 Yafang Liu
Yafang Liu Bin Chen1
Bin Chen1 Xing Yang
Xing Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 16 June 2023
Sec. Parasitology
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1218488
This article is part of the Research Topic Parasitism: The Good, The Bad and The Ugly View all 14 articles
Fleas are one of the most common ectoparasites in warm-blooded mammals and an important vector of zoonotic diseases with serious medical implications. We sequenced the complete mitochondrial genomes of Ceratophyllus anisus and Leptopsylla segnis for the first time using high-throughput sequencing and constructed phylogenetic relationships. We obtained double-stranded circular molecules of lengths 15,875 and 15,785 bp, respectively, consisting of 13 protein-coding genes, 22 transfer RNAs, 2 ribosomal RNAs, and two control regions. AT-skew was negative in both C. anisus (−0.022) and L. segnis (−0.231), while GC-skew was positive in both (0.024/0.248), which produced significant differences in codon usage and amino acid composition. Thirteen PCGs encoding 3,617 and 3,711 codons, respectively, isoleucine and phenylalanine were used most frequently. The tRNA genes all form a typical secondary structure. Construction of phylogenetic trees based on Bayesian inference (BI) and maximum likelihood (ML) methods for PCGs. The results of this study provide new information for the mitochondrial genome database of fleas and support further taxonomic studies and population genetics of fleas.
Fleas are the most notorious blood-sucking ectoparasites that transmit a variety of zoonotic pathogens to infect domestic and wild animals (1). Fleas can host a wide range of species, including human, domestic pets, rodents, and ungulates, resulting in a shortened host lifespan, while ecologically related hosts might share flea species, which increases the public health impact of fleas and causes serious economic losses (2). Fleas play a significant role in maintaining the plague’s natural foci (3). Several important pathogens have been identified in fleas, including plague, epidemic hemorrhagic fever, tularemia, leptospirosis, and murine typhus (4). Due to the significant medical, veterinary, and economic value of fleas, increasing attention has been placed on diseases transmitted by fleas, which currently cost global more than $15 billion annually (5). However, to date, phylogenetic relationships and clear classification among fleas have remained ambiguous, which has greatly hindered the control of fleas.
Ceratophyllus anisus belonging to the Siphonaptera order, Ceratophyllidae family, Ceratophyllus genus, is the main vector of plague. C. anisus can parasitize Rattus norvegecus, Rattus tanezumi, Apodemus agrarius, and Crocidura attenuata, and occasionally human. C. anisus has been found in the former Soviet Union, Korea, and Japan, and is most widely distributed in Yunnan Province, China (6). Leptopsylla segnis, in the genus Leptopsylla of the family Leptopsyllidae, has been reported in Libya and Cyprus and is common in numerous provinces of China, where wild rodents are most often infested by it (7, 8). Due to the large number of pathogens carried by fleas and the low resolution of traditional taxonomic features for fleas, the choice of a rapid and accurate identification method is necessary for the control of fleas and flea-borne diseases (9).
With the development of molecular and phylogenetic studies, the mitochondrial genome has been used in ectoparasites taxonomy, systematics, and population genetics, which to some extent make up for the limitations of traditional morphology (10, 11). The mitochondrion, the organelles of eukaryotic cells that maintain the structure of life, possess their genetic material and can replicate autonomously outside the nucleus. Mitochondrial DNA (mtDNA) has the advantages of simple structure, little recombination, fast evolution rate, and maintains inheritance in the process of evolution, which makes it an effective tool for studying species identification, relatedness, and phylogeny (12, 13). However, the mitochondrial genome data of fleas is still extremely scarce, and the phylogenetic relationship has not been established, which is a major obstacle to the prevention and control of fleas and flea-borne diseases.
This study is the first to sequence and analyze the mitochondrial genomes of C. anisus and L. segnis, with the aim of contributing to their correct identification and classification, enriching the mitochondrial genome database of fleas, facilitating the prevention and control of diseases caused by them to minimize the risk to hosts and humans, and providing new and useful markers for further species identification and molecular epidemiological studies. Genetic markers for further species identification and molecular epidemiological studies. We also studied the phylogenetic relationships with the gene sequences of other flea species in NCBI, which provides an important basis for population genetic, phylogenetic and evolutionary analysis.
The C. anisus samples (two females and one male) used in this study were collected in June 2022 from wild R. tanezumi in Laojun Mountain, Lijiang City, Yunnan Province of China (26°53′N, 99°58′E). The adult fleas L. segnis specimens were collected in August 2022 in Jianchuan, Yunnan Province from R. norvegicus (26°57′N, 99°90′E) (one female and one male). Preliminary identification of the collected flea specimens based on morphological diagnostic features (14). One specimen each was selected for subsequent DNA extraction and mitochondrial genome sequencing, while the others were placed in the Museum of Parasitology, Dali University, under voucher numbers DLUP2206 and DLUP2208, respectively. Specimens for experiments were rinsed in 0.9% saline, fixed in 96% alcohol and stored at −80°C until used for DNA extraction (11). DNA extraction was performed on C. anisus and L. segnis samples using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) and following the manufacturer’s instructions.
The mitochondrial genomes of C. anisus and L. segnis were amplified using two sets of overlapping long-fragment RCR primers. Specific PCR primers were constructed from the Xenopsylla cheopis (MW310242) and Pulex irritans (NC063709) mitochondrial genomes, which correspond to the COX1 and 12S rRNA genes, using Primer Premier 5.0 software (CA1: 5′-TTC CCT ACC TGT GCT TGC AG-3′; 3′-AAG AAT TGG ATC TCC CCC GC-5′; CA2: 5′-GCT TGA AAC TTA AAG AAT TTG GCG G-3′, 3′-AAG AGC GAC GGG CAA TAT GT-5′; LS1: 5′-GCA GGA GGG GGT GAT CCT AT-3′; 3′-ATC GTC GAG GTA TTC CTG CT-5′; LS2: 5′-CTT GAA ACT TAA AGA ATT TGG CGG T-3′, 3′-TCC AGT ACA TCT ACT ATG TTA CGA C-5′). The long-range PCR was performed in a total volume of 50 μL, consisting of 10 μL 5× PrimerSTAR GXL Buffer (Takara, Japan), 4 μL of each primer, 4 μL of dNTPs, 1 μL of PrimerSTAR GXL DNA Polymerase (Takara, Japan), 4 μL of DNA template and 23 μL of ddH2O under the following conditions: initial denaturation at 92°C for 2 min, followed by 30 cycles of denaturation at 92°C for 10 s, annealing at 68°C for 30 s and extension at 68°C for 10 min and the final extension step was subjected to 68°C for 10 min. The PCR products were tested using 1% agarose gel electrophoresis, which was purified and sequenced by Sangon Biotech Company (Shanghai, China).
Sequencing on the Illumina NovaSeq platform using AdapterRemoval software to eliminate low-quality data, assembly by software IDBA. The A5-miseq v20150522 program was used to assemble the complete mitochondrial genome and the MITOS WebServer1 was used for genome annotation (15). MITOZ tool for mitochondrial genome prediction and online site tRNAscan-SE2 for secondary structure prediction of transfer RNA (tRNA) (16, 17). Mitochondrial genome circle mapping with CGView Server.3 The software DNAStar V7.1 was used for nucleotide composition analysis and the program CodonW was used to calculate the relative synonymous codon usages (RSCU). The formulas GC-skew = [G – C]/[G + C] and AT-skew = [A – T]/[A + T] were used to measure the relative base content skewness. The total mitogenome informations of C. anisus and L. segnis have been deposited in NCBI.
Mitochondrial lineages from 15 fleas were determined by phylogeny based on the concatenated datasets of 13 PCGs (Table 1). Phylogenetic trees were constructed using the Bayesian inference (BI) and maximum likelihood (ML) methods in the Mrbayes v.3.2.7 (18) and MEGA 7.0 software, respectively. The BI approach selected GTR + G + I as the best model, running 10,000,000 generations in total, sampling every 1,000 generations. The ML analysis was based on 1,000 bootstrapped, which assessed branch reliability and nodal robustness. The resultant tree was visualized and edited with FigTree v1.4.2 (19).
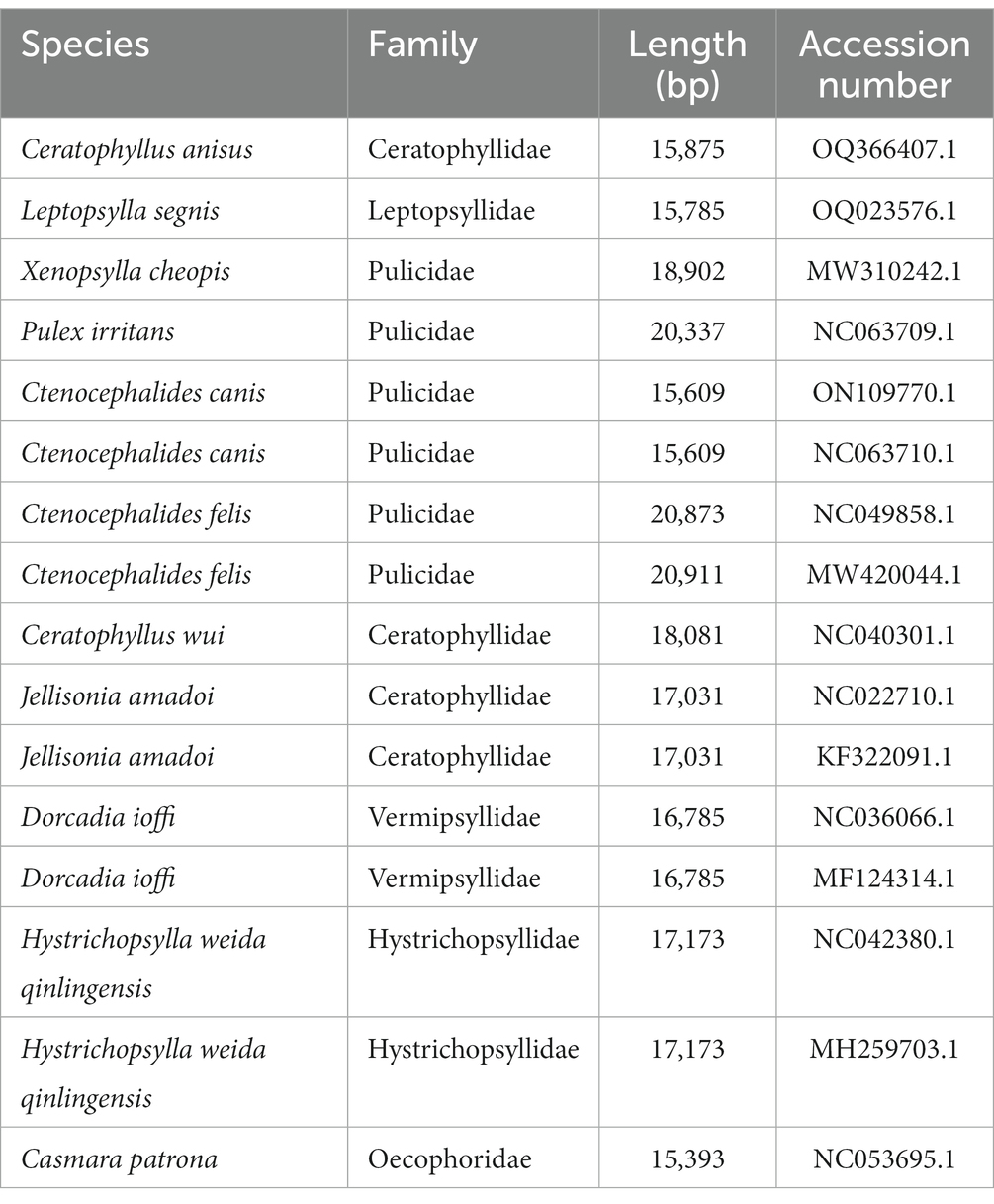
Table 1. List of the 15 flea species and Casmara patrona analyzed in this paper with their GenBank numbers.
The complete mitochondrial genomics of C. anisus and L. segnis were uploaded to Genbank in TBL format under the accession number OQ366407 and OQ023576. The C. anisus and L. segnis genomes are circular molecules of 15,875 bp and 15,785 bp in length, respectively, consisting of 13 protein-coding genes, 22 tRNAs, two rRNAs, and two D-loop (Figure 1). Fourteen tRNA genes and nine PCGs are located in the forward strand (+), and the remaining 14 genes are encoded in the reverse strand (−) (Table 2). The average AT content of C. anisus and L. segnis complete mitochondrial genome is 78.54% (78.89%) and GC content is 21.46% (21.11%), including A = 38.41% (40.37%), T = 40.14% (38.51%), G = 8.25% (13.17%) and C = 13.21% (7.94%; Table 3). The mitochondrial genome of C. anisus has 18 intergenic regions of 929 bp, accounting for 5.85% of the total length, and 13 overlapping regions totaling 28 bp. The genome of L. segnis has 19 spacer areas and 8 overlapping regions with a total of 894 bp and 21 bp (Table 2).
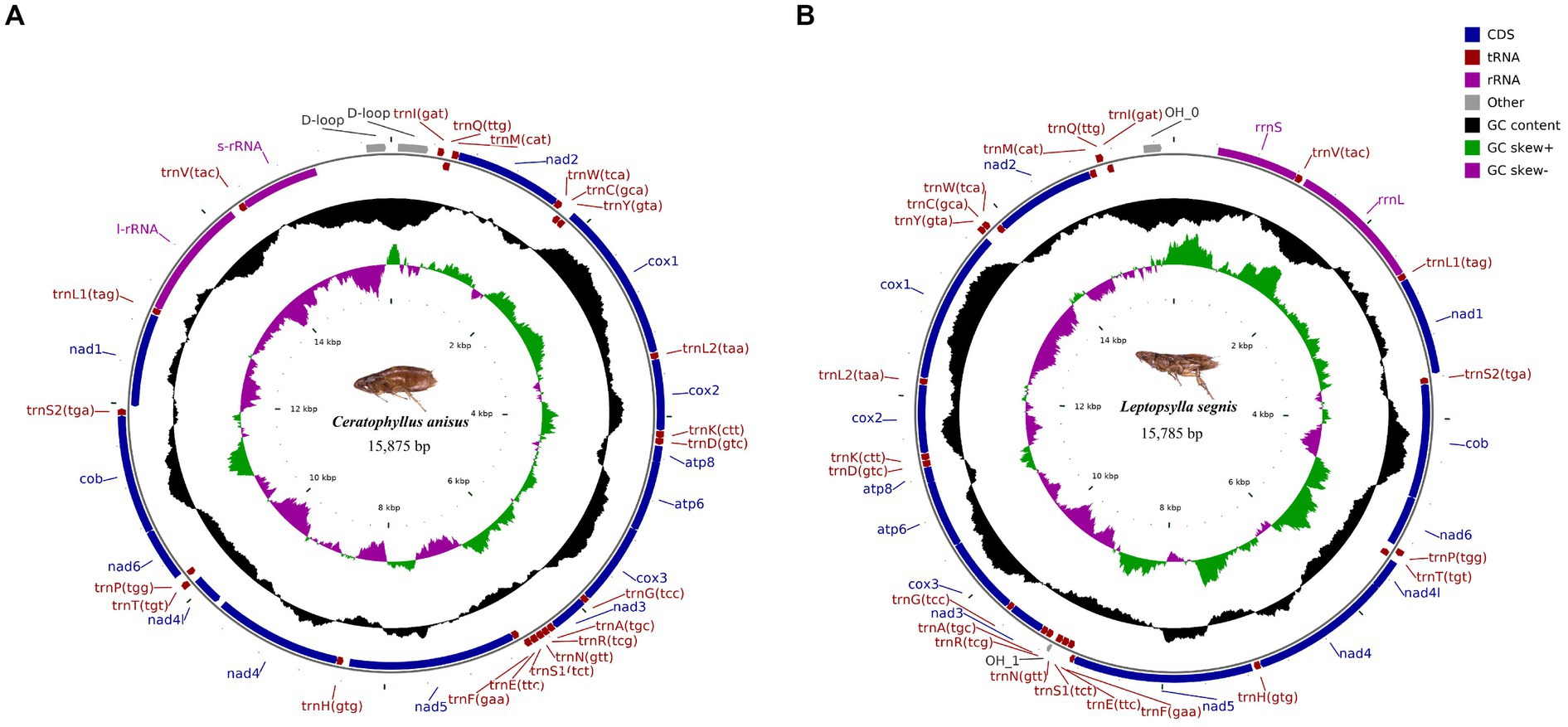
Figure 1. Circular map and organization of the mitochondrial genome of Ceratophyllus anisus (A) and Leptopsylla segnis (B).
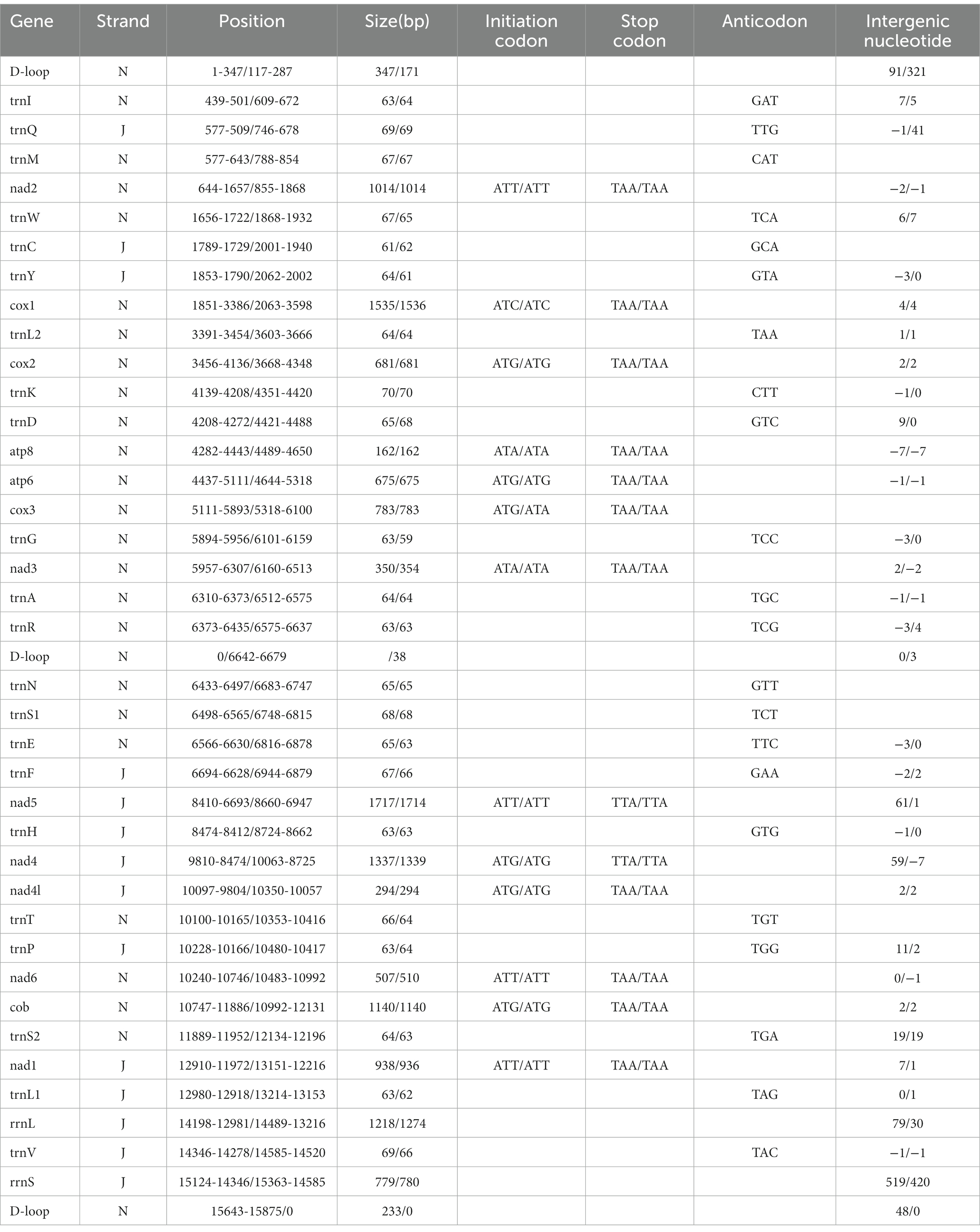
Table 2. Organization of the Ceratophyllus anisus and Leptopsylla segnis mitochondrial genomes.
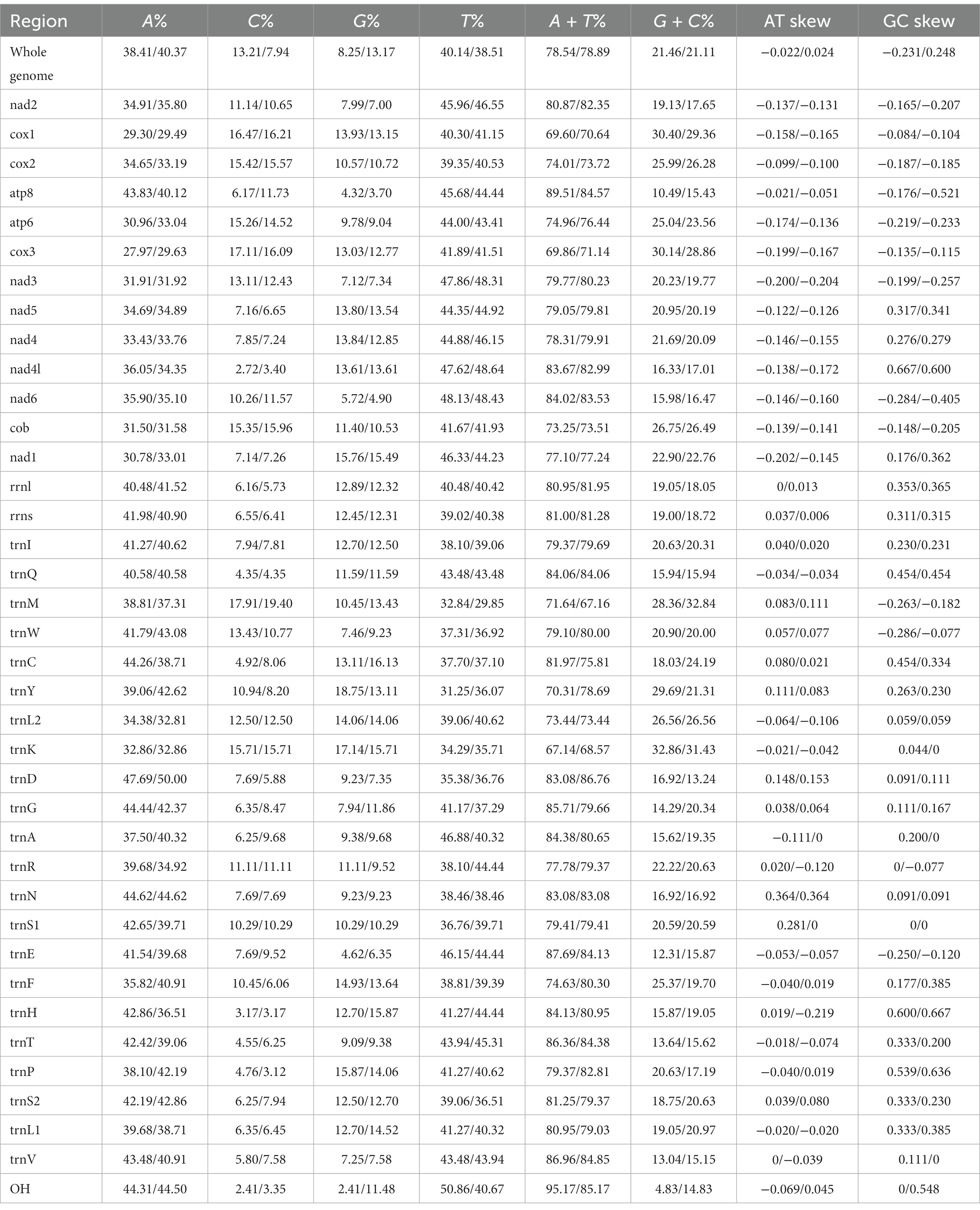
Table 3. Composition and skewness of Ceratophyllus anisus and Leptopsylla segnis mitogenome.
The mitochondrial genomes of C. anisus and L. segnis consist of 13 protein-coding genes, with a total length of 11,014 bp and 11,134 bp, accounting for 69.4% and 70.5% of the total length, respectively. The PCGs of C. anisus use the standard ATN as the initiation codon, and stop codons are TAA except for nad5 and nad4 (TTA). Amino acid utilization and RSCU were calculated for the PCGs of the mitochondrial genomic of C. anisus and L. segnis, encoding 3,617 and 3,711 amino acids, and the most abundant amino acid was found to be Isoleucine and Phenylalanine, accounting for 9.95% and 9.75%, respectively (Figure 2). The AT-skew and GC-skew of the PCGs of the C. anisus range from −0.202 (for NAD1) to −0.021 (for ATP8) and from −0.296 (for NAD3) to 0.320 (for NAD5), respectively. Among the 13 protein-coding genes, both C. anisus and L. segnis had nine genes in the forward chain (NAD2, COX1, COX2, ATP8, ATP6, COX3, NAD3, NAD6, Cob) and four genes in the reverse chain (NAD1, NAD5, NAD4, NAD4L), which are consistent with most metazoans such as fleas and ticks (5, 20).
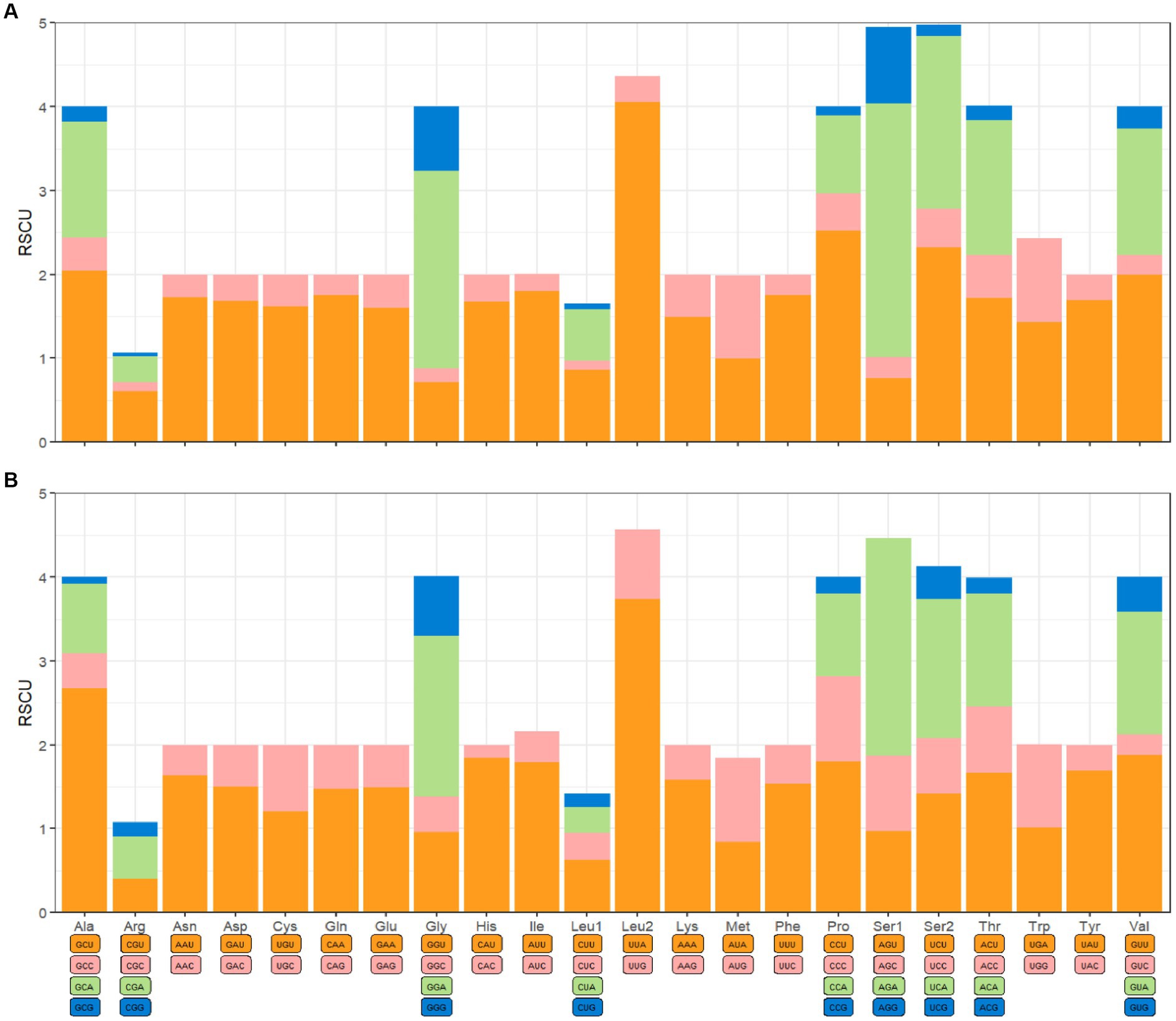
Figure 2. Relative synonymous codon usage (RSCU) of codons. (A) Ceratophyllus anisus and (B) Leptopsylla segnis. The horizontal coordinates represent all codons encoding each amino acid, and the vertical coordinates represent the sum of all RECU values.
The mitochondrial genomes of C. anisus and L. segnis have 22 tRNA genes and two rRNA genes. The length of 22 tRNAs ranged from 61 bp for tRNACys (59 bp for tRNAGly) to 70 bp for tRNALys (70 bp for tRNACys), with a total length of 1,433 bp (1,397 bp). The tRNA genes of both samples can form a complete typical canonical cloverleaf structure. There is an overlap between ATP8 and ATP6 with a length of 7 bp, which is typical of arthropods (21). The 16S rRNA and 12S rRNA genes of the C. anisus and L. segnis were separated by Valine, and were 1,218 bp (1,274 bp) and 779 bp (780 bp) in length, respectively (Table 2), a structure consistent with that reported in the mitogenome of other flea species (22).
To further analyze the phylogenetic relationships of fleas, we added the mitochondrial genomes of C. anisus and L. segnis to the analysis. Phylogenetic trees were constructed using the BI and ML methods for the concatenated nucleotide sequences of 13 PCGs of the mitochondrial genome of 15 fleas and Casmara patrona as an outgroup, and the topologies of the two methods were consistent. According to the topology analysis, C. anisus and Ceratophyllus wui are clustered in a branch with high statistical support and L. segnis is alone in a branch, forming a sister group with other families of fleas (Figure 3). The families Ceratophyllidae, Leptopsyllidae, Vermipsyllidae, Hystrichopsyllidae, and Pulicidae form monophyletic branches, which is consistent with the previous findings (23).
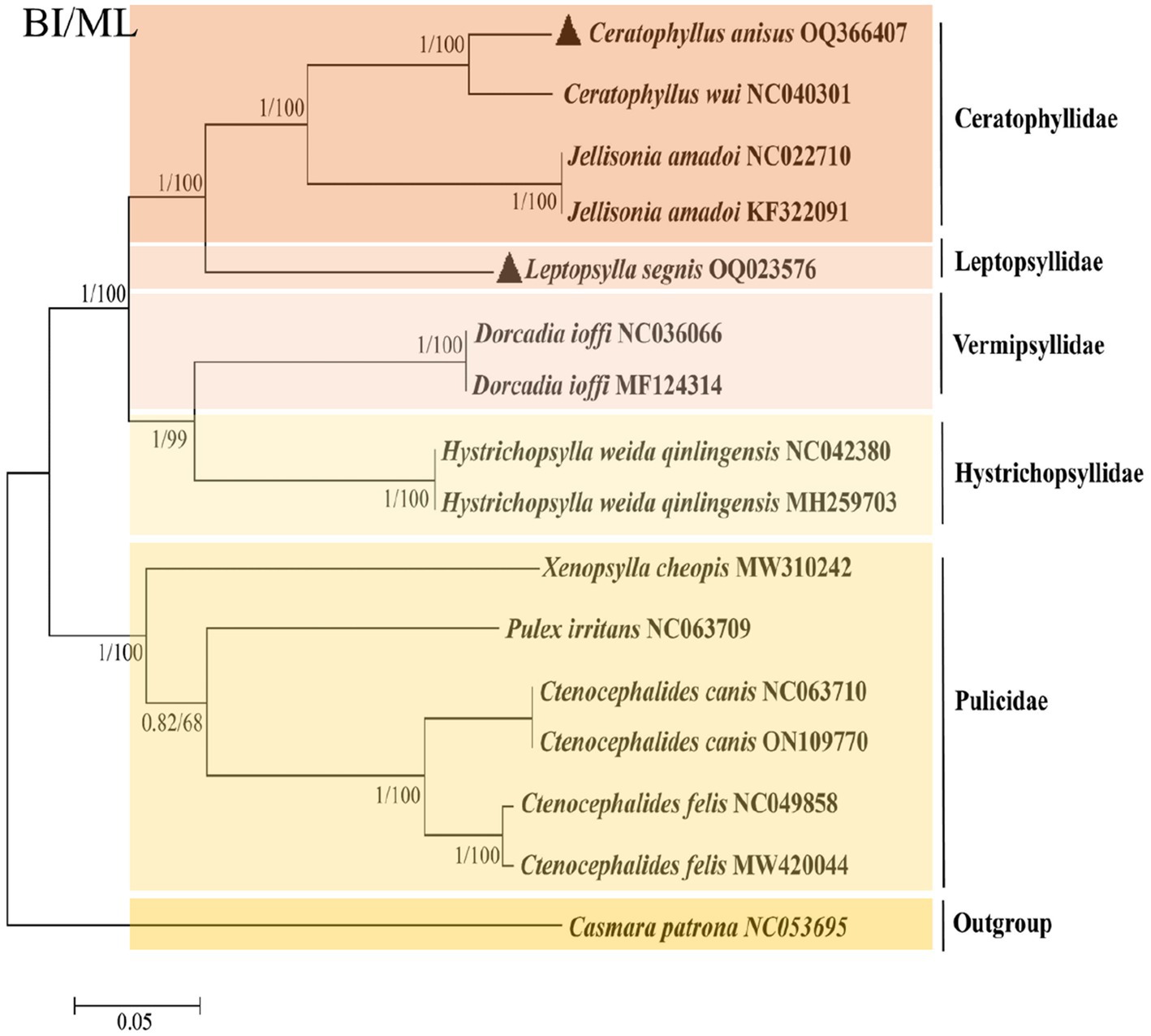
Figure 3. Phylogenetic analysis based on the nucleotide sequences of the 13 PCGs in the mitogenome. Each genus is represented by different colors. The solid black triangle represents the species in this study.
As a temporary host and vector of some important human infectious diseases such as plague and endemic typhus, fleas are early warning indicators for judging the prevalence of plague and other human-animal infectious diseases (24). In recent years, plague has rebounded in some regions, with an increasing trend in incidence, which has always been a persistent and difficult problem worldwide. C. anisus and L. segnis are common species of fleas that play an important role in the transmission of zoonotic diseases.
The D-loop region has low evolutionary pressure, a large number of gene rearrangements, and rapid base substitutions, making it an effective molecular marker for population genetic studies. The frequency and location of the D-loop vary from species to species and tissue to tissue, and its length is influenced by the number of tandem repeat copies, which in turn affects the length of the entire mitogenome (25). One D-loop region was found for X. cheopis, P. irritans, and C. wui, and two D-loops existed for Ctenocephalides felis, C. anisus, and L. segnis. The two control regions were also found in some ticks and sea cucumbers, and the mtDNA was replicated more efficiently, so it is speculated that the two D-loop regions acted synergistically during the evolutionary process (26). The mtDNAs of C. anisus and L. segnis have the same gene composition and arrangement as that of most flea species. Bases mismatch appears in most tRNA genes, and G-U wobble base pairs conform to the oscillating pairing principle, which is very important for maintaining the stability of the tRNA secondary structure (27).
The phylogenetic tree derived using all flea mitochondrial genomic data in the NCBI gene bank shows that the five families are divided into two distinct branches, with Ceratphyllidae, Leptosyllidae, Vermipsyllidae, and Hystrichopsyllidae clustered into one, and the Pulicidae family as the other branch. The branch where L. segnis is located and the C. anisus and C. wui branches form a sister group with high node support. The same species of fleas from different hosts and different geographical locations are clustered together in this phylogenetic tree with posterior probabilities and bootstrap values of 1 and 100, respectively, with a high degree of confidence.
The mtDNA is a valuable marker for population biology, species identification classification, and phylogenetic studies, especially for assessing genetic diversity and identifying cryptic species as well as population structure. There are still gaps in molecular data for C. anisus and L. segnis, which is a major obstacle to the development of flea species. In this study, we obtained the complete mitochondrial genome, which provides more accurate evidence for the phylogenetic relationships of flea species. To better understand the phylogenetic relationship among fleas, the mitochondrial genome study within the Siphonaptera order must be expanded. We expect that the complete mitogenomes of C. anisus and L. segnis will provide important genome information for molecular phylogenetic studies and contribute to clarifying the phylogeny and evolution of Siphonaptera.
In this study, the complete mitochondrial genomes of C. anisus and L. segnis are sequenced and annotated for the first time by long-range PCR combined with Illumina sequencing technology which will be helpful for future research on fleas. The results of this study contributed to the fleas, filling the flea mitochondrial genome database resources and laying the foundation for further understanding the phylogenetic relationships of the fleas. With the development of molecular biology, sequencing techniques using mitochondrial genomes as molecular markers have effectively bridged the morphological gap and have been widely used in species identification, kinship, and evolutionary studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The animal study was reviewed and approved by Laboratory Animal Management Committee of Dali University and First Affiliated Hospital of Chengdu Medical College.
YL conceived the study and wrote the manuscript. BC, XL, and DJ carried out the experiments and analysed the data. TW and LG contributed to the collection of C. anisus and L. segnis and discussions. QZ and XY is responsible for the interpretation of experimental data, critical revision of important knowledge content, and final approval of the version to be published. All authors contributed to the article and approved the submitted version.
This work was supported by the Yunnan Natural Science Foundation (2017FD139) and Scientific Research Fund of Yunnan Education Department (2022J0687).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Maleki-Ravasan, N, Solhjouy-Fard, S, Beaucournu, JC, Laudisoit, A, and Mostafavi, E. The fleas (Siphonaptera) in Iran: diversity, host range, and medical importance. PLoS Negl Trop Dis. (2017) 11:e5206. doi: 10.1317/journal.pntd.0005260
2. Devevey, G, and Christe, P. Flea infestation reduces the life span of the common vole. Parasitology. (2009) 136:1351–5. doi: 10.1017/S0031182009990746
3. Andrianaivoarimanana, V, Kreppel, K, Elissa, N, Duplantier, JM, Carniel, E, Rajerison, M, et al. Understanding the persistence of plague foci in Madagascar. PLoS Negl Trop Dis. (2013) 7:e2382. doi: 10.1371/journal.pntd.0002382
4. Brown, LD. Immunity of fleas (order Siphonaptera). Dev Comp Immunol. (2019) 98:76–9. doi: 10.1016/j.dci.2019.03.019
5. Zhang, Y, Nie, Y, Deng, YP, Liu, GH, and Fu, YT. The complete mitochondrial genome sequences of the cat flea Ctenocephalides felis felis (Siphonaptera: Pulicidae) support the hypothesis that C. felis isolates from China and USA were the same C. f. felis subspecies. Acta Trop. (2021) 217:105880. doi: 10.1016/j.actatropica.2021.105880
6. Shinozaki, Y, Shiibashi, T, Yoshizawa, K, Murata, K, Kimura, J, Maruyama, S, et al. Ectoparasites of the Pallas squirrel, Callosciurus erythraeus, introduced to Japan. Med Vet Entomol. (2004) 18:61–3. doi: 10.1111/j.0269-283x.2004.0475.x
7. Wu, AG, Zhong, YH, Li, YQ, Wu, HS, Pu, EN, Gao, ZH, et al. Survey of Ectoparasitic fleas on Rattus flavipectus and Rattus norvegicus from 34 counties in Yunnan Province, China. Sichuan J Zool. (2015) 34:53–8.
8. Christou, C, Psaroulaki, A, Antoniou, M, Toumazos, P, Ioannou, I, Mazeris, A, et al. Rickettsia typhi and Rickettsia felis in Xenopsylla cheopis and Leptopsylla segnis parasitizing rats in Cyprus. Am J Trop Med Hyg. (2010) 83:1301–4. doi: 10.4269/ajtmh.2010.10-0118
9. Zurita, A, Callejón, R, García-Sánchez, ÁM, Urdapilleta, M, Lareschi, M, and Cutillas, C. Origin evolution, phylogeny and taxonomy of Pulex irritans. Med Vet Entomol. (2019) 33:296–311. doi: 10.1111/mve.12365
10. Yssouf, A, Socolovschi, C, Leulmi, H, Kernif, T, Bitam, I, Audoly, G, et al. Identification of flea species using MALDI-TOF/MS. Comp Immunol Microbiol Infect Dis. (2014) 37:153–7. doi: 10.1016/j.cimid.2014.05.002
11. Lawrence, AL, Brown, GK, Peters, B, Spielman, DS, Morin-Adeline, V, and Šlapeta, J. High phylogenetic diversity of the cat flea (Ctenocephalides felis) at two mitochondrial DNA markers. Med Vet Entomol. (2014) 28:330–6. doi: 10.1111/mve.12051
12. Chen, SY, Qiu, QG, Mo, HL, Gong, TF, Li, F, He, JL, et al. Molecular identification and phylogenetic analysis of Ascarids in wild animals. Front Vet Sci. (2022) 9:891672. doi: 10.3389/fvets.2022.891672
13. Wang, HM, Li, R, Deng, YP, Liu, GH, and Fu, YT. Comparative mitochondrial genomic analysis robustly supported that cat tapeworm Hydatigera taeniaeformis (Platyhelminthes: Cestoda) represents a species complex. Front Vet Sci. (2022) 9:931137. doi: 10.3389/fvets.2022.931137
14. He, JH, Liang, Y, and Zhang, HY. A study on the transmission of plague though seven kinds of fleas in rat type and wild rodent type plague foci in Yunnan. Chin J Epidemiol. (1997) 4:45–9.
15. Meng, G, Li, Y, Yang, C, and Liu, S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. (2019) 47:e63. doi: 10.1093/nar/gkz173
16. Bernt, M, Donath, A, Jühling, F, Externbrink, F, Florentz, C, Fritzsch, G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. (2013) 69:313–9. doi: 10.1016/j.ympev.2012.08.023
17. Lowe, TM, and Eddy, SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. (1997) 25:955–64. doi: 10.1093/nar/25.5.955
18. Ronquist, F, and Huelsenbeck, JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. (2003) 19:1572–4. doi: 10.1093/bioinformatics/btg180
19. Chen, Y, Chen, S, Kang, J, Fang, H, Dao, H, Guo, W, et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PLoS One. (2014) 9:e107578. doi: 10.1371/journal.pone.0107578
20. Lu, XY, Zhang, QF, Jiang, DD, Liu, YF, Chen, B, Yang, SP, et al. Complete mitogenomes and phylogenetic relationships of Haemaphysalis nepalensis and Haemaphysalis yeni. Front Vet Sci. (2022) 9:1007631. doi: 10.3389/fvets.2022.1007631
21. Sun, ET, Li, CP, Nie, LW, and Jiang, YX. The complete mitochondrial genome of the brown leg mite, Aleuroglyphus ovatus (Acari: Sarcoptiformes): evaluation of largest non-coding region and unique tRNAs. Exp Appl Acarol. (2014) 64:141–57. doi: 10.1007/s10493-014-9816-9
22. Hornok, S, Beck, R, Farkas, R, Grima, A, Otranto, D, Kontschán, J, et al. High mitochondrial sequence divergence in synanthropic flea species (Insecta: Siphonaptera) from Europe and the Mediterranean. Parasit Vectors. (2018) 11:221. doi: 10.1186/s13071-018-2798-4
23. Wei, F, Jia, X, Wang, Y, Yang, Y, Wang, J, Gao, C, et al. The complete mitochondrial genome of Xenopsylla cheopis (Siphonaptera: Pulicidae). Mitochondrial DNA B. (2022) 7:170–1. doi: 10.1080/23802359.2021.2017368
24. Sebbane, F, and Lemaître, N. Antibiotic therapy of plague: a review. Biomol Ther. (2021) 11:724. doi: 10.3390/biom11050724
25. Nicholls, TJ, and Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. (2014) 56:175–81. doi: 10.1016/j.exger.2014.03.027
26. Shao, R, Aoki, Y, Mitani, H, Tabuchi, N, Barker, SC, and Fukunaga, M. The mitochondrial genomes of soft ticks have an arrangement of genes that has remained unchanged for over 400 million years. Insect Mol Biol. (2004) 13:219–24. doi: 10.1111/j.0962-1075.2004.00447.x
Keywords: fleas, Ceratophyllus anisus, Leptopsylla segnis, mitogenome, phylogenetic
Citation: Liu Y, Chen B, Lu X, Jiang D, Wang T, Geng L, Zhang Q and Yang X (2023) Complete mitogenomes characterization and phylogenetic analyses of Ceratophyllus anisus and Leptopsylla segnis. Front. Vet. Sci. 10:1218488. doi: 10.3389/fvets.2023.1218488
Edited by:
Guillermo Tellez-Isaias, University of Arkansas, United StatesReviewed by:
Roberto Senas Cuesta, University of Arkansas, United StatesCopyright © 2023 Liu, Chen, Lu, Jiang, Wang, Geng, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanfu Zhang, MjYyMjA0NTgwN0BxcS5jb20=; Xing Yang, eWFuZzA4MjIwMDEzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.