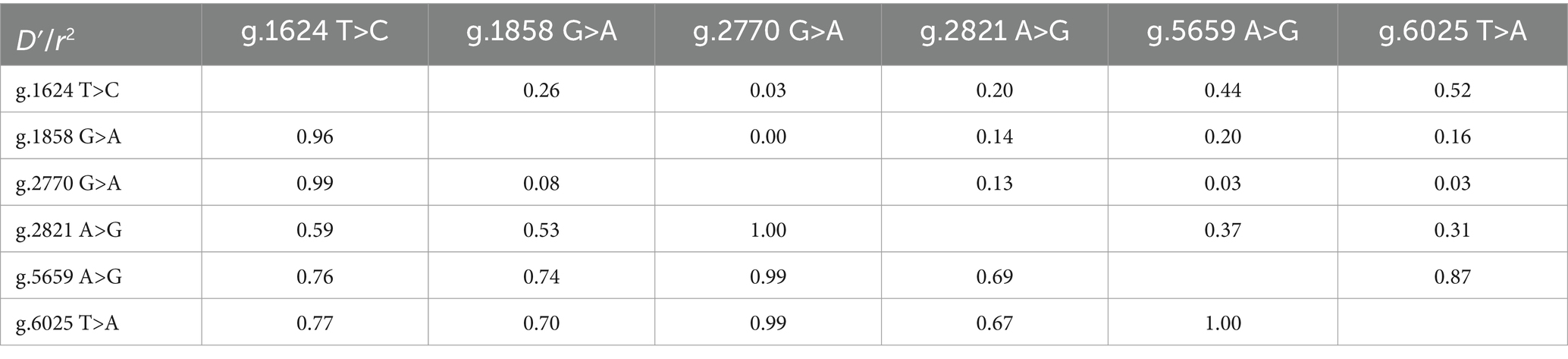
Table 2. Linkage disequilibrium coefficient between SNPs of NOBOX.
Haplotype analysis identified three dominant haplotypes with frequencies greater than 5% from the Xiangsu pig population. They included Hap1 (TAGAAT), Hap2 (TGGAAT), and Hap3 (CGGGGA), with the frequencies of 36.20, 18.30, and 18.10%, respectively (Table 3).
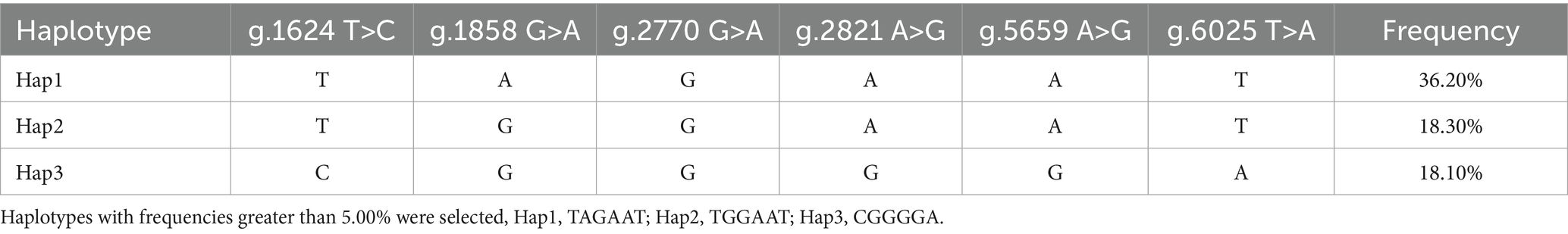
Table 3. Haplotype analysis and frequency of NOBOX SNPs.
3.8 Association analysis between NOBOX polymorphism and litter size traits in Xiangsu pigs
The correlation between the SNPs of pig NOBOX and litter size traits is shown in Table 4. The GG genotypes were significantly higher than AA genotypes at the g.1858 G>A locus and GG genotypes were significantly higher than AG genotypes at the g.5659 A>G locus in first-born TNB and NBA (p < 0.05). For g.6025 T>A, the TT and TA genotypes were significantly higher than AA genotype in the TNB and NBA of second-born sows (p < 0.05).
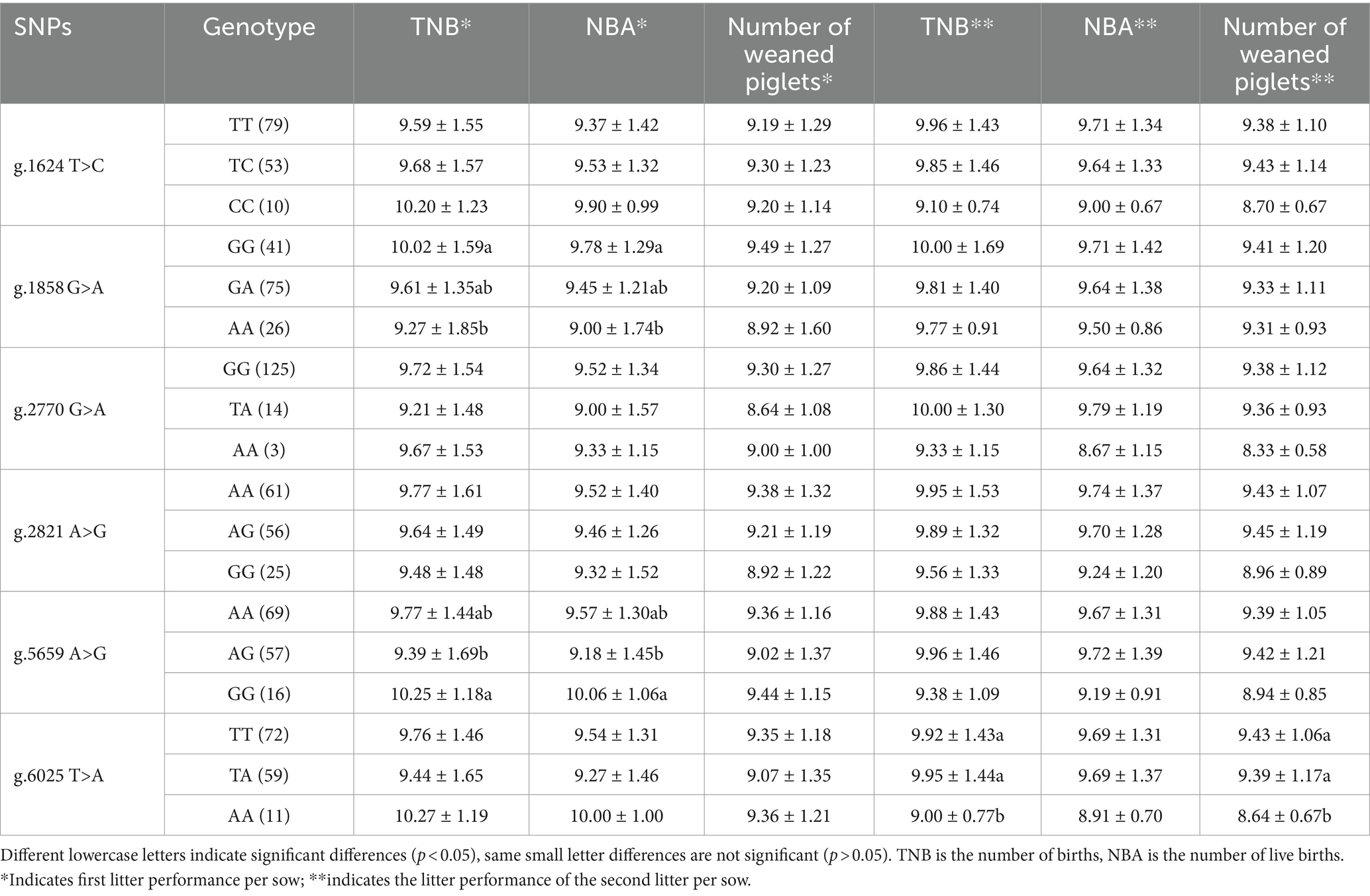
Table 4. Correlation analysis of NOBOX polymorphisms with litter size traits.
The association analysis between diplotype and litter size traits is summarized in Table 5. The TNB and number of weaned piglets in the first-born sows were significantly higher in diploid Hap1/1 (TTAAGGAAAATT) and Hap1/3 (TCGAGGAGAGTA) than in Hap1/2 (TTGAGGAAAATT) (p < 0.05). Diplotypes with <5.0% frequency were not considered.
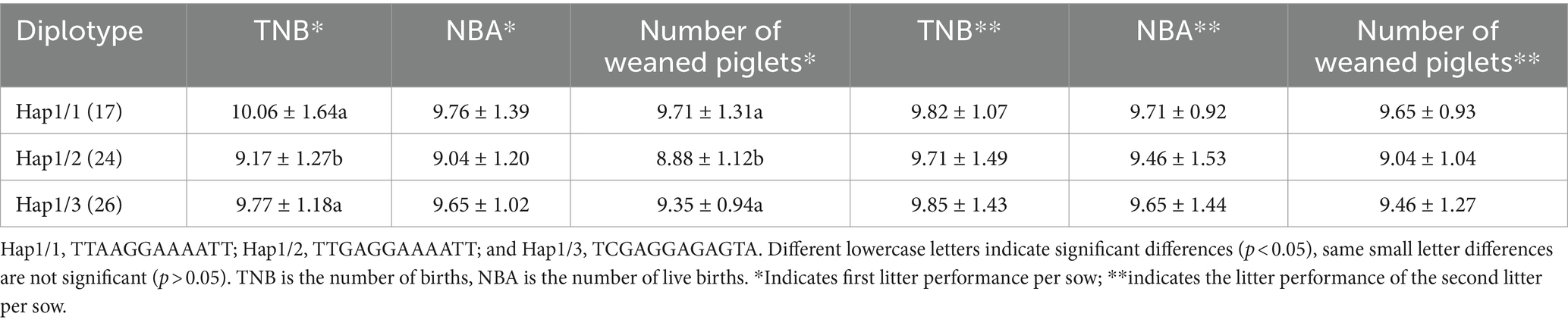
Table 5. Correlation analysis diplotype and litter size traits in Xiangsu pig.
4 Discussion
Guizhou is rich in genomically pure and small pig breeds, including the Congjiang Xiang pig, one of the most famous small pig breeds (33, 34). These small pig breeds have the disadvantages of small size and low farrowing rate, which seriously hinders the economic development of local pig farming (35). However, compared with the Congjiang Xiangsu pig, the Xiangsu pig has the advantages of delicious quality meat and strong disease resistance, with greatly improved body size and reproductive performance. Therefore, Xiangsu pig line breeding significantly promotes the overall pig breeding and economic development in Guizhou.
NOBOX is important for promoting ovarian differentiation and development, regulating early oogenesis to mature female follicles, and regulating germ cell development (36, 37). It is also a key factor in the development of various germ cells and the main regulator of key oocyte genes and is closely related to the number and quality of mature follicles produced by women (38, 39). Based on this, it is valuable to establish the relationship between NOBOX mutations and female reproductive performance. However, there are no relevant studies on this relationship. More importantly, it is of research significance to explore the correlation between the variation of SNPs in NOBOX and litter performance in Xiangsu pigs.
The differences in gene expression among different tissues are related to their corresponding functions. In this study, the qRT-PCR analysis in the different tissues of adult sows revealed different NOBOX expression levels in the heart, liver, kidney, and ovary tissues, with the highest expression in the ovary, consistent with previous reports (14, 40). In addition, the mutation at g.1858 G>A, a missense SNP, replaced valine (V) with methionine (M). Missense mutations can reduce protein stability and are associated with phenotype (41, 42). Herein, the missense mutation altered NOBOX protein structure and function, thereby greatly reducing the protein stability. In addition, the p.V82M mutation increased the α helix and β turn proportions but decreased the random coil, which altered the different protein components that regulate the protein function.
Predicting the NOBOX protein structure and function revealed that pigs and poultry had the farthest genetic relationship during species evolution. In addition, 15 significant amino acid sequences were found in eight species, implying that they had functional similarity at the super-secondary structure. However, NOBOX was less conserved in different species, with p.V82M, a non-conservative mutation. Further assessment of gene polymorphisms revealed that the mutant locus g.2770 G>A did not conform to the population HWE, contrary to the other five SNPs, which may be caused by long-term human intervention during breeding (43). An assessment of LD among the six loci revealed that the synonymous mutation g.5659A>G and the 3′-UTR mutation g.6025 T>A had a strong linkage relationship (D′ = 1.000, r2 = 0.874) with the strongest degree of interlocking, implying that the two SNPs may have a synergistic effect on sow litter size (44, 45).
The correlation between NOBOX polymorphisms and sow litter size revealed that the SNP loci g.1858 G>A, g.5659 A>G, and g.6025 T>A were significantly correlated with sow litter size traits. At the same time, the litter and weaned piglet sizes were significantly lower for diploid Hap1/2 than for Hap1/1 and Hap1/3. Intronic mutations mostly affect mRNA shearing and folding at the molecular level and do not directly affect phenotypic traits (46, 47). For example, two intronic NOBOX mutations in Chinese women with premature ovarian failure (POF) were not associated with the disease (48), consistent with the intronic mutations in the present study, where g.1624 T>C, g.2770 G>A, and g.2821 A>G were not significantly associated with sow litter size traits (49, 50). In the present study, the 3′-UTR mutation locus g.6025 T>A was significantly lower in the AA genotype, characterized by fewer litters and weaned piglets than in TT and TA genotypes in the second litter. Besides, there was a significant correlation between the number of live piglets produced in the first litter for the synonymous mutation loci g.5659 A>G. g.5659 A>G and g.6025 T>A that belong to a strong cascade, confirming that these two SNPs had a synergistic effect on the number of litters born. The mutation from GG to AA at the missense SNP locus g.1858 G>A was accompanied by a gradual decrease in litter size and number of live and weaned piglets born in the first and second litters, consistent with the NOBOX mutation that causes POF in females (12, 51). Therefore, NOBOX may be an important molecular co-marker gene associated with porcine reproductive performance, which is significant in future molecular breeding improvement efforts in pigs.
5 Conclusion
This study identified six new SNPs in the pig NOBOX, including g.1858 G>A, a missense SNP that alters the amino acid sequence structure. Additionally, g.1858 G>A, g.5659 A>G, and g.6025 T>A significantly correlated with the litter size traits. Hap1/1, a high yielding dominant diploid, had the highest and most stable litter size traits. At the same time, g.1858 G>A significantly reduced the protein stability and greatly affected protein function. The heterozygous and homozygous genotypes after g.1858 G>A mutation gradually decrease the litter performance; thus, NOBOX may be an important SNP molecular marker gene for improving the litter performance of sows.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Animal Welfare Committee of Guizhou University (EAE-GZU-2023-E015). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JH: Data curation, Methodology, Writing – original draft, Writing – review & editing. YR: Conceptualization, Supervision, Writing – review & editing. MX: Data curation, Methodology, Writing – review & editing. LD: Methodology, Software, Writing – review & editing. CJ: Methodology, Resources, Writing – review & editing. JL: Data curation, Resources, Writing – review & editing. JX: Investigation, Software, Writing – review & editing. XC: Methodology, Resources, Writing – review & editing. HX: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Guizhou Provincial Science and Technology Engineering Project [QKHFQ-2018,4007, (002)] and Guizhou Provincial Agricultural Major Industry Scientific Research Engineering Project (QKHKYZ-2019,011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1359312/full#supplementary-material
Footnotes
1. ^www.ncbi.nlm.nih.gov/protein
4. ^https://www.ebi.ac.uk/Tools/msa/clustalo/
5. ^https://consurf.tau.ac.il/consurf-old.php
6. ^https://snps.biofold.org/PhD-SNP/PhD-SNP.html
7. ^https://rostlab.org/services/snap2web/
8. ^http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi
9. ^http://mupro.proteomics.ics.uci.edu/
10. ^https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page1/4npsa%20_sopma.html
References
1. Koketsu, Y, and Iida, R. Sow housing associated with reproductive performance in breeding herds. Mol Reprod Dev. (2017) 84:979–86. doi: 10.1002/mrd.22825
2. Tomiyama, M, Kubo, S, Takagi, T, and Suzuki, K. Evaluation of genetic trends and determination of the optimal number of cumulative records of parity required in reproductive traits in a large white pig population. Anim Sci J. (2011) 82:621–6. doi: 10.1111/j.1740-0929.2011.00889.x
3. Meaburn, E, Butcher, LM, Schalkwyk, LC, and Plomin, R. Genotyping pooled DNA using 100k SNP microarrays: a step towards genomewide association scans. Nucleic Acids Res. (2006) 34:e27. doi: 10.1093/nar/gnj027
4. Zhu, H, Zhang, Y, Bai, Y, Yang, H, Yan, H, Liu, J, et al. Relationship between SNPs of POU1F1 gene and litter size and growth traits in Shaanbei white cashmere goats. Animals. (2019) 9:114. doi: 10.3390/ani9030114
5. Franca, MM, and Mendonca, BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract Res Clin Endocrinol Metab. (2022) 36:101594. doi: 10.1016/j.beem.2021.101594
6. Cloke, B, and Rymer, J. Premature ovarian insufficiency—the need for a genomic map. Climacteric. (2021) 24:444–52. doi: 10.1080/13697137.2021.1945025
7. Batiha, O, Alahmad, NA, Sindiani, A, Bodoor, K, Shaaban, S, and Al-Smadi, M. Genetics of female infertility: molecular study of newborn ovary homeobox gene in poor ovarian responders. J Hum Reprod Sci. (2019) 12:85–91. doi: 10.4103/jhrs.JHRS_112_18
8. Patton, BK, Madadi, S, Briley, SM, Ahmed, AA, and Pangas, SA. Sumoylation regulates functional properties of the oocyte transcription factors SOHLH1 and NOBOX. FASEB J. (2023) 37:e22747. doi: 10.1096/fj.202201481R
9. Yao, C, Lu, L, Ji, Y, Zhang, Y, Li, W, Shi, Y, et al. Hypo-hydroxymethylation of NOBOX is associated with ovarian dysfunction in rat offspring exposed to prenatal hypoxia. Reprod Sci. (2022) 29:1424–36. doi: 10.1007/s43032-022-00866-6
10. Sassi, A, Desir, J, Duerinckx, S, Soblet, J, Van Dooren, S, Bonduelle, M, et al. Compound heterozygous null mutations of NOBOX in sisters with delayed puberty and primary amenorrhea. Mol Genet Genomic Med. (2021) 9:e1776. doi: 10.1002/mgg3.1776
11. Li, L, Wang, B, Zhang, W, Chen, B, Luo, M, Wang, J, et al. A homozygous NOBOX truncating variant causes defective transcriptional activation and leads to primary ovarian insufficiency. Hum Reprod. (2017) 32:248–55. doi: 10.1093/humrep/dew271
12. Ferrari, I, Bouilly, J, Beau, I, Guizzardi, F, Ferlin, A, Pollazzon, M, et al. Impaired protein stability and nuclear localization of NOBOX variants associated with premature ovarian insufficiency. Hum Mol Genet. (2016) 25:5223–33. doi: 10.1093/hmg/ddw342
13. Lechowska, A, Bilinski, S, Choi, Y, Shin, Y, Kloc, M, and Rajkovic, A. Premature ovarian failure in NOBOX-deficient mice is caused by defects in somatic cell invasion and germ cell cyst breakdown. J Assist Reprod Genet. (2011) 28:583–9. doi: 10.1007/s10815-011-9553-5
14. Tripurani, SK, Lee, KB, Wang, L, Wee, G, Smith, GW, Lee, YS, et al. A novel functional role for the oocyte-specific transcription factor newborn ovary homeobox (NOBOX) during early embryonic development in cattle. Endocrinology. (2011) 152:1013–23. doi: 10.1210/en.2010-1134
15. Qin, M, Xie, Q, Wu, K, Zhou, X, and Ge, W. Loss of NOBOX prevents ovarian differentiation from juvenile ovaries in zebrafish. Biol Reprod. (2022) 106:1254–66. doi: 10.1093/biolre/ioac036
16. Xu, J, Ruan, Y, Sun, J, Shi, P, Huang, J, Dai, L, et al. Association analysis of PRKAA2 and MSMB polymorphisms and growth traits of Xiangsu hybrid pigs. Genes. (2023) 14:113. doi: 10.3390/genes14010113
17. Kumar, S, Stecher, G, Li, M, Knyaz, C, and Tamura, K. Mega X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
18. Bailey, TL, Boden, M, Buske, FA, Frith, M, Grant, CE, Clementi, L, et al. Meme suite: tools for motif discovery and searching. Nucleic Acids Res. (2009) 37:W202–8. doi: 10.1093/nar/gkp335
19. Sievers, F, Wilm, A, Dineen, D, Gibson, TJ, Karplus, K, Li, W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. (2011) 7:539. doi: 10.1038/msb.2011.75
20. Capriotti, E, Calabrese, R, and Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. (2006) 22:2729–34. doi: 10.1093/bioinformatics/btl423
21. Hecht, M, Bromberg, Y, and Rost, B. Better prediction of functional effects for sequence variants. BMC Genomics. (2015) 16:S1. doi: 10.1186/1471-2164-16-S8-S1
22. Capriotti, E, Fariselli, P, and Casadio, R. I-mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. (2005) 33:W306–10. doi: 10.1093/nar/gki375
23. Sahoo, NR, Kumar, P, Khan, MF, Mourya, R, Ravikumar, G, and Tiwari, AK. Sequence diversity of major histo-compatibility complex class II DQA1 in Indian Tharparkar cattle: novel alleles and in-silico analysis. HLA. (2019) 93:451–61. doi: 10.1111/tan.13521
24. Geourjon, C, and Deleage, G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. (1995) 11:681–4. doi: 10.1093/bioinformatics/11.6.681
25. Jumper, J, Evans, R, Pritzel, A, Green, T, Figurnov, M, Ronneberger, O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
26. Jin, JQ, and Sun, YB. AutoSeqMan: batch assembly of contigs for sanger sequences. Zool Res. (2018) 39:123–6. doi: 10.24272/j.issn.2095-8137.2018.027
27. Namipashaki, A, Razaghi-Moghadam, Z, and Ansari-Pour, N. The essentiality of reporting Hardy–Weinberg equilibrium calculations inpopulation-based genetic association studies. Cell J. (2015) 17:187–92. doi: 10.22074/cellj.2016.3711
28. Shi, YY, and He, L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. (2005) 15:97–8. doi: 10.1038/sj.cr.7290272
29. Ardlie, KG, Kruglyak, L, and Seielstad, M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. (2002) 3:299–309. doi: 10.1038/nrg777
30. Mishra, P, Singh, U, Pandey, CM, Mishra, P, and Pandey, G. Application of student’s t-test, analysis of variance, and covariance. Ann Card Anaesth. (2019) 22:407–11. doi: 10.4103/aca.ACA_94_19
31. Livak, KJ, and Schmittgen, TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
32. Barrett, JC, Fry, B, Maller, J, and Daly, MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. (2005) 21:263–5. doi: 10.1093/bioinformatics/bth457
33. Liu, C, Ran, X, Wang, J, Li, S, and Liu, J. Detection of genomic structural variations in Guizhou indigenous pigs and the comparison with other breeds. PLoS One. (2018) 13:e194282. doi: 10.1371/journal.pone.0194282
34. Xie, J, Li, R, Li, S, Ran, X, Wang, J, Jiang, J, et al. Identification of copy number variations in Xiang and Kele pigs. PLoS One. (2016) 11:e148565. doi: 10.1371/journal.pone.0148565
35. Noor, AU, Du, Z, Song, C, Lu, H, Zhou, X, Liu, X, et al. Gene cloning, tissue expression profiles and antiviral activities of interferon-beta from two Chinese miniature pig breeds. Vet Sci. (2022):190. doi: 10.3390/vetsci9040190
36. Belli, M, Cimadomo, D, Merico, V, Redi, CA, Garagna, S, and Zuccotti, M. The NOBOX protein becomes undetectable in developmentally competent antral and ovulated oocytes. Int J Dev Biol. (2013) 57:35–9. doi: 10.1387/ijdb.120125mz
37. Choudhury, A, and Khole, VV. HSP90 antibodies: a detrimental factor responsible for ovarian dysfunction. Am J Reprod Immunol. (2013) 70:372–85. doi: 10.1111/aji.12136
38. Choi, Y, and Rajkovic, A. Characterization of NOBOX DNA binding specificity and its regulation of Gdf9 and Pou5f1 promoters. J Biol Chem. (2006) 281:35747–56. doi: 10.1074/jbc.M604008200
39. Albertini, DF. NOBOX does right for the follicle reserve: insights into premature ovarian failure. J Assist Reprod Genet. (2011) 28:567–8. doi: 10.1007/s10815-011-9615-8
40. Suzumori, N, Yan, C, Matzuk, MM, and Rajkovic, A. NOBOX is a homeobox-encoding gene preferentially expressed in primordial and growing oocytes. Mech Dev. (2002) 111:137–41. doi: 10.1016/S0925-4773(01)00620-7
41. de Boer, E, Ockeloen, CW, Kampen, RA, Hampstead, JE, Dingemans, AJM, Rots, D, et al. Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein. Genet Med. (2022) 24:2051–64. doi: 10.1016/j.gim.2022.06.007
42. Stojiljkovic, M, Pérez, B, Desviat, LR, Aguado, C, Ugarte, M, and Pavlovic, S. The missense p.S231F phenylalanine hydroxylase gene mutation causes complete loss of enzymatic activity in vitro. Protein J. (2009) 28:294–9. doi: 10.1007/s10930-009-9194-z
43. Stark, AE. Stable populations and Hardy–Weinberg equilibrium. Hereditas. (2023) 160:19. doi: 10.1186/s41065-023-00284-x
44. Charita, B, Padma, G, Sushma, P, Deepak, P, and Padma, T. Estimation of risk and interaction of single nucleotide polymorphisms at angiotensinogen locus causing susceptibility to essential hypertension: a case control study. J Renin-Angiotensin-Aldosterone Syst. (2012) 13:461–71. doi: 10.1177/1470320312444650
45. Drabbels, J, Welleweerd, R, van Rooy, I, Johnsen, GM, Staff, AC, Haasnoot, GW, et al. HLA-G whole gene amplification reveals linkage disequilibrium between the HLA-G 3′UTR and coding sequence. HLA. (2020) 96:179–85. doi: 10.1111/tan.13909
46. Pohjanvirta, R, Wong, JM, Li, W, Harper, PA, Tuomisto, J, and Okey, AB. Point mutation in intron sequence causes altered carboxyl-terminal structure in the aryl hydrocarbon receptor of the most 2,3,7,8-tetrachlorodibenzo-p-dioxin-resistant rat strain. Mol Pharmacol. (1998) 54:86–93. doi: 10.1124/mol.54.1.86
47. Olsson, A, Lind, L, Thornell, LE, and Holmberg, M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet. (2008) 17:1666–72. doi: 10.1093/hmg/ddn057
48. Qin, Y, Shi, Y, Zhao, Y, Carson, SA, Simpson, JL, and Chen, ZJ. Mutation analysis of NOBOX homeodomain in Chinese women with premature ovarian failure. Fertil Steril. (2009) 91:1507–9. doi: 10.1016/j.fertnstert.2008.08.020
49. Yin, H, Du, X, Li, Q, Pan, Z, Wu, W, Liu, H, et al. Variants in BMP7 and BMP15 3′-UTRs associated with reproductive traits in a large white pig population. Animals. (2019) 9:905. doi: 10.3390/ani9110905
50. Zhang, Z, He, X, Liu, Q, Tang, J, Di, R, and Chu, M. TGIF1 and SF1 polymorphisms are associated with litter size in Small Tail Han sheep. Reprod Domest Anim. (2020) 55:1145–53. doi: 10.1111/rda.13753
Keywords: NOBOX gene, pig, polymorphism, litter size traits, missense mutation
Citation: Huang J, Ruan Y, Xiao M, Dai L, Jiang C, Li J, Xu J, Chen X and Xu H (2024) Association between polymorphisms in NOBOX and litter size traits in Xiangsu pigs. Front. Vet. Sci. 11:1359312. doi: 10.3389/fvets.2024.1359312
Edited by:
Kirsten E. Scoggin, University of Kentucky, United StatesReviewed by:
Alex Silva Da Cruz, Pontifícia Universidade Católica de Goiás, BrazilAshutosh Das, Chattogram Veterinary and Animal Sciences University, Bangladesh
Copyright © 2024 Huang, Ruan, Xiao, Dai, Jiang, Li, Xu, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Houqiang Xu, Z3pkeHhocUAxNjMuY29t