 Yanbin Zhu1,2,3,4
Yanbin Zhu1,2,3,4 Yangji Cidan1Guangming Sun1Xin Li2Muhammad Akbar Shahid5Zhaxi Luosang2Zhaxi Suolang2Lang Suo2Wangdui Basang1*
Yangji Cidan1Guangming Sun1Xin Li2Muhammad Akbar Shahid5Zhaxi Luosang2Zhaxi Suolang2Lang Suo2Wangdui Basang1*- 1Institute of Animal Husbandry and Veterinary Medicine, Tibet Academy of Agriculture and Animal Husbandry Sciences, Lhasa, China
- 2Linzhou Animal Husbandry and Veterinary Station, Lhasa, China
- 3Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, China
- 4Key Laboratory of Animal Genetics and Breeding on Tibetan Plateau, Ministry of Agriculture and Rural Affairs, Lhasa, China
- 5Department of Pathobiology, Faculty of Veterinary Sciences, Bahauddin Zakariya University, Multan, Pakistan
The yaks that inhabit the Tibetan plateau are a rare breed that is closely related to local economic development and human civilization. This ancient breed may have evolved a unique gut microbiota due to the hypoxic high-altitude environment. The gut microbiota is susceptible to external factors, but research regarding the effects of different feeding models on the gut fungal community in yaks remains scarce. In this study, we compared and analyzed the composition and variability of the gut fungal community among wild yaks (WYG), house-feeding domestic yaks (HFG), and grazing domestic yaks (GYG). The results revealed that Basidiomycota and Ascomycota were the most preponderant phyla in the gut fungal community, regardless of feeding models. Although the types of dominant fungal phyla did not change, their abundances did. Intergroup analysis of fungal diversity showed that the Shannon and Simpson indices of WYG and GYG were significantly higher than those of HFG. Fungal taxonomic analysis showed that there were 20 genera (Sclerostagonospora and Didymella) that were significantly different between WYG and GYG, and 16 genera (Thelebolus and Cystobasidium) that were significantly different between the WYG and HFG. Furthermore, the proportions of 14 genera (Claussenomyces and Papiliotrema) significantly decreased, whereas the proportions of eight genera (Stropharia and Lichtheimia) significantly increased in HFG as compared to GYG. Taken together, this study indicated that the gut fungal composition and structure differ significantly between yaks raised in different breeding groups.
Introduction
It is widely known that the gut microbiota is a complex micro-ecosystem involving a large number of different types of microorganisms, including bacteria, fungi, and viruses (1–3). Studies have shown that the gut microbiota can provide the host with nutrients and beneficial metabolites, such as amino acids, vitamins, and short-chain fatty acids, by fermenting sugars and carbohydrates (4–6). These metabolites play important roles in host health, immunity, intestinal homeostasis, and intestinal barrier (7, 8). Similar to the gut bacterial community, the gut fungal community is also an important component of the gut microbiota, which plays vital roles in host health by improving gut functions (9–12). Early investigations indicated that the gut fungal community could induce significant shifts in the gut bacterial structure and shape the gut microbiota during early life (13). The gut fungal community may also play a role in the maturation of the host immune system by interacting with the gut bacterial community to produce strong local and systemic immune responses (13, 14). Furthermore, some fungi are considered to be intestinal probiotics due to their potential role in alleviating inflammation, inhibiting pathogenic bacteria, degrading cellulose, and regulating digestion (15–17). For instance, administration of Candida kefyr has been demonstrated to alleviate gastrointestinal inflammation by changing the gut microbiota (18). Additionally, some fungi can synthesize and release neurotransmitters such as norepinephrine and histamine (19, 20). However, gut microbial homeostasis is easily affected by external factors such as age, sex, diet, and disease (21, 22). Moreover, recent studies have indicated that feeding methods, altitude, and habitat environment are also important driving forces for the development of gut microbiota (23, 24).
Yak is a rare species of cattle of the Tibetan plateau (above 3,000 m), which is characterized by adapting to high, cold, and oxygen-deficient environments (25). Statistically, approximately 90% of the world's yaks live in the Sichuan, Qinghai, Tibet, and Gansu provinces of China (26, 27). Yaks are also an important source of milk and meat products for local herdsmen and play an important role in economic development. Given the importance of yaks on the Tibetan plateau, any factors that threaten the health and development of this breed may lead to enormous economic losses. Previous studies indicated that cold and hypoxic environments could cause changes in the gut microbial structure (28–31). Therefore, the altitude hypoxia environments of the Tibetan Plateau may induce the accumulation of special gut microbiota in yaks compared with animals living in plains. Indeed, several studies have reported the unique composition and diversity of the gut microbiota in yaks (32, 33).
In the past, yaks were mainly raised in the open and were easily affected by the external environment. For instance, changeable weather and nutritional deficiencies can cause low production efficiency and a high disease rate of yaks (34, 35). To improve the productivity of yaks, a combination of free grazing and barn feeding is also implemented in some areas. Furthermore, there are still some wild yaks in the same area of the Tibetan Plateau. Although these yaks are the same species, they may have evolved specific microbial communities to adapt to different farming methods. A previous study has demonstrated significant differences in the gut bacterial community of yaks under different feeding models (36, 37). However, until now, little research has been conducted on the gut fungal community of yaks. Therefore, the aim of our study was to evaluate the composition and variability of the gut fungal community of yaks under different feeding models.
Materials and methods
Sample acquisition
In this study, fecal samples of grazing domestic yaks (GYG) and wild yaks (WYG) were obtained from Shuanghu County and Chang Tang Nature Reserve, China. The average altitude of this area exceeds 5,000 meters and is characterized by high temperatures, low precipitation, and strong wind speed in July and August. Moreover, the zonal vegetation includes Austrostipa pubescens as the dominant vegetation. We also collected fecal samples from house-fed domestic yaks (HFG), which were mainly fed green hay, to explore the changes in the gut fungal community under different feeding models. All samples were collected in July 2022, and each group contained five animals. Fecal samples from yaks of different feeding models were collected using dung samplers. Freshly rectal feces were selected and sub-sampled (approximately 100 g) from the central portion to minimize contamination from bedding and flooring. Subsequently, fecal samples from yaks of different feeding models were labeled (GYG: GYG1, GYG2, GYG3, GYG4, and GYG5; WYG: WYG1, WYG2, WYG3, WYG4, and WYG5; HFG: HFG1, HFG2, HFG3, HFG4, and HFG5) and placed at −80°C for further analysis.
DNA extraction and high-throughput sequencing
Fungal DNA was extracted from homogenized intestinal contents using a QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), based on the manufacturer's protocol. Subsequently, we amplified the ITS2 regions using primers (ITS5F: GGAAG TAAAAGTCGTAACAAGG and ITS2R: GCTGCGTTCTTCATCGA TGC) and synthesized them as per conserved regions. PCR amplification procedures were determined according to previous research (38–40). After purification and fluorescence quantification of the amplified products, the sequencing library was constructed using the TruSeq Nano DNA LT Library Prep Kit (Illumina, CA, USA). The initial libraries were subjected to sequence end repair, enrichment, and purification to improve their quality. The final libraries that passed the quality assessment were used to perform paired-end sequencing using a MiSeq sequencing machine. Raw data from the amplicon sequencing were further processed and modified using QIIME software (Qiime1.9.1) due to the presence of questionable sequences such as unqualified, short, mismatched, and chimera sequences. After quality inspection and filtering, the effective reads were applied for OTU clustering based on 97% similarity. Fungal OTU representative sequences were taxonomically classified using RDP Classifier v.2.2 based on the UNITE database (39, 41). Meanwhile, the Venn diagram was also generated to observe the common and unique OTUs among the groups. To study the variation in gut microbial diversity and abundance, we calculated multiple alpha diversity indices based on the number of OTUs in each sample using QIIME software (Qiime1.9.1). Moreover, PCoA plots reflecting beta diversity were also generated using QIIME software (Qiime1.9.1) to further compare and analyze the differences in gut microbial principal components. Statistical analysis of the data was conducted using R (v3.0.3) and GraphPad Prism (version 8.0c). LEfSe and meta-statistics analyses were employed to detect differential fungal taxa. The p-values (means ± SD) of < 0.05 were considered statistically significant.
Results
Sequencing data analysis
In this research, high-throughput sequencing generated 576,964 (WYG = 182,234, HFG = 193,448, and GYG = 201,282) valid sequences from the three groups with an average of 38,464 (ranging from 30,248 to 45,539) reads per sample (Appendix A). The high-quality sequences were clustered, and a total of 1,098 OTUs were identified, ranging from 500 to 552 OTUs per group (Figure 1A). Additionally, the numbers of unique OTUs in the WYG, HFG, and GYG were 255, 223, and 280, respectively. Notably, we also found 152 core OTUs in the three groups, which were not affected by the feeding models. To assess sequencing depth and evenness, we also generated rarefaction and species rank curves. The results indicated that the curves of all samples exhibited a saturation trend, suggesting the adequacy and reliability of the sequencing (Figures 1B, C).
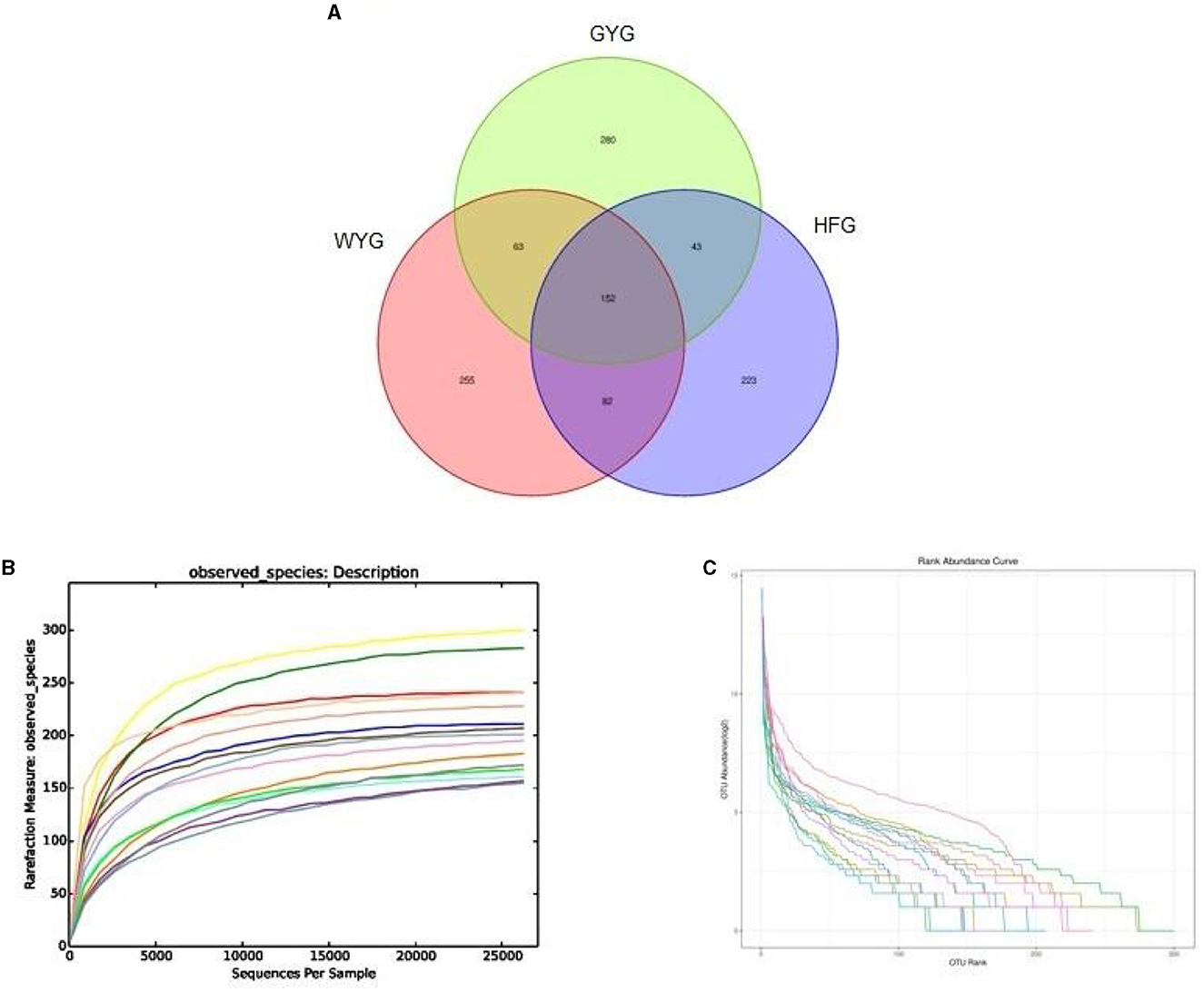
Figure 1. Sequencing data analysis and OTU distribution. Each colored curve in the rarefaction and rank abundance curves represents one sample. (A) The number of OTUs is indicated by different colored areas, and the middle area indicates the number of shared OTUs. (B) Rarefaction curve. (C) Rank abundance curve.
Comparative analysis of the gut fungal community in yaks with different feeding models
To explore the effects of different feeding models on the gut fungal alpha diversity of yaks, we calculated four indices, including Chao1, ACE, Simpson, and Shannon indices (Figures 2A–D). The Chao1 and ACE indices of the gut fungal community, from high to low, are WYG, HFG, and GYG. Moreover, the GYG had the highest Simpson and Shannon indices, followed by WYG and HFG. There were statistically distinct differences in Simpson (0.71 ± 0.11 vs. 0.38 ± 0.097, P < 0.01) and Shannon (3.17 ± 0.73 vs. 2.08 ± 0.64, P < 0.05) indices, whereas the Chao1 (233.69 ± 33.47 vs. 215.94 ± 62.69, P > 0.05) and ACE (224.71 ± 40.33 vs. 216.11 ± 57.06, P > 0.05) indices were not significantly different between the WYG and HFG. Similarly, we also found that the Simpson (0.80 ± 0.11 vs. 0.38 ± 0.097, P < 0.001) and Shannon (3.92 ± 1.33 vs. 2.08 ± 0.64, P < 0.05) indices of the GYG were significantly higher than those of the HFG, while there was no difference in the Chao1 (211.04 ± 32.59 vs. 215.94 ± 62.69, P > 0.05) and ACE (210.71 ± 31.78 vs. 216.11 ± 57.06, P > 0.05) indices. The comparative analysis between WYG and GYG indicated that there were no significant differences in the four indices. These results indicated that the gut fungal diversity in WYG and GYG was significantly higher than that in HFG, while there was no difference in the gut fungal abundance among WYG, HFG, and GYG. The results of the beta analysis showed that the samples of different groups were separated from each other, indicating significant differences in the principal components of the gut fungal community (Figures 2E, F).
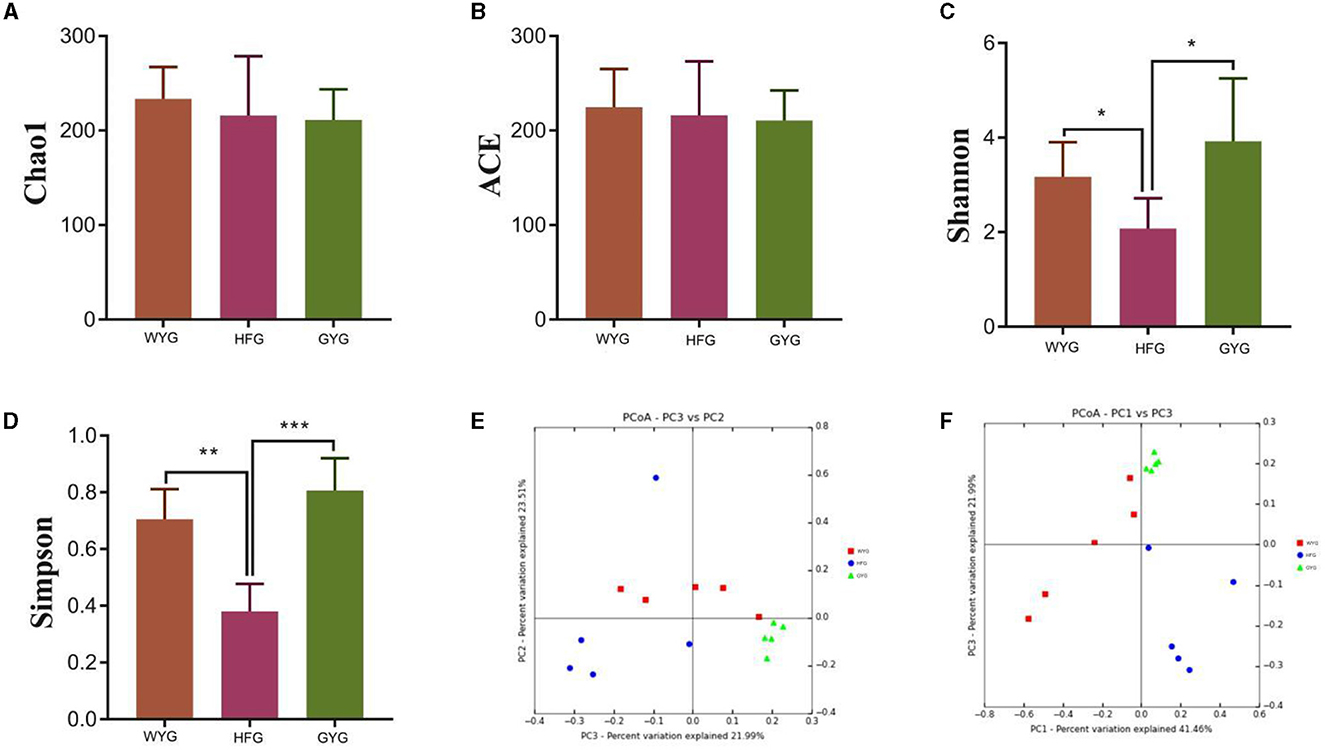
Figure 2. Comparative analysis of gut fungal diversity in yaks under different feeding models. The four indices including Chao1, ACE, Shannon, and Simpson were used to assess alpha diversity. (A) Chao1 index. (B) ACE index. (C) Shannon index. (D) Simpson index. (E, F) Gut fungal beta diversity was assessed by PCoA plots. Data are presented as means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Composition and differences of the gut fungal community in yaks with different feeding models
The phyla Ascomycota (69.90, 92.02, and 90.18%) and Basidiomycota (28.33, 5.47, and 5.86%) were abundant in the WYG, HFG, and GYG, accounting for more than 95% of the total fungal composition, respectively (Figure 3A). Moreover, other phyla, such as Mucoromycota (0.058, 0.57, and 0.11%), Rozellomycota (0.078%, 0.39%, and 0.19%), Mortierellomycota (0.058, 0.26, and 0.21%), Anthophyta (0.011, 0.13, and 0.082%), GS19 (0.14, 0.012, and 0.067%), Olpidiomycota (0.00, 0.086, and 0.02%), Neocallimastigomycota (0.017, 0.0068, and 0.069%), Cercozoa (0.012, 0.00, and 0.059%), Blastocladiomycota (0.00, 0.00, and 0.044%), and Rotifera (0.022, 0.0053, and 0.012%), were detected in WYG, HFG, and GYG in low abundances. Thelebolus (54.27%), Naganishia (25.43%), and Cutaneotrichosporon (1.29%) were the most predominant genera in the WYG (Figure 3B). Moreover, the dominant genera found in the HFG were Thelebolus (84.52%), Naganishia (3.08%), Cutaneotrichosporon (1.11%), and Lecanicillium (1.03%). The fungal genera with an abundance of more than 1% in the GYG were Thelebolus (31.22%), Naganishia (1.71%), Candida (1.65%), Acremonium (1.72%), and Vishniacozyma (1.00%). Furthermore, the gut fungal compositions and changes in WYG, HFG, and GYG could also be observed through a visualized clustering heatmap (Figure 4).
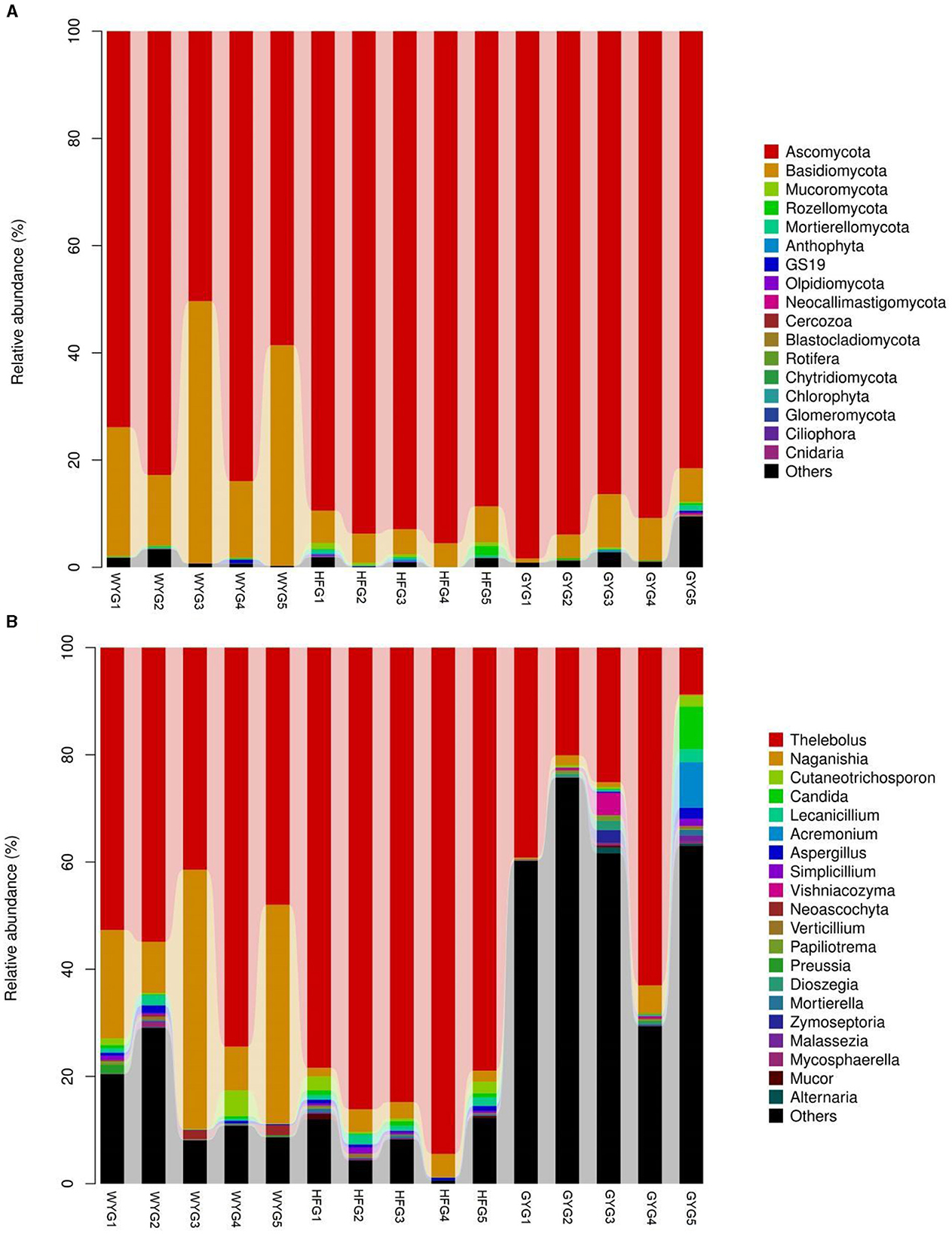
Figure 3. The abundance of dominant fungal phyla and genera in the gut fungal community of yaks under different feeding models. (A) The abundance of dominant fungal phyla. (B) The abundance of dominant fungal genera. The abundance of different fungal phyla or genera is represented by different colors and the height of the histogram.
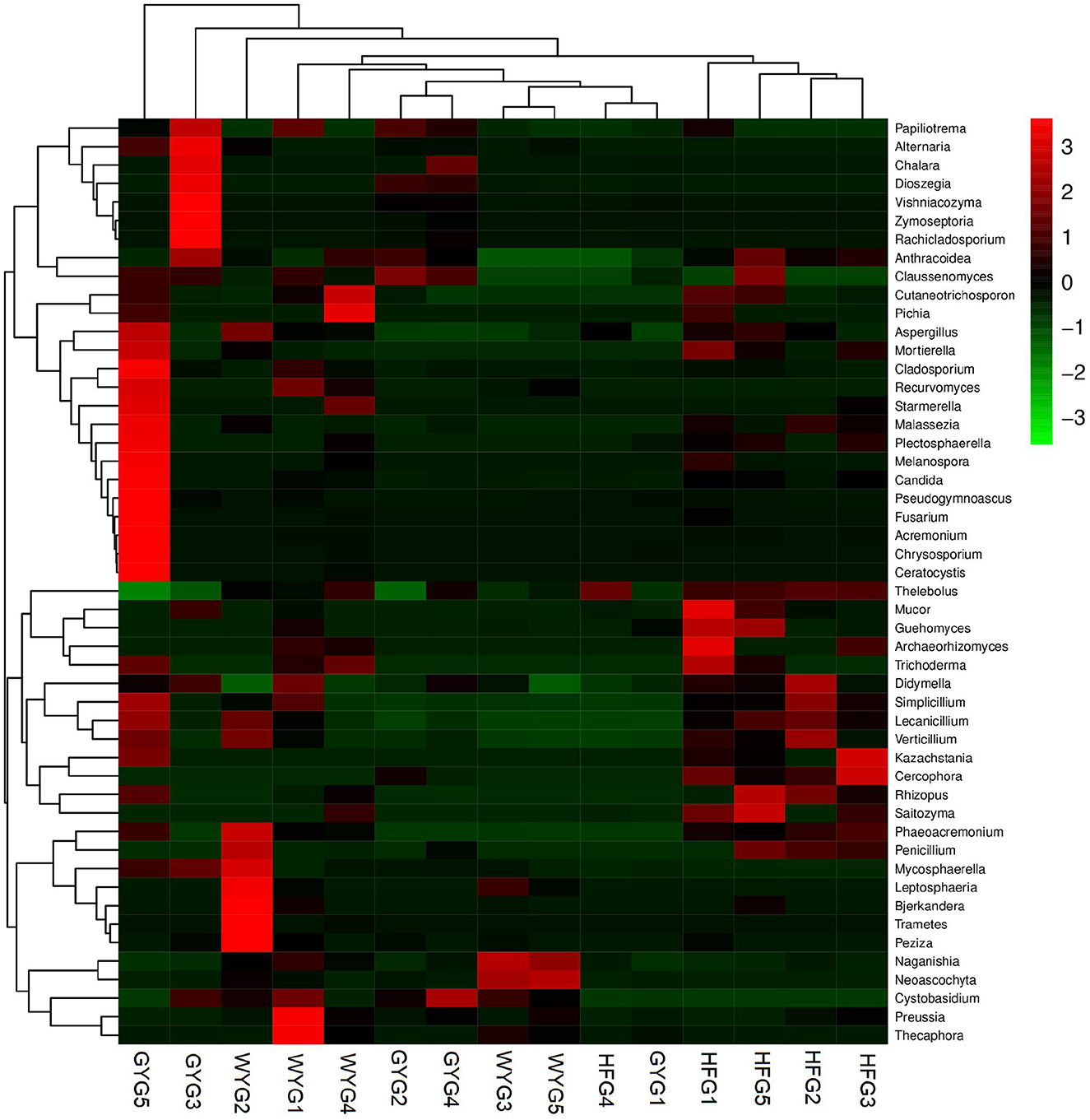
Figure 4. Heatmap of the abundant fungal genera in the WYG, HFG, and GYG. The abundance of different fungal genera is represented by different colors.
We also conducted meta-statistics analysis to identify differences in the gut fungal community at different taxonomic levels. At the phyla level, the HFG indicated significantly higher levels of Ascomycota and Mucoromycota, while the WYG enriched for Basidiomycota (Table 1). A comparison of the GYG and WYG showed an obvious increase in the levels of Ascomycota and Cercozoa and a distinct reduction in the level of Basidiomycota. Additionally, the abundance of Cercozoa in GYG was significantly more dominant than that in HFG, whereas that of Mucoromycota was lower. Compared with the HFG, the fungal community in the WYG displayed an obvious increase in the relative abundances of Cystobasidium, Naganishia, Sclerostagonospora, Physalospora, Neoascochyta, Recurvomyces, Schizonella, Orpinomyces, Herpotrichia, Sphaerulina, and Podospora, while Thelebolus, Cercophora, Stropharia, Lichtheimia, and Rhizopus decreased dramatically. The GYG indicated dramatically higher proportions of Didymella, Claussenomyces, Anthracoidea, Neoascochyta, Papiliotrema, Mastigosporium, Dioszegia, Heyderia, Ramularia, Cladonia, Urocystis, and Parapenidiella, whereas the WYG was dramatically enriched for Naganishia, Sclerostagonospora, Kurtzmanomyces, Thermoascus, Physalospora, Herpotrichia, Panaeolus, and Archaeorhizomyces. Moreover, the abundances of Cystobasidium, Claussenomyces, Papiliotrema, Mycosphaerella, Neoascochyta, Mastigosporium, Dioszegia, Urocystis, Humicola, Physalospora, Heyderia, Sphaerulina, Ramularia, and Cladonia in the GYG were significantly preponderant compared to the HFG, while the abundances of Stropharia, Lichtheimia, Saitozyma, Penicillium, Pyrenochaetopsis, Thermomyces, Knufia, and Melanocarpus were lower. Given that this discriminant analysis did not distinguish the predominant taxon, LEfSe was used to generate a cladogram to identify the specific bacteria associated with different feeding models (Figure 5). In addition to the above-mentioned differential taxa, the GYG also showed significantly higher abundances of Humicola, Vishniacozyma, Rachicladosporium, and Alternaria as compared to the WYG and HFG.
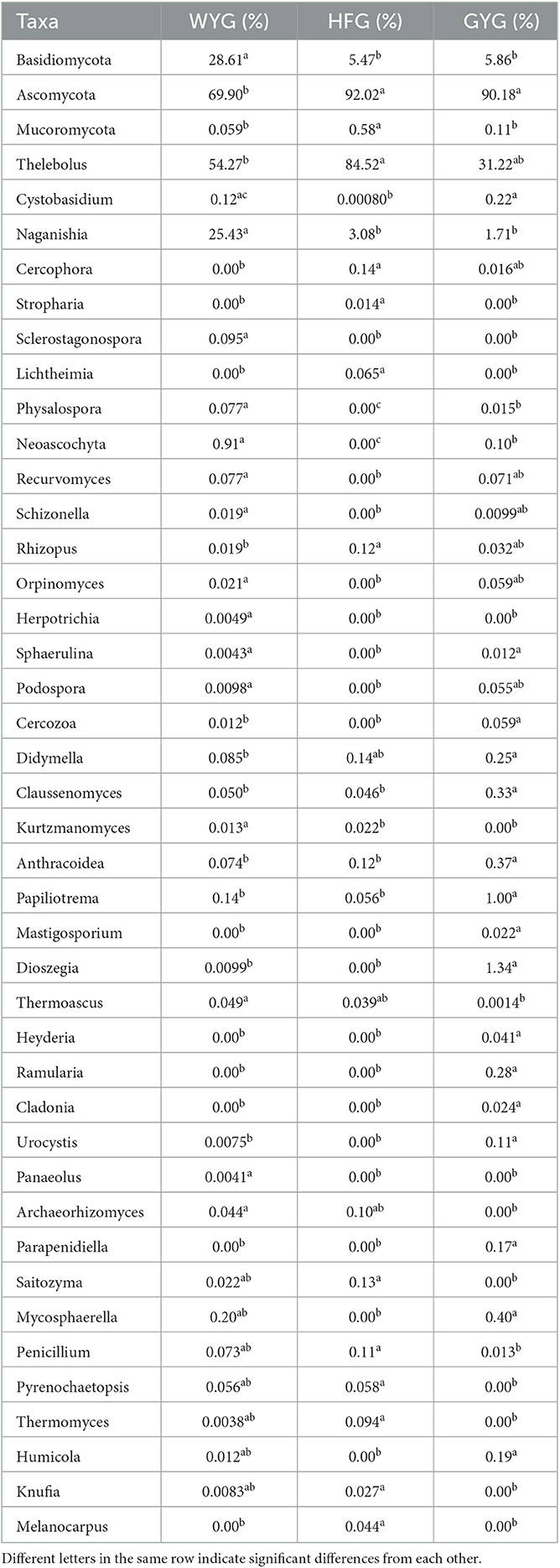
Table 1. Statistical analysis of differential fungi between different groups.
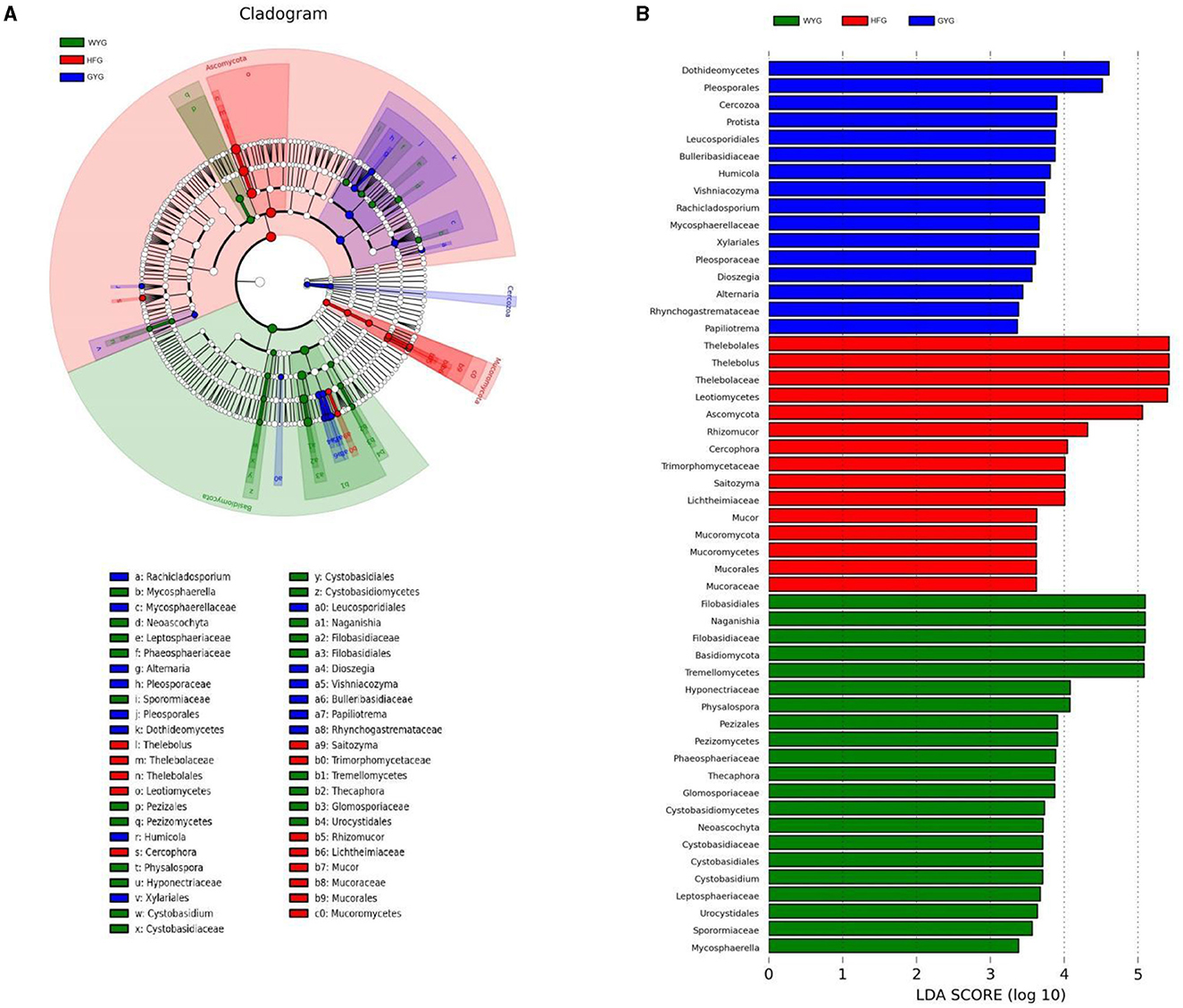
Figure 5. Detection of differentially abundant taxon using LEfSe and LDA scores. (A) Differential fungi and phylogenies are represented by a cladogram. (B) Differential fungi of yaks under different feeding models are represented by LDA scores.
Discussion
The wild yak is an endangered breed that has been listed as a first-class protected wild animal in China. Compared with WYG, domestic yaks can be artificially intervened and raised. Currently, there are two main feeding modes for domestic yaks: free range and artificial breeding. Growing studies have indicated that the gut fungal community plays a role in host health, metabolism, and the immune system (7, 11, 42). Recent studies on the gut fungal community have also revealed its important driving roles in the development of diarrhea, inflammatory bowel disease, and liver cirrhosis (3, 43). Currently, studies of the gut fungal community have involved many species, such as mice, pigs, and dairy cattle, and revealed their importance in host health and development (19, 39, 44). However, research on the characteristics and differences of the gut fungal community in yaks under different feeding models is still insufficient to date. Analyzing the gut microbiota of yaks under different feeding models may contribute to revealing the differences in various characteristics of these yaks. In this study, we dissected the compositions and differences of the gut fungal community among WYG, HFG, and GYG.
Microbes that inhabit the intestine can interact in symbiotic or antagonistic relationships, which are an important basis for the maintenance of gut microbial homeostasis (45, 46). Gut microbial homeostasis is the precondition for maintaining host metabolism, digestion, and absorption (47, 48). Inversely, the disruption of gut microbial homeostasis may affect intestinal barrier function and immune function, seriously threatening host health (49, 50). Gut microbial homeostasis could be assessed by diversity indices such as Chao1, ACE, Shannon, and Simpson. Typically, the diversity index representing gut microbial homeostasis is in a relatively stable state due to the plasticity of the gut microbiota (51, 52). However, multiple factors, such as exercise, diet, age, and environmental pollutants, can affect the composition and structure of the gut microbiota, thereby affecting gut microbial homeostasis (53–55). In this study, we observed that the gut fungal Shannon and Simpson indices of WYG and GYG were higher than those of HFG, indicating that WYG and GYG have higher microbial diversity. Early surveys indicated that the higher diversity represents stronger gut microbial plasticity, which contributes to improving the gut microbiota's response to external stress, thereby maintaining gut microbial homeostasis (56, 57). Furthermore, increased gut microbial diversity also contributes to the maintenance and improvement of intestinal functions such as energy utilization and nutrient intake (58, 59). Studies have shown that primitive yaks existed in the Pleistocene more than 3 million years ago and were widely distributed in northeast Eurasia. Later, primitive yaks moved south to the Tibetan Plateau of China due to crustal movement and climate change, adapted to the Alpine climate, and continued to evolve into modern yaks. WYG inhabits plateau meadows, shrubs, and deserts at an altitude of 4,000 to 5,000 meters, which have strong adaptability to the environmental conditions of alpine grasslands. Environmental factors, such as altitude hypoxia and low temperatures, are important players in the evolution of gut microbiota (60, 61). Compared with domestic yaks, WYGs inhabit higher altitudes and harsher living environments. Therefore, WYG may have evolved a more complex gut microbial structure to adapt to complex environments. Compared with HFG, WYG, and GYG will constantly migrate in search of food and water supplies, which inevitably increases the amount of exercise for yaks. Numerous studies have indicated that the frequency of exercise is closely related to gut microbial composition and structure (62, 63). For instance, physical exercise could significantly alter the composition and diversity of the gut microbiota in mice (54). Compared with HFG, WYG, and GYG have more complicated and irregular diets. In addition, some plants in the wild environment may be polluted or contain pathogenic microorganisms, which also contribute to the evolution of a more complex gut microbial community. Therefore, we speculated that exercise, complex diet, and harsh environment may be some of the reasons for the more diverse gut microbiota of WYG and GYG.
The gut fungal community, as a vital constituent of the gut microbiota, is critical for host health (64, 65). The gut fungal community has been shown to be involved in the development of intestinal barrier function and intestinal inflammation (14, 66). Furthermore, recent investigations of the gut fungal community have also provided evidence that it is closely associated with diarrhea (3, 34). However, the importance and role of the gut fungal community in the host have long been overlooked because of their low proportion in the gut microbiota. To further explore the effects of different feeding models on the yaks, we also assessed the composition and variability of the gut fungal community in these populations. The results demonstrated that the phyla Ascomycota and Basidiomycota were abundantly present in the wild yak, HFG, and GYG, indicating that the different feeding models could not affect the species of the main dominant phyla. Moreover, Ascomycota and Basidiomycota have also been demonstrated to be major dominant fungal phyla in other animals, such as giraffes, sheep, and cows, suggesting their importance in the ruminant fungal community and intestinal function (3). Notably, although the species of the main dominant fungal phyla did not change, their abundances did. For instance, the abundance of Basidiomycota was significantly higher in WYG than in GYG and HFG. Basidiomycota is the highest phylum of fungi with more than 20,000 species, characterized by wide distribution, large number, and variety (67). Among these three types of yaks, WYG possessed the most complicated habitat environment and diet structure, which may change the gut microbial composition of yaks. Meanwhile, this may also be one of the reasons for the increase in Basidiomycota in WYG. In this study, we also observed significant changes in the abundance of some fungal genera between different feeding models. Previous investigations showed that shifts in some specific microbial communities could affect host phenotypes and intestinal functions (34, 68). The ecological environment and dietary structure are important driving factors for gut microbial succession, and the gut microbiota will change appropriately under various external stimuli to adapt to the habitat environment.
Conclusions
This research explores the dynamic changes of the gut fungal community in yaks under different feeding models. The results indicated that feeding models could significantly alter the gut fungal composition and structure of yaks, including significant changes in some dominant phyla (Ascomycota and Basidiomycota) and fungal genera (Thelebolus, Naganishia, and Vishniacozyma). Moreover, WYG and GYG had a higher gut fungal diversity compared to HFG. This investigation elucidated the characteristics of the gut fungal community in WYG, HFG, and GYG and filled a gap in the effects of different feeding models on the gut fungal community. Meanwhile, this research also indicated that different feeding models are important drivers of changes in the gut fungal community. However, this study has some limitations that need to be acknowledged, such as the small sample size and lack of information on age and dietary habits.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA948534.
Ethics statement
The study was conducted under the guidance and approval of the Animal Welfare and Ethics Committee of Institute of Animal Husbandry and Veterinary Medicine, Tibet Academy of Agriculture and Animal Husbandry Sciences.
Author contributions
YZ and WB provided the research idea. YC, GS, XL, ZL, LS, and ZS contributed reagents, materials, and analytical tools. YZ wrote the manuscript. MS, YZ, and WB revised the manuscript. All authors participated in the writing and review of the manuscript.
Funding
The study was supported by the Yak Seed Industry Innovation and Healthy Breeding (XZ202101ZD0002N), the Gesangtang Yak Breeding and Efficient Multiplication in Linzhou County (QYXTZX-LS2020-01), and the National Beef Yak Industry Technology System (CARS-37).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liao J, Liu Y, Yi J, Li Y, Li Q, Li Y, et al. Gut microbiota disturbance exaggerates battery wastewater-induced hepatotoxicity through a gut-liver axis. Sci Total Environ. (2022) 809:152188. doi: 10.1016/j.scitotenv.2021.152188
2. Li AY, Wang YL, Kulyar M, Iqbal M, Lai RH, Zhu HS, et al. Environmental microplastics exposure decreases antioxidant ability, perturbs gut microbial homeostasis and metabolism in chicken. Sci Total Environ. (2023) 856:159089. doi: 10.1016/j.scitotenv.2022.159089
3. Li A, Liu B, Li F, He Y, Wang L, Fakhar-E-Alam KM, et al. Integrated bacterial and fungal diversity analysis reveals the gut microbial alterations in diarrheic giraffes. Front Microbiol. (2021) 12:712092. doi: 10.3389/fmicb.2021.712092
4. Wu Y, Xu H, Tu X, Gao Z. The Role of Short-Chain Fatty Acids of Gut Microbiota Origin in Hypertension. Front Microbiol. (2021) 12:730809. doi: 10.3389/fmicb.2021.730809
5. Lou XR, Xue JF, Shao RF, Yang Y, Ning DY, Mo CY, et al. Fecal microbiota transplantation and short-chain fatty acids reduce sepsis mortality by remodeling antibiotic-induced gut microbiota disturbances. Front Immunol. (2023) 13:1063543. doi: 10.3389/fimmu.2022.1063543
6. Lu YB, Li ZY, Peng X. Regulatory effects of oral microbe on intestinal microbiota and the illness. Front Cell Infect Microbiol. (2023) 13:1093967. doi: 10.3389/fcimb.2023.1093967
7. Gao J, Xu K, Liu HN, Liu G, Bai MM, Peng C, et al. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front Cell Infect Microbiol. (2018) 8:13. doi: 10.3389/fcimb.2018.00013
8. Liu YJ, Chen HN, Feng LP, Zhang J. Interactions between gut microbiota and metabolites modulate cytokine network imbalances in women with unexplained miscarriage. NPJ Biofilms Microbomes. (2021) 7:24. doi: 10.1038/s41522-021-00199-3
9. Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. (2014) 14:405–16. doi: 10.1038/nri3684
10. Chiaro TR, Soto R, Zac W, Kubinak JL, Petersen C, Gogokhia L, et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci Transl Med. (2017) 9:9044. doi: 10.1126/scitranslmed.aaf9044
11. Paterson MJ, Oh S, Underhill DM. Host-microbe interactions: commensal fungi in the gut. Curr Opin Microbiol. (2017) 40:131–7. doi: 10.1016/j.mib.2017.11.012
12. Jiang TT, Shao T, Ang W, Kinder JM, Turner LH, Pham G, et al. Commensal Fungi Recapitulate the Protective Benefits of Intestinal Bacteria. Cell Host Microbe. (2017) 22:809–816. doi: 10.1016/j.chom.2017.10.013
13. van Tilburg BE, Pettersen VK, Gutierrez MW, Laforest-Lapointe I, Jendzjowsky NG, Cavin JB, et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat Commun. (2020) 11:2577. doi: 10.1038/s41467-020-16431-1
14. Leonardi I, Li X, Semon A, Li DL, Doron I, Putzel G, et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science. (2018) 359:232. doi: 10.1126/science.aao1503
15. Kumamoto CA, Gresnigt MS, Hube B. The gut, the bad and the harmless: candida albicans as a commensal and opportunistic pathogen in the intestine. Curr Opin Microbiol. (2020) 56:7–15. doi: 10.1016/j.mib.2020.05.006
16. Markey L, Shaban L, Green ER, Lemon KP, Mecsas J, Kumamoto CA. Pre-colonization with the commensal fungus candida albicans reduces murine susceptibility to clostridium difficile infection. Gut Microbes. (2018) 9:497–509. doi: 10.1080/19490976.2018.1465158
17. Buts JP, Dekeyser N, Stilmant C, Delem E, Smets F, Sokal E. Saccharomyces boulardii produces in rat small intestine a novel protein phosphatase that inhibits escherichia coli endotoxin by dephosphorylation. Pediatr Res. (2006) 60:24–9. doi: 10.1203/01.pdr.0000220322.31940.29
18. Takata K, Tomita T, Okuno T, Kinoshita M, Koda T, Honorat J, et al. Dietary yeasts reduce inflammation in central nerve system via microflora. Ann Clin Transl Neurol. (2015) 2:56–66. doi: 10.1002/acn3.153
19. Kong G, Le Cao KA, Hannan AJ. Alterations in the gut fungal community in a mouse model of huntington's disease. Microbiol Spectr. (2022) 10:e219221. doi: 10.1128/spectrum.02192-21
20. Tsavkelova EA, Botvinko IV, Kudrin VS, Oleskin AV. Detection of neurotransmitter amines in microorganisms with the use of high-performance liquid chromatography. Dokl Biochem. (2000) 372:115–7.
21. Li A, Yang Y, Qin S, Lv S, Jin T, Li K, et al. Microbiome analysis reveals gut microbiota alteration of early-weaned Yimeng black goats with the effect of milk replacer and age. Microb Cell Fact. (2021) 20:78. doi: 10.1186/s12934-021-01568-5
22. Fan QJ, Yi M, Liu H, Wang YS, Li XK, Yuan JL, et al. The Impact of Age and Pathogens Type on the Gut Microbiota in Infants with Diarrhea in Dalian, China. Can J Infect Dis Med Microbiol. (2020) 2020:8837156. doi: 10.1155/2020/8837156
23. Ma Y, Ma S, Chang L, Wang HJ, Ga Q, Ma L, et al. Gut microbiota adaptation to high altitude in indigenous animals. Biochem Biophys Res Commun. (2019) 516:120–6. doi: 10.1016/j.bbrc.2019.05.085
24. Liu G, Li C, Liu Y, Zheng CM, Ning Y, Yang HG, et al. Highland adaptation of birds on the Qinghai-Tibet Plateau via gut microbiota. Appl Microbiol Biotechnol. (2022) 106:6701–11. doi: 10.1007/s00253-022-12171-y
25. Li K, Jiang JH, Shahzad M, Zhang H, Mehmood K, Jiang X, et al. Revealing the parasitic infection in diarrheic yaks by piloting high-throughput sequencing. Microb Pathog. (2018) 117:153–6. doi: 10.1016/j.micpath.2018.02.033
26. Han Z, Li K, Shahzad M, Zhang H, Luo H, Qiu G, et al. Analysis of the intestinal microbial community in healthy and diarrheal perinatal yaks by high-throughput sequencing. Microb Pathog. (2017) 111:60–70. doi: 10.1016/j.micpath.2017.08.025
27. Li J, Li K, Shahzad M, Han Z, Nabi F, Gao J. Seroprevalence of Bluetongue virus in domestic yaks (Bosgrunniens) in Tibetan regions of China based on circulating antibodies. Trop Anim Health Prod. (2015) 47:1221–3. doi: 10.1007/s11250-015-0853-0
28. Li AY, Wang M, Zhang Y, Lin ZR, Xu ME, Wang L, et al. Complete genome analysis of Bacillus subtilis derived from yaks and its probiotic characteristics. Front Vet Sci. (2023) 9:1099150. doi: 10.3389/fvets.2022.1099150
29. Han Y, Xu JY, Yan Y, Zhao XJ. Dynamics of the gut microbiota in rats after hypobaric hypoxia exposure. PeerJ. (2022) 10:14090. doi: 10.7717/peerj.14090
30. Huang L, Li TY, Zhou M, Deng MY, Zhang LD, Yi L, et al. Hypoxia improves endurance performance by enhancing short chain fatty acids production via gut microbiota remodeling. Front Microbiol. (2022) 12:820691. doi: 10.3389/fmicb.2021.820691
31. Zhang L, Jiang X, Li A, Waqas M, Gao X, et al. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe. (2020) 61:102115. doi: 10.1016/j.anaerobe.2019.102115
32. Su YX, Su JH, Li FL, Tian XJ, Liu ZW, Ding GT, et al. Yak gut microbiota: a systematic review and meta-analysis. Front Vet Sci. (2022) 9:889594. doi: 10.3389/fvets.2022.889594
33. Liu WW, Wang Q, Song JJ, Xin JW, Zhang SS, Lei YH, et al. Comparison of gut microbiota of yaks from different geographical regions. Front Microbiol. (2021) 12:666940. doi: 10.3389/fmicb.2021.666940
34. Liu J, Wang X, Zhang W, Kulyar MF, Ullah K, Han Z, et al. Comparative analysis of gut microbiota in healthy and diarrheic yaks. Microb Cell Fact. (2022) 21:111. doi: 10.1186/s12934-022-01836-y
35. Dong HL, Liu BX, Li AY, Iqbal M, Mehmood K, Jamil T, et al. Microbiome analysis reveals the attenuation effect of lactobacillus from yaks on diarrhea via modulation of gut microbiota. Front Cell Infect Microbiol. (2021) 10:610781. doi: 10.3389/fcimb.2020.610781
36. Zhu Y, Li X, Sun G. House feeding pattern increased male yak fertility by improving gut microbiota and serum metabolites. Front Vet Sci. (2022) 9:989908. doi: 10.3389/fvets.2022.989908
37. Zhang XL, Xu TW, Wang XG, Geng YY, Liu HJ, Hu LY, et al. The effect of transitioning between feeding methods on the gut microbiota dynamics of yaks on the qinghai-tibet plateau. Animals. (2020) 10:1641. doi: 10.3390/ani10091641
38. Li A, Wang Y, Wang Y, Dong H, Wu Q, Mehmood K, et al. Microbiome analysis reveals soil microbial community alteration with the effect of animal excretion contamination and altitude in Tibetan Plateau of China. Int Soil Water Conserv Res. (2021) 9:639–48. doi: 10.1016/j.iswcr.2021.04.011
39. Hu J, Nie Y, Chen J, Zhang Y, Wang Z, Fan Q, et al. Gradual Changes of Gut Microbiota in Weaned Miniature Piglets. Front Microbiol. (2016) 7:1727. doi: 10.3389/fmicb.2016.01727
40. Hu L, Geng S, Li Y, Cheng S, Fu X, Yue X, et al. Exogenous Fecal Microbiota Transplantation from Local Adult Pigs to Crossbred Newborn Piglets. Front Microbiol. (2017) 8:2663. doi: 10.3389/fmicb.2017.02663
41. Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S, et al. The UNITE database for molecular identification of fungi–recent updates and future perspectives. New Phytol. (2010) 186:281–5. doi: 10.1111/j.1469-8137.2009.03160.x
42. Pérez J. (2021). Fungi of the human gut microbiota: roles and significance. Int J Med Microbiol. 311:151490. doi: 10.1016/j.ijmm.2021.151490
43. Bajaj JS, Liu EJ, Kheradman R, Fagan A, Heuman DM, White M, et al. Fungal dysbiosis in cirrhosis. Gut. (2018) 67:1146–54. doi: 10.1136/gutjnl-2016-313170
44. Li MH, Meng JX, Wang W, He M, Zhao ZY, Ma N, et al. Dynamic description of temporal changes of gut microbiota in broilers. Poult Sci. (2022) 101:37. doi: 10.1016/j.psj.2022.102037
45. Chen W, Liu D, Ren CH, Su XM, Wong CK, Yang RC. A Special Network Comprised of Macrophages, Epithelial Cells, and Gut Microbiota for Gut Homeostasis. Cells. (2022) 11:307. doi: 10.3390/cells11020307
46. Arenas-Gomez CM, Garcia-Gutierrez E, Escobar JS, Cotter PD. Human gut homeostasis and regeneration: the role of the gut microbiota and its metabolites. Crit Rev Microbiol. (2022) 2022:1–22. doi: 10.1080/1040841X.2022.2142088
47. Delzenne NM, Knudsen C, Beaumont M, Rodriguez J, Neyrinck AM, Bindels LB. Contribution of the gut microbiota to the regulation of host metabolism and energy balance: a focus on the gut-liver axis. Proc Nutr Soc. (2019) 78:319–28. doi: 10.1017/S0029665118002756
48. Uebanso T, Kano S, Yoshimoto A, Naito C, Shimohata T, Mawatari K, et al. Effects of Consuming Xylitol on Gut Microbiota and Lipid Metabolism in Mice. Nutrients. (2017) 9:756. doi: 10.3390/nu9070756
49. Zhou X, Li WY, Wang S, Zhang PL, Wang Q, Xiao J, et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. (2019) 27:1176. doi: 10.1016/j.celrep.2019.03.028
50. Lu J, Jin X, Yang SJ, Li YJ, Wang XY, Wu M. Immune mechanism of gut microbiota and its metabolites in the occurrence and development of cardiovascular diseases. Front Microbiol. (2022) 13:1034537. doi: 10.3389/fmicb.2022.1034537
51. Santo CE, Caseiro C, Martins MJ, Monteiro R, Brandao I. Gut Microbiota, in the Halfway between Nutrition and Lung Function. Nutrients. (2021) 13:1716. doi: 10.3390/nu13051716
52. Li ZX, Zhou J, Liang H, Ye L, Lan LY, Lu F, et al. Differences in Alpha Diversity of Gut Microbiota in Neurological Diseases. Front Neurosci. (2022) 16:879318. doi: 10.3389/fnins.2022.879318
53. Greenhill C. Exercise affects gut microbiota and bone. Nat Rev Endocrinol. (2018) 14:322–322. doi: 10.1038/s41574-018-0014-4
54. Yang WQ, Liu YQ, Yang G, Meng BL, Yi ZC, Yang G, et al. Moderate-Intensity Physical Exercise Affects the Exercise Performance and Gut Microbiota of Mice. Front Cell Infect Microbiol. (2021) 11:712381. doi: 10.3389/fcimb.2021.712381
55. Wu SY, Wang YJ, Iqbal M, Mehmood K, Li Y, Tang ZX, et al. Challenges of fluoride pollution in environment: Mechanisms and pathological significance of toxicity-A review. Environ Pollut. (2022) 304:119241. doi: 10.1016/j.envpol.2022.119241
56. Larsen O, Claassen E. The mechanistic link between health and gut microbiota diversity. Sci Rep. (2018) 8:2083. doi: 10.1038/s41598-018-20141-6
57. Li AY, Wang YL, Hao JY, Wang L, Quan LT, Duan K, et al. Long-term hexavalent chromium exposure disturbs the gut microbial homeostasis of chickens. Ecotox Environ Safe. (2022) 237:113532. doi: 10.1016/j.ecoenv.2022.113532
58. Laitinen K, Mokkala K. Overall Dietary Quality Relates to Gut Microbiota Diversity and Abundance. Int J Mol Sci. (2019) 20:1835. doi: 10.3390/ijms20081835
59. You ZQ, Deng J, Liu JL, Fu JH, Xiong H, Luo W, et al. Seasonal variations in the composition and diversity of gut microbiota in white-lipped deer (Cervus albirostris). PeerJ. (2022) 10:13753. doi: 10.7717/peerj.13753
60. Ramos-Romero S, Santocildes G, Pinol-Pinol D, Roses C, Pages T, Hereu M, et al. Implication of gut microbiota in the physiology of rats intermittently exposed to cold and hypobaric hypoxia. PLoS ONE. (2020) 15:686. doi: 10.1371/journal.pone.0240686
61. Olson CA, Iniguez AJ, Yang GE, Fang P, Pronovost GN, Jameson KG, et al. Alterations in the gut microbiota contribute to cognitive impairment induced by the ketogenic diet and hypoxia. Cell Host Microbe. (2021) 29:1378. doi: 10.1016/j.chom.2021.07.004
62. Zhu QW, Jiang SF, Du GK. Effects of exercise frequency on the gut microbiota in elderly individuals. Microbiologyopen. (2020) 9:1053. doi: 10.1002/mbo3.1053
63. Cerda B, Perez M, Perez-Santiago JD, Tornero-Aguilera JF, Gonzalez-Soltero R, Larrosa M. Gut microbiota modification: another piece in the puzzle of the benefits of physical exercise in health? Front Physiol. (2016) 7:51. doi: 10.3389/fphys.2016.00051
64. Mahtab N, Zhou LZ, Zhang FL, Wang W. Seasonal Variations in the Gut Fungal Communities of Hooded Crane (Grus monacha) at Wintering and Stopover Sites in China. Animals. (2021) 11:941. doi: 10.3390/ani11040941
65. Wu YN, Li ZH, Zhao JR, Chen Z, Xiang XJ. Significant differences in intestinal fungal community of hooded cranes along the wintering periods. Front Microbiol. (2022) 13:991998. doi: 10.3389/fmicb.2022.991998
66. Sciavilla P, Strati F, Di Paola M, Modesto M, Vitali F, Cavalieri D, et al. Gut microbiota profiles and characterization of cultivable fungal isolates in IBS patients. Appl Microbiol Biotechnol. (2021) 105:3277–88. doi: 10.1007/s00253-021-11264-4
67. He MQ, Zhao RL, Liu DM, Denchev TT, Begerow D, Yurkov A, et al. Species diversity of basidiomycota. Fungal Divers. (2022) 114:281–325. doi: 10.1007/s13225-021-00497-3
68. Li A, Wang Y, He Y, Liu B, Iqbal M, Mehmood K, et al. Environmental fluoride exposure disrupts the intestinal structure and gut microbial composition in ducks. Chemosphere. (2021) 277:130222. doi: 10.1016/j.chemosphere.2021.130222
Appendix
Appendix A. Fungal sequence information for each sample.
Keywords: yak, wild, Qinghai-Tibet plateau, gut microbiota, gut fungal community
Citation: Zhu Y, Cidan Y, Sun G, Li X, Shahid MA, Luosang Z, Suolang Z, Suo L and Basang W (2023) Comparative analysis of gut fungal composition and structure of the yaks under different feeding models. Front. Vet. Sci. 10:1193558. doi: 10.3389/fvets.2023.1193558
Received: 25 March 2023; Accepted: 09 May 2023;
Published: 15 June 2023.
Edited by:
Isa Ozaydin, Kafkas University, TürkiyeReviewed by:
Rao Zahid Abbas, University of Agriculture, Faisalabad, PakistanNahla Mohammed Saeed, Sulaimania University, Iraq
Anum Ali Ahmad, Lanzhou University, China
Copyright © 2023 Zhu, Cidan, Sun, Li, Shahid, Luosang, Suolang, Suo and Basang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wangdui Basang, YncwODkxQDE2My5jb20=