 Sayed Zamiti
Sayed Zamiti Moez Mhadhbi1
Moez Mhadhbi1 Mohamed Aziz Darghouth
Mohamed Aziz Darghouth Mourad Ben Said
Mourad Ben Said- 1Laboratory of Parasitology, National School of Veterinary Medicine of Sidi Thabet, University of Manouba, Manouba, Tunisia
- 2Department of Basic Sciences, Higher Institute of Biotechnology of Sidi Thabet, University of Manouba, Manouba, Tunisia
Introduction: Hyalomma and Rhipicephalus ticks are important genera that can transmit diseases to both animals and humans, including Crimean-Congo hemorrhagic fever, tick-borne encephalitis, and several types of spotted fever. The accurate identification of tick species is essential for the effective control and prevention of tick-borne diseases. However, traditional identification methods based on morphology can be challenging and subjective, leading to errors. The development of DNA markers has provided more precise and efficient methods for tick species identification, but the currently available markers have limitations in their discriminatory power and sensitivity. To address this need for more sensitive and specific markers, this study aimed to identify two minimum sequence fragments required for tick Hyalomma and Rhipicephalus species identification using the Bm86 cDNA marker, which has previously been shown to be in perfect agreement with the current taxonomy of hard ticks based on its complete sequence.
Methods: Based on our in silico determination that a minimum sequence of 398 bp for Rhipicephalus spp. (from 1487 to 1884) and 559 bp for Hyalomma species (from 539 to 1097) was necessary for species delineation, two distinct PCR assays were developed to apply these sequences in practice.
Results and discussion: Discrimination between species within each genus was achieved through sequence homology and phylogenetic analysis following the sequencing of the two PCR products. Subsequently, their performance was evaluated by testing them on the field-collected ticks of the Hyalomma and Rhipicephalus genera obtained from various host animals in different geographic regions of Tunisia. The use of shorter partial sequences specific to the tick genera Rhipicephalus and Hyalomma, which target the tick's RNA banks, could represent a significant advance in the field of tick species identification, providing a sensitive and discriminatory tool for interspecific and intraspecific diversity analysis.
1. Introduction
Rhipicephalus is a genus of ticks that includes several species known to transmit a range of pathogens to both animals and humans (1). Some of the most common species of Rhipicephalus ticks include R. sanguineus sensu lato, R. microplus, and R. annulatus (2). These ticks are distributed in different regions worldwide, with R. sanguineus s.l. found in the temperate and tropical regions and R. microplus and R. annulatus being more commonly found in tropical and subtropical regions (3). Rhipicephalus ticks are known to transmit various pathogens, including bacteria, viruses, and protozoans, such as Crimean-Congo hemorrhagic fever virus, Babesia, Anaplasma, Ehrlichia, and Rickettsia species, which can cause diseases such as Crimean-Congo hemorrhagic fever, Lyme disease, Rocky Mountain spotted fever, and other tick-borne illnesses (3, 4).
The genus Hyalomma comprises several species of hard ticks that are widely distributed in Africa, Asia, and Europe (5). These ticks are known to transmit a number of pathogens, including viruses, bacteria, and protozoa, some of which can cause severe diseases in humans and animals (3). Hyalomma ticks are important vectors of the Crimean-Congo hemorrhagic fever virus, a highly pathogenic virus that can cause severe hemorrhagic fever in humans (6, 7). They are also known to transmit several other viral pathogens, such as the Nairobi sheep disease virus and the Alkhumra virus (8). In addition, Hyalomma ticks can transmit bacterial pathogens such as Rickettsia, Ehrlichia, and Anaplasma, as well as protozoan pathogens such as Theileria and Babesia (9–12). Some Hyalomma species are also associated with the transmission of tick-borne encephalitis virus, a flavivirus that causes a range of neurological symptoms in humans (13).
The identification of Rhipicephalus and Hyalomma species using morphological diagnosis keys might be constrained by several factors (14). For instance, species from the R. sanguineus group have very similar morphologies, making their distinction quite laborious (15) and requiring therefore advanced taxonomic knowledge (i.e., male sclerites) for reliable species identification. In addition, tick species morphological diagnosis could be rendered problematic due to the alteration of some key morphological traits during the course of engorgement, in particular, for female ticks. Moreover, some species may exhibit significant variations in morphology depending on their geographic location or the host they are feeding on (16). Finally, in comparison to adult ticks, species identification is more problematic with juvenile stages of Rhipicephalus and Hyalomma due to the absence of expression of some key morphological features present in adults. Accordingly, morphological diagnosis can be time-consuming, requiring specialized knowledge and training, and these taxonomic competences may not be readily available in some regions (17). These challenges underscore the importance of incorporating additional approaches to morphological diagnosis, including molecular and genetic techniques, to identify and classify tick species more accurately (17).
The use of DNA markers has significantly improved the reliability of the identification of tick species, including those of the genera Hyalomma and Rhipicephalus (18). Compared to morphological diagnosis, DNA-based identification was shown, for several tick species, to be highly accurate and able to differentiate between closely related species (19). Some of the DNA markers used for species identification in these genera include mitochondrial DNA (mtDNA) genes, such as cytochrome c oxidase subunit 1 (CO1) and 16S rRNA, as well as nuclear DNA markers like the internal transcribed spacer (ITS) region (20–22). These markers were successfully applied to species identification for instance with Hyalomma dromedarii and Rhipicephalus microplus (23–25).
However, and despite their usefulness, there are limitations to the use of existing DNA markers for tick species identification (26, 27). Indeed, there are still gaps in the available DNA sequence databases for some tick species, emphasizing accordingly, the need to design more reliable molecular markers for their identification (28). Moreover, the existence of hybridization and introgression events between different tick species can further complicate the identification process. Therefore, the development of more comprehensive DNA sequence databases and the use of multiple molecular markers can help to increase the accuracy and reliability of tick species identification (29).
In this context, the present study targeted an mRNA-based marker as a complementary tool to morphological diagnosis and to genomic and/or mitochondrial DNA markers. Unlike genomic DNA markers, which have several limitations, mRNA markers provide more specific and sensitive information for species identification. mRNA is a complementary copy of a gene that is used as a template for protein synthesis (30). mRNA-based markers are highly conserved among species and are often more species-specific than genomic markers (31, 32). These markers can be used in combination with traditional morphological methods and other molecular markers to improve the accuracy of tick species identification.
Our study focused on identifying and validating mRNA-based markers for the diagnosis of Hyalomma and Rhipicephalus species. The rational base of our study was to determine the minimum length of partial Bm86 cDNA fragments required for species identification in two tick genera, namely Rhipicephalus and Hyalomma. The decision to use Bm86 cDNA encoding the vaccine target gut protein is justified by the fact that the complete cDNA sequence perfectly aligns with the recent classification of hard ticks (31). This strategy offers several expected benefits, including the potential to detect several tick species using a single molecular marker (33). Although changes in climate and in the length of the different seasons will directly affect tick survival, activity, and development, our mRNA-based markers can be applied to both engorged and non-engorged ticks, making it useful for analyzing ticks in various developmental stages and environmental settings (34, 35).
Therefore, the aims of this study were to conduct an in silico analysis to identify the minimum partial Bm86 cDNA sequence required for the identification of species within the Hyalomma and Rhipicephalus genera, to develop a molecular method based on the amplification and sequencing of these two minimal sequences, and to apply this technique for the identification of tick field specimens belonging to the two genera collected from several hosts located in different Tunisian regions.
2. Materials and methods
2.1. Sequence and data acquisition from GenBank
In this study, a selection of minimum length partial Bm86 cDNA fragments required for species delineation within Rhipicephalus and Hyalomma tick genera was performed. This selection was based on all complete or nearly complete Bm86 cDNA sequences available in GenBank on 1 January 2023. All sequences were screened and downloaded from GenBank (http://www.ncbi.nlm.nih.gov/GenBank/) through a BLAST analysis (maximum and minimum recovery of 100 and 68%, respectively). In particular, 175 isolates or strains belonging to the six classified Rhipicephalus species and 19 isolates or strains belonging to the five classified Hyalomma species, which is available in the GenBank® database of NCBI (http://www.ncbi.nlm.nih.gov/nuccore/), were analyzed in this study (36–45) (Supplementary Tables 1–3).
2.2. Multiple sequence alignments
Multiple sequence alignments and sequence similarities were calculated using the CLUSTAL W 1.81. The DNAMAN software (Version 5.2.2; Lynnon Biosoft, Que., Canada) was also used to create two Bm86 sequences' alignment profiles. Partial Bm86 sequences that ensure effective discrimination between species of Rhipicephalus and Hyalomma genera were selected to investigate interspecific diversity positions with the analysis of sequence alignments (Figures 1, 2).

Figure 1. Multiple alignment profiles allowing the selection of minimum length Bm86 cDNA fragments required for species delineation within Rhipicephalus tick genera. 1Rhipicephalus species analyzed in each alignment profile. 2Alignment profiles of all available genetic variants isolated from each Rhipicephalus species. 3Average percent identity between genetic variants of each Rhipicephalus species.

Figure 2. Multiple alignment profiles allowing the selection of minimum length Bm86 cDNA fragments required for species delineation within Hyalomma tick genera. 1Hyalomma species analyzed in each alignment profile. 2Alignment profiles of all available genetic variants isolated from each Hyalomma species. 3Average percent identity between genetic variants of each Hyalomma species.
2.3. Collection and species identification of tick samples
Between June and August 2020, adult tick specimens were collected from four different bioclimatic areas and five governorates (Table 1). Specimens were hand-picked from live domestic animals (e.g., cattle, camels, goats, sheep, and dogs). Tick specimens were identified based on morphological characteristics according to the identification keys of Walker et al. (46). Tick samples were preserved in 1 ml of TRIzol reagent (Invitrogen life technologies, Invitrogen Corporation, California, USA) and stored at −80°C for subsequent uses.
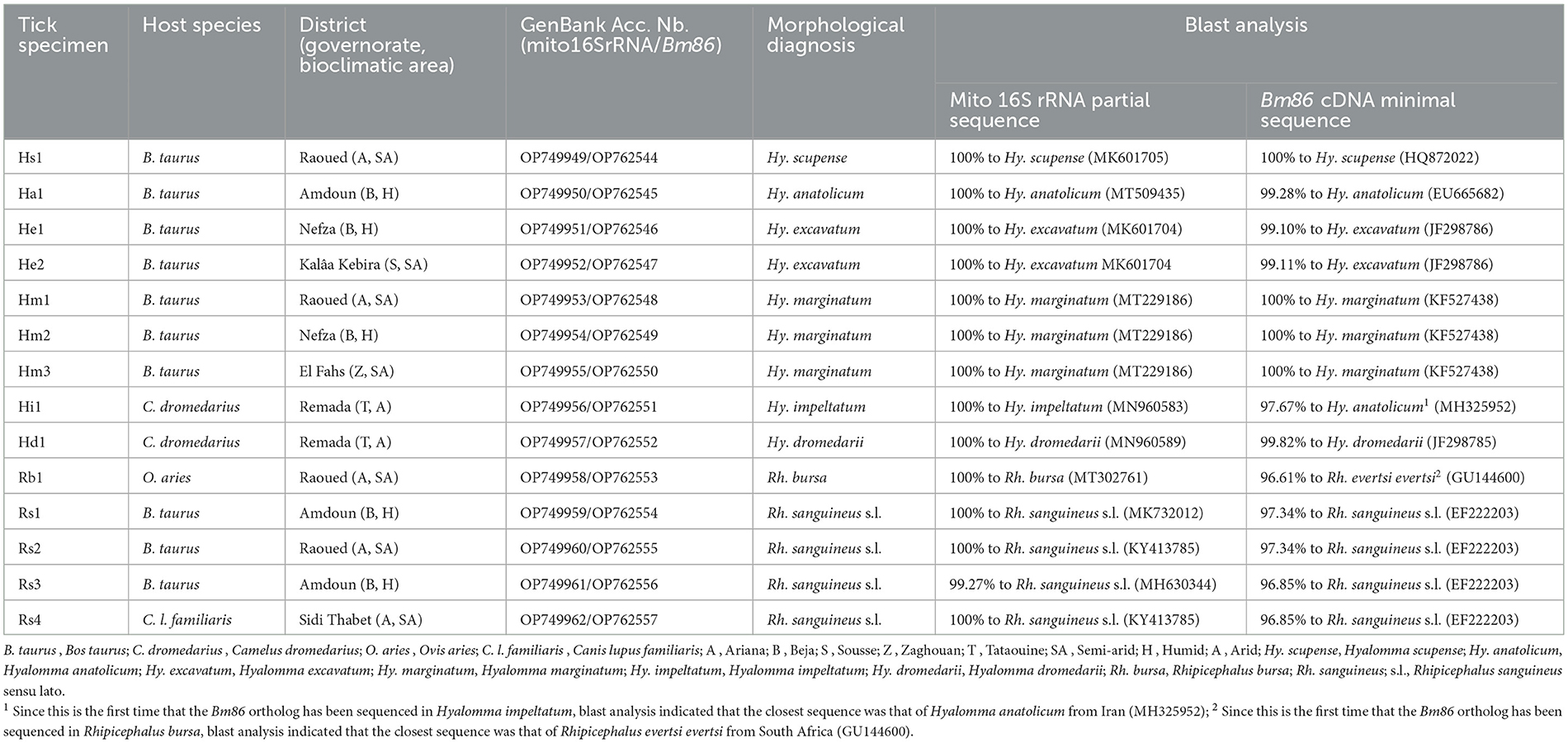
Table 1. Information about origins and hosts of studied field tick specimens belonging to Hyalomma and Rhipicephalus genera from Tunisia and blast analysis of sequenced mitochondrial 16S rRNA and Bm86 cDNA fragments.
2.4. Total RNA and genomic DNA co-extraction
A total of 14 field tick specimens belonging to Rhipicephalus and Hyalomma genera were selected for the field evaluation of minimum length Bm86 cDNA fragments. DNA and RNA co-extraction from a single adult tick specimen were performed using TRIzol reagent (Invitrogen, CA, USA). Each whole tick was crashed and homogenized in TRIzol reagent (Invitrogen, CA, USA) (1 mL/100 mg tissue) using a pestle and liquid nitrogen. Phases' separation was performed by adding chloroform in a TRIzol to chloroform ratio of 3:1 according to the manufacturer's instructions. For each sample, the aqueous phase was separated from the interphase/organic phase and subjected to RNA isolation according to the TRIzol reagent protocol. The final resuspension of total RNA was performed in 50 μl of RNase-free water, followed by incubation at 56°C for 10 min for complete resolubilization of the nucleic acid. RNA samples were immediately stored at −80°C until further use. The DNA samples present in the interphase/organic phase were purified according to the TRIzol reagent according to the manufacturer's instructions. DNA isolation was carried out with a final resuspension in 0.1–0.2 ml of 8 mM NaOH. Insoluble materials were pelleted by centrifugation for 10 min at 12,000 × g at 4°C. The supernatant was transferred to a new 1.5 ml tube and stored at −20°C until use.
2.5. Amplification of mitochondrial 16s rRNA partial sequences
To confirm the results of the morphological diagnosis, a partial mitochondrial 16S rRNA sequence (273 bp) was amplified from all analyzed tick specimens by using primers TQ16S+1F (5′-CTGCTCAATGATTTTTTAAATTGCTGTGG-3′) and TQ16S-2R (5′-ACGCTGTTATCCCTAGAG−3′), as previously described by Black and Piesman (47).
2.6. cDNA synthesis and amplification of Bm86 partial sequences
The first-strand synthesis reaction was carried out using the SuperScript First-strand Synthesis System for the RT-PCR kit (Invitrogen USA) following the manufacturer's instructions. Two primer sets employed to amplify the selected minimum length partial Bm86 cDNA fragments needed for species delineation within Rhipicephalus and Hyalomma tick genera were designed, and the position of each primer was obtained with respect to the Bm86 cDNA sequence isolated from the R. (B.) australis (formerly R. microplus) Yeerongpilly strain (GenBank Accession number M29321) and the Ha98 cDNA sequence isolated from the H. anatolicum India strain (GenBank accession number AF347079), for Rhipicephalus and Hyalomma tick genera, respectively (Table 2).
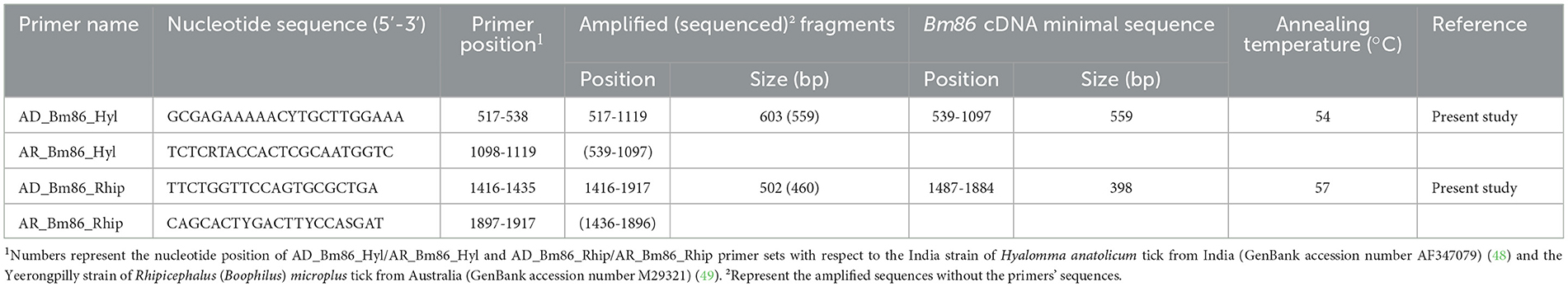
Table 2. Primers employed to amplify and sequence Bm86 cDNA minimal sequence used for species delineation within Hyalomma and Rhipicephalus genera.
All PCRs were performed in a final volume of 50 μl containing 0.125 U/μl of Taq DNA polymerase, 1 × PCR buffer 10 × , 2.5 mM of MgCl2, 0.2 mM of dNTPs, 4 μl (50 to 150 ng) cDNA, and 0.5 μM of primers. PCR reactions were performed in an automated DNA thermal cycler. The selected minimum length partial Bm86 cDNA fragment (603 bp) for Hyalomma species delineation was amplified using the following conditions: an initial denaturation step of 5 min at 94°C followed by 40 cycles of 30 s at 94°C, 30 s at 54°C, 50 s at 72°C, and a final extension step of 72°C for 10 min. However, the thermal profile used for the amplification of partial Bm86 cDNA fragment required for the Rhipicephalus spp. discrimination was as follows: an initial denaturation step of 5 min at 94°C followed by 35 cycles of 30 s at 94°C, 30 s at 57°C, 1 min at 72°C, and a final extension step of 72°C for 15 min. PCR products were observed after electrophoretic migration on 1% agarose gels stained with ethidium bromide and under UV transillumination (Figure 3).
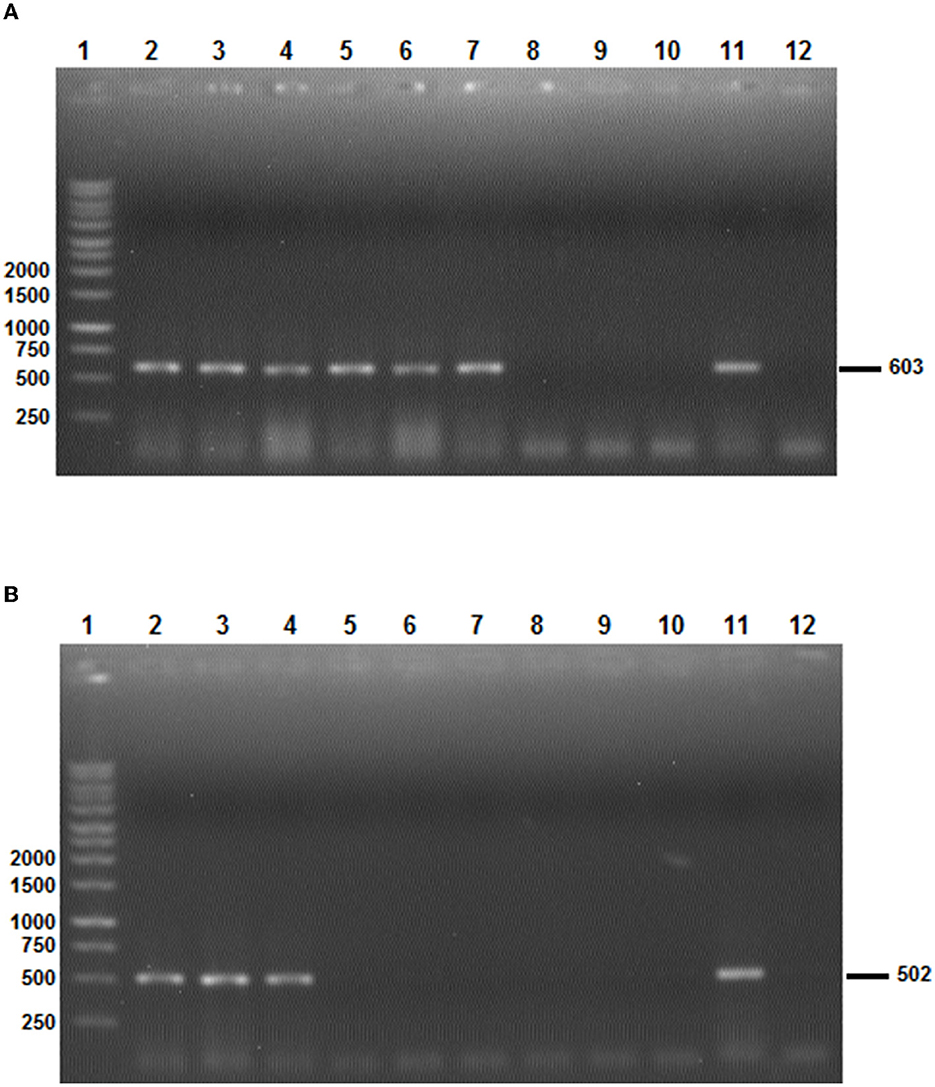
Figure 3. The results of the two Bm86 minimal partial fragments' amplification specific to Hyalomma (A) and Rhipicephalus (B) species. (A) Line 1: 1 Kb ladder; lines 2–7: PCR products after the amplification of the Hyalomma spp. partial fragment based on Bm86 cDNA orthologs isolated from Hy. scupense, Hy. marginatum, Hy. excavatum, Hy. anatolicum, Hy. Dromedarii, and Hy. impeltatum, respectively; lines 8–10: PCR products showing the absence of amplification of the Hyalomma spp. partial fragment based on Bm86 cDNA orthologs isolated from Rh. bursa, Rh. sanguineus s.l (Rs1 from cattle), and Rh. sanguineus s.l (Rs4 from a dog), respectively; and lines 11 and 12: positive and negative controls, respectively. (B) Line 1: 1 Kb ladder; lines 2–4: PCR products after the amplification of Rhipicephalus spp. partial fragment based on Bm86 cDNA orthologs isolated from Rh. bursa, Rh. sanguineus s.l (Rs1 from cattle), and Rh. sanguineus s.l (Rs4 from a dog); lines 5–10: PCR products showing the absence of amplification of the Rhipicephalus spp. partial fragment based on Bm86 cDNA orthologs isolated from Hy. scupense, Hy. marginatum, Hy. excavatum, Hy. anatolicum, Hy. Dromedarii, and Hy. impeltatum, respectively; and lines 11 and 12: positive and negative controls, respectively.
2.7. Specificity and sensitivity of PCR assays
To determine the specificity of the PCR reactions, we utilized two sets of primers—AD-Bm86-Hyl/AR-Bm86-Hyl and AD-Bm86-Rhip/AR-Bm86-Rhip—to analyze samples of Rhipicephalus and Hyalomma spp. through PCR. In addition, to evaluate the sensitivity of the assay, we conducted serial dilutions of a quantified cDNA and calculated the PCR sensitivity in terms of the amount of cDNA per μl (Figure 4).
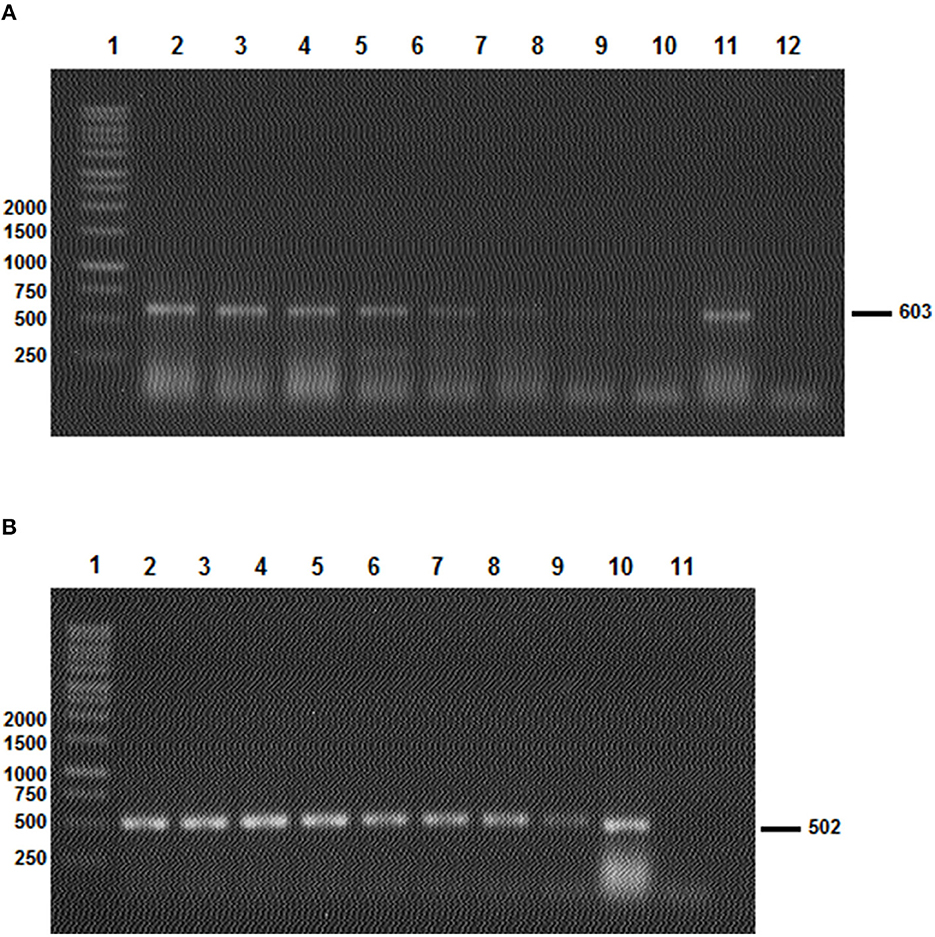
Figure 4. The results of the two Bm86 minimal partial fragments' amplification sensibility test to Hyalomma (A) and Rhipicephalus (B) species. (A) Line 1: 1 Kb ladder; lines 2–10: PCR products after the amplification of the Hyalomma spp. partial fragment based on a serial dilution of a quantified Bm86 cDNA (from 1.4 μg/μl to 1.4 × 10−8 μg/μl) isolated from Hy. scupense and lines 11 and 12: positive and negative controls, respectively. (B) Line 1: 1 Kb ladder; lines 2–4: PCR products after the amplification of Rhipicephalus spp. partial fragment based on a serial dilution of a quantified Bm86 cDNA (from 1.9 μg/μl to 1.9 × 10−8 μg/μl) isolated from Rh. sanguineus s.l (Rs1 from cattle) and lines 10 and 11: positive and negative controls, respectively.
2.8. DNA sequencing, sequence analysis, and phylogenetic trees' construction
PCR products generated from mitochondrial 16S rRNA and Bm86 partial sequences were purified and sequenced on both forward and reverse strands by using the same primers employed in PCR to obtain maximal data accuracy. The Big Dye Terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, USA) and an ABI3730XL automated DNA sequencer (Macrogen Europe, Amsterdam, The Netherlands) were employed.
The chromatograms were evaluated with Chromas Lite v 2.01 (http://www.technelysium.com.au/chromas_lite.html). Obtained raw sequences were edited, primer region sequences were manually removed, and the overlapping parts were selected. BLAST analysis was performed to assess the level of similarity with previously reported sequences (http://blast.ncbi.nlm.nih.gov/).
Nucleotide sequences of the partial Bm86 orthologs were compared with previously reported sequences isolated from Hyalomma and Rhipicephalus spp. ticks using the DNAMAN program (Version 5.2.2; Lynnon Biosoft, Quebec, Canada). By using the same software, genetic distances were computed by the maximum composite likelihood method (50) and were used to construct neighbor-joining trees (50). Statistical support for the internal branches of trees was established by bootstrap analysis with 1,000 reiterations (51).
The mitochondrial 16S rRNA and Bm86 partial sequences of Hyalomma and Rhipicephalus spp. isolates were deposited in the GenBank under the accession numbers OP749949-OP749962 and OP762544-OP762557, respectively.
3. Results
3.1. Selection of minimum length partial Bm86 cDNA fragments needed for species delineation
3.1.1. Identification of genetic variants
Based on all available complete and nearly complete sequences of Bm86 cDNA isolated from Rhipicephalus and Hyalomma species, we precisely selected the minimal length partial sequences that will allow us to discriminate between all classified species within each tick genus. All genetic variants of Bm86 cDNA belonging to each Rhipicephalus and Hyalomma species were included in our data set. These genetic variants differ from each other by at least one mutation and represent complete or nearly complete sequences of all strains and isolates submitted in GenBank (Supplementary Tables 1–3). For all available Rhipicephalus and Hyalomma species, the two genetic variants (each representing one classified species) that showed the lowest diversity were selected, and the two sequences' alignment profiles are shown in Figures 1, 2.
3.1.2. Identification of conserved and variable sequences
First, we identified the Bm86 regions that are conserved at least between the two closest genetic variants belonging to the two closest classified species within Rhipicephalus and Hyalomma genera to eliminate them when selecting the minimal partial sequences required for species delineation (Figures 1, 2).
Analysis of all possible combinations of two sequence alignments for all selected variants belonging to the Rhipicephalus species revealed that only the alignment profile resulting from Rh. microplus and Rh. annulatus variants showed one conserved region of 786 bp (from 465 to 1,250) and two variable regions. The first variable region is located from position 33 to position 464 by having a size of 432 pb, while the second variable region begins just after the conserved region and continues until the end of the Bm86 cDNA sequence (from position 1,251 to position 2,054, 804 bp).
Moreover, the same analysis was performed for the selected genetic variants of each Hyalomma species. The two sequences' alignments showed that only the alignment between the two selected genetic variants of Hy. excavatum and Hy. anatolicum showed a conserved region having a size of 455 bp (from 1,092 to 1,546) and two variable regions of 999 bp (from 93 to 1,091) and 367 bp (from 1,547 to 1,913).
3.1.3. Selection of partial sequences required for species delineation
For the two genera and only within these two variable regions, successive phylogenetic analyses were performed using different window lengths by transferring approximately 50 nucleotides from one assay to another. After evaluating several fragment sizes, we found that the minimum size to delineate species in each genus was located for Rhipicephalus spp. in the second variable region from the position 1,487 to 1,884 with a size of 398 bp (Figure 1) and in the first variable region from the position 539 to 1,097 with a size of 559 bp for Hyalomma species (Figure 2).
3.2. Morphological and molecular identification of tick species
Based on the morphological characteristics, collected tick specimens were classified into two genera and eight species: Hyalomma scupense, Hy. anatolicum, Hy. excavatum, Hy. marginatum, Hy. impeltatum, Hy. dromedarii, Rhipicephalus sanguineus sensu lato, and Rh. bursa. It is relevant to note that this is the first time that H. anatolicum has been recorded in Northern Tunisia in the humid zone. Tick species identification was also confirmed by the analysis of mitochondrial 16S rRNA partial sequences (272 bp, GenBank accession numbers OP749949-OP749962). Blast analysis showed 99–100% identity of our sequences isolated from Hyalomma and Rhipicephalus spp. with those genetically closest published in GenBank, which thus confirms the morphological diagnosis (Table 1).
3.3. Amplification of minimal length Bm86 cDNA partial sequences
Selected tick field samples, nine from Hyalomma spp. and five from Rhipicephalus, were amplified using the specific set of primers AD-Bm86-Hyl and AR-Bm86-Hyl as well as AD-Bm86-Rhip and AR-Bm86-Rhip with the optimized PCR conditions. All Hyalomma samples gave an amplicon of the expected size (603 bp) when amplified with primers specific for Hyalomma spp. and reacted negative when amplified with primers specific for Rhipicephalus species (Figure 3A). Similarly, all Rhipicephalus samples gave an amplicon of the expected size (502 bp) only when amplified using the set of primers specific for Rhipicephalus species (Figure 3B). Using the newly designed primers, the cDNA Bm86 was not amplified for any of the Rhipicephalus spp. tick samples using the specific primers for the Hyalomma genus, and similarly, cDNA Bm86 was not amplified for any of the Hyalomma spp. tick samples using the specific primers for the Rhipicephalus genus. Our results clearly indicate that the newly developed PCR assays were highly specific and capable of accurately distinguishing between closely related tick genera, such as Hyalomma and Rhipicephalus. Additionally, the sensitivity of the assays was found to be 1.4 pg/μl and 1.9 pg/μl for the genus-specific PCRs for Hyalomma and Rhipicephalus, respectively (Figure 4).
3.4. Genetic analysis of minimal length Bm86 cDNA partial sequences
To assess the taxonomic interest of Bm86 orthologs, the amplified fragments of 559 bp and 460 bp containing partial targeted sequences of the Bm86 transcript were sequenced for Hyalomma and Rhipicephalus spp., respectively. All sequences were subjected to a Basic Local Alignment Search Tool (BLAST) search to confirm tick species and estimate the homology rate of each sequence with the closest sequence already published in GenBank.
Blast analysis performed on the sequenced Hy. scupense ortholog showed 100% sequence homology with the previously published Hy. scupense strain Beja (GenBank accession number HQ872022) recovered from cattle in Tunisia. Only one genotype HmBm86G1 was revealed from three Hy. marginatum isolates which also shared 100% homology with the Hy. marginatum strain Kutahya from Turkey (GenBank accession number KF527438). Two distinct and novel genotypes (HeBm86G1 and HeBm86G1) were identified (GenBank accession numbers OP762546 and OP762546) from two Hy. excavatum isolates which shared 99% homology with Hy. excavatum strain Sousse recovered from cattle in Tunisia (GenBank accession number JF298786). For Hy. anatolicum and Hy. dromedarii orthologs, nucleotide sequence identities were superior to 99% in comparison with other previously published Bm86 Hyalomma species, resulting in one novel genotype each (HaBm86G1 and HdBm86G1, respectively; Table 1). Since there is no published sequence in GenBank of the Bm86 gene from Hy. impeltatum, one genotype HiBm86G1 was revealed as novel, and blast analysis performed on the Bm86 ortholog isolated from Hy. impeltatum (GenBank accession number OP762551) showed that this sequence has 97.6% sequence homology to the closest sequence that was isolated from the Hy. anatolicum isolate Alborz found in an Iranian sheep (GenBank accession number MH325952).
Based on nucleotide alignments of Bm86 partial nucleotidic sequences (413 bp) of four Tunisian Rhipicephalus sanguineus sensu lato isolates, a total of three distinct and novel genotypes RsBm86G1-3 were identified (GenBank accession numbers OP762554 to OP762557). The revealed genotypes were 96.8% to 97.3% similar to Rh. sanguineus s.l. (GenBank accession number EF222203; Table 1). Since this is the first time that the Bm86 ortholog has been sequenced in Rhipicephalus bursa, one genotype RbBm86G1 was revealed as novel, and the blast analysis indicated that the closest sequence was that of Rhipicephalus evertsi evertsi from South Africa (GU144600) with 96.6% homology (Table 1).
3.5. Comparative analysis of phylogenetic trees built for species delineation
Phylogenetic trees based on the alignment of studied minimum length Bm86 fragments belonging to both Hyalomma and Rhipicephalus species were compared with those generated from all complete or nearly complete Bm86 gene sequences of all Hyalomma and Rhipicephalus species found in GenBank.
Phylogenetic analysis based on the alignment of Tunisian genotypes with different minimal partial sequences of the Bm86 gene of all classified species of the genus Hyalomma obtained from the GenBank generated various clades (Figures 5A, B). The phylogenetic tree demonstrated the discriminatory power of the minimal Bm86 fragment between Hyalomma species. Both revealed Hy. excavatum Tunisian strains were classified separately with the Tunisian ortholog strain Sousse isolated from cattle (GenBank accession number JF298786) within Hy. excavatum cluster. This latter strain was closely related to the Tunisian Hy. anatolicum isolate TunHa1Bm86 found on cattle (GenBank accession number OP762545), which was clustered within the Hy. anatolicum cluster containing both orthologs, strain India and isolate Izatnagar, isolated from Hyalomma anatolicum in India (GenBank accession numbers AF347079 and EU665682, respectively).
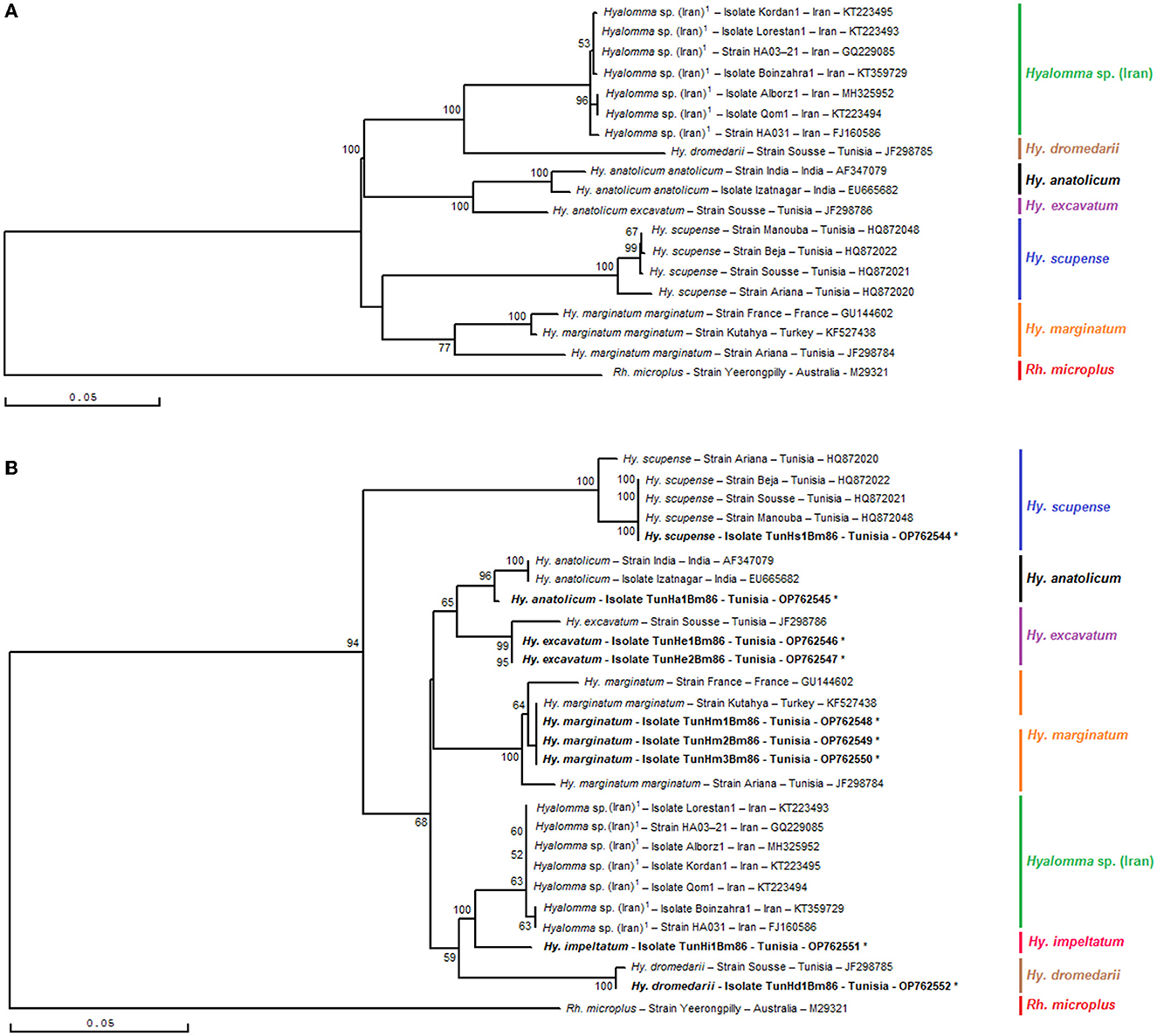
Figure 5. Phylogenetic tree inferred with complete or nearly complete Bm86 gene sequences (A) and selected minimal partial Bm86 sequences (559 bp) (B) required for species delineation within Hyalomma tick genera using the neighbor-joining method. Numbers associated with nodes represent the percentage of 1,000 bootstrap iterations supporting the nodes (only percentages >50% are show). The host or vector, strain or isolate name, the country of origin, and the GenBank accession number are indicated. The Yeerongpilly strain of Rhipicephalus (Boophilus) microplus tick from Australia (M29321) was added as an out-group.
Moreover, the studied minimal length Bm86 fragment allowed the same isolates' discrimination within each one of the Hyalomma species than that obtained with the complete or nearly complete sequences. In particular, as shown by the Hyalomma tree generated by complete or nearly complete sequences, all Hy. marginatum Tunisian strains were grouped into one Hy. marginatum sub-cluster with the Kutahya strain isolated from Turkey and closely related to Hy. marginatum strain France and strain Ariana isolated from Tunisian cattle (GenBank accession numbers GU144602 and JF298784, respectively) in the tree based on our minimal length Bm86 fragment. Furthermore, the studied minimal length Bm86 fragment allowed the discrimination within Hy. scupense isolates resembling that made by the complete or nearly complete sequences. In fact, the Tunisian Hy. scupense isolate TunHs1Bm86 found on cattle (GenBank accession number OP762544) was grouped into one of two Hy. scupense sub-clusters containing three ortholog strains, namely, Beja, Sousse, and Manouba from Tunisia and thereby leaving the Hy. scupense strain Ariana from Tunisia (GenBank accession number HQ872020) in a different separate Hy. scupense sub-cluster (Figures 5A, B).
As shown in Figures 6, 7, the phylogenetic tree based on selected minimal partial Bm86 Rhipicephalus spp. fragments exhibited almost the same topology found by the analysis of complete or nearly complete Bm86 gene sequences from all available Rhipicephalus species available in GenBank. Almost no changes in phylogenetic relationships were observed. All revealed Tunisian Rhipicephalus spp. isolates were classified into the Rh. sanguineus s.l. cluster which was closely related to the USA strain (GenBank accession number EF222203) except for the novel Tunisian Rh. bursa isolate which was closely related to Rh. evertsi evertsi strain from South Africa (GenBank accession number GU144600).
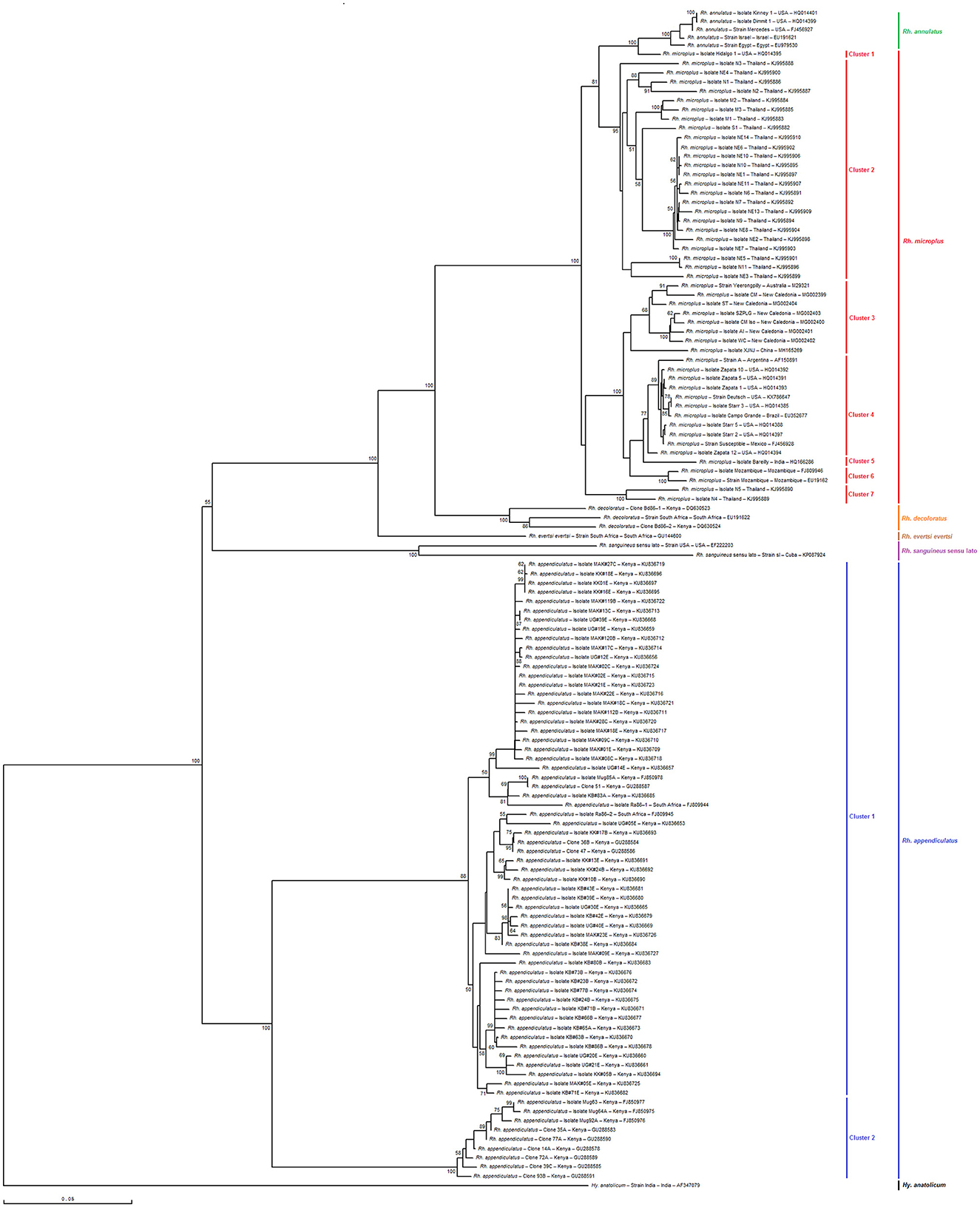
Figure 6. Phylogenetic tree inferred with complete or nearly complete Bm86 gene sequences required for species delineation within Rhipicephalus tick genera using the neighbor-joining method. Numbers associated with nodes represent the percentage of 1,000 bootstrap iterations supporting the nodes (only percentages >50% are show). The host or vector, strain or isolate name, the country of origin, and the GenBank accession number are indicated. The India strain of Hyalomma anatolicum tick from India (AF347079) was added as an out-group.
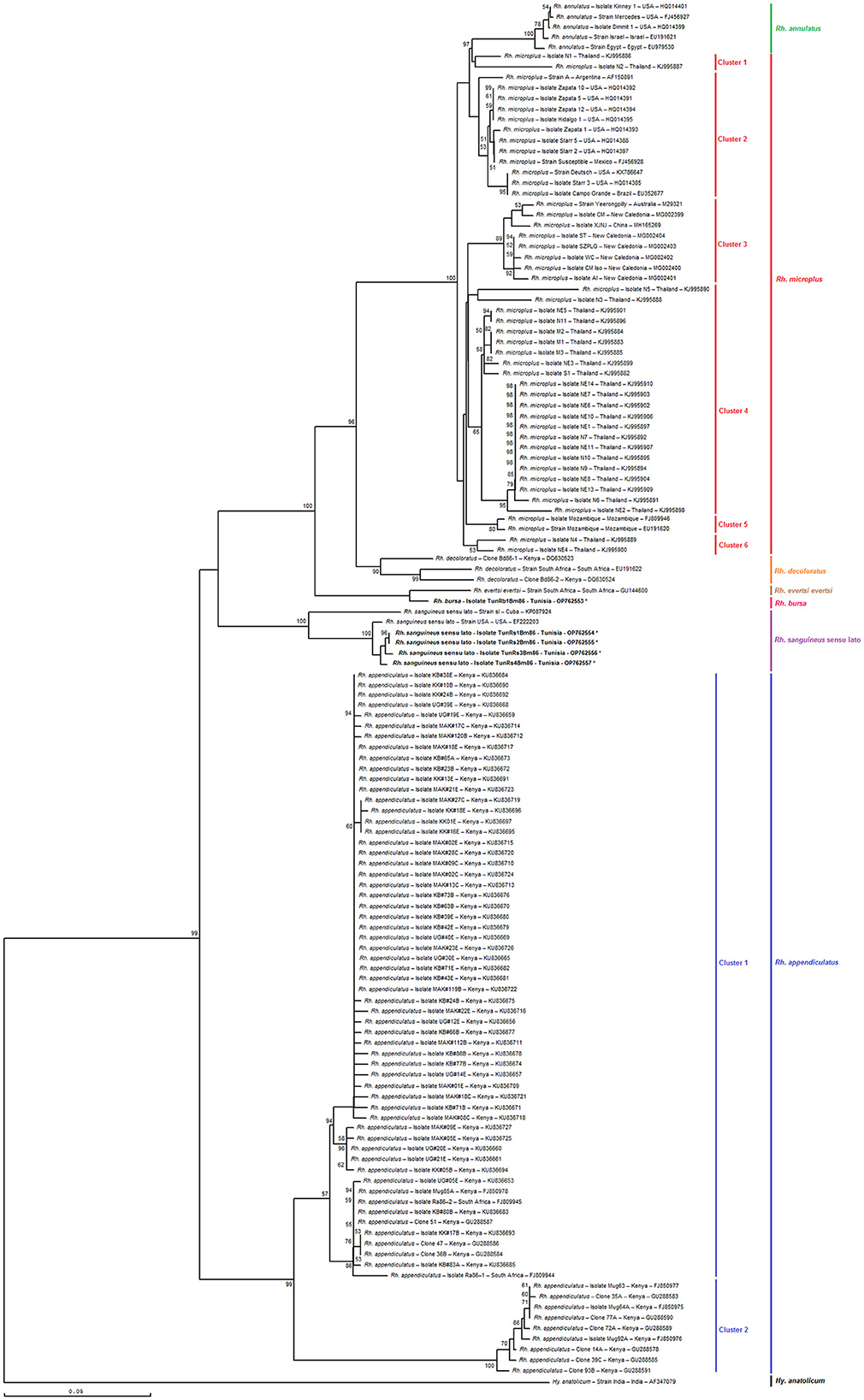
Figure 7. Phylogenetic tree inferred with selected minimal partial Bm86 gene sequences (398 bp) required for species delineation within Rhipicephalus tick genera using the neighbor-joining method. Numbers associated with nodes represent the percentage of 1,000 bootstrap iterations supporting the nodes (only percentages >50% are shown). The host or vector, strain or isolate name, the country of origin, and the GenBank accession number are indicated. The India strain of Hyalomma anatolicum tick from India (AF347079) was added as an out-group.
The results also revealed that the minimal length Bm86 transcript sequence specific to the Rhipicephalus genus was effective in discriminating between different groups of Rh. microplus and Rh. appendiculatus isolates. Specifically, phylogenetic analysis based on the minimal length Bm86 transcript sequence generated groups of Rh. microplus isolates based on the geographic regions similar to those found by the phylogenetic analysis based on the complete or nearly complete sequence. In fact, the groups of isolates from the North and Latin America group (USA, Mexico, and Brazil), Asia-Pacific group (New Caledonia, Australia, China, and India), Thailand, and Mozambique were revealed in both phylogenetic trees based on the two types of sequences (Figures 6, 7). In addition, our partial sequence was able to distinguish between two exceptionally divergent homologs (Ra86) of Rh. appendiculatus isolates in a manner similar to the complete or nearly complete transcript Bm86 sequence, as indicated by the two types of generated trees (Figures 6, 7).
Both phylogenetic trees, constructed by aligning the studied minimum length Bm86 fragments with complete or nearly complete Bm86 gene sequences of all Rhipicephalus species available in GenBank, clearly showcased the remarkable discriminatory ability of the minimal Bm86 fragment in distinguishing Rhipicephalus species at a phylogeographic level. In particular, as shown by the Rhipicephalus tree generated by minimum length Bm86 fragments, all Rh. microplus sequences isolated from Thailand were grouped into Rh. microplus clusters 1, 4, and 6, which were also grouped into Rh. microplus clusters 2 and 7 regarding the Rhipicephalus tree generated by complete or nearly complete Bm86 gene sequences. Nonetheless, the studied minimal length Bm86 fragment allowed the discrimination within Rh. microplus sequences isolated from countries of the American continent (USA, Mexico, Brazil, and Argentina) which were grouped into Rh. microplus cluster 2 and Rh. microplus sequences isolated from countries of the African continent, specifically from Mozambique, which were grouped into Rh. microplus cluster 5. A consistent discrimination profile was created similar to that made by the Rhipicephalus phylogenetic tree generated by complete or nearly complete Bm86 gene sequences, in which Rh. microplus sequences isolated from countries of the American continent were grouped into Rh. microplus clusters 1 and 4, and Rh. microplus sequences isolated from Mozambique were grouped into Rh. microplus cluster 6 (Figures 6, 7).
Furthermore, the studied minimal length Bm86 fragment allowed the discrimination within Rh. annulatus isolates similar to that made by the complete or nearly complete sequences. In particular, five isolates were grouped into Rh. annulatus cluster that differ by the country of origin, a total of three Rh. annulatus isolates from the USA, one Rh. annulatus strain from Palestine (GenBank accession number EU191621), and one Rh. annulatus strain from Egypt (GenBank accession number EU979530) (Figures 6, 7).
4. Discussion
The identification of tick species has traditionally relied on morphological methods, but these methods can be difficult and subjective. Difficulties may arise during the morphological diagnosis of tick species due to the presence of significant morphological variations related to the stages of development, the state of engorgement of ticks, and the possible presence of hybrid specimens resulting from crossovers between two closely related species (52–54). In recent years, the use of DNA markers for determining tick species has gained widespread popularity due to its precision and efficiency compared to traditional morphological methods (17) and its ability to provide more detailed information about the evolutionary relationships between different species (55).
In recent years, several DNA markers such as the internal transcribed spacer (ITS) region, the cytochrome c oxidase subunit I (COI) gene, and the 16S rRNA gene have been used for tick species identification (15, 52, 56, 57). However, despite their wide use, these markers have been found to have limitations in terms of their discriminatory power and sensitivity (15, 27, 58, 59). For example, the ITS region has been shown to have limited resolution in differentiating between closely related species (27), while the COI gene has been found to have low sensitivity in detecting intraspecific genetic variations (27). Similarly, the 16S rRNA gene has limitations in its ability to differentiate between tick species that belong to different genera (60). In light of these limitations, there is a need for more effective DNA markers that can provide better tick species identification. These limitations are the reason where the proposed study comes in, as it aims to identify the minimum length of partial Bm86 cDNA fragments needed for species delineation in the tick genera Rhipicephalus and Hyalomma.
The Bm86 cDNA marker represents a new and innovative way of species delineation in ticks as it targets the tick's RNA banks that have been previously utilized in various research studies. In many research laboratories, these RNA libraries are for the most part widely studied for their potential in the development of anti-tick vaccines mainly based on proteins from the intestine and salivary glands of ticks (61–65) and in the search for RNA viruses that infect ticks such as tick-borne encephalitis virus (TBEV) and Kyasanur forest disease virus (KFDV) (66–71). In fact, the Bm86 cDNA marker could represent a significant advance in the field of delineating tick species that make up these banks, providing a sensitive and discriminatory tool that researchers can use in interspecific and intraspecific diversity analyses. Its ability to target the tick's RNA banks, which have already been utilized in previous research, offers new avenues for a more accurate and efficient method for tick species and isolate identification, which will have important implications for ticks and tick-borne disease research and control.
The selection of the Bm86 transcript as a species identification marker for ticks in the genera Hyalomma and Rhipicephalus was motivated by several factors. First, this transcript had been previously used in commercial and experimental anti-tick vaccines (48, 49, 72–75), making it a readily available target in tick RNA banks (64, 76). Second, research showed that the phylogeny based on the complete or almost complete sequence of this transcript was in perfect agreement with the recent taxonomy of hard ticks (31, 77). This made it an ideal candidate for species identification as it had already demonstrated its utility in representing the genetic diversity of these ticks (78–81). Furthermore, using this transcript as a marker for species identification would help to better understand the evolution and ecology of these ticks, which are important vectors of various diseases affecting livestock and humans.
However, the purpose of our study was to identify the minimum length partial Bm86 cDNA fragments required to differentiate between species within the tick genera Rhipicephalus and Hyalomma. Using a shorter sequence has practical benefits as it only requires one primer pair for amplification and a single primer for sequencing. This reduces the cost of the test compared to analyzing the complete or nearly complete sequence, which requires multiple primer pairs.
Through a combination of genetic analysis and phylogenetic analysis, the study identified the conserved and variable regions within the Bm86 cDNA of these tick species. The crucial factor in choosing the partial sequences specific to Hyalomma and Rhipicephalus genera was the alignment of two genetically closest species Hy. excavatum and Hy. anatolicum (96.9% identity rate) and Rh. microplus and Rh. annulatus (98.6%), respectively. As a result of these alignments, one conserved and two variable regions were identified in each genus, as shown in Figure 2 for Hyalomma and Figure 1 for Rhipicephalus. Therefore, to ensure accurate results in future molecular diagnosis studies of Hyalomma and Rhipicephalus spp., we suggest avoiding the use of partial sequences within these conserved regions and instead focusing on amplifying and sequencing fragments found in the two variable regions of each genus (Figures 1, 2). By evaluating different fragment sizes within the variable regions, we determined that the minimum size for species delineation was 398 bp for Rhipicephalus spp. (from 1,487 to 1,884) and 559 bp for Hyalomma species (from 539 to 1,097).
The study also confirmed tick species identification through morphological characteristics and mitochondrial 16S rRNA partial sequences, with a 99.27–100% identity with the closest published Hyalomma and Rhipicephalus sequences in GenBank. Interestingly, we identified a Hyalomma anatolicum tick specimen based on its morphological characteristics, and its identity was validated by mitochondrial 16S rRNA partial sequences. The validation revealed a 100% match with the closest published Hyalomma anatolicum sequence in GenBank. Notably, this finding marks the second report of this tick species since the discovery of a single Hyalomma anatolicum specimen in 1970 by Van Den Ende in Southern Tunisia (82). This discovery highlights the risk that this tick becomes a vector of the apicomplexan hemoparasite Theileria annulata in Tunisia as already advanced by Gharbi et al. (83).
Furthermore, the field evaluation study successfully amplified and genetically analyzed the minimal length Bm86 cDNA partial sequences for the identified tick species, with 99.10–100% and 96.85–97.34% homology rates with previously published Hyalomma and Rhipicephalus sequences, respectively. These findings have important implications for the accurate identification and classification of tick species, which is crucial for understanding the epidemiology and transmission of tick-borne diseases.
In addition, our results also support the utility of Bm86 orthologs as a potential marker for tick isolate or strain identification. In particular, the results suggest that the studied minimal length Bm86 fragment is just as effective at discriminating between isolates and strains within each Hyalomma and Rhipicephalus species as the complete or nearly complete sequences. The phylogenetic inference based on the two partial sequences that were chosen showed the same pattern as previously observed when using the complete or nearly complete sequence (Figures 5, 6). No changes in the relationships between the isolates and strains of each species were detected, which suggests that there is no incongruity in the phylogeny. The trees generated using the neighbor-joining method had high support values at the terminal nodes and most of the deeper branches. There were also no instances of polytomies, which is likely because the selected fragments contained the most informative sites, resulting in a conservation of the phylogenetic signal. These findings were consistent with previous studies (84, 85). This is interesting because it suggests that this shorter fragment could be a more efficient and cost-effective reliable method for identifying and characterizing Hyalomma isolates and strains. In fact, our findings indicate that the two Hyalomma trees generated using the complete or nearly complete sequences and the minimal sequence showed similar results. Specifically, the genetic analysis revealed that the Hy. marginatum strain from Turkey (Kutahya) (86) is more closely related to the Hy. marginatum strain from France (80) than to all the Hy. marginatum Tunisian strains (31). Additionally, the isolate Ariana from Hy. scupense was relatively more distant from the isolates from Sousse, Manouba, and Beja (31).
Additionally, our result suggests that the minimal length Bm86 transcript sequence selected in this study can be as effective as the complete or nearly complete sequence in discriminating between isolates and strains within Rh. microplus and Rh. appendiculatus species. First, the phylogenetic analysis based on the minimal length sequence was able to generate groups of isolates based on their geographic regions, which were similar to the groups generated by the analysis based on the complete or nearly complete sequence. Second, the results showed that the minimal length Bm86 transcript sequence specific to the Rhipicephalus genus was also effective in discriminating between the two genospecies groups of Rh. appendiculatus isolates (87) in a similar way than with the complete or nearly complete transcript Bm86 sequence (Figures 6, 7). This finding indicates that the shorter fragment could also be a useful tool for identifying and characterizing Rhipicephalus strains. Overall, these results highlight the potential benefits of using shorter sequence fragments to identify and distinguish between different strains and isolates of tick species belonging to these two genera, which could have important implications for tick control and disease prevention strategies. However, it is important to note that further research may be necessary to confirm these findings and to determine whether they are applicable to other tick species.
However, the primers we designed for the amplification of each Bm86 minimum partial sequence specific to Hyalomma and Rhipicephalus were found to be sensitive and specific for each targeted tick genus. We were able to confirm this by successfully obtaining an amplicon of the expected size for all samples when amplified with primers specific for their respective genus. This indicates that the two sets of primers we created are suitable for amplifying and sequencing our cDNA marker and are capable of differentiating between the two closely related tick genera.
The use of a minimal Bm86 cDNA sequence for tick species differentiation offers advantages such as the use of a single pair of genus-specific primers for amplification and only a single primer for sequencing, followed by a simple blast analysis to determine the species within each genus. However, it is important to acknowledge the limitations associated with cDNA, as researchers need to extract total RNA and obtain cDNA through reverse transcription. Nonetheless, the decision to exclusively use cDNA for both the Hyalomma and Rhipicephalus genera was made due to the presence of a large 2,888 bp intron at position 1,752 bp in Bm86 cDNA (88), which is located within the minimal sequence used to discriminate Rhipicephalus species. Consequently, amplification of gDNA using specific primers for the Rhipicephalus genus is not feasible. To maintain consistency, we chose to utilize cDNA exclusively for both Hyalomma and Rhipicephalus genera.
Furthermore, this cDNA marker is expected to be particularly useful in identifying tick specimens that are difficult to recognize morphologically, for instance, with fully engorged tick immature females, in case of occurrence of malformations, or in the occurrence of hybrids of two species of ticks that are taxonomically closely related (31, 89, 90). Our marker, which is able to accurately identify tick species that may be otherwise difficult to differentiate, could be used as an additional taxonomic tool for morphological diagnosis for improving the reliability of tick species identification, particularly when considering the increased risks for changes in tick fauna and the emergence of new tick species driven by the combination of climate change with anthropogenic factors.
5. Conclusion
Genomic and mitochondrial DNA markers have been shown to be effective in identifying tick species compared to traditional morphological methods. In addition to these existing markers, we have identified here a novel Bm86 cDNA marker that provides a new approach to tick species identification and can target tick RNA banks. In this study, we developed a molecular method based on the minimum length of partial Bm86 cDNA fragments needed for identifying accurately tick species within the Rhipicephalus and Hyalomma genera, increasing, therefore, the range of taxonomic tools applied to identify reliably hard tick species.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The collection of ticks from cattle was done in accordance with the Tunisian National School of Veterinary Medicine's guidelines for animal care and handling and required no ethical approval. Consent was sought from the farmers before sampling. The animals were gently restrained by their owners in the same way as during routine clinical examinations, and invasive sampling or tranquilizers were not used. Moreover, the sampling was overseen by veterinarians and veterinary technicians from the National School of Veterinary Medicine in Sidi Thabet.
Author contributions
MBS conceived the idea, conducted in silico analysis, and designed the experiments. SZ, MM, and MD performed the experiments. SZ and MBS wrote the manuscript. MAD collaborated with MBS in developing the study concept and in editing and finalizing the manuscript. All authors have reviewed and approved the current version of the manuscript.
Funding
This study was supported by the research laboratory Epidemiology of enzootic infections in herbivores in Tunisia: application to the control (LR02AGR03) and the research project Application of artificial intelligence and DNA barcoding in the identification of tick species of the genus Hyalomma of medical and economic interest (21P2ES-D4P2), which are funded by the Ministry of Higher Education and Scientific Research of Tunisia.
Acknowledgments
The authors are thankful to Dr. Rachid Selmi, Rihab Romdhane, Meriem Ben Abdallah, Hayet Ben Yedem, and Limam Sassi for their help in tick collection and to Dr. Mohamed Ridha Rjeibi and Dr. Safa Amairia for their help during the experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1209210/full#supplementary-material
References
1. Dantas-Torres F. Biology and ecology of the brown dog tick Rhipicephalus Sanguineus. Parasit Vectors. (2010) 3:26. doi: 10.1186/1756-3305-3-26
2. Walker A. The Genus Rhipicephalus (Acari, Ixodidae): a guide to the brown ticks of the world. Trop Anim Health Prod. (2000) 32:417. doi: 10.1017/CBO9780511661754
3. Perveen N, Muzaffar SB, Al-Deeb MA. Ticks and tick-borne diseases of livestock in the middle east and north africa: a review. Insects. (2021) 12:1. doi: 10.3390/insects12010083
4. Brites-Neto J, Duarte KM, Martins TF. Tick-borne infections in human and animal population worldwide. Vet World. (2015) 8:301–15. doi: 10.14202/vetworld.2015.301-315
5. Kumar B, Manjunathachar HV, Ghosh S. A review on hyalomma species infestations on human and animals and progress on management strategies. Heliyon. (2020) 6:e05675. doi: 10.1016/j.heliyon.2020.e05675
6. Bouaicha F, Eisenbarth A, Elati K, Schulz A, Ben Smida B, Bouajila M, et al. Epidemiological investigation of crimean-congo haemorrhagic fever virus infection among the one-humped camels (Camelus Dromedarius) in Southern Tunisia. Ticks Tick Borne Dis. (2021) 12:101601. doi: 10.1016/j.ttbdis.2020.101601
7. Bonnet SI, Vourc'h G, Raffetin A, Falchi A, Figoni J, Fite J. The control of hyalomma ticks, vectors of the crimean-congo hemorrhagic fever virus: where are we now and where are we going? PLoS Negl Trop Dis. (2022) 16:e0010846. doi: 10.1371/journal.pntd.0010846
8. Shi J, Hu Z, Deng F, Shen S. Tick-borne viruses. Virol Sin. (2018) 33:21–43. doi: 10.1007/s12250-018-0019-0
9. Bouattour A, Darghouth MA, Ben Miled L. Cattle infestation by Hyalomma ticks and prevalence of theileria in H. Detritum Species in Tunisia. Vet Parasitol. (1996) 65:233–45. doi: 10.1016/S0304-4017(96)00951-X
10. d'Oliveira C, van der Weide M, Jacquiet P, Jongejan F. Detection of Theileria Annulata by the Pcr in Ticks (Acari:Ixodidae) collected from cattle in mauritania. Exp Appl Acarol. (1997) 21:279–91.
11. KT F. Tick-born disease of sheep and goats caused by Babesia, Theileria or Anaplasma Spp. Parassitologia. (1997) 39:99–109.
12. Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. Reorganization of Genera in the Families Rickettsiaceae and Anaplasmataceae in the Order Rickettsiales: Unification of Some Species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and 'hge agent' as subjective synonyms of Ehrlichia Phagocytophila. Int J Syst Evol Microbiol. (2001) 51:2145–65. doi: 10.1099/00207713-51-6-2145
13. Estrada-Pena A, Ayllon N, de la Fuente J. Impact of climate trends on tick-borne pathogen transmission. Front Physiol. (2012) 3:64. doi: 10.3389/fphys.2012.00064
14. Estrada-Pena A, D'Amico G, Palomar AM, Dupraz M, Fonville M, Heylen D, et al. A comparative test of ixodid tick identification by a network of european researchers. Ticks Tick Borne Dis. (2017) 8:540–6. doi: 10.1016/j.ttbdis.2017.03.001
15. Abouelhassan EM, El-Gawady HM, Abdel-Aal AA, El-Gayar AK, Esteve-Gassent MD. Comparison of some molecular markers for tick species identification. J Arthropod Borne Dis. (2019) 13:153–64. doi: 10.18502/jad.v13i2.1241
16. Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin North Am. (2008) 22:195–215. doi: 10.1016/j.idc.2007.12.006
17. Dantas-Torres F, Latrofa MS, Annoscia G, Giannelli A, Parisi A, Otranto D. Morphological and genetic diversity of Rhipicephalus Sanguineus sensu lato from the new and old worlds. Parasit Vectors. (2013) 6:213. doi: 10.1186/1756-3305-6-213
18. Schindel DE, Miller SE. Barcoding a useful tool for taxonomists. Nature. (2005) 435:17. doi: 10.1038/435017b
19. Lv J, Wu S, Zhang Y, Zhang T, Feng C, Jia G, et al. Development of a DNA barcoding system for the Ixodida (Acari: Ixodida). Mitochondrial DNA. (2014) 25:142–9. doi: 10.3109/19401736.2013.792052
20. Dobson SJ, Barker SC. Phylogeny of the hard ticks (Ixodidae) inferred from 18s RRNA indicates that the genus Aponomma is paraphyletic. Mol Phylogenet Evol. (1999) 11:288–95. doi: 10.1006/mpev.1998.0565
21. Norris DE, Klompen JSH, Black WC. Comparison of the mitochondrial 12s and 16s ribosomal DNA genes in resolving phylogenetic relationships among hard ticks (Acari: Ixodidae). Ann Entomol Soc Am. (1999) 92:117–29. doi: 10.1093/aesa/92.1.117
22. Guglielmone AA, Estrada-Pena A, Mangold AJ, Barros-Battesti DM, Labruna MB, Martins JR, et al. Amblyomma Aureolatum (Pallas, 1772) and Amblyomma Ovale Koch, 1844 (Acari: Ixodidae): hosts, distribution and 16s Rdna Sequences. Vet Parasitol. (2003) 113:273–88. doi: 10.1016/S0304-4017(03)00083-9
23. Abdigoudarzi M, Noureddine R, Seitzer U, Ahmed J. Rdna-Its2 Identification of Hyalomma, Rhipicephalus, Dermacentor and Boophilus Spp.(Acari: Ixodidae) collected from different geographical regions of Iran. Adv Stu Biol. (2011) 3:221–38.
24. Latrofa MS, Dantas-Torres F, Annoscia G, Cantacessi C, Otranto D. Comparative analyses of mitochondrial and nuclear genetic markers for the molecular identification of Rhipicephalus Spp. Infect Genet Evol. (2013) 20:422–7. doi: 10.1016/j.meegid.2013.09.027
25. Abdullah HH, El-Molla A, Salib FA, Allam NA, Ghazy AA, Abdel-Shafy S. Morphological and molecular identification of the brown dog tick Rhipicephalus Sanguineus and the camel tick Hyalomma Dromedarii (Acari: Ixodidae) vectors of rickettsioses in Egypt. Vet World. (2016) 9:1087–101. doi: 10.14202/vetworld.2016.1087-1101
27. Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, et al. Assessment of Four DNA fragments (Coi, 16s Rdna, Its2, 12s Rdna) for species identification of the ixodida (Acari: Ixodida). Parasit Vectors. (2014) 7:93. doi: 10.1186/1756-3305-7-93
28. Amin MHFA, Andriyani AP, Sari ST, Pratiwi IA, Suhargo L, Irawan B. First Report of Amblyomma Sp. Collected from Varanus Salvator in Baluran National Park Identified by DNA Barcoding Ecology, Environment and Conservation. (2019) 25:S90–S5.
29. Jinbo U, Kato T, Ito M. Current progress in DNA barcoding and future implications for entomology. Entomol Sci. (2011) 14:107–24. doi: 10.1111/j.1479-8298.2011.00449.x
30. Charrier NP, Hermouet A, Hervet C, Agoulon A, Barker SC, Heylen D, et al. A transcriptome-based phylogenetic study of hard ticks (Ixodidae). Sci Rep. (2019) 9:12923. doi: 10.1038/s41598-019-49641-9
31. Ben Said M, Galai Y, Mhadhbi M, Jedidi M, de la Fuente J, Darghouth MA. Molecular characterization of Bm86 gene orthologs from Hyalomma Excavatum, Hyalomma Dromedarii and Hyalomma Marginatum Marginatum and comparison with a vaccine candidate from Hyalomma Scupense. Vet Parasitol. (2012) 190:230–40. doi: 10.1016/j.vetpar.2012.05.017
32. Gergen J, Petsch B. mRNA-based vaccines and mode of action. Curr Top Microbiol Immunol. (2022) 440:1–30. doi: 10.1007/82_2020_230
33. Almazan C, Blas-Machado U, Kocan KM, Yoshioka JH, Blouin EF, Mangold AJ, et al. Characterization of three Ixodes Scapularis CDNAs protective against tick infestations. Vaccine. (2005) 23:4403–16. doi: 10.1016/j.vaccine.2005.04.012
34. Gray JS. Ixodes Ricinus seasonal activity: implications of global warming indicated by revisiting tick and weather data. Int J Med Microbiol. (2008) 298:19–24. doi: 10.1016/j.ijmm.2007.09.005
35. Ben Said M, Galai Y, Ben Ahmed M, Gharbi M, de la Fuente J, Jedidi M, et al. Hd86 Mrna expression profile in Hyalomma Scupense life stages, could it contribute to explain anti-tick vaccine effect discrepancy between adult and immature instars? Vet Parasitol. (2013) 198:258–63. doi: 10.1016/j.vetpar.2013.07.035
36. Yong-Hong LIU, Kai-Rui LI, Bo HE, Fei LI, Zhang LY, Jiao-Jiao PAN, et al. Preliminary study of high efficiency vaccine of Rhipicephalus (Boophilus) Microplus in south Xinjiang, China. Kafkas Üniversitesi Veteriner Fakültesi Dergisi. (2019) 25:4.
37. Hue T, Petermann J, Bonnefond R, Mermoud I, Rantoen D, Vuocolo T. Experimental efficacy of a vaccine against Rhipicephalus Australis. Exp Appl Acarol. (2017) 73:245–56. doi: 10.1007/s10493-017-0184-0
38. Kaewmongkol S, Kaewmongkol G, Inthong N, Lakkitjaroen N, Sirinarumitr T, Berry CM, et al. Variation among Bm86 sequences in Rhipicephalus (Boophilus) Microplus ticks collected from cattle across Thailand. Exp Appl Acarol. (2015) 66:247–56. doi: 10.1007/s10493-015-9897-0
39. Canales M, Almazan C, Naranjo V, Jongejan F, de la Fuente J. Vaccination with recombinant Boophilus Annulatus Bm86 Ortholog protein, Ba86, protects cattle against B. Annulatus and B. Microplus infestations. BMC Biotechnol. (2009) 9:29. doi: 10.1186/1472-6750-9-29
40. Canales M, de la Lastra JM, Naranjo V, Nijhof AM, Hope M, Jongejan F, et al. Expression of recombinant Rhipicephalus (Boophilus) Microplus, R. Annulatus and R. Decoloratus Bm86 orthologs as secreted proteins in pichia pastoris. BMC Biotechnol. (2008) 8:14. doi: 10.1186/1472-6750-8-14
41. Andreotti R, Pedroso MS, Caetano AR, Martins NF. Comparison of Predicted Binders in Rhipicephalus (Boophilus) Microplus intestine protein variants bm86 campo grande Strain, Bm86 and Bm95. Rev Bras Parasitol Vet. (2008) 17:93–8. doi: 10.1590/S1984-29612008000200006
42. Shahein YE, Abouelella AM, Hussein NA, Hamed RR, El-Hakim AE, Abdel-Shafy S, et al. Identification of four novel Rhipicephalus annulatus upregulated salivary gland proteins as candidate vaccines. Protein J. (2013) 32:392–8. doi: 10.1007/s10930-013-9498-x
43. Kamau LM, Skilton RA, Githaka N, Kiara H, Kabiru E, Shah T, et al. Extensive polymorphism of ra86 genes in field populations of Rhipicephalus Appendiculatus from Kenya. Ticks Tick Borne Dis. (2016) 7:772–81. doi: 10.1016/j.ttbdis.2016.03.011
44. Azhahianambi P, De La Fuente J, Suryanarayana VV, Ghosh S. Cloning, Expression and Immunoprotective Efficacy of Rhaa86, the Homologue of the Bm86 tick vaccine antigen, from Hyalomma Anatolicum Anatolicum. Parasite Immunol. (2009) 31:111–22. doi: 10.1111/j.1365-3024.2008.01082.x
45. Mohammadi A, Aghaeipour K, Keyvanfar H, Paykari H, Tajbakhsh H, Jalali AH, et al. Ha03 as an Iranian candidate concealed antigen for vaccination against Hyalomma anatolicum anatolicum: comparative Structural and in Silico Studies. Archives Razi Inst. (2013) 68:131–8.
46. Walker AR. Ticks of Domestic Animals in Africa: A Guide to Identification of Species. Edinburgh: Bioscience Reports (2003).
47. Black WC, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16s RDNA sequences. Proc Natl Acad Sci U S A. (1994) 91:10034–8. doi: 10.1073/pnas.91.21.10034
48. de Vos S, Zeinstra L, Taoufik O, Willadsen P, Jongejan F. Evidence for the utility of the Bm86 antigen from Boophilus Microplus in vaccination against other tick species. Exp Appl Acarol. (2001) 25:245–61. doi: 10.1023/A:1010609007009
49. Rand KN, Moore T, Sriskantha A, Spring K, Tellam R, Willadsen P, et al. Cloning and expression of a protective antigen from the cattle tick Boophilus Microplus. Proc Natl Acad Sci U S A. (1989) 86:9657–61. doi: 10.1073/pnas.86.24.9657
50. Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. (1993) 10:512–26.
51. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. (1985) 39:783–91. doi: 10.1111/j.1558-5646.1985.tb00420.x
52. Araya-Anchetta A, Busch JD, Scoles GA, Wagner DM. Thirty years of tick population genetics: a comprehensive review. Infect Genet Evol. (2015) 29:164–79. doi: 10.1016/j.meegid.2014.11.008
53. Che Lah EF, Yaakop S, Ahamad M, George E, Md Nor S. precise identification of different stages of a tick, Ixodes Granulatus Supino, 1897 (Acari: Ixodidae). Asian Pac J Trop Biomed. (2016) 6:597–604. doi: 10.1016/j.apjtb.2016.05.003
54. Ondrejicka DA, Morey KC, Hanner RH, DNA. Barcodes identify medically important tick species in Canada. Genome. (2017) 60:74–84. doi: 10.1139/gen-2015-0179
55. Caporale DA, Rich SM, Spielman A, Telford SR, Kocher TD. Discriminating between ixodes ticks by means of mitochondrial DNA sequences. Mol Phylogenet Evol. (1995) 4:361–5. doi: 10.1006/mpev.1995.1033
56. Andrews RH, Chilton NB, Beveridge I, Spratt D, Mayrhofer G. Genetic markers for the identification of three australian tick species at various stages in their life cycles. J Parasitol. (1992) 5:366–8. doi: 10.2307/3283491
57. Dergousoff SJ, Chilton NB. Differentiation of three species of ixodid tick, Dermacentor Andersoni, D. Variabilis and D Albipictus, by Pcr-based approaches using markers in ribosomal DNA. Mol Cell Probes. (2007) 21:343–8. doi: 10.1016/j.mcp.2007.04.003
58. Zhang RL, Zhang B. Prospects of using DNA barcoding for species identification and evaluation of the accuracy of sequence databases for ticks (Acari: Ixodida). Ticks Tick Borne Dis. (2014) 5:352–8. doi: 10.1016/j.ttbdis.2014.01.001
59. Al-Deeb MA, Enan MR. Genetic diversity in the camel tick Hyalomma Dromedarii (Acari: Ixodidae) based on mitochondrial cytochrome c oxidase subunit I (Coi) and randomly amplified polymorphic DNA polymerase chain reaction (Rapd-Pcr). Adv Entomol. (2018) 6:285–94. doi: 10.4236/ae.2018.64021
60. Caudill MT, Brayton KA. The use and limitations of the 16s rrna sequence for species classification of Anaplasma Samples. Microorganisms. (2022) 10:3. doi: 10.3390/microorganisms10030605
61. Xu Y, Bruno JF, Luft BJ. Identification of novel tick salivary gland proteins for vaccine development. Biochem Biophys Res Commun. (2005) 326:901–4. doi: 10.1016/j.bbrc.2004.11.127
62. Zivkovic Z, Esteves E, Almazan C, Daffre S, Nijhof AM, Kocan KM, et al. Differential expression of genes in salivary glands of male Rhipicephalus (Boophilus)Microplus in response to infection with Anaplasma Marginale. BMC Genomics. (2010) 11:186. doi: 10.1186/1471-2164-11-186
63. Rego ROM, Trentelman JJA, Anguita J, Nijhof AM, Sprong H, Klempa B, et al. Counterattacking the tick bite: towards a rational design of anti-tick vaccines targeting pathogen transmission. Parasit Vectors. (2019) 12:229. doi: 10.1186/s13071-019-3468-x
64. Martins LA, Bensaoud C, Kotal J, Chmelar J, Kotsyfakis M. Tick salivary gland transcriptomics and proteomics. Parasite Immunol. (2021) 43:e12807. doi: 10.1111/pim.12807
65. Kasaija PD, Contreras M, Kirunda H, Nanteza A, Kabi F, Mugerwa S, et al. Inspiring anti-tick vaccine research, development and deployment in tropical africa for the control of cattle ticks: review and insights. Vaccines. (2022) 11:1–14. doi: 10.3390/vaccines11010099
66. Labuda M, Nuttall PA. Tick-borne viruses. Parasitology. (2004) 129:S221–45. doi: 10.1017/S0031182004005220
67. Durmisi E, Knap N, Saksida A, Trilar T, Duh D, Avsic-Zupanc T. Prevalence and molecular characterization of tick-borne encephalitis virus in Ixodes Ricinus ticks collected in slovenia. Vector Borne Zoonotic Dis. (2011) 11:659–64. doi: 10.1089/vbz.2010.0054
68. Brackney DE, Armstrong PM. Transmission and evolution of tick-borne viruses. Curr Opin Virol. (2016) 21:67–74. doi: 10.1016/j.coviro.2016.08.005
69. Hoffman T, Lindeborg M, Barboutis C, Erciyas-Yavuz K, Evander M, Fransson T, et al. Alkhurma hemorrhagic fever virus Rna in Hyalomma Rufipes ticks infesting migratory birds, europe and asia minor. Emerg Infect Dis. (2018) 24:879–82. doi: 10.3201/eid2405.171369
70. Shah SZ, Jabbar B, Ahmed N, Rehman A, Nasir H, Nadeem S, et al. Epidemiology, pathogenesis, and control of a tick-borne disease- kyasanur forest disease: current status and future directions. Front Cell Infect Microbiol. (2018) 8:149. doi: 10.3389/fcimb.2018.00149
71. Zhang Y, Hu B, Agwanda B, Fang Y, Wang J, Kuria S, et al. Viromes and surveys of rna viruses in camel-derived ticks revealing transmission patterns of novel tick-borne viral pathogens in Kenya. Emerg Microbes Infect. (2021) 10:1975–87. doi: 10.1080/22221751.2021.1986428
72. Rodriguez M, Massard CL, da Fonseca AH, Ramos NF, Machado H, Labarta V, et al. Effect of vaccination with a recombinant Bm86 antigen preparation on natural infestations of Boophilus Microplus in grazing dairy and beef pure and cross-bred cattle in Brazil. Vaccine. (1995) 13:1804–8. doi: 10.1016/0264-410X(95)00119-L
73. Garcia-Garcia JC, Montero C, Redondo M, Vargas M, Canales M, Boue O, et al. Control of ticks resistant to immunization with bm86 in cattle vaccinated with the recombinant antigen Bm95 isolated from the cattle tick, Boophilus Microplus. Vaccine. (2000) 18:2275–87. doi: 10.1016/S0264-410X(99)00548-4
74. Odongo D, Kamau L, Skilton R, Mwaura S, Nitsch C, Musoke A, et al. Vaccination of cattle with tickgard induces cross-reactive antibodies binding to conserved linear peptides of Bm86 homologues in Boophilus Decoloratus. Vaccine. (2007) 25:1287–96. doi: 10.1016/j.vaccine.2006.09.085
75. Azhahianambi P, Ray DD, Chaudhuri P, Gupta R, Ghosh S. Vaccine efficacy of Bm86 Ortholog of H. A. Anatolicum, Rhaa86 expressed in prokaryotic expression system. J Parasitol Res. (2009) 10:1–27. doi: 10.1155/2009/165812
76. Abbas MN, Jmel MA, Mekki I, Dijkgraaf I, Kotsyfakis M. Recent advances in tick antigen discovery and anti-tick vaccine development. Int J Mol Sci. (2023) 24:969. doi: 10.3390/ijms24054969
77. Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci. (2009) 14:2857–77. doi: 10.2741/3418
78. Nijhof AM, Taoufik A, de la Fuente J, Kocan KM. Gene silencing of the tick protective antigens, bm86, bm91 and subolesin, in the one-host tick Boophilus Microplus by RNA interference. Int J Parasitol. (2007) 37:653–62. doi: 10.1016/j.ijpara.2006.11.005
79. Bastos RG, Ueti MW, Knowles DP, Scoles GA. The Rhipicephalus (Boophilus) Microplus Bm86 gene plays a critical role in the fitness of ticks fed on cattle during acute Babesia Bovis infection. Parasit Vectors. (2010) 3:111. doi: 10.1186/1756-3305-3-111
80. Nijhof AM, Balk JA, Postigo M, Rhebergen AM, Taoufik A, Jongejan F. Bm86 homologues and novel ataq proteins with multiple epidermal growth factor (Egf)-like domains from hard and soft ticks. Int J Parasitol. (2010) 40:1587–97. doi: 10.1016/j.ijpara.2010.06.003
81. Rodriguez-Mallon A. The Bm86 discovery: a revolution in the development of anti-tick vaccines. Pathogens. (2023) 12:2. doi: 10.3390/pathogens12020231
82. Van En Ende M. Les Tiques (Ixodidae) Des animaux domestiques en tunisie et leur biologie. Archives de l'Institut Pasteur de Tunis. (1970) 47:253–64.
83. Gharbi M, Darghouth MA, Elati K, Al-Hosary AAT, Ayadi O, Salih DA, et al. Current status of tropical theileriosis in northern africa: a review of recent epidemiological investigations and implications for control. Transbound Emerg Dis. (2020) 67 Suppl 1:8–25. doi: 10.1111/tbed.13312
84. Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: The beginning of incongruence? Trends Genet. (2006) 22:225–31. doi: 10.1016/j.tig.2006.02.003
85. Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Worheide G, et al. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. (2011) 9:e1000602. doi: 10.1371/journal.pbio.1000602
86. Iça A, Pekcan M, Vatansever Z, Öktem IMA, Carlos S. Bm86 Orthologs in Hyalomma Marginatum as Vaccine Candidate. Immunity to Veterinary Pathogens: Informing Vaccine Development. Denver, United States Of America (2015).
87. Kamau L, Skilton RA, Odongo DO, Mwaura S, Githaka N, Kanduma E, et al. Differential Transcription of Two Highly Divergent Gut-Expressed Bm86 Antigen Gene Homologues in the Tick Rhipicephalus Appendiculatus (Acari: Ixodida). Insect Mol Biol. (2011) 20:105–14. doi: 10.1111/j.1365-2583.2010.01043.x
88. De la Fuente J, Garcia-Garcia JC, González DM, Izquierdo G, Ochagavia ME. Molecular analysis of Boophilus spp. (Acari: Ixodidae) tick strains. Vet Parasitol. (2000) 92:209–22. doi: 10.1016/S0304-4017(00)00308-3
89. Fulton RE, Salasek ML, DuTeau NM, Black WC. Sscp analysis of cdna markers provides a dense linkage map of the Aedes Aegypti genome. Genetics. (2001) 158:715–26. doi: 10.1093/genetics/158.2.715
Keywords: in silico selection, minimum length partial sequences, Bm86 cDNA, Hyalomma and Rhipicephalus spp., species delineation
Citation: Zamiti S, Mhadhbi M, Dhibi M, Darghouth MA and Ben Said M (2023) Development and field evaluation of PCR assays based on minimum length Bm86 cDNA fragments required for Rhipicephalus and Hyalomma tick species delineation. Front. Vet. Sci. 10:1209210. doi: 10.3389/fvets.2023.1209210
Received: 20 April 2023; Accepted: 06 June 2023;
Published: 29 June 2023.
Edited by:
Nicola Pugliese, University of Bari Aldo Moro, ItalyReviewed by:
Alberto Moraga-Fernández, Spanish National Research Council (CSIC), SpainReginaldo G. Bastos, Washington State University, United States
Copyright © 2023 Zamiti, Mhadhbi, Dhibi, Darghouth and Ben Said. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Aziz Darghouth, ZGFtYXppekB5YWhvby5mcg==; Mourad Ben Said, YmVuc2FpZG1vdXJhZDgzQHlhaG9vLmZy