 Tom Jukier
Tom Jukier Amanda Gross
Amanda Gross Dawn Boothe3
Dawn Boothe3- 1Department of Clinical Sciences, College of Veterinary Medicine, Auburn University, Auburn, AL, United States
- 2Scott Ritchey Research Center, College of Veterinary Medicine, Auburn University, Auburn, AL, United States
- 3Department of Anatomy, Physiology, and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL, United States
Introduction: Phenobarbital has been used for many decades in both human and veterinary epileptic patients. Many formulations for a particular drug exist, most of which are marketed for humans. Recently a veterinary specific phenobarbital product has been introduced to the market in the United States. Utilizing a specific formulation to treat patients may help decrease the issue of bioequivalence between one pharmaceutical product to another. Therefore, the goal of this study was to determine single and multiple dosing pharmacokinetics and tolerability of a veterinary specific phenobarbital product over a 4-week time period.
Materials and methods: 8 Healthy dogs from a canine research colony were used in the study.
Results: Overall, this phenobarbital formulation was well tolerated in the dogs in this study. Cmax, Tmax, half-life, and AUC after single 12 mg/kg oral dose were 23.5 μg/mL, 4.2 h, 94 h, and 2,758 h*μg/mL. Following chronic dosing, these parameters were 29.1 μg/mL, 3.4 h, 70 h, and 2,971 h*μg/mL, respectively.
Discussion: This formulation demonstrated a mean absolute bioavailability of 100%, with similar pharmacokinetic properties to previously published data.
1 Introduction
The use of barbiturates in the dog dates to the early twentieth century, when the first barbiturate drug, diethyl barbituric acid was used to induce sleep in dogs (1). This barbiturate’s effect led to its marketing under the trade name Veronal®. Subsequent derivations of this compound lead to the development of phenobarbital in 1911, which was marketed as Luminal®. Although it’s intended use was to induce sleep in human patients, phenobarbital’s antiseizure properties were first noted and published in 1912 by serendipitous discovery in a ward of patients with epilepsy (2). By the end of the decade, the first report recommending its use in people with epilepsy was published in 1919 (3). Since then, phenobarbital has been used to help treat epileptic patients in both humans, dogs, cats, and other species.
Phenobarbital has the longest historical use to treat epileptic seizures in dogs (4), and its popularity is still evident based on a survey of neurologists and emergency clinicians in the United States (5). From an efficacy perspective, two consensus reports (4, 6), and a systematic analysis (7) demonstrate evidence for efficacy of phenobarbital, as an antiseizure medication (ASM). Additionally, of the antiseizure medications used in veterinary medicine, only phenobarbital and bromide have established reference intervals to help guide clinicians in making therapeutic decisions (8, 9).
Although there are several previous studies describing the oral pharmacokinetics of phenobarbital in dogs (10–13), none involved approved products (14). Save for a single IV preparation (15), in the United States, no phenobarbital is approved in either humans or dogs because it was among the drugs already on the market when the 1938 Food and Drug Act was enacted, and thus exempt from a new drug status under a grandfather clause (Food and Drug Administration, Compliance Guideline Sec 440 Marketed New Drugs Without Approved NDAs and ANDAs). DailyMed, which in contrast to the Orange Book includes unapproved drugs, lists over 20 phenobarbital products that are unapproved. Any one of these products might be used to treat epileptic seizures in dogs. There is a need for an approved canine product, which would be accompanied by relevant pharmacokinetic information supporting a dosing regimen for the targeted indication.
The purposes of this study were: (1) Describe the pharmacokinetics and oral bioavailability of a new veterinary specific phenobarbital formulation. (2) Describe the pharmacokinetics and oral bioavailability after 4 weeks of once daily therapy at dose designed to maintain therapeutic concentrations; and (3) Assess tolerability after a 4 week dosing period.
2 Materials and methods
This study was implemented as a randomized cross-over study using healthy dogs (7 male, 1 female) from a canine colony at the Scott Ritchie Research Center at Auburn University. Health status was determined based on physical examination, neurological examination, complete blood count, and serum biochemistry. The study was conducted in three phases; all dogs were studied in each phase. Phase I consisted of a crossover single 12 mg/kg (or the nearest tablet size for oral administration) dose of phenobarbital administered either orally (NOBATOL® Mizner Bioscience LLC, Boca Raton, FL) to 4 dogs, or intravenously (Hikma Pharmacceuticals PLC, London, United Kingdom, and Cameron Pharmaceuticals, Lousiville, Kentucky) to four dogs. The route of administration was crossed over after a 3-week wash-out period from the last sample collection. A 12 mg/kg dose was calculated to achieve plasma drug concentration (PDC) of 20 μg/mL (4, 8, 16), a concentration considered in the mid therapeutic reference interval, based on the equation Dose = PDC * volume of distribution (16). Phase II commenced immediately after collection of the last sample from phase I. During this time all dogs were treated with 6 mg/kg (to the nearest half tablet size) PO q24h for 4 weeks with dosing occurring in the morning. Complete blood counts and serum biochemistry were obtained for each dog immediately prior to starting this phase, 2 weeks into treatment, and at the end of phase II (week 4). Blood samples for phenobarbital analysis were also collected at 2 (considered peak) and 24 h (considered trough) at the end of weeks, 1, 2, 3, and 4. After the last dose of Phase II, a 7-day wash-out period was allowed during which a blood sample was collected once a day at 24-h intervals. Lastly, phase III commenced on day 7 of the washout period following phase II. Phase III was conducted in a similar manner to phase I at the same mg/kg dose, with the exception that only 7 days elapsed between routes of administration. The funder of this project had no influence on study design.
3 Sample collection and handling
One day before blood collection for each phase, jugular catheters (MILA International INC., Florence, Kentucky, United States) were placed in each dog to facilitate blood collection. Dogs were sedated for jugular placement using 375 μg/m2 dexmedetomidine (Dexdomitor® 0.5 mg/mL, Orion Corporation, Espoo, Finland), and if additional sedation was required, 5-10 mg/kg of ketamine (Ketamine hydrochloride injection 100 mg/mL, Covetrus North America, Dublin OH). Dogs were fasted for approximately 14 h prior to phenobarbital administration and remained fasted for 12 h after drug administration. For phase I 3 mL of blood was collected at each time point and placed into a red top tube. For PO administration, samples were collected (hours) at: 0 (prior to drug administration) 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 72, 96, 120, 144, and 168. For IV administration, timing was the same with the addition of 0.083 and 0.156 h after dose administration. For phase III, blood samples were at the same time, but only to hour 144. For Phase II, as drug concentrations declined after multiple dosing, during the 7-day washout period, samples were collected at 24, 48, 72, 96, 120, 144, and 168 h. Table 1 shows timeline of the study design. Upon collection, all samples were centrifuged within 2 h at 2500 g for 10 min. Serum was harvested, stored in CRYO.S tubes (greiner BIO ONE, Germany) tubes at −80°C until analysis.

Table 1. Representation of dose administered and time points where blood samples were collected.
Serum phenobarbital was quantitated using a Siemens (Malvern, PA) Dimension® PHNO homogenous particle enhanced turbidimetric inhibition immunoassay (PETINIA) (10444933), carried out on a Siemens Dimension® Xpand® Plus integrated clinical chemistry system. This assay is approved by the US Food and Drug Administration for use in human medicine. The assay is calibrated using the Siemens Dimension® DRUG CAL (10445014) serum calibrator set with concentrations ranging from 0-80 μg/mL. The lower limit of quantitation is 1 mcg/ml and the CV is 4.42% at 14 μg/mL in pooled serum and 4.72% at 28.4 μg/mL in pooled plasma (n = 40).
4 Pharmacokinetic and statistical analysis
Serum phenobarbital concentrations versus time curves were analyzed by non-compartmental analysis using pharmacokinetic software (Phoenix WinNonLin®, CERTARA, United States). Non-compartmental analysis was performed using the linear-log up down trapezoidal option for determination of the area under the time-concentration curve (AUC) versus time (Phoenix WinNonlin®). From this, the following were determined: mean residence time (MRT), disappearance rate constant (kel), terminal half-life (t1/2, determined from the relationship t1/2 = 0.693/kel), area under the curve to infinity (AUC∞), percent of the AUC that was extrapolated from the terminal component of the curve (AUC ext), and, in the absence of IV administration (the decline of phenbobarbital concentrations during Phase II), the apparent volume of distribution (Vz) and clearance (Cl). Maximum plasma drug concentration (Cmax), and time to maximum concentration (Tmax) were observed. Data was reported both as a mean and standard deviation (SD) and as median and range (minimum and maximum values) with the exception of half-life, which was reported as harmonic mean ± pseudostandard deviation. Absolute bioavailability was calculated based on (AUCoral*DOSEIV/AUCIV*DOSEoral) × 100.
Kolmogorov–Smirnov test was performed to assess normality of the data. Comparison of oral Tmax, Cmax, absolute bioavailability, terminal half-life, were compared using paired student t-test or Mann–Whitney test, while blood work parameters were compared using One-Way Analysis of Variance (ANOVA) or Friedman’s test was used to assess parameters from all groups, followed by multiple comparison (Tukey’s for parametric data, Dunn’s test for nonparametric data). Bonferroni correction was applied when assessing for statistical significance within blood work parameters. Statistical significance was set at an α <0.05. Statistical analysis was performed using GraphPad Prism 9.5.1 (Dotmatics, San Diego, CA).
5 Results
Mean ± SD of dog weights included in the study was 13.4 ± 3.2, and mean age was 6.0 ± 2.9 years. Table 2 shows demographic information about each dog in this study. Mean phenobarbital dose was as follows: Phase I PO 11.9 ± 0.1, phase I IV 12.1 ± 0.4, phase II 6.2 ± 0.4 q24h, phase III PO 12.2 ± 0.5, and phase III IV 12.3 ± 0.4 mg/kg.
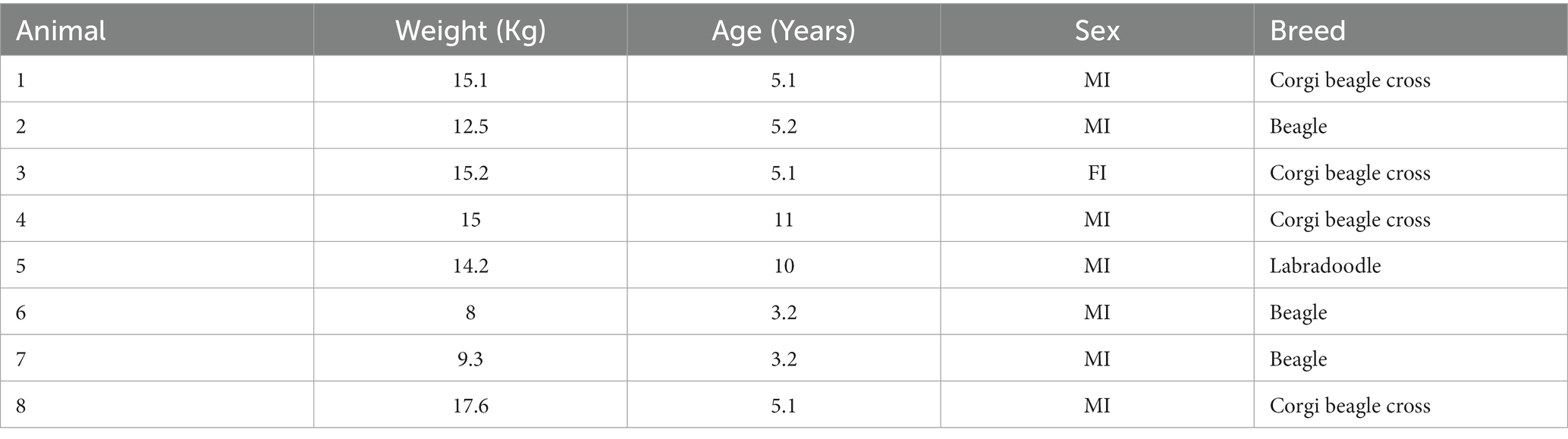
Table 2. Demographics of dogs in the study.
5.1 Phase I vs. phase III – single dose pharmacokinetics across time
Descriptive statistics for pharmacokinetic parameters and oral bioavailability are presented in Table 3. Administration of the calculated loading dose yielded a Cmax of 23.5 ± 3.2 μg/mL at phase I which was lower than that achieved after phase III, 29.1 ± 4.1 μg/mL (p = 0.0002). The Tmax (hr) of phase I was 4.2 ± 2.7 compared to 3.4 ± 1.9 for Phase III (p = 0.45). For IV administration, the C0 after Phase I was 17.3 ± 2.7 μg/mL compared to 20.6 ± 1.9 μg/mL for phase III (p = 0.02). Clearance for IV administration of phase I (6.2 ± 1.5 mL/kg/h) and phase III (6.3 ± 1.3 mL/kg/h) was not statistically different (p = 0.81). Mean absolute bioavailability (F) was 1.0 ± 0.1 for Phase I and 1.1 ± 0.1 for phase III. Figure 1 shows mean ± SD phenobarbital concentration vs. time for all dogs.
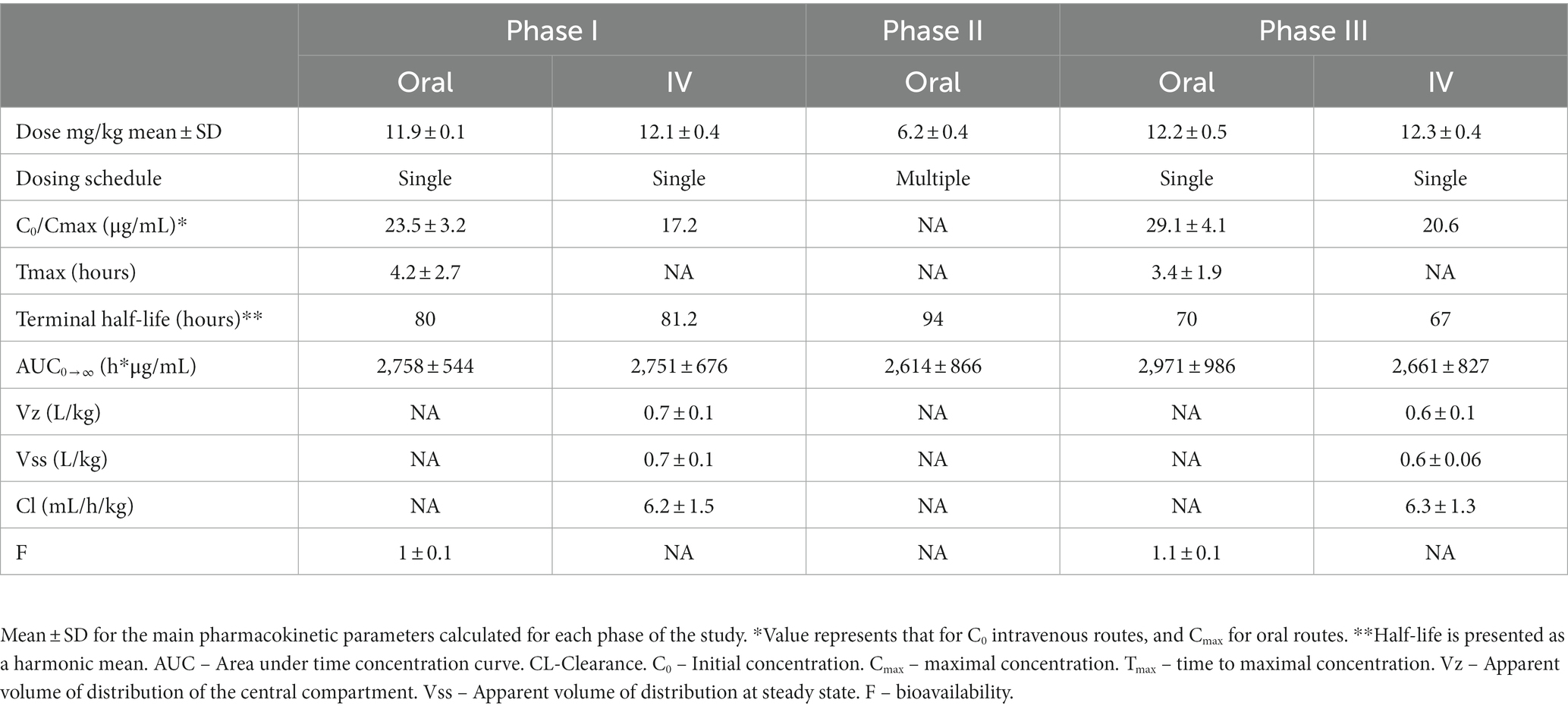
Table 3. Pharmacokinetic parameters for single, multiple, and single-post multiple dosing, of the veterinary specific phenobarbital product used in this study (Table 1).
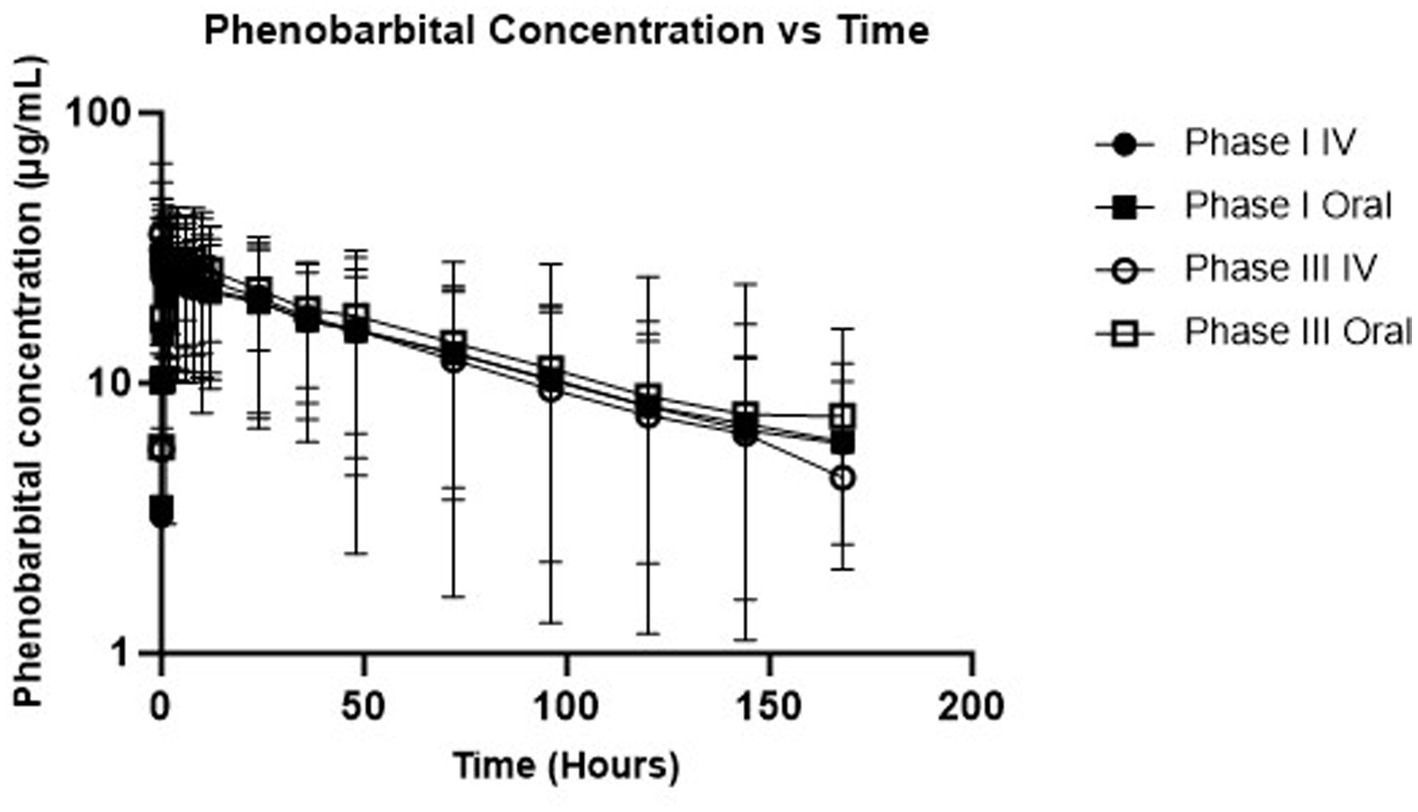
Figure 1. Semi-logarithmic depiction of phenobarbital concentration (with standard deviation bars) vs. time for single dose administration (phases I and III).
5.2 Phase II – multiple dosing
Specific pharmacokinetic parameters for this phase of the study are presented in Table 3. Weekly mean ± SD phenobarbital concentrations and percentage fluctuation during the 24-h dosing interval are presented in Table 4. Approximately 25% fluctuation in plasma drug concentrations occurred over the 24-h dosing interval.
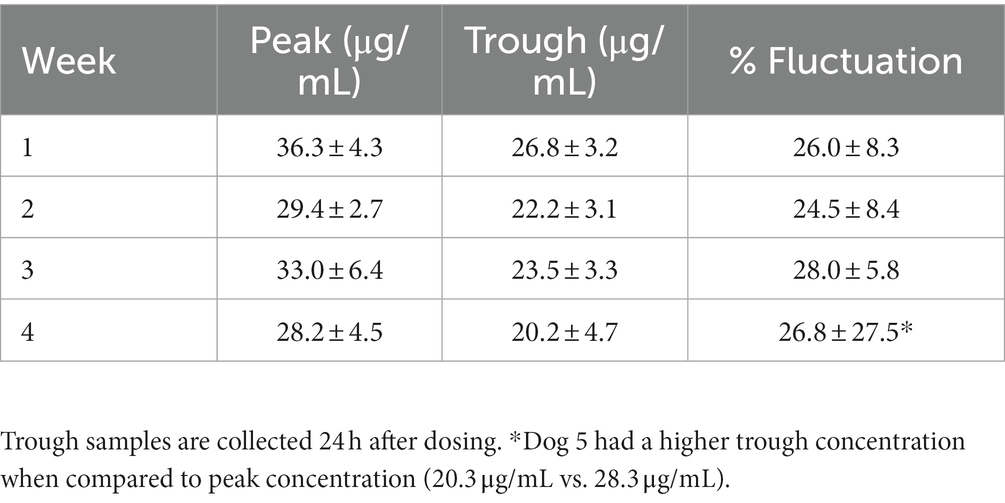
Table 4. Mean ± SD peak and trough concentrations for dogs during the Phase II daily administration of oral phenobarbital.
5.3 Serum biochemical analysis and complete blood counts
During the study period, dogs there were seldom notes about singular episodes of diarrhea. More frequently, slower or no interest in food in the mornings was noted. However, comparison of weight prior to the onset of the study and at the end did not reveal as statistically significant difference. Descriptive statistics including statistical comparisons are presented in Table 5 (serum biochemistry) and Table 6 (completed blood count). Both ALP and ALT remained within the reference interval during the 4-week daily administration. However, a statistically significant difference (p = 0.0014) was found between medians between the pre-ALP (36 IU, range 17–136) and median post ALP (81.5 IU/L, range 36–258). A statistically significant difference could not be found in when comparing medians for ALT (p = 0.07). All means at each time point were all within laboratory reference intervals with the exception for glucose at week 4. Mean glucose concentrations for the 8 dogs was below reference values. Hypoglycemia was ruled out using an in-house glucose test that confirmed euglycemia. For each dog with a hypoglycemic result, an in-house glucose test was performed to ensure euglycemia.
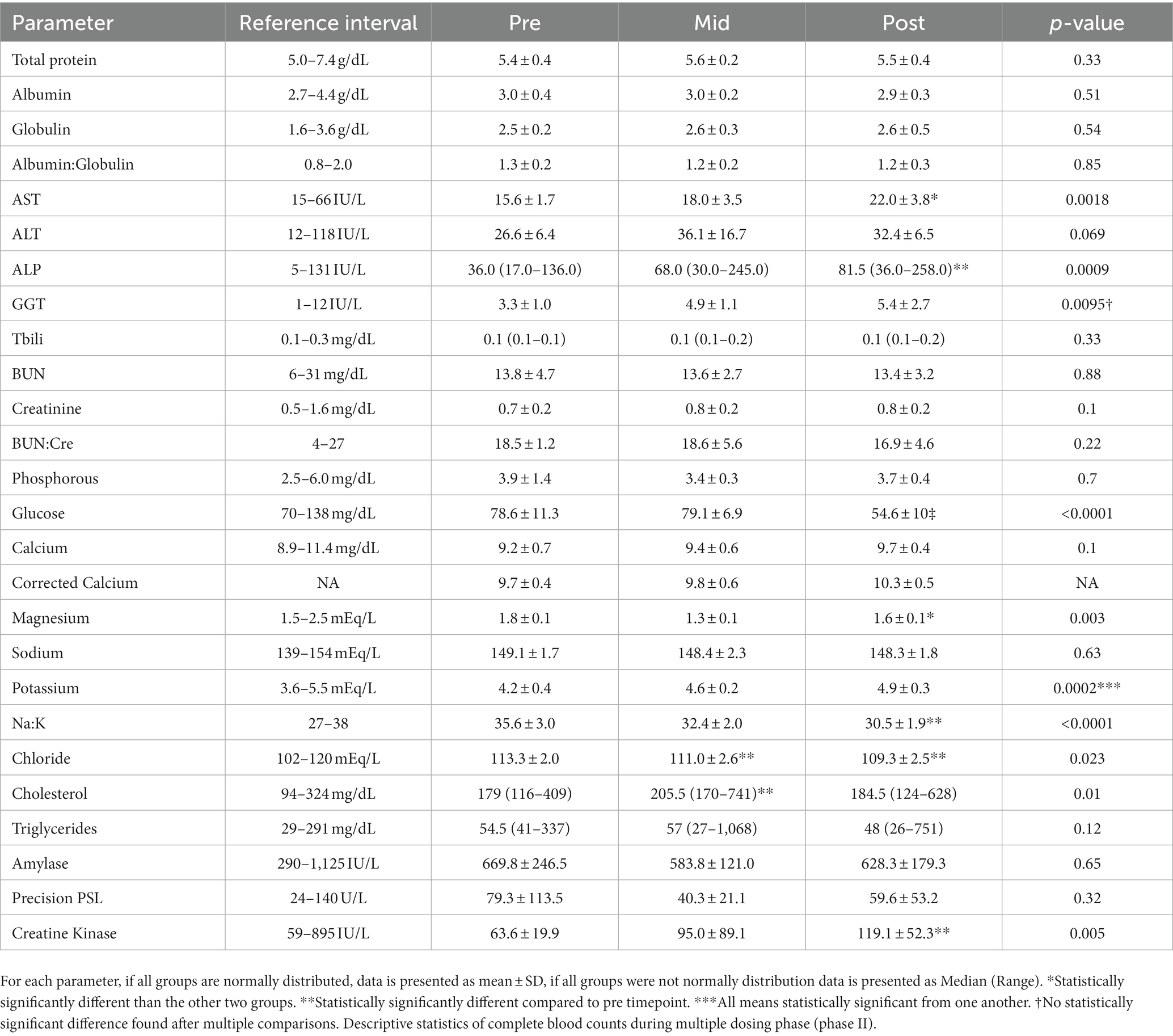
Table 5. Serum biochemistry results (and reference intervals from the laboratory) from dogs prior to 4-week administration (pre), 2 weeks into administration (mid), and at the termination of 4 weeks of administration (post).
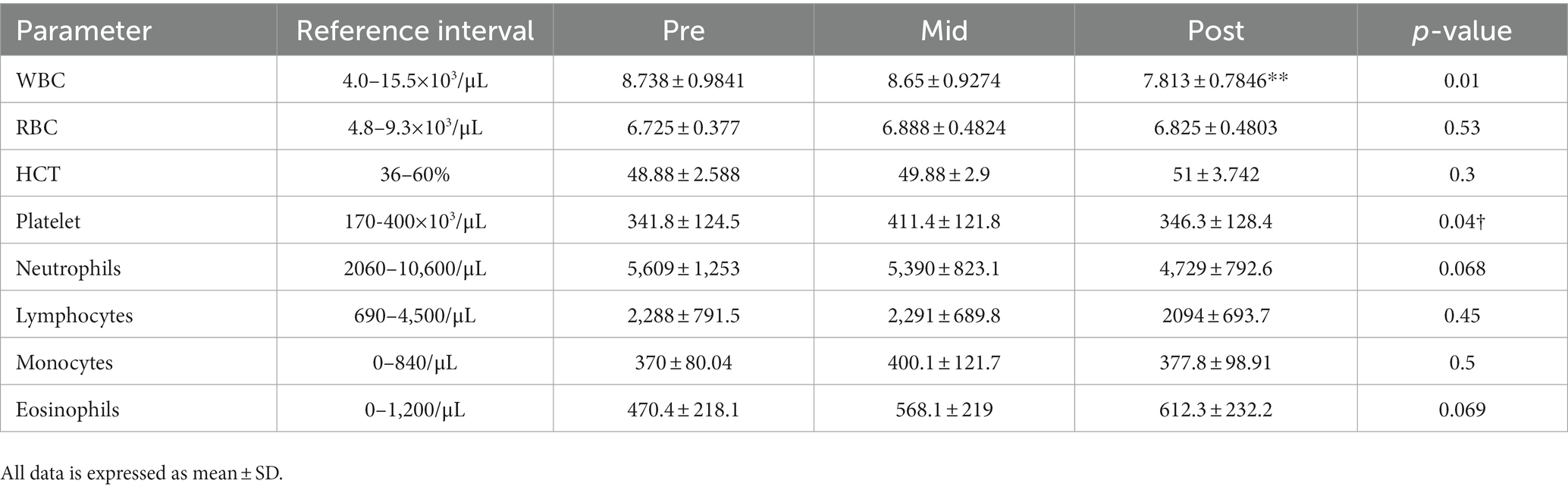
Table 6. Mean ± SD of selected complete blood counts results (and reference intervals from the laboratory) from dogs prior to 4-week administration (pre), 2 weeks into administration (mid), and at the termination of 4 weeks of administration (post).
6 Discussion
Several decades have elapsed since the pharmacokinetics of phenobarbital have been described (10–13, 17, 18). Table 7 shows a summary of previously reported pharmacokinetic parameters for phenobarbital in dogs. This study demonstrates that the veterinary specific phenobarbital product shows excellent bioavailability (F) in dogs with near complete absorption through the oral route of administration, and at 6 mg/kg/d drug concentrations within the reference interval should be achieved. For intravenous administration, the apparent volume of distribution of the central compartment (Vz) and the apparent volume of distribution at steady-state (Vss) for phase III was 0.6 ± 0.1 L/kg and 0.6 ± 0.06 L/kg compared to 0.7 ± 1 and 0.7 ± 0.1 L/kg for phase I, and difference found to be statistically significantly different (p = 0.04), which may account for the higher Cmax seen during phase III of the study. This appears to be the case given that weights did not differ prior to the study and at completion, and that AUC were similar.
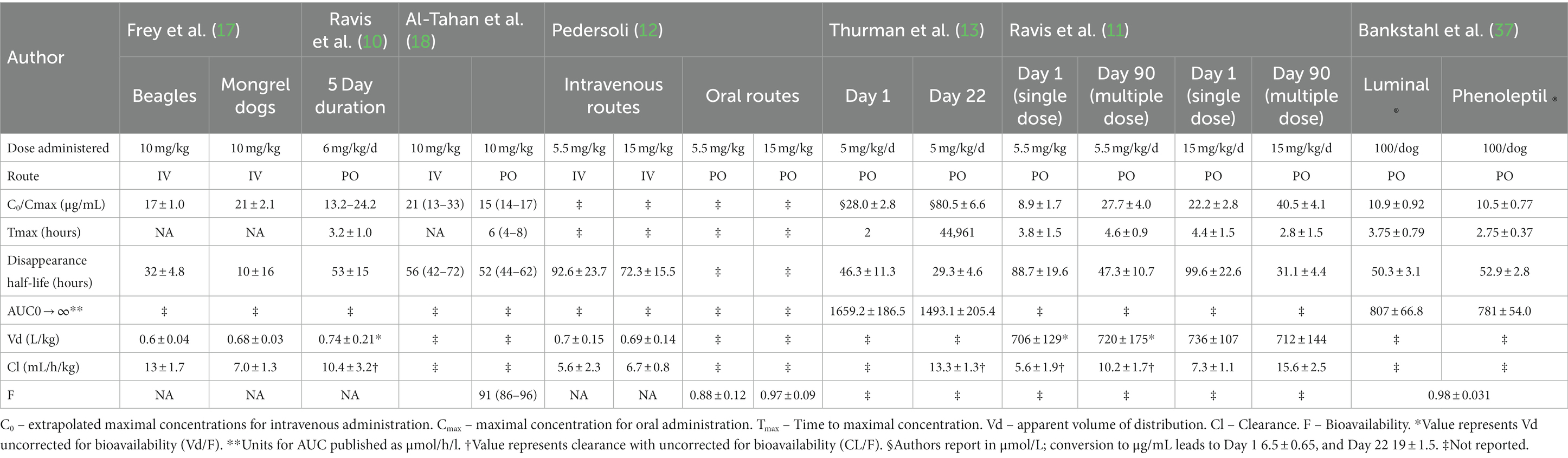
Table 7. Previously reported pharmacokinetic data in dogs.
Mean harmonic half-lives were not statistically different between the phases and routes and ranged from 67–94 h. When assessing peak and trough concentration between each week, approximately 25% fluctuation in PDC occurred during a 24-dosing interval. This suggests that a q24 hour dosing interval may be insufficient for phenobarbital given its longer half-life, and that a q12h dosing interval is justified.
We calculated a loading dose of ~12 mg/kg for phase I, single dose studies based on a previously reported Vd of 0.7 L/kg for dogs and targeting a PDC of 20 μg/mL. Our loading dose appears to have achieved the concentrations within the reported reference interval of 15-45 μg/mL (8) for both IV and PO administration. Furthermore, the maintenance dose used here (6 mg/kg/d) maintained concentrations within the reference interval, and a 3 mg/kg dose q12h may serve as a good starting point for a patient with epilepsy. As such, this dose could be used when a clinician is aiming to achieve a concentration that should produce a therapeutic effect in a patient that requires a useful concentration more immediately. Having said that, although the reference interval is 15-45 μg/mL, concentrations >35 μg/mL may be associated with an increased risk of hepatotoxicity (4, 19). When this dose was administered to the dogs in this study, minimal side effects were noted. However, it would be important to acknowledge that none of these patients had epileptic seizures, a patient that has experienced recent epileptic seizures, or a patient with a structural cause for epilepsy may be more prone to display an adverse effect from such high doses given over a short period of time.
Despite reports of autoinduction associated with phenobarbital induction of cytochrome 450 metabolism, we were not able to demonstrate an expected increased clearance or shortened elimination half-life of phenobarbital over the 4-week treatment period. Indeed, clearance was decreased at 4-weeks compared to baseline. It has been previously demonstrated that clearance was higher in dogs receiving a higher phenobarbital dose (escalating doses starting at ~1.2 mg/kg/d and ending at 32 mg/kg/d) (20), and in dogs treated with phenobarbital over a longer period of time (11). In the latter study, a significantly higher clearance was found 30, 60, and 90 days from initial phenobarbital therapy when compared to the single dose. Furthermore, in a study investigating the lasting effects of liver enzyme induction from a 10 mg/kg/d dose of phenobarbital after a 34 day administration, activity remained enhanced for 4 weeks after cessation of phenobarbital administration in dogs administered antipyrine (21). In this study, initial clearance of 6.2 ± 1.5 mL/kg/h was not statistically different (p = 0.8) from that at the end of the study at 6.3 ± 1.3 mL/kg/h It is possible that dogs in this study were not treated with phenobarbital long enough to demonstrate an increase in clearance or there may be individual variation in autoinduction.
Overall phenobarbital was well tolerated in all dogs in both forms. Slight ataxia was noted after the intravenous administration in a couple of dogs, but no dog was profoundly sedated. On blood work evaluation, all parameters except for glucose concentrations remained within laboratory reference intervals. The 4-week glucose concentrations had a mean consistent with hypoglycemia. Since an outside laboratory (ANTECH®, Antech diagnostics, Inc) was used for this diagnostic, the hypoglycemia most likely represents improper sample handling and processing prior to submission into the lab, rather than true hypoglycemia. In all dogs that had a hypoglycemic value at any time point, an in-house glucose test was performed as soon as possible to ensure that the laboratory value was spurious. Liver enzymes remained within laboratory reference intervals at each time point. However, an increase in serum ALP, and AST was noted at the 4-week mark when compared to the pre-and mid-point values. The increases in ALP have previously been demonstrated to be largely from induction of corticosteroid induced alkaline phosphatase activity (22, 23).
There are several reports assessing bone marrow suppression in relation to the use of phenobarbital (24, 25). All CBC parameters remained within laboratory reference intervals throughout the 4-week dosing period. The only statistically significant difference found involved total white blood cell counts, with the 4-week time point being lower than the mid-point (p = 0.01).
Of important note for therapeutics is the individual patient. This study did not address the potential role of pharmacogenomics and its role in variable phenobarbital pharmacokinetics. Phenobarbital has been demonstrated to induce multiple types for CYP enzymes including CYP1A, CYP2B, CYP2C, and CYP3A (26). With evidence for polymorphisms in various CYP enzymes in the dog (27–35), it is reasonable to think that this may be important when considering therapeutic outcome (or development of adverse effects). As such, therapeutic drug monitoring might serve as a powerful tool to help guide the clinician when making decisions regarding dose and dosing interval. An example involving phenobarbital therapy in dogs demonstrated improved control of epileptic seizures in dogs switching to a from a q12h to q8h dosing schedule (36).
A study assessing bioequivalence between two European approved phenobarbital products has been done (37). This study determined that the two products showed similar relative bioavailability and pharmacokinetic parameters including Cmax, Tmax, elimination half-life, and AUC. This suggests that the two specific products used are bioequivalent to eachother, which is important for therapeutic success, and preventing toxicity. Direct comparison between pharmacokinetic data cannot be made as a set dose per dog of 100 mg was administered. This would affect AUC and Cmax values obtained. Having said that, calculated elimination half-life and Tmax were more closely aligned between the two studies.
Other limitations of the study included a lack of female dogs in the study (7 males to 1 female). Pharmacokinetics and adverse drug reactions can be different between males and females (38). Although from an ASM efficacy perspective, no definitive differences exist between sexes (39).
Lastly, during the writing of this manuscript a different veterinary specific phenobarbital product was given conditional approval from the FDA. However, to the author’s knowledge, this is the only veterinary specific phenobarbital product with pharmacokinetic data in dogs.
7 Conclusion
The veterinary specific phenobarbital formulation to be marketed as NOBATOL® shows near complete systemic absorption, and pharmacokinetic parameters similar to those previously published. The formulation appears to be safe in the dog, the utilization of a consistent formulation may allow for better dose titrations and prevent variations in plasma drug concentration in relation to when one generic product is changed to another within individual patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Animal Care and Use, Auburn University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TJ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. AG: Data curation, Project administration, Resources, Supervision, Writing – review & editing. DB: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from Mizner Bioscience LLC. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
Acknowledgments
The authors would like to acknowledge Benjamin Driggers, Stephanie Maples, and Jordan Doss for assisting with this project.
Conflict of interest
AG is a beneficiary of a licensing agreement with Sio Gene Therapies (New York City).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yasiry, Z, and Shorvon, SD. How phenobarbital revolutionized epilepsy therapy: the story of phenobarbital therapy in epilepsy in the last 100 years. Epilepsia. (2012) 53:26–39. doi: 10.1111/epi.12026
3. Hauptmann, A . rfahrungen aus der behandlung der epilepsie mitluminal. Munch Med Wochenschr. (1919) 46:1319–21.
4. Podell, M, Volk, HA, Berendt, M, Loscher, W, Munana, K, Patterson, EE, et al. 2015 ACVIM Small Animal Consensus Statement on Seizure Management in Dogs. J Vet Intern Med. (2016) 30:477–90. doi: 10.1111/jvim.13841
5. Meland, T, Carrera-Justiz, S, and Buckley, GJ. Antiepileptic drug use patterns in suspect epileptic dogs among neurology and emergency specialists. J Am Anim Hosp Assoc. (2019) 55:138–43. doi: 10.5326/JAAHA-MS-6795
6. Bhatti, SF, De Risio, L, Munana, K, Penderis, J, Stein, VM, Tipold, A, et al. International veterinary epilepsy task force consensus proposal: medical treatment of canine epilepsy in Europe. BMC Vet Res. (2015) 11:176. doi: 10.1186/s12917-015-0464-z
7. Charalambous, M, Brodbelt, D, and Volk, HA. Treatment in canine epilepsy – a systematic review. BMC Vet Res. (2014) 10:257. doi: 10.1186/s12917-014-0257-9
8. Farnbach, GC . Serum concentrations and efficacy of phenytoin, phenobarbital, and primidone in canine epilepsy. J Am Vet Med Assoc. (1984) 184:1117–20.
9. Trepanier, LA, Van Schoick, A, Schwark, WS, and Carrillo, J. Therapeutic serum drug concentrations in epileptic dogs treated with potassium bromide alone or in combination with other anticonvulsants: 122 cases (1992-1996). J Am Vet Med Assoc. (1998) 213:1449–53.
10. Ravis, WR, Nachreiner, RF, Pedersoli, WM, and Houghton, NS. Pharmacokinetics of phenobarbital in dogs after multiple oral administration. Am J Vet Res. (1984) 45:1283–6.
11. Ravis, WR, Pedersoli, WM, and Wike, JS. Pharmacokinetics of phenobarbital in dogs given multiple doses. Am J Vet Res. (1989) 50:1343–7.
12. Pedersoli, WM, Wike, JS, and Ravis, WR. Pharmacokinetics of single doses of phenobarbital given intravenously and orally to dogs. Am J Vet Res. (1987) 48:679–83.
13. Thurman, GD, McFadyen, ML, and Miller, R. The pharmacokinetics of phenobarbitone in fasting and non-fasting dogs. J S Afr Vet Assoc. (1990) 61:86–9.
14. https://dailymed.nlm.nih.gov/dailymed/search.cfm?labeltype=all&query=PHENOBARBITAL&pagesize=20&page=1.
15. Administration FaD . Orange book: Approved drug products with therapeutic equivalence evaluations: U/S/Food and Drug Administration; (2023) [updated 8/11/2023 September 2023]. Available at: https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm.
16. Boothe, DM . Small animal clinical pharmacology and therapeutics. Second ed. Riverport Lane, St. Louis Missouri: Elsevier Saunders (2012). 1348 p.
17. Frey, HH, Gobel, W, and Loscher, W. Pharmacokinetics of primidone and its active metabolites in the dog. Arch Int Pharmacodyn Ther. (1979) 242:14–30.
18. Al-Tahan, F, and Frey, HH. Absorption kinetics and bioavailability of phenobarbital after oral administration to dogs. J Vet Pharmacol Ther. (1985) 8:205–7. doi: 10.1111/j.1365-2885.1985.tb00946.x
19. Dayrell-Hart, B, Steinberg, SA, Van Winkle, TJ, and Farnbach, GC. Hepatotoxicity of phenobarbital in dogs: 18 cases (1985-1989). J Am Vet Med Assoc. (1991) 199:1060–6.
20. Abramson, FP . Autoinduction of phenobarbital elimination in the dog. J Pharm Sci. (1988) 77:768–70. doi: 10.1002/jps.2600770910
21. Fukunaga, K, Saito, M, Matsuo, E, Muto, M, Mishima, K, Fujiwara, M, et al. Long-lasting enhancement of CYP activity after discontinuation of repeated administration of phenobarbital in dogs. Res Vet Sci. (2009) 87:455–7. doi: 10.1016/j.rvsc.2009.03.018
22. Gaskill, CL, Hoffmann, WE, and Cribb, AE. Serum alkaline phosphatase isoenzyme profiles in phenobarbital-treated epileptic dogs. Vet Clin Pathol. (2004) 33:215–22. doi: 10.1111/j.1939-165X.2004.tb00376.x
23. Gaskill, CL, Miller, LM, Mattoon, JS, Hoffmann, WE, Burton, SA, Gelens, HC, et al. Liver histopathology and liver and serum alanine aminotransferase and alkaline phosphatase activities in epileptic dogs receiving phenobarbital. Vet Pathol. (2005) 42:147–60. doi: 10.1354/vp.42-2-147
24. Bersan, E, Volk, HA, Ros, C, and De Risio, L. Phenobarbitone-induced haematological abnormalities in idiopathic epileptic dogs: prevalence, risk factors, clinical presentation and outcome. Vet Rec. (2014) 175:247. doi: 10.1136/vr.102158
25. Scott, TN, Bailin, HG, Jutkowitz, LA, Scott, MA, and Lucidi, CA. Bone marrow, blood, and clinical findings in dogs treated with phenobarbital. Vet Clin Pathol. (2021) 50:122–31. doi: 10.1111/vcp.13013
26. Hojo, T, Ohno, R, Shimoda, M, and Kokue, E. Enzyme and plasma protein induction by multiple oral administrations of phenobarbital at a therapeutic dosage regimen in dogs. J Vet Pharmacol Ther. (2002) 25:121–7. doi: 10.1046/j.1365-2885.2002.00385.x
27. Karakus, E, Prinzinger, C, Leiting, S, and Geyer, J. Sequencing of the canine cytochrome P450 CYP2C41 gene and Genotyping of its polymorphic occurrence in 36 dog breeds. Front Vet Sci. (2021) 8:663175. doi: 10.3389/fvets.2021.663175
28. Tenmizu, D, Endo, Y, Noguchi, K, and Kamimura, H. Identification of the novel canine CYP1A2 1117 C > T SNP causing protein deletion. Xenobiotica. (2004) 34:835–46. doi: 10.1080/00498250412331285436
29. Mise, M, Hashizume, T, Matsumoto, S, Terauchi, Y, and Fujii, T. Identification of non-functional allelic variant of CYP1A2 in dogs. Pharmacogenetics. (2004) 14:769–73. doi: 10.1097/00008571-200411000-00008
30. Scherr, MC, Lourenco, GJ, Albuquerque, DM, and Lima, CS. Polymorphism of cytochrome P450 A2 (CYP1A2) in pure and mixed breed dogs. J Vet Pharmacol Ther. (2011) 34:184–6. doi: 10.1111/j.1365-2885.2010.01243.x
31. Whiterock, VJ, Morgan, DG, Lentz, KA, Orcutt, TL, and Sinz, MW. Phenacetin pharmacokinetics in CYP1A2-deficient beagle dogs. Drug Metab Dispos. (2012) 40:228–31. doi: 10.1124/dmd.111.041848
32. Blaisdell, J, Goldstein, JA, and Bai, SA. Isolation of a new canine cytochrome P450 CDNA from the cytochrome P450 2C subfamily (CYP2C41) and evidence for polymorphic differences in its expression. Drug Metab Dispos. (1998) 26:278–83.
33. Roussel, F, Duignan, DB, Lawton, MP, Obach, RS, Strick, CA, and Tweedie, DJ. Expression and characterization of canine cytochrome P450 2D15. Arch Biochem Biophys. (1998) 357:27–36. doi: 10.1006/abbi.1998.0801
34. Paulson, SK, Engel, L, Reitz, B, Bolten, S, Burton, EG, Maziasz, TJ, et al. Evidence for polymorphism in the canine metabolism of the cyclooxygenase 2 inhibitor, celecoxib. Drug Metab Dispos. (1999) 27:1133–42.
35. Lankford, SM, Bai, SA, and Goldstein, JA. Cloning of canine cytochrome P450 2E1 cDNA: identification and characterization of two variant alleles. Drug Metab Dispos. (2000) 28:981–6.
36. Stabile, F, Barnett, CR, and De Risio, L. Phenobarbital administration every eight hours: improvement of seizure management in idiopathic epileptic dogs with decreased phenobarbital elimination half-life. Vet Rec. (2017) 180:178. doi: 10.1136/vr.104051
37. Bankstahl, M, Bankstahl, JP, and Loscher, W. Is switching from brand name to generic formulations of phenobarbital associated with loss of antiepileptic efficacy?: a pharmacokinetic study with two oral formulations (luminal((R)) vet, Phenoleptil((R))) in dogs. BMC Vet Res. (2013) 9:202. doi: 10.1186/1746-6148-9-202
38. Zucker, I, and Prendergast, BJ. Sex differences in pharmacokinetics. Handb Exp Pharmacol. (2023). doi: 10.1007/164_2023_669
Keywords: phenobarbital, dog, pharmacokinetics, seizure, epilepsy
Citation: Jukier T, Gross A and Boothe D (2024) Pharmacokinetics and tolerability of a veterinary phenobarbital product in healthy dogs. Front. Vet. Sci. 10:1307888. doi: 10.3389/fvets.2023.1307888
Edited by:
Holger Andreas Volk, University of Veterinary Medicine Hannover, GermanyReviewed by:
Melanie Hamann, University of Giessen, GermanyPierre-Louis Toutain, Ecole Nationale Vétérinaire de Toulouse (ENVT), France
Copyright © 2024 Jukier, Gross and Boothe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tom Jukier, dHpqMDAzNEBhdWJ1cm4uZWR1