 Keon Kim1†
Keon Kim1† Jung Eun Song1,2†Jae Beom Joo1Hyeon A Park1Chang Hyeon Choi1Chang Yun Je1
Jung Eun Song1,2†Jae Beom Joo1Hyeon A Park1Chang Hyeon Choi1Chang Yun Je1 Ock Kyu Kim1
Ock Kyu Kim1 Sin Wook Park1Yoon Jung Do3Tai-Young Hur3Sang-Ik Park4*
Sin Wook Park1Yoon Jung Do3Tai-Young Hur3Sang-Ik Park4* Chang-Min Lee1*
Chang-Min Lee1*- 1Department of Veterinary Internal Medicine, College of Veterinary Medicine and BK21 FOUR Program, Chonnam National University, Gwangju, Republic of Korea
- 2Gwangju Animal Medical Center, Gwangju, Republic of Korea
- 3Division of Animal Diseases and Health, National Institute of Animal Science, Rural Development Administration, Wanju-gun, Republic of Korea
- 4Department of Veterinary Pathology, College of Veterinary Medicine and BK21 FOUR Program, Chonnam National University, Gwangju, Republic of Korea
Background: A genome-wide association study (GWAS) is a valuable tool for investigating genetic and phenotypic variation in many diseases.
Objective: The objective of this study was to identify variations in the genomes of Maltese dogs that are associated with the mammary gland tumor (MGT) phenotype and to assess the association between each biological condition and MGT phenotype in Maltese dogs.
Methods: DNA was extracted from 22 tumor samples and 11 whole blood samples from dogs with MGTs. Genome-wide single-nucleotide polymorphism (SNP) genotyping was performed, and the top 20 SNPs associated with various conditions and genetic variations were mapped to their corresponding gene locations.
Results: The genotyping process successfully identified 173,662 loci, with an overall genotype completion rate of 99.92%. Through the quality control analysis, 46,912 of these SNPs were excluded. Allelic tests were conducted to generate Manhattan plots, which showed several significant SNPs associated with MGT phenotype in intergenic region. The most prominent SNP, located within a region associated with transcription and linked to the malignancy grade of MGT, was identified on chromosome 5 (p = 0.00001) though there may be lack of statistical significance. Other SNPs were also found in several genes associated with oncogenesis, including TNFSF18, WDR3, ASIC5, STAR, and IL1RAP.
Conclusion: To our knowledge, this is the first GWAS to analyze the genetic predisposition to MGT in Maltese dogs. Despite the limited number of cases, these analyzed data could provide the basis for further research on the genetic predisposition to MGTs in Maltese dogs.
Introduction
Mammary gland tumors (MGTs) are the most common neoplastic disease in female dogs, and approximately 50% of MGTs are malignant (1–3). Genetic predisposition as well as other factors, such as hormonal influence, diet, and obesity, are known to contribute to the risk of developing MGTs (4). The breeds with an increased MGT risk can be variable according to each study and geographic location, but several breeds such as Maltese, Poodles, Yorkshire Terriers have been reported to have increased incidence of MGTs (5–7). Clinical staging of canine MGTs may be performed using three factors according to the World Health Organization staging system: (1) size evaluation, (2) assessment of regional lymph node involvement, and (3) identification of distant metastasis (8).
A genome-wide association study (GWAS) is a useful tool for understanding genetic and phenotypic variation. However, its usefulness is dependent on the level of linkage disequilibrium (LD) as only a limited number of markers can be genotyped (9, 10). In one study, canine LD was found to be 20–50 times more extensive than that in humans, indicating that fewer SNPs are required for genotype analysis in dogs than in humans (11). Furthermore, dogs with a history of inbreeding are estimated to show low levels of genetic variation and a high degree of LD within breeds (10, 12). Due to the small genetic variation, a homogenous origin within a single breed may allow for easier identification of risk factors for canine MGTs.
There are limited studies of genetic mutations involved in the development of canine MGTs. In one study, one single-nucleotide polymorphism (SNP) in the BRCA1/2 genes, in which mutations are known to be associated with an 84% increased risk of breast cancer in human medicine, was confirmed to be significantly associated with canine MGTs (1, 9). In another study, the significance of 10 genes associated with human breast cancer was analyzed in dogs with MGT (1). However, most of previous studies have been based on gene mutations in human breast cancer.
So far, only a limited number of GWAS studies have performed to examine the association between specific SNPs and the susceptibility to MGT in dogs. Melin et al. (13) were the first to conduct GWAS analysis in English Springer Spaniels and identified significant candidate genes for canine MGT development (13). Furthermore, more recently, Mucha et al. (14) conducted a preliminary GWAS study in female dogs with MGT, leading to the discovery of different significant SNPs and candidate genes compared to previous research (15). Therefore, further genetic analysis including other breeds needs to be performed at the whole-genome level to elucidate the genetic factors for the development of canine MGTs.
The purpose of the present study is to identify genomic variations in Maltese dogs that affect the development of canine MGT and to investigate the association of genetic polymorphisms with MGT conditions. To our knowledge, this study is the first GWAS of canine MGTs in Maltese dogs.
Materials and methods
Animals
Thirty-three Maltese dogs (22 in the MGT group and 11 controls) were included in the study. All cases and controls were selected from dogs referred to the veterinary medical teaching hospital of Chonnam National University and local animal hospitals between July 2021 and June 2022. The medical records, physical examinations, diagnostic imaging, and cytologic and histopathologic findings of all Maltese dogs were evaluated retrospectively. The criteria for inclusion as a control included healthy Maltese dogs without other neoplastic lesions confirmed by physical examination and diagnostic imaging. Furthermore, all patients with reproductive or hormonal disorders were excluded. The experimental design of the study was approved by the Chonnam National University Institutional Animal Care and Use Committee (approval No. CNU IACUC-YB-2021-70).
Distribution of MGTs
The macroscopic distribution of MGTs was classified according to whether the tumors had a bilateral distribution (involvement of both sides of the mammary chain) or a unilateral distribution (involvement of one mammary chain). The evaluation of the distribution was based on physical examination records and photographs from clinical charts.
Histopathological classification of MGTs
All histological evaluations of MGTs was performed by only one pathological specialist. In this study, as a condition for analysis, mammary tumors were classified into benign tumors and malignant tumors.
Malignancy grade
The MGT group was classified histopathologically into benign tumors and malignant tumors, and among the malignant tumors, the patients who were diagnosed with carcinoma were graded by the “Elston and Ellis grading method” used in human medicine (16). The grades for the association analysis were benign, Grade 1, Grade 2, and Grade 3.
Obesity
In the MGT group, the body condition score (BCS) system was used to distinguish between the obese and normal groups (17, 18). The obese group consisted of the dogs with MGTs with scores of 8 to 9 out of 9 based on the BCS system. The normal group was composed of the dogs with MGTs with scores of 4 to 6 out of 9 on the BCS system.
Sample collection and DNA preparation
After surgical removal of tumors in the MGT group, neoplastic tissues were fixed in 10% neutralized formalin solution. The pathological material was processed with “Pathcentre” and embedded into paraffin. The 3-μm slices were prepared and stained with hematoxylin and eosin (HE). For DNA extraction, residual neoplastic tissues were soaked in a 1.5-ml microcentrifuge tube containing RNAlater. Tissues soaked in RNAlater were stored at −80°C until DNA extraction. Fasting venous blood samples were collected for genomic DNA extraction in the control group. A 1-mL aliquot of the sample was added to an ethylenediaminetetraacetic acid (EDTA) anticoagulant tube, and the tubes were maintained on ice during transport and stored at −80°C.
Genomic DNA was extracted from neoplastic tissues and EDTA-treated whole blood using commercial kits (QIAamp DNA Mini Kit and QIAamp DNA Blood Mini Kit; Qiagen, Germany) according to the manufacturer’s instructions. DNA concentrations were measured using a spectrometer. The A260/A280 ratio of all extracted samples was within the normal range of 1.7–1.9. The extracted DNA was stored at −20°C until genotyping of SNPs.
Genome-wide SNP genotyping and association study
Genotyping of SNPs was conducted in a commercial laboratory using the BeadChip system (CanineHD BeadChip; Illumina, USA). DNA from 22 dogs with MGTs and 11 control dogs was analyzed. These samples were processed for 3 days using a reading system (InfiniumR HD Assay Ultra system; Illumina). According to the manufacturer’s recommendations, the preparation and quantification of DNA samples were performed. After that, the samples were amplified and incubated for 20 h. The next day, the amplified DNA was fragmented by using restriction enzymes, and the fragmented DNA was precipitated and resuspended. Prepared DNA samples were pipetted into the BeadChip and incubated at 48°C for 20 h for hybridization. On day 3, the hybridized samples were extended and stained using the BeadChip. Visualization and scanning of the stained samples were achieved using a commercially available platform (Illumina iScan platform; Illumina).
Quality control
Specialized software (Genome Studio software; Illumina) was used to analyze the output from iScan. The samples from both the MGT and control groups were combined to optimize genotype clustering. Polymorphic genotype data for each group were analyzed separately to assess the levels of heterozygosity and deviation from the Hardy–Weinberg equilibrium (<0.01).
A total of 173,662 SNPs were found to have polymorphisms within the combined sample set, with greater than 99% of the SNPs included in acceptable genotype clusters. In our samples, SNPs with call rates below 95%, those that were monomorphic, and those with minor allele frequencies of less than 5% in our samples were excluded. We also excluded SNPs on the X chromosome due to allelic imbalance. Principal component analysis (PCA) was performed to determine if the distribution of the dog samples between the MGT and control groups was tendentious.
Association and statistical analysis
We performed six association analyses of 126,750 SNPs using specialized program (Variation Suite v8 software; Golden Helix, USA). Statistical analysis on these SNPs was conducted, and a nominal value of p less than 0.05 was considered significant. Fisher’s exact tests were used to compare the proportions of samples with different characteristics such as MGT phenotype, distribution, histopathology, obesity, and spayed or not. We also used multivariate linear regressions to examine the relationships between different groups, including malignancy grade.
Interpretation of GWAS results
In GWAS analysis, p-values are obtained by testing the association between each SNP and a trait. To determine the significance of these associations, a significance level is typically used, which is often derived from the Bonferroni correction, known as the genome-wide significance level. As we are using a number of SNPs in GWAS analysis, using a significance level of 0.05 for each individual test would increase the probability of Type I errors. To address this issue, Bonferroni correction was conducted by dividing the overall significance level by the total number of tests, denoted as κ. The adjusted significance level (α′) after Bonferroni correction is calculated as follows: α′ = α/κ. Here, α represents the overall significance level for the entire analysis, and κ corresponds to the number of tests conducted, which, in the context of GWAS, is equivalent to the number of SNPs used in the analysis. In this study, we used a total significance level of 0.05, and with 126,750 SNPs analyzed, we calculated the significance level for each test, resulting in a genome-wide significance level of α′ = 3.94×10−7. This adjusted significance level is commonly used in a single test during GWAS analysis to control for the multiple comparisons issue.
Identification of SNP genes and markers
We obtained data on SNP loci from the Ensembl genome browser (European Bioinformatics Institute, Cambridge, U.K.) to identify genes associated with independent SNPs in each analysis. To predict how the variants may affect gene transcripts, we used gene annotations from the Ensembl database. Our findings revealed the location of variants, including coding sequences or introns of alternatively spliced gene transcripts.
Results
Case and association condition group
The study involved a total of 33 female Maltese dogs; of these, 11 were spayed, and 22 were intact. The average age was 10.3 ± 2.72 years. Table 1 summarizes the fundamental characteristics of the Maltese dogs. For the association study, six different condition groups were formed based on clinical manifestations of MGT, as shown in Table 2. One Maltese dog was excluded from the conditional analysis as any information regarding the MGT-associated condition could not be obtained except for neutering. Histopathological evaluation showed that all malignant tumors were classified as carcinomas. The benign group (n = 6) consisted of simple adenoma (n = 3), complex adenoma (n = 1), intraductal papillary adenoma (n = 1), and lobular hyperplasia (n = 1). The group of carcinoma grade 1 (n = 6) was composed of 4 cases of tubular carcinoma, 1 case of tubulopapillary carcinoma, and 1 case of In situ carcinoma. Within the carcinoma grade 2 group (n = 6), there were 3 cases of complex carcinoma, 1 case of tubulopapillary carcinoma, 1 case of carcinoma combined with malignant myoepithelioma, and 1 case of intraductal papillary carcinoma. The carcinoma grade 3 group (n = 3) included 1 case of tubular carcinoma, 1 case of carcinoma along with malignant myoepithelioma, and 1 case of complex carcinoma. To prepare for variation analysis, DNA was extracted from MGT tissue for the case group, while for the control group, DNA was extracted from leukocytes in whole blood samples.
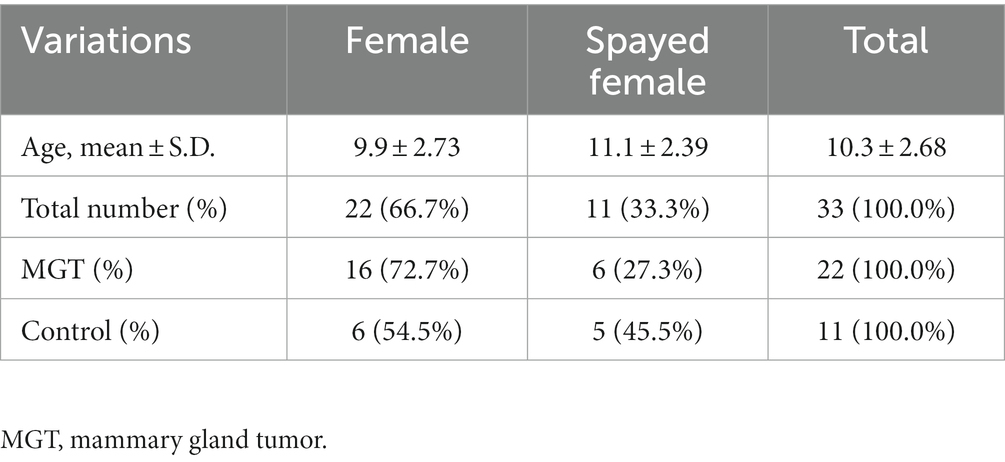
Table 1. Characteristics of the dogs in the present study.
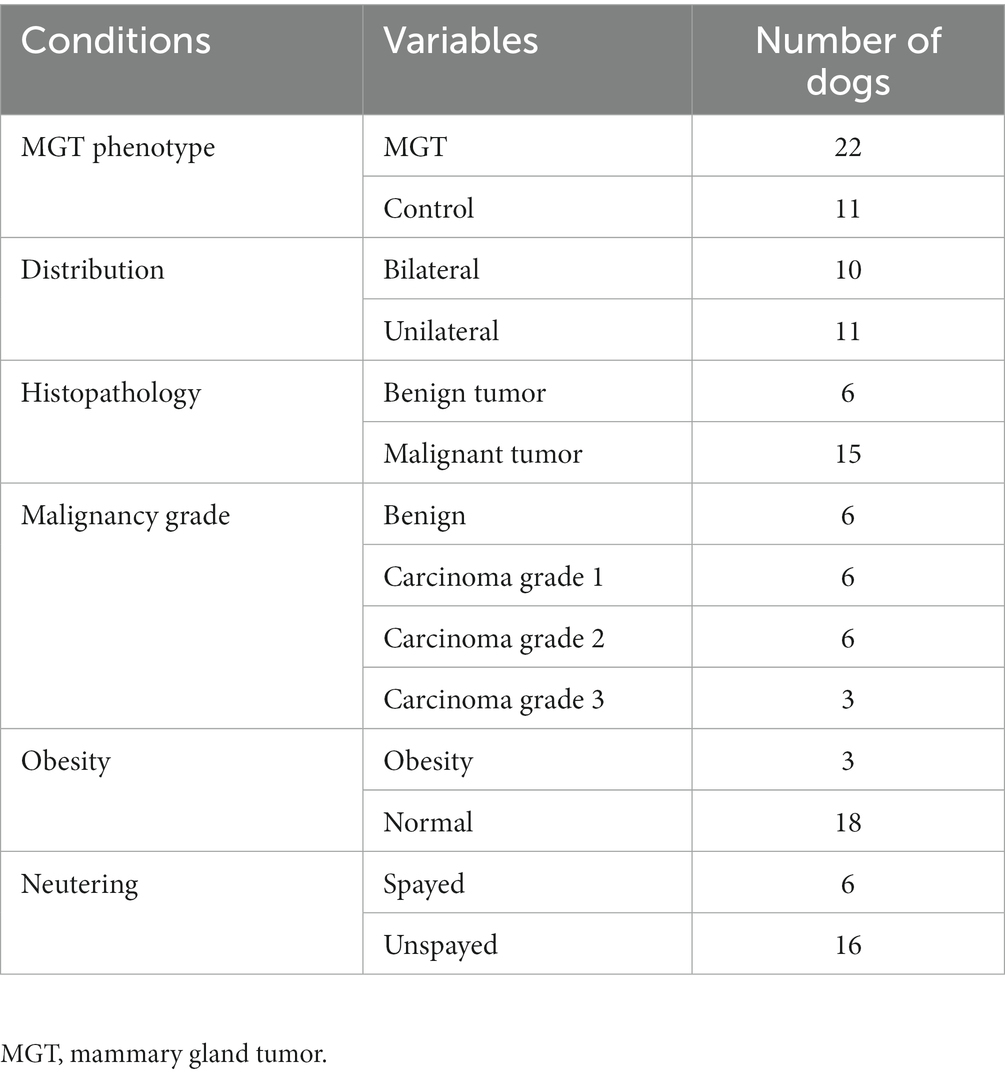
Table 2. Number of dogs for association studies classified as each condition associated with MGT.
Principal component analysis
The use of PCA showed that all dogs included in the study were grouped together in one cluster, as illustrated in Figure 1. The results indicate that there was no population stratification within the sample.
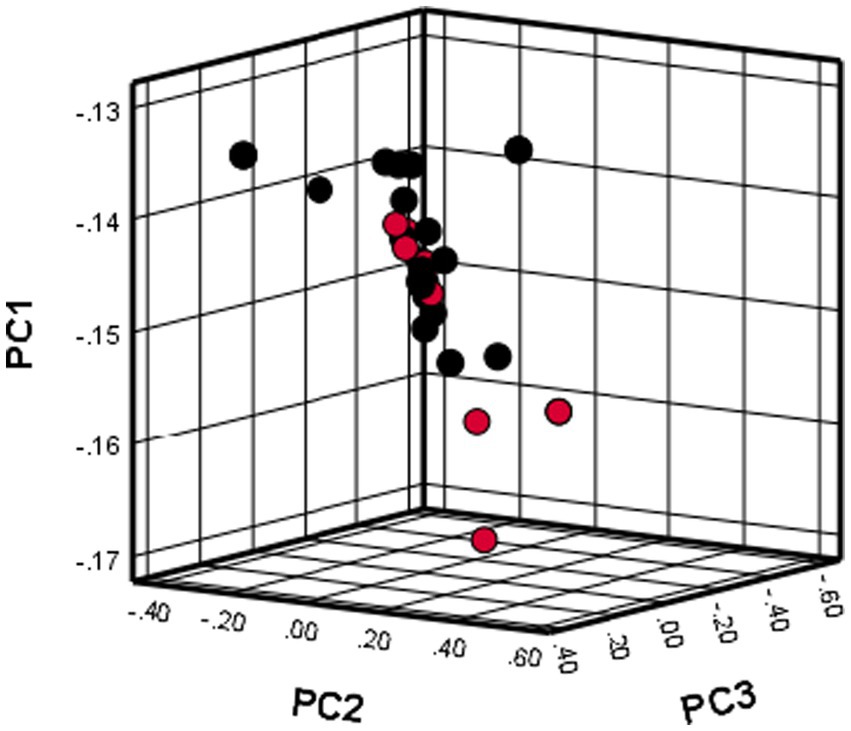
Figure 1. PCA results showing the distribution of cases and controls in the present study.
Genotyping result and association analysis
In this study, a total of 173,662 loci were genotyped in the samples, and of these, 46,912 SNPs were excluded, resulting in 126,750 SNPs that were used for subsequent analysis. The overall genotype completion rate was 99.92%.
Based on the SNP-chip results using the allelic test, Manhattan plots were produced to identify major loci associated with various conditions such as MGT phenotypes, macroscopic distribution of MGT, histopathological classification, malignancy grade, obesity, and neutering. The physical map position of the SNPs was plotted against the −log10 p-values. No SNPs that were significantly associated with MGT phenotype following Bonferroni correction were identified. However, a few significant peaks of association that reached the 5% threshold for genome-wide significance (10−5) were identified, as reported in a previous study (19). Three significant SNPs associated with MGT phenotype were found and all were located in the intergenic region (BICF2G630778783 in chromosome 35, BICF2P118387 in chromosome 34 and BICF2S23451480 in chromosome 21) (Figure 2). Similarly, two significant SNPs were identified for the malignancy grade of MGT (BICF2P660085 in chromosome 18 and BICF2S2324173 in chromosome 11) (Figure 3). Likewise, all these SNPs were located in the intergenic region. Although there may be no statistical significance, the most significant SNP which is located in transcription-related region as well as associated with MGT malignancy grade was found on chromosome 5 (p = 0.00001).
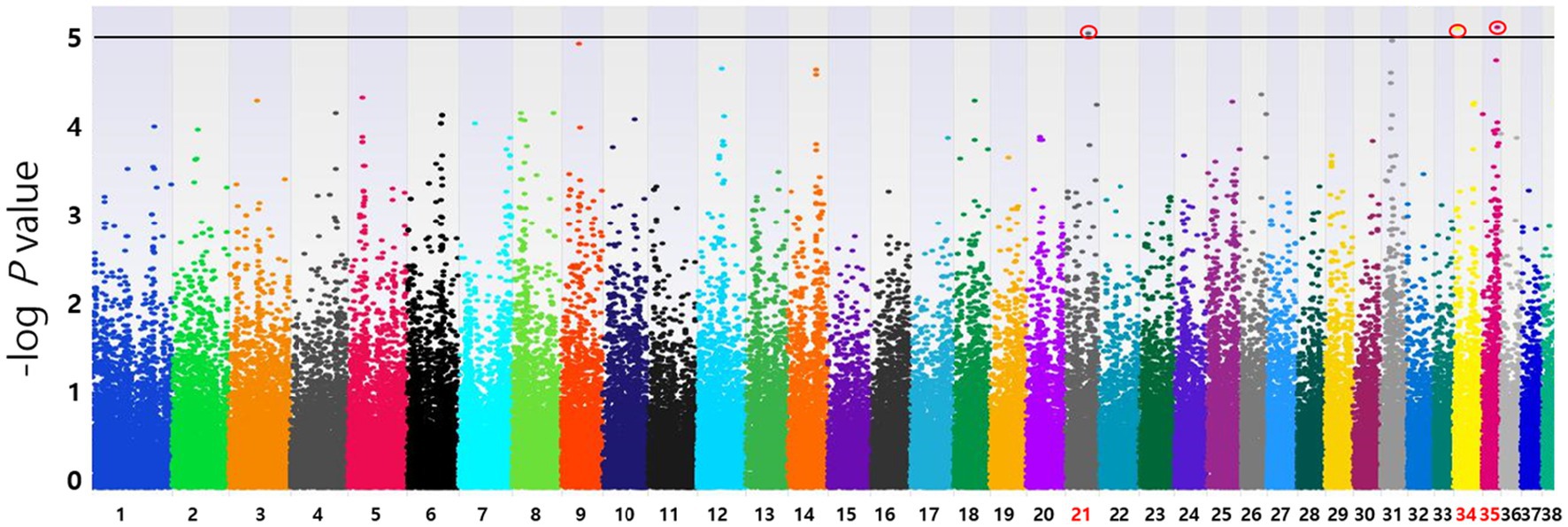
Figure 2. Manhattan plots of the GWAS for MGT phenotype. Fisher’s exact test was performed to confirm the statistical significance of SNPs. Chromosome numbers are displayed on the X-axis, and the results for each chromosome are shown in the diagram as individual colors for each chromosome. The Y-axis is the –log10 of the calculated p-values. Three significant SNPs associated with MGT phenotype were found in chromosome 21(p = 9.20×10−6), 34(p = 7.96×10−6), and 35(p = 7.81×10−6). All statistically significant SNPs have been marked with a red circle.
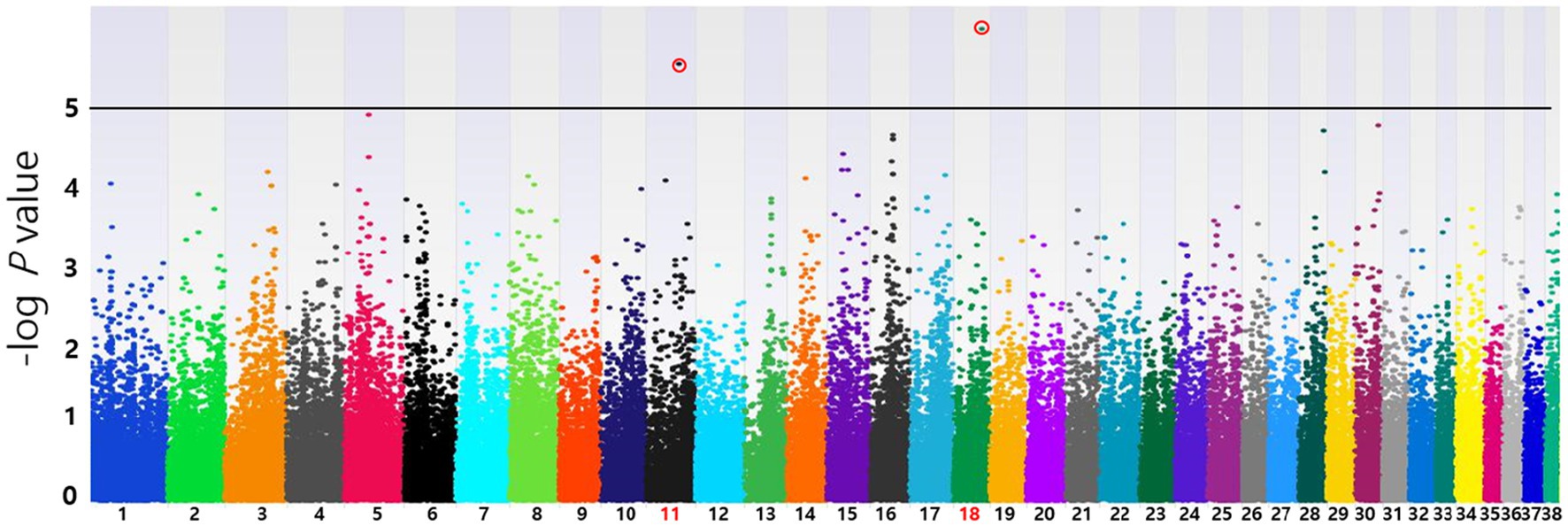
Figure 3. Manhattan plots of the GWAS for histopathological malignancy grade of MGT. Linear regression test was performed to confirm the statistical significance of SNPs. Chromosome numbers are displayed on the X-axis, and the results for each chromosome are shown in the diagram as individual colors for each chromosome. The Y-axis is the –log10 of the calculated p-values. Two significant SNPs associated with MGT phenotype were identified in chromosome 11(p = 2.39×10−6) and 18(p = 8.53×10−7). All statistically significant SNPs have been marked with a red circle.
In this study, the top 20 SNPs based on their p-values were identified, and their corresponding gene regions were determined and are listed in Supplementary Tables S1–S6. The majority of SNPs for each condition were located in the intergenic region. Considering the difficulty in elucidating the roles of these SNPs during transcription and translation processes, we have excluded them from the table even if they meet the statistical significance criteria (p < 10−5). The SNPs with a high possibility of affecting gene transcription are summarized in Tables 3–5. Tables 3, 4 contain the GWAS results of Fisher’s exact tests for conditions that comprise two groups, while Table 5 presents the GWAS results of linear regression tests for conditions with more than two groups.
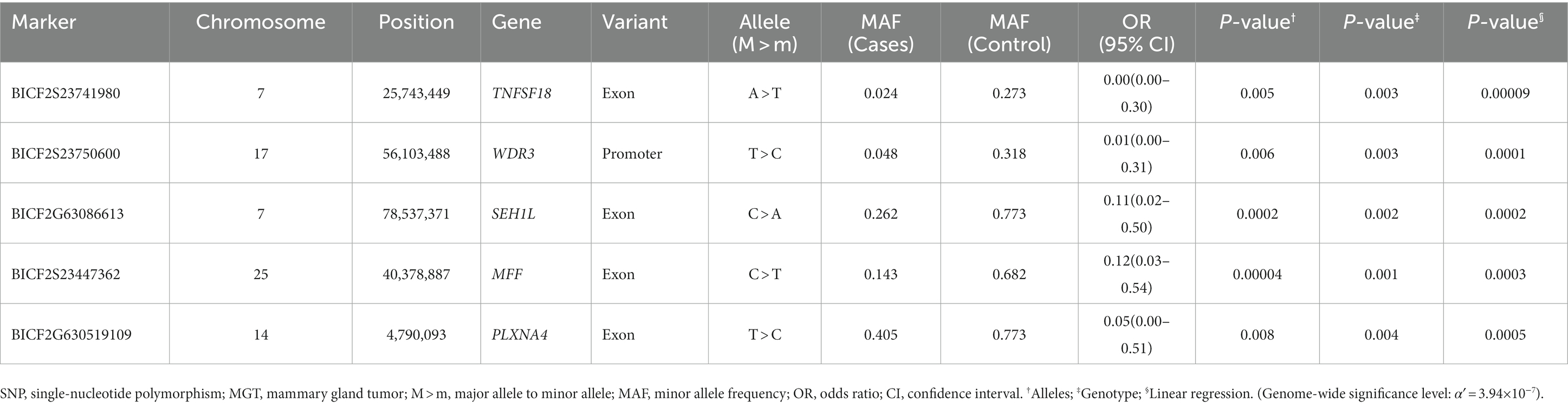
Table 3. Candidate SNP markers associated with the MGT phenotype.
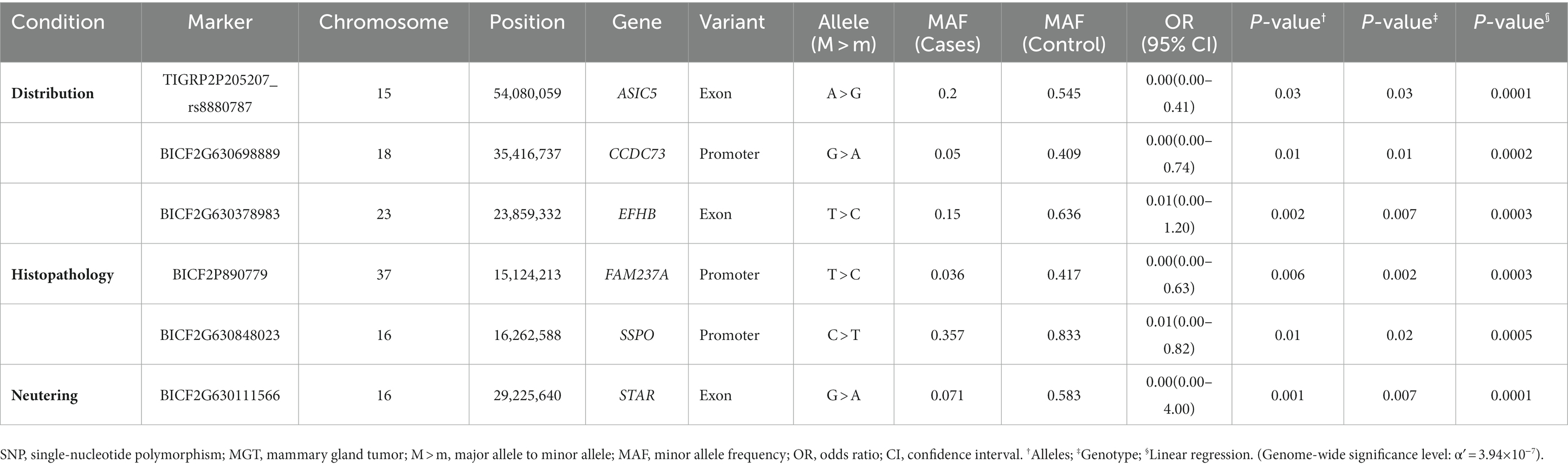
Table 4. Candidate SNP markers associated with various conditions of MGT (distribution, histopathology, and neutering) derived from Fisher’s exact tests.
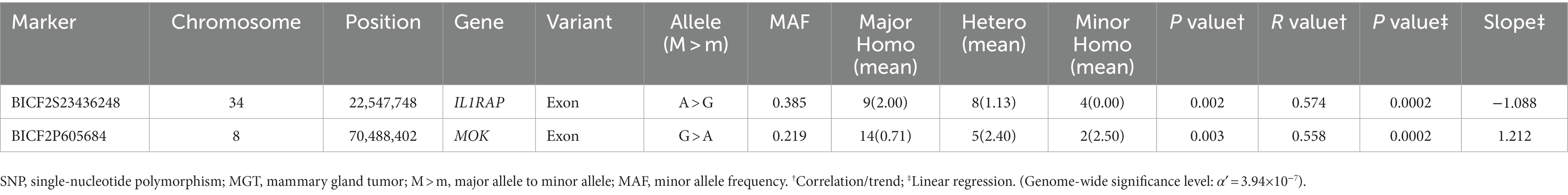
Table 5. Candidate SNP markers associated with the malignancy grade of MGT derived from the linear regression test.
Discussion
Breeding practices of purebred dogs through selection pressure have led to decreased genetic variation within breeds (20). Due to this characteristic of dogs, they are being recognized as a model for studying various genetic diseases, and even a small number of gene analyses are valuable for evaluating their risks (21, 22). In this study, we selected Maltese dogs, which are known to be a breed at high risk of MGTs and are one of the most commonly bred dogs in South Korea (5–7). Canine MGTs and human breast cancer have many similarities in several aspects. Furthermore, since dogs spend considerable time with humans, they are very likely to be exposed to the same environmental factors that contribute to tumorigenesis (23). Considering these features, this study could establish a foundation for discovering candidate genes that may affect MGTs in dogs based on whole-genome analysis as well as extend research on breast cancer genes in humans.
Oncogenesis of MGTs is known to be multifactorial, but a high incidence of MGTs in specific dog breeds supports an important role of genetic factors (24). Several candidate genes, such as BRCA1, BRCA2, BRIP1, CHECK2, ERBB2, STK11, TOX3, TP53, and PTEN, have been reported to be associated with canine MGT risk based on previous research (1, 24). Furthermore, according to the first GWAS study on canine MGT by Melin et al. (13) the CDK5RAP2 gene exhibited a significant association in English Springer Spaniels (13). In this study, no SNP showed a significant association (p < 10−5) with the MGT phenotype in Maltese breeds except to located in intergenic region. As seen in the results, there is a possibility of the absence of statistically significant associations with SNPs, and there is also a likelihood that the results may have been affected by the small sample size. Therefore, further analysis with a larger sample size will be necessary in the future. Despite the results that no SNP showed association with the MGT phenotype, the most notable SNP, situated in a region related to transcription and linked to the malignancy grade of MGT, was discovered on intronic regions of candidate genes. The involvement of introns in regulation of tissue-specific gene expression, mRNA transcription, and translation has been well established (25). SNPs located in intron regions have the potential to generate splice variants of transcripts and promote or disrupt the function of long non-coding RNAs (lncRNAs) (26–28). Consequently, the genetic susceptibility to cancer could be influenced by SNPs located within introns (25).
In the MGT malignancy grade condition, genes such as MAP2K4, NRG1, and PPFIA2 were found to be associated with the identified SNPs (Supplementary Table S3). MAP2K4 is a member of the mitogen-activated protein kinase (MAPK) activator family in humans, and MAPK signaling is known to play a significant role in cell proliferation and differentiation (29). In cancer, MAP2K4 has the potential to activate phosphoinositide-3-kinase (PI3K)/AKT signaling, which can result in cell proliferation, migration, and invasion. Liu et al. (30) confirmed the possibility of MAP2K4 serving as an oncogene in breast cancer (30). NRG1 encodes ligands for the ERBB2-ERBB3 heterodimeric receptor tyrosine kinase, and gene fusions are believed to lead to autocrine receptor stimulation (31). ERBB2 is an important factor in breast cancer in human medicine. Patients with ERBB2-overexpressing breast cancer have lower overall survival rates and shorter disease-free intervals than patients whose cancer does not overexpress ERBB2. Moreover, ERBB2 overexpression has been linked to increased breast cancer metastasis (32). This study suggests that the genes related to tumor biology and the prognosis of breast cancer in humans may also be applicable to canine MGTs. The variations in the candidate genes that are associated with the MGT phenotype based on the tumor grade could ultimately affect their expression.
There were several SNPs that were found in important transcript sequences, which could have an impact on the structure and function of proteins. Of these SNPs, five were particularly significant in relation to the MGT phenotype and were found in transcripts of the TNFSF18, WDR3, SEHIL, MFF, and PLXNA4 genes (Table 3). TNF superfamily member 18 (TNFSF18) is known as common immune checkpoint gene. In the field of human medicine, blocking immune checkpoints has been shown to improve the immune response against cancer, and several agents targeting immune checkpoints have been identified and applied clinically to treat cancer. In addition, Li et al. (33) confirmed significant overexpression of TNFSF18 in breast carcinomas. Based on previous reports, the findings of this study suggest that TNFSF18, identified as a candidate gene, may have an expression abnormality associated with SNP variations in canine MGTs. This could also expand as another therapeutic option for MGTs in dogs, which are currently not actively targeted. WD repeat domain 3 (WDR3) has been shown to participate in different cellular processes including gene regulation, signal transduction, cell cycle progression, and apoptosis (34). Furthermore, the inhibition of WDR3 has been demonstrated to decrease breast carcinoma cell proliferation by McMahon et al. (35). Research on the correlation between WDR3 overexpression and the development of MGTs has not been conducted in both human and veterinary medical fields, but this study suggests a potential area for future research through its findings. SEH1-like nucleoporin (SEH1L) is associated with glycolysis, a complex reaction that initiates the catabolism of most carbohydrates (36). Cancer cells are characterized by the reprogramming of metabolism, and aerobic glycolysis is the primary energy source for these cells (37). In fact, it has been shown that the dysregulation of glycolysis occurs in several types of tumors (38–40).
The most significant SNPs associated with macroscopic distribution included ASIC5, CCDC73 and EFHB (Table 4). Acid-sensing ion channel (ASIC) is a member of the degenerin/epithelial Na + channel subfamily and is primarily expressed in the central and peripheral nervous system (41). The tumor microenvironment is typically acidic due to disorganized tumor vasculature, heterogeneous blood flow, and increased glycolysis in tumor cells (42). Previous studies have demonstrated that an acidic microenvironment contributes to breast tumor invasion and metastasis (43–45). In addition, Gupta et al. (41) suggest that ASIC1 contributes to breast cancer pathogenesis in response to acidic tumor microenvironments (41). Nonetheless, since the function of ASIC5 remains unknown, further studies are necessary to establish the specific characteristics in breast cancer. EFHB is a significant regulator of store-operated calcium entry (SOCE), a major mechanism that enables calcium entry from the extracellular region through the plasma membrane (45). SOCE supports a variety of breast cancer hallmarks, including cell viability, proliferation, migration, and metastasis (45).
One SNP that was located in the exon of the STAR gene was found to be among the most significant SNPs associated with neutering (Table 4). The STAR gene encodes the steroidogenic acute regulatory (STAR) protein, which plays a crucial role in the biosynthesis of steroid hormones by regulating the transportation of cholesterol, the substrate for all steroid hormones, from the outer to the inner mitochondrial membrane (46, 47). In humans, estrogens, particularly 17b-estradiol (E2), are known to promote breast cancer (48). Furthermore, dysfunction in the biosynthesis of androgens and/or estrogens has been linked to the development and growth of various cancers that are responsive to hormones (49). Manna et al. have confirmed that the STAR protein of MGT cells is expressed abundantly compared with normal mammary epithelial cells and that it is associated with increased levels of E2. It is possible that the abundant expression of STAR facilitates the delivery of cholesterol to the inner mitochondrial membrane, leading to the generation of additional precursors for E2, which could contribute to the promotion of breast tumorigenesis (48).
Obesity is known to be a risk factor for adverse effects of breast tumors due to numerous and complex reasons (50). However, in this study, significant SNPs were not identified according to the obesity conditions in the MGT group. Obesity could mainly be affected by acquired components such as environmental factors. However, this does not necessarily mean that genetic factors are not involved. Furthermore, only very few cases were classified as obese in this study.
In human breast cancer, it is known that the cancer can be classified into five molecular subtypes (Luminal A, Luminal B, Basal-like, ErbB2+, and Normal-like), and these classifications have been found to have significant associations with survival and clinical outcomes (51). When comparing this molecular subtype classification in human breast cancer to that of canine MGTs, it was confirmed that they share considerable similarities while there are a few differences (23). On that note, if the likelihood of developing a specific subtype of breast cancer is influenced by genetic factors, it would be anticipated that the novel discovered susceptibility genes exhibit varying levels of expression in each tumor subclasses. Additionally, it is expected that their transcription is regulated in cis by SNPs within them (52). In other words, some of the candidate genes associated with SNPs in crucial positions for transcription (identified through the GWAS study) may exhibit varying expressions across varying tumor subclasses. Therefore, it is important to remember that in future studies, the results of mRNA expression levels of these candidate genes should take into consideration the molecular subtypes.
The present study has several limitations. First, the study had a small sample size. This limited sample size may have had a restrictive effect on obtaining statistically significant results through Bonferroni correction. In addition, the possibility that the results might have been due to the small number of samples leading to the significant association between genomic variation and phenotype cannot be excluded. Second, as the GWAS analysis for confirming major SNPs associated with phenotype is based on the principle of common variant-common disease, next-generation sequencing (NGS) may be necessary for a rigorous whole-genome analysis. Finally, this study did not verify the identified candidate genes at the translation level. In this study, only functional predictions regarding candidate genes associated with MGTs were based on existing research. Further studies are needed to verify the expression of candidate genes that affect protein levels related to the tumorigenesis.
In conclusion, this study examined the genetic correlation between SNPs that are significantly linked to the MGT phenotype and various associated conditions. A number of genetic variations have been identified for the MGT phenotype and conditions, and the most significantly associated condition was the histological malignancy grade of MGT. To our knowledge, this is the first GWAS to analyze a genetic predisposition to the MGT phenotype and associated conditions in Maltese dogs. Despite the limited number of cases, all the analyzed data could be the basis for further research on the genetic predisposition to MGTs in Maltese dogs.
Data availability statement
Original datasets are available in a publicly accessible repository: The original contributions presented in the study are publicly available. This data can be found here: https://www.ebi.ac.uk/eva/?eva-study=PRJEB65760.
Ethics statement
The animal studies were approved by Chonnam National University Institutional Animal Care and Use Committee (approval No. CNU IACUC-YB-2021-70). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
KK: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. JS: Conceptualization, Resources, Writing – original draft, Writing – review & editing. JJ: Data curation, Resources, Writing – original draft. HP: Data curation, Visualization, Writing – original draft. CC: Data curation, Visualization, Writing – original draft. CJ: Data curation, Visualization, Writing – original draft. OK: Data curation, Project administration, Visualization, Writing – original draft. SP: Data curation, Project administration, Visualization, Writing – original draft. YD: Data curation, Funding acquisition, Visualization, Writing – review & editing. T-YH: Data curation, Funding acquisition, Visualization, Writing – review & editing. S-IP: Conceptualization, Data curation, Methodology, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. C-ML: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Rural Development Administration (RDA) under grant number RS-2023-00232301 and Korea Technology & Information Promotion Agency for SMEs (TIPA) under Grant number S3247434.
Acknowledgments
The authors acknowledge the valuable contributions of the advisory task from Cheong.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1255981/full#supplementary-material
References
1. Rivera, P, Melin, M, Biagi, T, Fall, T, Häggström, J, Lindblad-Toh, K, et al. Mammary tumor development in dogs is associated with BRCA1 and BRCA2. Cancer Res. (2009) 69:8770–4. doi: 10.1158/0008-5472.CAN-09-1725
2. Gilbertson, S, Kurzman, I, Zachrau, R, Hurvitz, A, and Black, M. Canine mammary epithelial neoplasms: biologic implications of morphologic characteristics assessed in 232 dogs. Vet Pathol. (1983) 20:127–42. doi: 10.1177/030098588302000201
3. Moulton, J, Taylor, D, Dorn, C, and Andersen, A. Canine mammary tumors. Pathol Vet. (1970) 7:289–320. doi: 10.1177/030098587000700401
4. Sorenmo, K. Canine mammary gland tumors. Vet Clin. (2003) 33:573–96. doi: 10.1016/S0195-5616(03)00020-2
5. Goldschmidt, MH, Peña, L, and Zappulli, V. Tumors of the mammary gland. In: Meuten DJ, editor. Chapter 17: Tumors in domestic animals. Fifth edn. (2016).
6. Kurzman, ID, and Gilbertson, SR. Prognostic factors in canine mammary tumors. Semin Vet Med Surg Small Anim. (1986) 1:25–32.
7. Yamagami, T, Kobayashi, T, Takahashi, K, and Sugiyama, M. Prognosis for canine malignant mammary tumors based on TNM and histologic classification. J Vet Med Sci. (1996) 58:1079–83. doi: 10.1292/jvms.58.11_1079
8. Owen, LN, Organization WH. TNM classification of Tumours in domestic animals. Veterinary Public Health Unit & WHO Collaborating Center for Comparative Oncolog (1980).
9. Fesseha, H. Mammary tumours in dogs and its treatment option-a review. Biomed J Sci Tech Res. (2020) 30:23552–61. doi: 10.26717/BJSTR.2020.30.004980
10. Sutter, NB, Eberle, MA, Parker, HG, Pullar, BJ, Kirkness, EF, Kruglyak, L, et al. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. (2004) 14:2388–96. doi: 10.1101/gr.3147604
11. Ostrander, EA, and Wayne, RK. The canine genome. Genome Res. (2005) 15:1706–16. doi: 10.1101/gr.3736605
12. Lindblad-Toh, K, Wade, CM, Mikkelsen, TS, Karlsson, EK, Jaffe, DB, Kamal, M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature (2005) 438:803–19.
13. Melin, M, Rivera, P, Arendt, M, Elvers, I, Murén, E, Gustafson, U, et al. Genome-wide analysis identifies germ-line risk factors associated with canine mammary Tumours. PLoS Genet. (2016) 12:e1006029. doi: 10.1371/journal.pgen.1006029
14. Mucha, A, Nowak, B, Dzimira, S, Liszka, B, and Zatoń-Dobrowolska, M. Identification of SNP markers for canine mammary gland tumours in females based on a genome-wide association study– preliminary results. J. Vet Res. (2023) 67. doi: 10.2478/jvetres-2023-0040
15. Lindblad-Toh, K, Wade, CM, Mikkelsen, TS, Karlsson, EK, Jaffe, DB, Kamal, M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. (2005) 438:803–19. doi: 10.1038/nature04338
16. Elston, CW, and Ellis, IO. Pathological prognostic factors in breast cancer. I. the value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. (1991) 19:403–10. doi: 10.1111/j.1365-2559.1991.tb00229.x
17. Laflamme, D. Development and validation of a body condition score system for dogs. Canine Practice (1997) 22:10–15.
18. Kronfeld, DS, Donoghue, S, and Glickman, LT. Body condition and energy intakes of dogs in a referral teaching hospital. J Nutr. (1991) 121:S157–8. doi: 10.1093/jn/121.suppl_11.S157
19. French, AT, Ogden, R, Eland, C, Hemani, G, Pong-Wong, R, Corcoran, B, et al. Genome-wide analysis of mitral valve disease in cavalier king Charles spaniels. Vet J. (2012) 193:283–6. doi: 10.1016/j.tvjl.2011.09.011
20. Hedhammar, ÅA, Malm, S, and Bonnett, B. International and collaborative strategies to enhance genetic health in purebred dogs. Vet J. (2011) 189:189–96. doi: 10.1016/j.tvjl.2011.06.018
21. Karlsson, EK, Baranowska, I, Wade, CM, Salmon Hillbertz, NH, Zody, MC, Anderson, N, et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. (2007) 39:1321–8. doi: 10.1038/ng.2007.10
22. Karlsson, EK, and Lindblad-Toh, K. Leader of the pack: gene mapping in dogs and other model organisms. Nat Rev Genet. (2008) 9:713–25. doi: 10.1038/nrg2382
23. Bergholtz, H, Lien, T, Lingaas, F, and Sørlie, T. Comparative analysis of the molecular subtype landscape in canine and human mammary gland tumors. J Mammary Gland Biol Neoplasia. (2022) 27:171–83. doi: 10.1007/s10911-022-09523-9
24. Borge, K, Børresen-Dale, A, and Lingaas, F. Identification of genetic variation in 11 candidate genes of canine mammary tumour. Vet Comp Oncol. (2011) 9:241–50. doi: 10.1111/j.1476-5829.2010.00250.x
25. Deng, N, Zhou, H, Fan, H, and Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget. (2017) 8:110635:–110649. doi: 10.18632/oncotarget.22372
26. Pagenstecher, C, Wehner, M, Friedl, W, Rahner, N, Aretz, S, Friedrichs, N, et al. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. (2006) 119:9–22. doi: 10.1007/s00439-005-0107-8
27. Li, L, Sun, R, Liang, Y, Pan, X, Li, Z, Bai, P, et al. Association between polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of colorectal cancer. J Exp Clin Cancer Res. (2013) 32:1–7. doi: 10.1186/1756-9966-32-104
28. Xiong, HY, Alipanahi, B, Lee, LJ, Bretschneider, H, Merico, D, Yuen, RK, et al. The human splicing code reveals new insights into the genetic determinants of disease. Science. (2015) 347:1254806. doi: 10.1126/science.1254806
29. Cuenda, A. Mitogen-activated protein kinase kinase 4 (MKK4). Int J Biochem Cell Biol. (2000) 32:581–7. doi: 10.1016/S1357-2725(00)00003-0
30. Liu, S, Huang, J, Zhang, Y, Liu, Y, Zuo, S, and Li, R. MAP2K4 interacts with vimentin to activate the PI3K/AKT pathway and promotes breast cancer pathogenesis. Aging (Albany NY). (2019) 11:10697–710. doi: 10.18632/aging.102485
31. Howarth, KD, Mirza, T, Cooke, SL, Chin, S-F, Pole, JC, Turro, E, et al. NRG1 fusions in breast cancer. Breast Cancer Res. (2021) 23:1–12. doi: 10.1186/s13058-020-01377-5
32. Tan, M, and Yu, D. Molecular mechanisms of erbB2-mediated breast cancer chemoresistance. Adv Exp Med Biol. (2007) 608:119–29. doi: 10.1007/978-0-387-74039-3_9
33. Li, K, Ma, L, Sun, Y, Li, X, Ren, H, Tang, S-C, et al. The immunotherapy candidate TNFSF4 may help the induction of a promising immunological response in breast carcinomas. Sci Rep. (2021) 11:18587. doi: 10.1038/s41598-021-98131-4
34. Su, W, Zhu, S, Chen, K, Yang, H, Tian, M, Fu, Q, et al. Overexpressed WDR3 induces the activation of hippo pathway by interacting with GATA4 in pancreatic cancer. J Exp Clin Cancer Res. (2021) 40:1–15. doi: 10.1186/s13046-021-01879-w
35. McMahon, M, Ayllon, V, Panov, KI, and O’Connor, R. Ribosomal 18 S RNA processing by the IGF-I-responsive WDR3 protein is integrated with p53 function in cancer cell proliferation. J Biol Chem. (2010) 285:18309–18. doi: 10.1074/jbc.M110.108555
36. Yu, J, Liu, T-T, Liang, L-L, Liu, J, Cai, H-Q, Zeng, J, et al. Identification and validation of a novel glycolysis-related gene signature for predicting the prognosis in ovarian cancer. Cancer Cell Int. (2021) 21:1–14. doi: 10.1186/s12935-021-02045-0
37. Hanahan, D, and Weinberg, RA. Hallmarks of cancer: the next generation. Cells. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
38. Pavlova, NN, and Thompson, CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
39. Munkley, J, and Elliott, DJ. Hallmarks of glycosylation in cancer. Oncotarget. (2016) 7:35478–89. doi: 10.18632/oncotarget.8155
40. Liu, Y, Zhang, Z, Wang, J, Chen, C, Tang, X, Zhu, J, et al. Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. Onco Targets Ther. (2019) 12:1195–204. doi: 10.2147/OTT.S189687
41. Gupta, SC, Singh, R, Asters, M, Liu, J, Zhang, X, Pabbidi, MR, et al. Regulation of breast tumorigenesis through acid sensors. Oncogene. (2016) 35:4102–11. doi: 10.1038/onc.2015.477
42. Gatenby, RA, and Gillies, RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. (2004) 4:891–9. doi: 10.1038/nrc1478
43. Abbey, CK, Borowsky, AD, McGoldrick, ET, Gregg, JP, Maglione, JE, Cardiff, RD, et al. In vivo positron-emission tomography imaging of progression and transformation in a mouse model of mammary neoplasia. Proc Natl Acad Sci. (2004) 101:11438–43. doi: 10.1073/pnas.0404396101
44. Gupta, SC, Singh, R, Pochampally, R, Watabe, K, and Mo, Y-Y. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-κB pathway. Oncotarget. (2014) 5:12070–82. doi: 10.18632/oncotarget.2514
45. Jardin, I, Nieto-Felipe, J, Alvarado, S, Diez-Bello, R, Lopez, JJ, Salido, GM, et al. SARAF and EFHB modulate store-operated Ca2+ entry and are required for cell proliferation, migration and viability in breast Cancer cells. Cancers. (2021) 13:4160. doi: 10.3390/cancers13164160
46. Miller, WL, and Bose, HS. Early steps in steroidogenesis: intracellular cholesterol trafficking: thematic review series: genetics of human lipid diseases. J Lipid Res. (2011) 52:2111–35. doi: 10.1194/jlr.R016675
47. Manna, PR, Stetson, CL, Slominski, AT, and Pruitt, K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine. (2016) 51:7–21. doi: 10.1007/s12020-015-0715-6
48. Manna, PR, Ahmed, AU, Vartak, D, Molehin, D, and Pruitt, K. Overexpression of the steroidogenic acute regulatory protein in breast cancer: regulation by histone deacetylase inhibition. Biochem Biophys Res Commun. (2019) 509:476–82. doi: 10.1016/j.bbrc.2018.12.145
49. Chumsri, S, Howes, T, Bao, T, Sabnis, G, and Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol. (2011) 125:13–22. doi: 10.1016/j.jsbmb.2011.02.001
50. Lee, K, Kruper, L, Dieli-Conwright, CM, and Mortimer, JE. The impact of obesity on breast cancer diagnosis and treatment. Curr Oncol Rep. (2019) 21:1–6. doi: 10.1007/s11912-019-0787-1
51. Sørlie, T, Perou, CM, Tibshirani, R, Aas, T, Geisler, S, Johnsen, H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
Keywords: canine, genome-wide association study, Maltese, mammary gland tumor, single-nucleotide polymorphism
Citation: Kim K, Song JE, Joo JB, Park HA, Choi CH, Je CY, Kim OK, Park SW, Do YJ, Hur T-Y, Park S-I and Lee C-M (2023) Genome-wide association study of mammary gland tumors in Maltese dogs. Front. Vet. Sci. 10:1255981. doi: 10.3389/fvets.2023.1255981
Edited by:
Naoki Miura, Kagoshima University, JapanReviewed by:
Helga Bergholtz, Oslo University Hospital, NorwayMd Mahfuzur Rahman, University of Wisconsin–Madison, United States
Copyright © 2023 Kim, Song, Joo, Park, Choi, Je, Kim, Park, Do, Hur, Park and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang-Ik Park, c2lwYXJrQGpudS5hYy5rcg==; Chang-Min Lee, Y21sZWUxMTIyQGpudS5hYy5rcg==
†These authors have contributed equally to this work and share first authorship