 Nicola Palmieri
Nicola Palmieri Ilias Apostolakos
Ilias Apostolakos Surya Paudel
Surya Paudel Michael Hess
Michael Hess
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 21 June 2023
Sec. Veterinary Infectious Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1195585
Introduction: Colibacillosis is a worldwide prevalent disease in poultry production linked to Escherichia coli strains that belong to the avian pathogenic E. coli (APEC) pathotype. While many virulence factors have been linked to APEC isolates, no single gene or set of genes has been found to be exclusively associated with the pathotype. Moreover, a comprehensive description of the biological processes linked to APEC pathogenicity is currently lacking.
Methods: In this study, we compiled a dataset of 2015 high-quality avian E. coli genomes from pathogenic and commensal isolates, based on publications from 2000 to 2021. We then conducted a genome-wide association study (GWAS) and integrated candidate gene identification with available protein-protein interaction data to decipher the genetic network underlying the biological processes connected to APEC pathogenicity.
Results: Our GWAS identified variations in gene content for 13 genes and SNPs in 3 different genes associated with APEC isolates, suggesting both gene-level and SNP-level variations contribute to APEC pathogenicity. Integrating protein-protein interaction data, we found that 15 of these genes clustered in the same genetic network, suggesting the pathogenicity of APEC might be due to the interplay of different regulated pathways. We also found novel candidate genes including an uncharacterized multi-pass membrane protein (yciC) and the outer membrane porin (ompD) as linked to APEC isolates.
Discussion: Our findings suggest that convergent pathways related to nutrient uptake from host cells and defense from host immune system play a major role in APEC pathogenicity. In addition, the dataset curated in this study represents a comprehensive historical genomic collection of avian E. coli isolates and constitutes a valuable resource for their comparative genomics investigations.
Escherichia coli infection in poultry might cause a wide variety of pathologies such as salpingitis, peritonitis, airsacculitis, femoral head necrosis, cellulitis or omphalitis, which are collectively called colibacillosis (1). This disease is one of the major concerns in poultry production due to high economic losses and the requirement of antibiotic use for their treatment, which has public health importance as well. In recently published prevalence studies in broiler and layer chickens, E. coli was found to be the leading pathogen (2, 3). Despite being one of the major diseases in poultry, options for prevention by vaccination are still limited largely due to the high genomic heterogeneity and complex pathogenicity mechanisms of avian E. coli isolates (4). Although colibacillosis might exhibit varying clinical manifestations in different avian species (1), the underlying molecular mechanisms of pathogenicity are thought to be common (2, 3).
Historically, isolates collected from the systemic organs of diseased birds are classified as avian pathogenic E. coli (APEC), a subset of extra-intestinal pathogenic E. coli (ExPEC) responsible for causing colibacillosis. In contrast, commensal strains originating from the gut are classified as avian fecal E. coli (AFEC). APEC strains can arise through four distinct evolutionary mechanisms (5): (1) the extra-intestinal spread of pathogenic clones possessing specific virulence factors; (2) host-mediated selection that favors highly pathogenic strains; (3) plasmid-mediated extra-intestinal spread, which involves the transfer of plasmids among lineages; and (4) horizontal gene transfer that generates novel APEC strains through the reassortment of plasmids and virulence factors.
However, the genetic distinction between APEC and AFEC remains unclear, as researchers have yet to define a unique set of features that can reliably identify APEC strains. Numerous attempts have been made to pinpoint marker genes specific to APEC strains [e.g., (6–8)]. The most widely used typing method focuses on five genes (iutA, hlyF, iss, iroN, ompT) (7), which are located on the ColV plasmid (9). This method has been employed to characterize APEC strains in over 60 genomic reports. However, a large-scale study involving 568 isolates questioned the discriminatory power of these five genes, revealing their widespread presence in commensal isolates as well (5). Additionally, a recent analysis of 3,479 isolates demonstrated that no single gene can be uniquely associated with APEC strains (8). Consequently, high-resolution studies are necessary to uncover the intricate genomic traits that contribute to avian E. coli pathogenicity.
A promising approach to unravel the genetic basis of the APEC pathotype is the Genome-Wide Association Study (GWAS). A recent GWAS analysis, involving 568 E. coli isolates from healthy and diseased birds on commercial poultry farms across various European regions, identified 143 APEC-associated genes. These genes play roles in metabolism, lipopolysaccharide synthesis, heat shock response, antimicrobial resistance, and toxicity (5). However, a simulation study demonstrated that the power of detecting associations in bacterial GWAS significantly increases as the sample size approaches 1,000 genomes, reaching a plateau at around 3,000 genomes (10). In light of this information, we conducted an extensive literature and database search to collect all available avian E. coli genomes. This effort resulted in a comprehensive dataset comprising 2,015 high-quality genomes. We then integrated GWAS analysis with existing protein–protein interaction data to identify candidate genes associated with the APEC pathotype. This approach allowed us to propose a systematic overview of the cellular processes underlying APEC pathogenicity.
Literature screening was performed to collect studies that reported genome sequences of E. coli isolated from domestic avian species (chickens, turkeys, ducks, and geese). The search was focused on field studies where (a) commensal E. coli retrieved from the intestinal tract of apparently healthy birds (AFEC) and (b) pathogenic isolates retrieved from clinical cases of avian colibacillosis (APEC) were reported. Other types of studies such as those describing experimental infection models were not included. Literature searches were performed in the PubMed and Web of Science databases during November-December 2021, using the following Boolean search string:
(chicken* OR poultry* OR broiler* OR turkey* OR hen* OR fowl* OR geese OR goose OR duck*) AND (“Escherichia coli” OR “E. coli” OR APEC OR “pathogenic E. coli” OR “pathogenic Escherichia coli” OR ExPEC OR colibacillosis OR colisepticaemia OR colisepticemia OR peritonitis OR salpingitis OR cellulitis) AND (genom* OR wgs)
Studies published before 2000 or written in languages other than in English were excluded. The studies (n = 730) meeting the above set criteria were downloaded, uploaded to the Rayyan systematic review program (11) and deduplicated. In the first phase, studies were screened at the title and abstract level, and those not complying with our inclusion criteria were excluded. In the second phase, studies passing phase one and those marked as unclear were evaluated at the full-text level. Ultimately, 48 studies reporting 2,209 E. coli genomes fulfilled the eligibility criteria and were included in our analyses.
From each publication, the corresponding genome assemblies were downloaded from the NCBI assembly database based on the accession numbers provided. If genome sequences were not available, reads data from the NCBI SRA database were assembled using the Comprehensive Genome Analysis service from the PATRIC web application (12) with default parameters (minimum contig length = 300 bp) by directly providing the SRA accession to the PATRIC interface. The quality of the assemblies was assessed with the checkM software using the default settings (13). Low-quality genomes (n = 185) were filtered out based on the following criteria: completeness ≤ 95%, contamination ≥ 5%, number of contigs ≥ 800, N50 < 20 Kbp and genomes with no marker genes detected. Moreover, genomes were annotated with PROKKA (14) using the default parameters. Finally, E. coli phylogroups and multi-locus sequence types (MLSTs) were computed with the ClermontTyping tool (15) and the pubMLST (https://pubmlst.org/) database, respectively. In addition to the genome sequences, a metadata table describing the 2015 high-quality isolates was generated (Supplementary Table 1).
A schematic representation of the pipeline is summarized in Supplementary Figure 1. Initially, a gene presence/absence matrix for the 2015 high-quality genomes was produced using Panaroo (16) (parameters –clean-mode strict -a core –aligner mafft –core_threshold 0.95) by providing the gene annotations from PROKKA as input (14). Next, a phylogenetic tree was generated using FastTree (17) (parameters -nt -nopr -cat 20 -nosupport -fastest) from the Panaroo core-gene alignment. For the gene-based GWAS, the gene presence/absence matrix and phylogenetic tree were used as input to the R package treeWAS (18) (parameters p-value = 0.05), together with a phenotype binary vector in which the phenotype of the 2015 strains was encoded with 0 for AFEC and 1 for APEC. The program returned a list of genes that were significantly enriched in one class over the other. For each of the significant genes, its prevalence (in percentage) among APEC and AFEC isolates was calculated. Then, significant genes with prevalence > 50% in APEC and APEC vs. AFEC prevalence ratio > 1 were retained for further processing. For the SNP-based GWAS, the 2015 strains were aligned with parsnp (19) (default parameters) using the APEC O1 strain as a reference (GenBank accession GCA_902880315.1). From the parsnp output, a VCF file with SNPs was generated and separated into biallelic SNPs and multiallelic SNPs using bcftools (20) (parameters view m2 -M2). Biallelic SNPs were filtered by removing sites where the alternative allele was equal to N and converted to a binary matrix, encoding the reference allele as 1 and the alternative allele as 0; finally, each SNP was named using the convention snp_xxxxxx where xxxxxx is the genomic position in the parsnp alignment. Multiallelic SNPs were also filtered by removing sites where one of the alternative alleles was equal to N and converted into a binary matrix using a multi-line representation, following the suggestions from Saund et al. (21). Recoded multiallelic SNPs were named using the convention snp_xxxxxx_y where xxxxxx is the genomic location in the parsnp alignment and y is a progressive number (1, 2, 3) uniquely identifying the alternative allele. The whole set of biallelilc SNPs and recoded multiallelic SNPs was given as input to treeWAS (parameters p-value = 0.05) together with the phylogenetic tree and phenotype binary vector that were used for the gene-based GWAS. The program returned a list of significant SNPs associated to APEC isolates. The corresponding gene associated to each significant SNP was retrieved from the parsnp output. To further validate the significant genes obtained from both gene-based and SNP-based GWAS, we conducted a unitig-based GWAS. We generated unitigs for all genomes with unitig caller (4) (default parameters) and assessed their association with the APEC phenotype using the same approach as in the previous GWAS methods. We then aligned the significant unitigs to the candidate genes from the gene-based and SNP-based GWAS analyses using BLASTN (parameters -word_size 7 -evalue 1e-05). Only candidate genes with at least 5 aligned unitigs were retained as final candidates, ensuring a more robust validation of the identified genes.
Through a comprehensive literature screening, we identified an initial set of 730 publications reporting E. coli genomes from different avian hosts. After deduplication and extensive manual curation, we refined our list to 48 publications that all together describe the genome sequences of 2209 avian E. coli genomes divided into APEC and AFEC isolates. After filtering out low quality genomes, we generated a final database of 2015 E. coli avian genomes including 1,089 APEC isolates and 926 AFEC isolates. Host distribution for this dataset includes chicken (n = 1,732), turkey (n = 152), duck (n = 56), wildfowl (n = 12), gull (n = 5), goose (n = 4) and poultry unknown (n = 54). A summary table containing the number of isolates in each study and the corresponding publication reference is shown in Table 1, while the metadata table for the 2015 isolates is provided in Supplementary Table 1.
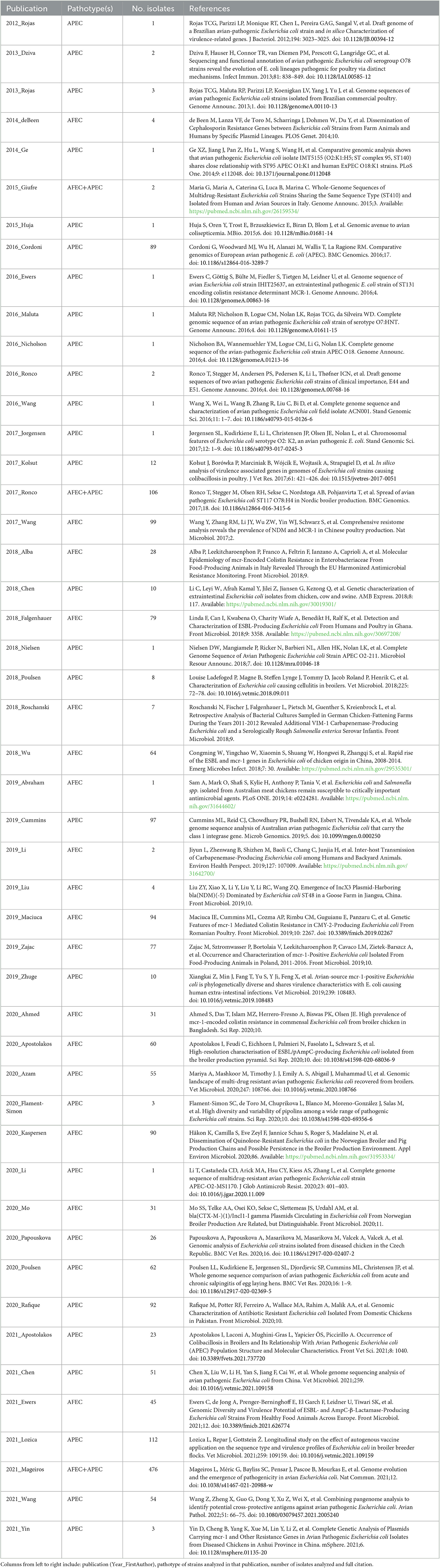
Table 1. List of publications used to construct the avian E. coli genomic database.
A K-mer tree in form of a circular cladogram for the curated 2015 avian E. coli isolates is depicted in Figure 1 highlighting the connection of pathotype with phylogroups in different colors. Isolates from each pathotype were unequally distributed in each phylogroup (Chi-Square test – P = 1.04 × 10−77) (Figure 2A), with phylogroups B2, C, and G significantly enriched in APEC isolates (Chi-Square post-hoc test – PB2 = 0, PC = 0 and PG = 0) and phylogroups A and F significantly enriched in AFEC isolates (Chi-Square post-hoc test – PA = 0 and PF = 0.0001). Genome length distribution is significantly different between APEC and AFEC isolates (Mann-Whitney U test – P = 0.0225), with APEC genomes being significantly shorter than AFEC genomes (median length APEC = 5.127 Mbp, median length AFEC = 5.142 Mbp). However, when splitting the data by phylogroup, APEC genomes were significantly shorter than AFEC genomes for phylogroups B1, E and G only (Mann-Whitney U test – PB1 = 8.99 × 10−12, PE = 8.16 × 10−6, PG = 9.09 × 10−8), indicating that shorter genome length is not an exclusive feature of APEC isolates. Gene number distribution is also significantly different between APEC and AFEC isolates (Mann-Whitney U test – P = 0.0095), with APEC isolates having significantly less genes than AFEC isolates (median no. genes APEC = 4888, median no. genes AFEC = 4916). When splitting the data by phylogroup, this pattern holds only for phylogroups B1, E, G (Mann-Whitney U test – PB1 = 8.23 × 10−11, PE = 1.25 × 10−5, PG = 3.79 × 10−8) implying that a lower number of genes might simply reflect a shorter genome length and is not a general feature of APEC isolates. In Figure 2B we report the top 10 sequence types with at least 10 isolates, sorted by prevalence in APEC isolates (top table) and AFEC isolates (bottom table), which provides additional insights into the genetic diversity of the isolates. The pan-genome of the 2015 E. coli isolates consisted of 29,262 genes, which were divided into 2,899 core and 26,363 accessory genes (Figure 2C).
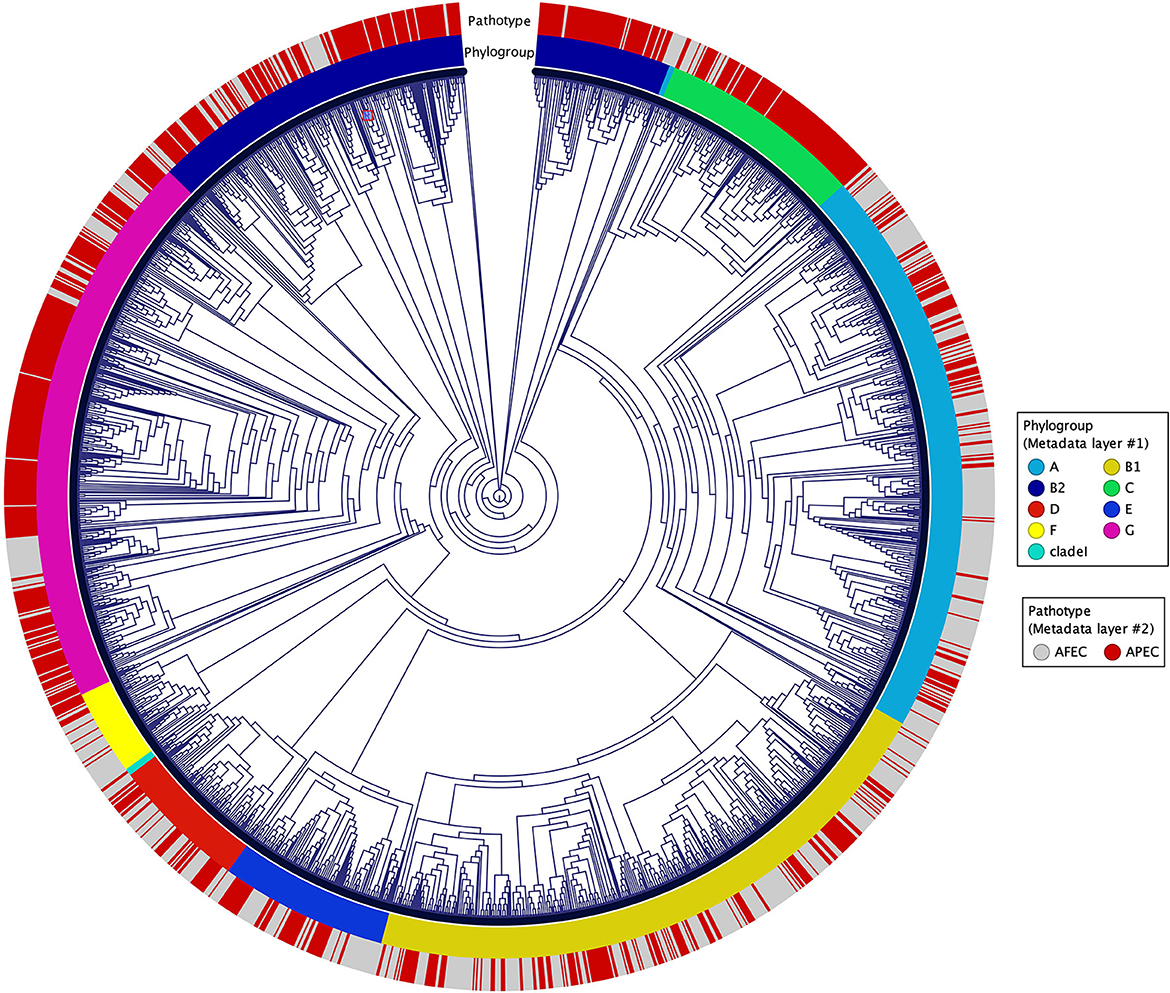
Figure 1. Circular cladogram for the 2015 avian E. coli isolates. The circular cladogram shows the relationships between the 2015 avian E. coli isolates, with the outermost circle indicating the pathotypes, and the second circle indicating the phylogroups highlighted in different colors.
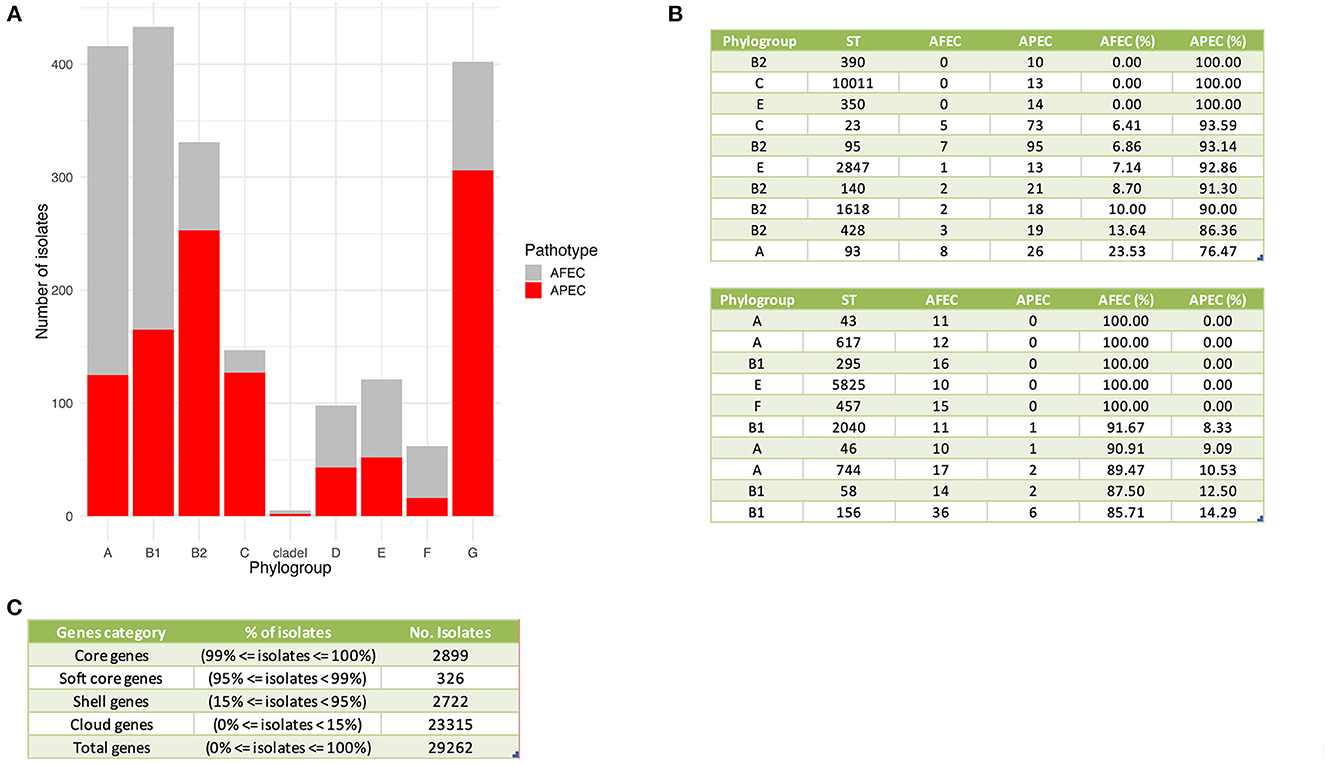
Figure 2. General genomic features of APEC and AFEC isolates. (A) Distribution of APEC and AFEC isolates by phylogroup. (B) Top 10 sequence types with at least 10 isolates associated with APEC isolates (top table) and AFEC isolates (bottom table). Columns from left to right indicate: phylogroup, sequence type (ST), number of AFEC isolates, number of APEC isolates, and the percentage of each group with that ST. (C) Division of genes in the pan-genome of 2015 E. coli isolates based on their prevalence among the isolates. The genes are categorized as core, soft core, shell, or cloud genes, depending on the percentage of isolates in which they are present. The table displays the percentage of isolates and the number of isolates for each gene category, as well as the total number of genes in the pan-genome.
To assess the relative contributions of gene-level and SNP-level genomic variation associated with APEC isolates, we initially performed two independent GWAS: (1) a gene-based GWAS to identify associations between the phenotypic vector of each strain (1 = APEC, 0 = AFEC) and variation in gene content defined by a gene presence/absence matrix; (2) an SNP-based GWAS to find associations between the phenotypic vector of each strain (1 = APEC, 0 = AFEC) and an SNPs matrix derived from the whole-genome alignment of our 2015 isolates against the APEC O1 reference strain. Out of a total of 29,262 genes in the pan-genome, the gene-based GWAS identified 16 genes significantly associated with APEC isolates. From a total of 22,920 SNPs in the core genome, the SNP-based GWAS found 7 SNPs significantly associated with APEC isolates, located in 6 different genes. To further validate these candidate genes, we employed a unitig-based GWAS. This approach validated 13 of the 16 genes from the gene-based GWAS (Figure 3A) and 3 of the 6 genes from the SNP-based GWAS (Figure 3B). In total, we identified 16 final candidate genes linked to APEC isolates after considering the results from both gene-based and SNP-based GWAS, along with the unitig-based validation.
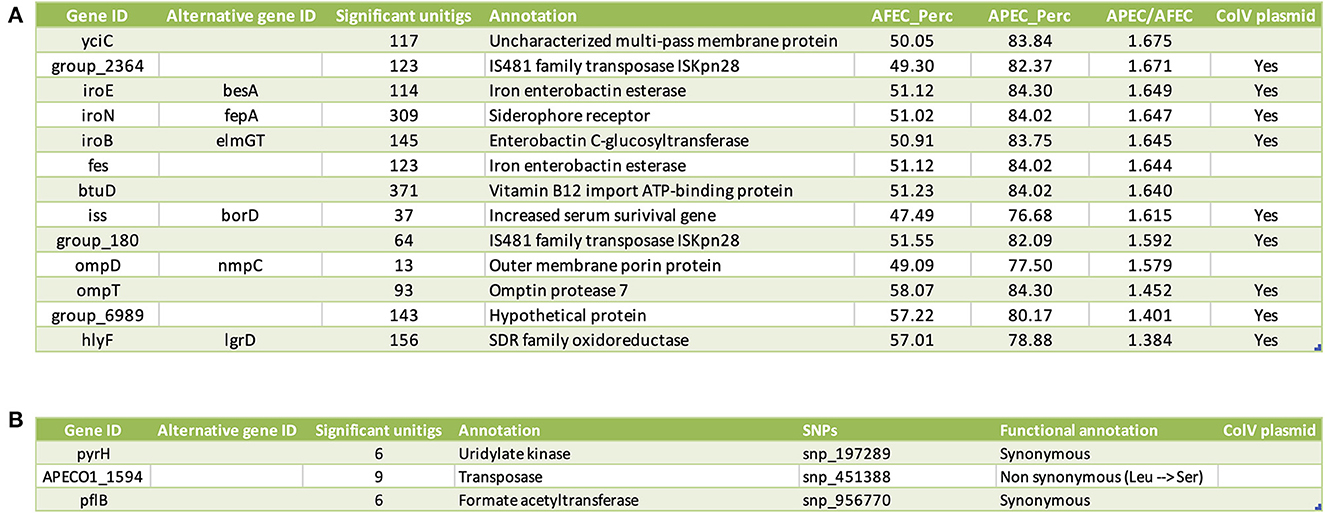
Figure 3. List of candidate genes linked to the APEC pathotype. (A) Candidate genes identified through the gene-based GWAS. Columns from left to right indicate: gene ID, alternative gene ID(s), functional annotation of the gene, prevalence of the gene (in %) among AFEC isolates (AFEC_Perc), prevalence of the gene (in %) among APEC isolates (APEC_Perc), and the prevalence ratio of APEC_Perc over AFEC_Perc. Additionally, a column indicates whether the gene is located on the ColV plasmid. (B) Candidate genes identified through the SNP-based GWAS. columns from left to right indicate: gene ID, alternative gene ID(s), functional annotation of the gene, and significant SNPs, which are encoded as snp_xxxxxx_y. Here, xxxxxx represents the position of the SNP in the genome whereas y stands for a distinct numerical value (1, 2, 3) that uniquely distinguishes each alternative allele.
To further investigate the 16 candidate genes associated with APEC isolates, we utilized protein–protein interaction networks from the STRING database (22). As these data are only available for genes with standard 3 or 4 letter gene names, we excluded the 4 candidate genes with generic gene names (group_2364, group_180, group_6989, and APECO1_1594) from this analysis. Among the remaining 12 candidate genes, protein–protein interaction data were identified for 11 of them (all of them except hlyF). These included 7 genes that belonged to the primary genetic network (Figure 4—main network) derived from strain K12 MG1655, 3 genes related to subnetwork A (Figure 4) derived from strain CFT073, and 1 gene linked to subnetwork B (Figure 4) derived from strain UMN026. It is important to note that subnetwork B shares two genes (ompT and borD) in common with the primary genetic network; however, they are not circled in bold in subnetwork B to avoid confusion, as they are already highlighted in the primary network. Altogether, these interactions encompassed a total of 99 genes. In order to unambiguously describe this network, we applied MCL clustering with inflation parameter = 3, colored each node based on the membership to the respective cluster, and numbered the clusters from 1 to 8. To avoid ambiguities, we will report the alternative gene names throughout the manuscript in case of genes with multiple gene names.
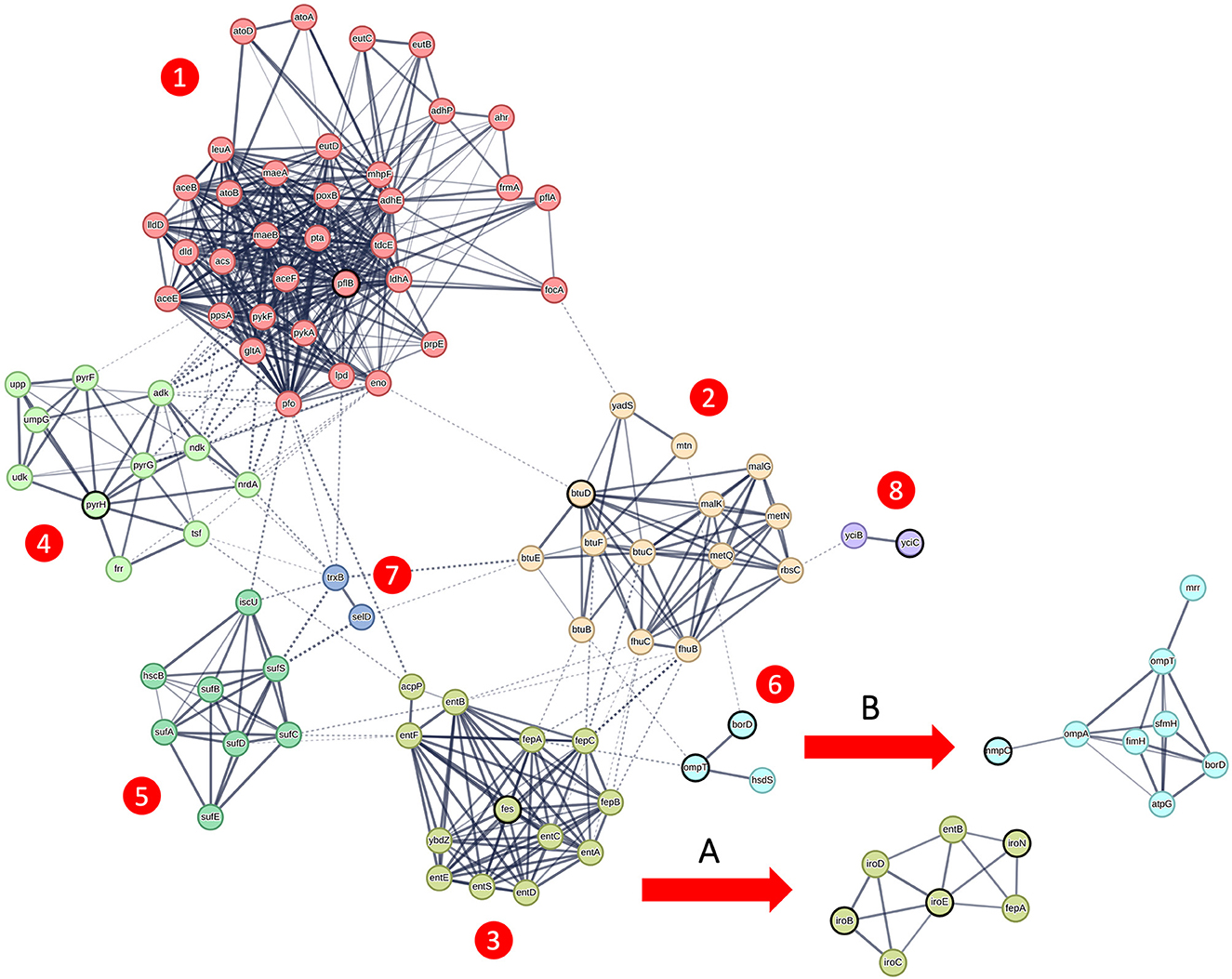
Figure 4. Genetic network of candidate genes linked to the APEC pathotype. Genetic network obtained from the STRING database (22), featuring candidate genes emphasized in bold. The core genetic network, originating from strain K12 MG1655, displays genes color-coded based on MCL clustering using an inflation parameter of 3. These genes are organized into seven distinct clusters, numbered 1 through 7. Additionally, subnetwork A, indicated by the arrow, is derived from strain CFT073, while subnetwork B originates from strain K12 MG1655.
Cluster 1 includes the gene formate acetyltransferase (pflB) and is linked to pyruvate metabolism (P = 2.43e-17), acetate metabolic process and ethanol biosynthetic process (P = 4.68e-05). Cluster 2 includes the vitamin B12 import ATP-binding protein (btuD) and is linked to cobalamin transport (P = 2.34e-05), nitrogen compound transport (P = 0.0043) and organic substance transport (P = 0.00019).
Cluster 3 together with subnetwork A includes the genes iron enterobactin esterase (fes) enterobactin C-glucosyltransferase (iroB), iron enterobactin esterase (iroE) and siderophore receptor (iroN) and is enriched for enterobactin metabolic process (P = 3.35e-14), siderophore-dependent iron import in the cell (P = 1.38e-05) and colicin transport (P = 0.0034). Cluster 4 includes the gene (pyrH) and is related to CTP metabolic process (P = 8.50e-07) and ribonucleoside monophosphate biosynthetic process (P = 2.11e-06). Cluster 5 does not contain any candidate gene but it connects clusters 1 and 3. This cluster is enriched for sulfur incorporation into metallo-sulfur cluster (P = 0.0042) and iron-sulfur cluster assembly.
Cluster 6 together with subnetwork B includes the genes omptin protease (ompT), increased serum survival gene (borD – more commonly known as iss), and outer membrane porin (nmpC, also known as ompD) and is linked to restriction system, and Bor protein (P = 2.45e-05) and signal peptide (P = 0.0040). Cluster 7 do not contain candidate genes and it is connected to selenocompound metabolism (P = 0.0023). Cluster 8 contains the uncharacterized multi-pass membrane protein (yciC) and is linked to intracellular septation protein A and uncharacterized protein family (P = 0.0033).
The remaining candidate gene with standard gene names without protein–protein interaction data encodes for a SDR family oxidoreductase (hlyF). Further characterization using the KEGG database did not provide additional functional details for this gene. However, it is known that this gene encodes for a virulence factor involved in outer membrane vesicle biogenesis in ExPEC strains (5).
Finally, the 4 candidate genes with generic names encode for three transposase genes (group_2364, group_180, APECO1_1594) and for a hypothetical protein (group_6989). For these genes, we looked at their genomic location on the APEC O1 reference strain and observed that 3 out of 4 are located on the ColV plasmid. More in detail, the IS418 family transposases group_2364 and group_180 are located in the region upstream to the candidate gene iss (Supplementary Figure 2A); while the hypothetical protein group_6989 is adjacent to the candidate gene hlyF (Supplementary Figure 2B).
In an effort to elucidate the underlying genetic network of avian E. coli involved in colibacillosis, we collected all available genomes of relevant isolates reported in the period 2000-2021 and constructed a comprehensive dataset including 2015 avian E. coli isolates covering a wide range of APEC and AFEC strains. Our aim was to construct a sufficiently large database in order to conduct a robust GWAS analysis (23).
Two major trends emerged from the exploratory analysis of this dataset. First, we found that APEC strains can emerge from any phylogroup; however, phylogroups B2, C, and G were significantly enriched with APEC strains, consistent with the results from Johnson et al. (8). Second, we observed that APEC genomes are significantly shorter than AFEC genomes. This is in line with the observation that many pathogenic bacteria have usually smaller genomes compared to their non-pathogenic counterpart (24). This might be an effect of host adaptation, for which some genes could either become less necessary for survival or could be selectively lost if they encode for immune targets. This finding adds another unexplored layer in explaining the evolution of APEC pathogenicity.
Our GWAS analysis identified variation in gene content for 13 genes and SNPs in 3 different genes as linked to APEC isolates. Thus, our findings suggest that both variations at gene-level and SNP-level contribute to the APEC pathotype. By integrating available protein–protein interaction data for these 16 genes, we showed that 15 of them clustered in the same genetic network connecting a total of 99 genes. This result suggests that APEC pathogenicity might be the result of the interplay of different tightly regulated pathways.
Based on the results from our genetic network analysis, we classified the candidate genes into four categories: (1) genes associated with nutrient uptake; (2) genes involved in defense against the host immune system; (3) genes co-selected due to their proximity to other candidate genes; and (4) previously unidentified genes that are now linked to APEC isolates.
In the first category of candidate genes, iron uptake seems to play a major role in APEC pathogenicity. The role of iron uptake in APEC pathogenicity is well documented (25): the genes from the iroBCDEN operon can be located on the ColV plasmid (9) or chromosomally encoded and are involved in glycosylation (iroB), processing (iroE) and transport (iroN) of the salmocheline siderophore (26), a protein able to sequester iron from host cells. These proteins were functionally investigated in the APEC O78 strain and found to be essential for APEC pathogenicity (26). The fes gene is also involved in biosynthesis of salmocheline and its deletion in mutants of the APEC strain χ7122 caused a reduction of salmocheline production (27), but not a complete ablation of the product. The role of iron uptake was also confirmed in a recent GWAS study on ExPEC strains (28).
The genetic network further suggests that the uptake of other nutrients might be also important for APEC pathogenicity: these include vitamin B12 uptake, connected to the vitamin B12 import ATP-binding protein (btuD) and acetate assimilation, linked to the formate acetyltransferase (pflB) gene. While the role of vitamin B12 in APEC pathogenicity was never reported before, a study performed on two highly virulent APEC strains highlighted that the deletion of three genes directly connected to the pflB gene (acs, yjcH and actP) caused a decrease of cytotoxicity in macrophages, which was linked to the ablation of the acetate assimilation system (29). Thus, the authors concluded that acetate assimilation conferred a fitness advantage during APEC early colonization. Based on these results, it appears that ability to uptake iron, vitamin B12 and acetate from the host promotes the emergence of pathogenicity in avian E. coli.
The second category of candidate genes includes genes involved in the defense from host-immune system, among which the ompT, borD (iss), and hlyF genes. The omptin protease (ompT) is located on the ColV plasmid (9) and is involved in the degradation of host proteins and antimicrobial peptides (30). It was reported as a standard APEC marker gene (7) and its role in APEC pathogenicity has been confirmed in a genetic deletion study in the APEC strain TW-XM, where it affected adhesion, invasion, colonization, and proliferation capacities (31). Closely linked to ompT in the genetic network, lays the borD gene, more commonly known as increased serum survival gene (iss), also located on the ColV plasmid (9) and expressed on the outer membrane (32). The iss has been also extensively used as APEC marker (7) and a deletion in the APEC O78-9 strain affected its ability to grow in serum (33), but did not provide a detailed functional characterization. Only recently, a functional study in ExPEC strains revealed its role in group 4 capsule synthesis (34), which protects the bacteria from the complement proteins of the host immune system. The SDR family oxidoreductase (hlyF) gene is also located on the ColV plasmid and one of the standard APEC markers (32). This gene was originally thought to be a hemolysin, however a functional study showed that it is involved in the regulation of outer membrane vesicle biogenesis (35), which prevents the autophagosome-lysosome fusion during the immune response, amplifying the pathogenic potential of strains containing this gene (36). These results suggest that a combination of host-specific immune-defense mechanisms contribute to the appearance of pathogenicity in avian E. coli.
The third category of candidate genes includes genes that might be co-selected due to proximity to other candidate genes. These include: (1) two transposase genes (group_2364, group_180), which could be involved in horizontal gene transfer of other candidate genes; (2) the hypothetical protein (group_6989), which might be simply co-transposed together with hlyF due to their adjacent position.
In addition to already known genes, we also revealed three previously unidentified genes associated with APEC isolates: (1) an uncharacterized multi-pass membrane protein (yciC), encoding for a hypothetical protein containing six transmembrane domains; remarkably, this gene had the highest APEC/AFEC prevalence ratio in the gene-based GWAS analysis, pointing to a potential but unknown pivotal role in APEC pathogenicity; (2) the outer membrane porin (nmpC, more commonly known as ompD), that was previously found to be overexpressed in a mutant of the APEC strain DE17 deleted for the pfs gene (37), suggesting a possible role in antibiotic resistance; (3) a significant SNP in the pyrH gene, which is a synonymous mutation. This gene encodes uridylate kinase (UMPK), an enzyme involved in pyrimidine nucleotide metabolism. Since this mutation does not impact the protein sequence, we initially hypothesized that the association of this SNP with APEC isolates could potentially be explained by genetic hitchhiking, where the mutation's frequency in the population increases due to its close linkage to another candidate gene linked to APEC. However, upon screening the chromosomal locations of candidate genes in the APEC O1 strain, we found that none of them were in close proximity to pyrH, making this hypothesis less likely. Alternatively, the linkage of this SNP to APEC isolates might be explained by genetic epistasis: the mutation in pyrH could interact with other genetic variants elsewhere in the genome, leading to a combined effect that results in an association with APEC isolates.
Two other GWAS analyses were previously published to address the question of finding genes linked to the APEC pathotype. The first GWAS, by Mageiros et al. (5), was performed on 568 isolates, which are also included in our dataset. Their GWAS analysis identified 143 genes involved in metabolism, lipopolysaccharide synthesis, heat shock response, antimicrobial resistance and toxicity. From the subset of 10 candidate genes with standard names from our gene-based GWAS, only 1 (hlyF) was also detected by Mageiros et al. (5). This may be explained by their approach to conduct the analysis separately for each phylogroup, thus drastically reducing GWAS statistical power. The second GWAS, by Johnson et al. (8), was published after we terminated our data collection phase, and was conducted on 959 clinical and caecal isolates from turkeys, which are part of a larger dataset including a total of 3,479 isolates. Their estimate of 430 candidate genes is higher compared to our 13 genes from the gene-based GWAS. This may be explained by the less conservative filtering criteria applied by Johnson et al. (8). Remarkably, 8 of the 10 candidate genes with standard names from our gene-based GWAS were also found by Johnson et al. (yciC, iroE, iroN, iroB, btuD, iss, ompT, hlyF), providing independent validation for our findings. This overlap further confirms the hypothesis that the genes underlying APEC pathogenesis are common between chicken and turkey, as corroborated by Johnson's GWAS conducted specifically on turkey strains. Interestingly, this list includes the uncharacterized multi-pass membrane protein (yciC), which we proposed as an attractive novel candidate for functional testing. This discovery suggests the importance of investigating the potential role of yciC in APEC pathogenesis and its potential as a therapeutic target for controlling APEC infections in poultry. In conclusion, our results suggest that the interplay between nutrient uptake and the ability to escape host immune defenses enable the emergence of pathogenicity in avian extra-intestinal E. coli.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
NP and IA conceived, designed the study, and performed the data analysis. SP and MH contributed to data interpretation and provided critical insights. All authors participated in drafting the manuscript, revising it for important intellectual content, and provided final approval for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1195585/full#supplementary-material
Supplementary Figure 1. Pipeline for the identification of genes linked to the APEC pathotype. Genome assemblies were downloaded from NCBI (unfiltered genomes), and reads data were assembled using the PATRIC web application if the genome sequences were not available. The quality of the assemblies was assessed using checkM, and low-quality genomes were filtered out based on several criteria (filtered genomes). The genomes were annotated using PROKKA (Genes annotation) and a Maximum Likelihood tree was constructed with FastTree. For the gene-based GWAS, a gene presence/absence matrix was produced with Panaroo, and treeWAS was used to identify significant genes associated with APEC isolates. Candidate genes with a prevalence > 50% in APEC and APEC vs. AFEC prevalence ratio > 1 were selected. For the SNP-based GWAS, parsnp was used to align the strains and generate a VCF file of SNPs, which were filtered and processed using bcftools. Significant SNPs associated with APEC isolates were identified using treeWAS. A unitig-based GWAS was conducted to further validate the significant genes identified from gene-based and SNP-based GWAS. The unitigs were generated and assessed for association with APEC phenotype, and the significant unitigs were aligned to the candidate genes using BLASTN. Final candidates were selected by retaining only the genes with at least 5 aligned unitigs.
Supplementary Figure 2. Genomic locations of candidate genes with generic gene names. Each panel contains two tracks from top to bottom: genes and candidate genes. (A) Locations of group_2364 and group_180 on the ColV plasmid from the APEC O1 strain with respect to other candidate genes; (B) Locations of group_6989 on the ColV plasmid from the APEC O1 strain with respect to other candidate genes.
Supplementary Table 1. Metadata for the 2015 avian E. coli isolates. Columns from left to right indicate: name of the isolate, publication (Year_FirstAuthor), host, pathotype (APEC/AFEC), genome length (bp), number of genes, phylogroup, sequence type (ST), name of the allele variant for the 7 genes used to define the sequence type by the ClermontTyping tool (15).
1. Nolan LK, John BH, Vaillancourt JP, Abdul-Aziz T, Logue CM. Colibacillosis. Diseases of Poultry. 13th ed. Chichester: John Wiley & Sons, Ltd. (2017). p. 751–805.
2. Kaufmann-Bat M, Hoop RK. Diseases in chicks and laying hens during the first 12 years after battery cages were banned in Switzerland. Vet Record. (2009) 164:203–7. doi: 10.1136/vr.164.7.203
3. Grafl B, Gaußmann B, Sulejmanovic T, Hess C, Hess M. Risks and disease aetiologies of compromised performance in commercial broilers kept at lower stocking density and limited antimicrobial use. Avian Pathol. (2020) 49:621–30. doi: 10.1080/03079457.2020.1805411
4. Optimising Poultry Flock Health | Sjaak de Wit | Taylor & Francis eBoo. Available online at: https://www.taylorfrancis.com/pdfviewer/ (accessed September 26, 2022).
5. Mageiros L, Méric G, Bayliss SC, Pensar J, Pascoe B, Mourkas E, et al. Genome evolution and the emergence of pathogenicity in avian Escherichia coli. Nat Commun. (2021) 12:765. doi: 10.1038/s41467-021-20988-w
6. Ewers C, Janßen T, Kießling S, Philipp HC, Wieler LH. Rapid detection of virulence-associated genes in avian pathogenic Escherichia coli by multiplex polymerase chain reaction. Avian Dis. (2005) 49:269–73. doi: 10.1637/7293-102604R
7. Johnson TJ, Wannemuehler Y, Doetkott C, Johnson SJ, Rosenberger SC, Nolan LK. Identification of minimal predictors of avian pathogenic Escherichia coli virulence for use as a rapid diagnostic tool. J Clin Microbiol. (2008) 46:3987–96. doi: 10.1128/JCM.00816-08
8. Johnson TJ, Miller EA, Flores-Figueroa C, Munoz-Aguayo J, Cardona C, Fransen K, et al. Refining the definition of the avian pathogenic Escherichia coli (APEC) pathotype through inclusion of high-risk clonal groups. Poult Sci. (2022) 101:102009. doi: 10.1016/j.psj.2022.102009
9. Johnson TJ, Siek KE, Johnson SJ, Nolan LK. DNA sequence of a ColV plasmid and prevalence of selected plasmid-encoded virulence genes among avian Escherichia coli strains. J Bacteriol. (2006) 188:745–58. doi: 10.1128/JB.188.2.745-758.2006
10. Saber MM, Jesse Shapiro B. Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes. Microb Genom. (2020) 6:e000337. doi: 10.1099/mgen.0.000337
11. Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan-a web and mobile app for systematic reviews. Syst Rev. (2016) 5:1. doi: 10.1186/s13643-016-0384-4
12. Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, et al. The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res. (2020) 48:D606–D612. doi: 10.1093/nar/gkz943
13. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. (2015) 25:1043–55. doi: 10.1101/gr.186072.114
14. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
15. Beghain J, Bridier-Nahmias A, Nagard H le, Denamur E, Clermont O. ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb Genom. (2018) 4:e000192. doi: 10.1099/mgen.0.000192
16. Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. (2020) 21:1–21. doi: 10.1186/s13059-020-02090-4
17. Price MN, Dehal PS, Arkin AP. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. (2009) 26:1641–50. doi: 10.1093/molbev/msp077
18. Collins C, Didelot X. A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination. PLoS Comput Biol. (2018) 14:e1005958. doi: 10.1371/journal.pcbi.1005958
19. Treangen TJ, Ondov BD, Koren S, Phillippy AM. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. (2014) 15:1–15. doi: 10.1186/s13059-014-0524-x
20. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
21. Saund K, Lapp Z, Thiede SN, Pirani A, Snitkin ES. Prewas: data pre-processing for more informative bacterial gwas. Microb Genom. (2020) 6:1–8. doi: 10.1099/mgen.0.000368
22. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–12. doi: 10.1093/nar/gkab835
23. Power RA, Parkhill J, de Oliveira T. Microbial genome-wide association studies: lessons from human GWAS. Nat Rev Genet. (2016) 18:41–50. doi: 10.1038/nrg.2016.132
24. Weinert LA, Welch JJ. Why might bacterial pathogens have small genomes? Trends Ecol Evol. (2017) 32:936–47. doi: 10.1016/j.tree.2017.09.006
25. Gao Q, Wang X, Xu H, Xu Y, Ling J, Zhang D, et al. Roles of iron acquisition systems in virulence of extraintestinal pathogenic Escherichia coli: Salmochelin and aerobactin contribute more to virulence than heme in a chicken infection model. BMC Microbiol. (2012) 12:1–2. doi: 10.1186/1471-2180-12-143
26. Caza M, Lépine F, Milot S, Dozois CM. Specific roles of the iroBCDEN genes in virulence of an avian pathogenic Escherichia coli O78 strain and in production of salmochelins. Infect Immun. (2008) 76:3539–49. doi: 10.1128/IAI.00455-08
27. Caza M, Garénaux A, Lépine F, Dozois CM. Catecholate siderophore esterases Fes, IroD and IroE are required for salmochelins secretion following utilization, but only IroD contributes to virulence of extra-intestinal pathogenic Escherichia coli. Mol Microbiol. (2015) 97:717–32. doi: 10.1111/mmi.13059
28. Galardini M, Clermont O, Baron A, Busby B, Dion S, Schubert S, et al. Major role of iron uptake systems in the intrinsic extra-intestinal virulence of the genus Escherichia revealed by a genome-wide association study. PLoS Genet. (2020) 16:e1009065. doi: 10.1371/journal.pgen.1009065
29. Zhuge X, Sun Y, Jiang M, Wang J, Tang F, Xue F, et al. Acetate metabolic requirement of avian pathogenic Escherichia coli promotes its intracellular proliferation within macrophage. Vet Res. (2019) 50:1–8. doi: 10.1186/s13567-019-0650-2
30. Kukkonen M, Korhonen TK. The omptin family of enterobacterial surface proteases/adhesins: from housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int J Med Microbiol. (2004) 294:7–14. doi: 10.1016/j.ijmm.2004.01.003
31. Hejair HMA, Ma J, Zhu Y, Sun M, Dong W, Zhang Y, et al. Role of outer membrane protein T in pathogenicity of avian pathogenic Escherichia coli. Res Vet Sci. (2017) 115:109–16. doi: 10.1016/j.rvsc.2017.01.026
32. Lynne AM, Skyberg JA, Logue CM, Nolan LK. Detection of Iss and Bor on the surface of Escherichia coli. J Appl Microbiol. (2007) 102:660–6. doi: 10.1111/j.1365-2672.2006.03133.x
33. Huja S, Oren Y, Trost E, Brzuszkiewicz E, Biran D, Blom J, et al. Genomic avenue to avian colisepticemia. mBio. (2015) 6:e01681–14. doi: 10.1128/mBio.01681-14
34. Biran D, Sura T, Otto A, Yair Y, Becher D, Ron EZ. Surviving serum: the Escherichia coli is gene of extraintestinal pathogenic E. coli is required for the synthesis of group 4 capsule. Infect Immunity. (2021) 89:e00316–21. doi: 10.1128/IAI.00316-21
35. Murase K, Martin P, Porcheron G, Houle S, Helloin E, Pénary M, et al. HlyF produced by extraintestinal pathogenic Escherichia coli is a virulence factor that regulates outer membrane vesicle biogenesis. J Infect Dis. (2015) 212:856–65. doi: 10.1093/infdis/jiv506
36. David L, Taieb F, Pénary M, Bordignon PJ, Planès R, Bagayoko S, et al. Outer membrane vesicles produced by pathogenic strains of Escherichia coli block autophagic flux and exacerbate inflammasome activation. Autophagy. (2022) 18:2913–25. doi: 10.1080/15548627.2022.2054040
Keywords: GWAS, APEC, pathogenicity (infectivity), Escherichia coli (E. coli), protein–protein interaction (PPI) network
Citation: Palmieri N, Apostolakos I, Paudel S and Hess M (2023) The genetic network underlying the evolution of pathogenicity in avian Escherichia coli. Front. Vet. Sci. 10:1195585. doi: 10.3389/fvets.2023.1195585
Received: 29 March 2023; Accepted: 05 June 2023;
Published: 21 June 2023.
Edited by:
Orhan Sahin, Iowa State University, United StatesReviewed by:
Dipak Kathayat, Cornell University, United StatesCopyright © 2023 Palmieri, Apostolakos, Paudel and Hess. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Palmieri, bmljb2xhLnBhbG1pZXJpQHZldG1lZHVuaS5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.