
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 05 July 2023
Sec. Veterinary Infectious Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1191497
 Amanda Beylefeld*
Amanda Beylefeld* Celia Abolnik
Celia AbolnikIntroduction: Salmonella enterica subspecies enterica serovar Gallinarum biovar Gallinarum (SG) is associated with fowl typhoid fever, and the attenuated rough strain SG9R is widely used as a vaccine in many regions. Reversion to virulence of vaccine strains was suspected as the cause during recent fowl typhoid fever outbreaks in poultry in South Africa and Eswatini.
Methods: To compare nine field isolates with global wild-type SG9 strains and the two commercial SG9R vaccines in use, Nobilis® SG9R and Cevac®-SG, we used whole-genome comparison with single-nucleotide polymorphism (SNP) detection.
Results: SNP phylogenic analysis showed that all the southern African field isolates were more closely related to the vaccine strains than wild-type SG9 strains. Furthermore, SNPs in the pyruvate dehydrogenase (aceE) and/or lipopolysaccharide 1,2-glucosyltransferase (rfaJ) genes, which are known markers of attenuation, were found in four of the field isolates along with intact spv, SPI-1, and SPI-2 gene clusters, providing conclusive evidence that these four isolates were originally vaccine strains that reverted to virulence. Five other field isolates lacked the SG9R attenuation markers, but variant analysis identified an SNP in the yihX gene, insertions in the ybjX and hydH genes, and deletions in the ftsK and sadA genes that were shared between the field isolates and vaccine strains but absent in wild-type SG9, indicating that these field isolates were also likely revertant vaccines.
Discussion: Overall, this study highlights different mechanisms of reversion of two commercial vaccines, where virulence caused by field isolates closely related to the Nobilis® SG9R vaccine was associated with the restoration of intact virulence gene clusters, and those derived from the Cevac®-SG vaccine were characterized by point mutations resulting in restored aceE and rfaJ genes. A possible new marker of attenuation was identified as a point mutation in the yihX gene, as well as four new candidate genes that could potentially be used to distinguish current vaccine strains from wild-type strains using PCR assays.
Salmonella enterica subspecies enterica serovar Gallinarum biovar Gallinarum (S. Gallinarum; SG) is associated with fowl typhoid fever, an acute or chronic septicemic disease, which leads to large economic losses in poultry due to reduced production and high mortality rates (1). SG is reported to be under control or eradicated in many developed countries, but it is still a common disease in many developing countries including South Africa (2). A recent review of the prevalence of SG between 1945 and 2021 revealed that SG is more prevalent in Europe and Africa, and Salmonella enterica subspecies enterica serovar Gallinarum biovar Pullorum (S. Pullorum; SP) is more prevalent in North and South America and Asia (2). There has also been an increase in the prevalence of both in the last decade (2). Clinical signs of infection include depression, anorexia, droopy wings, dehydration, and diarrhea (1). SG is a non-motile Gram-negative bacterium with slender rod morphology, appearing as small, smooth, blue-gray, or grayish-white colonies on standard beef agar (3). The size of the SG genome is between 4.2 and 4.9 megabase pair (Mbp) with an average GC content of 52%, 4,272 predicted coding sequences (CDSs), and seven rRNA operons (4). S. Gallinarum harbors a large virulent plasmid of 85 kilobase pair (kbp) and a small plasmid of 2.5 kbp, with the large plasmid encoding the spvRABCD virulence genes (5, 6).
Good management procedures can keep flocks free of SG and, along with eradication programs, have been used successfully to eradicate fowl typhoid in developed countries. However, the eradication of SG is unrealistic in developing countries including South Africa, where SG persists and is listed as a reportable and controlled animal disease (7). Antimicrobials used in the management of SG are ineffective in preventing and controlling the infection, with survivors eventually becoming asymptomatic carriers that perpetuate the persistence and spread of SG (8). Vaccination is common, and both inactivated and live attenuated vaccines have been developed, with the latter more widely used and the preferred method of disease control. Inactivated vaccines are whole-killed bacteria that can elicit a strong antibody response, but these types of vaccines do not elicit a cell-mediated response, resulting in failure to clear the pathogen from the host (9). A few subunit vaccines, which consist of single multiple defined antigens, have also been developed against other Salmonella species, but these vaccines have not yet been successful in providing protection and are still being investigated (10). Live-attenuated vaccines consist of live bacteria that contain mutations or deletions resulting in the loss of functions essential to the metabolism, host-survivability, or virulence of the pathogen. These types of vaccines can elicit both antibody and cell-mediated immune responses, but they can persist in the host and carry the risk of reversion to virulence (11). Three live vaccines are currently registered for use in South Africa, namely the Onderstepoort Biological Product (OBP) Fowl Typhoid vaccine which is an attenuated rough SG strain 5503; Nobilis® SG9R produced by MSD Animal Health, and Cevac® S. Gallinarum 1000D produced by Ceva Animal Health. The latter two vaccines are based on the attenuated rough SG strain 9R (SG9R) and are also the most widely used globally. Strain SG9R was developed in 1956; however, the molecular basis for its attenuation was not known at the time. It has been proven to be safe for use in adult chickens protecting against both mortality and colonization of the organs (12). There is currently no validated polymerase chain reaction (PCR) assay available to distinguish between the field and vaccine strains of SG (1), but a triplex PCR assay to differentiate between the two Salmonella gallinarum serovars, SG and SP, and SG9R is described (13). The glgC and speC genes were used to differentiate between SG and SP, and single-nucleotide polymorphisms (SNPs) found in a hypothetical protein were used to differentiate SG9R (13, 14); however, this PCR method was not validated against the SG9 parental or wild-type SG9R strains (13).
Reversion to virulence of live SG vaccines is a concern in many countries and has been the subject of numerous studies since the advent of DNA sequencing. Kwon and Cho (15) used Sanger DNA sequencing to compare specific genes between the SG9R vaccine strain and SG9R-like field isolates from a fowl typhoid outbreak in Korea. A nonsense mutation in the lipopolysaccharide (rfa) gene cluster was found to be a likely attenuation and they hypothesized that any residual virulence in the vaccine was due to intact SPI-2 and spv genes (15). A comparative proteome and transcriptome analysis of the SG9R vaccine strain with two wild-type strains, 287/91 and 06Q110, found that the lack of the phage shock protein (PspA), an ABC transporter protein, and a flagellar component FliM, contributed to the avirulent nature of the vaccine strain and that the impaired expression of the SPI-1 or SPI-2 type III secretion system (TTSS) is a key factor (16). It was also reported that numerous mutations are required for reversion to virulence unless the mutation completely restored the regulation of the SPI-1 and SPI-2 gene clusters (16). Van Immerseel et al. (17) used whole-genome sequencing and SNP detection to show that the SG9R vaccine was closely related to a field isolate, MB4523, from a fowl typhoid outbreak in Belgium, specifically that two SNPs in the pyruvate dehydrogenase (aceE) and/or lipopolysaccharide 1,2-glucosyltransferase (rfaJ) genes were the most likely cause of reversion to virulence (17). SNP detection and whole-genome comparison were similarly applied to determine that multiple fowl typhoid outbreaks in Brazil were caused by field strains and not by the reversion of the Nobilis®SG9R and Cevac® SG9R vaccine strains (18).
There has been a concerning increase in anecdotal reports from the field in South Africa and the neighboring country of Eswatini of suspected reversion of the SG9R vaccines to virulence, especially among layer hens. Flocks vaccinated with SG9R vaccines have developed symptoms typically associated with fowl typhoid fever, especially in immune-compromised flocks co-infected with other pathogens. Such disease could stem from the residual virulence of the vaccine or reversion to virulence through a single-point mutation (19). Immune-compromised flock or flocks with nutrition deficiency allows the SG9R strain to proliferate resulting in vertical transmission and re-isolation during routine testing (15, 20). The recommended method of administration of the vaccine is subcutaneous for optimal protection, but it can be administered orally for 60% protection (21), and the application of SG vaccines in drinking water is a common practice in the local industry. In this study, whole-genome sequencing and comparative genomics with SNP detection were used to determine whether recent fowl typhoid outbreaks had been caused by wild-type field SG strains introduced into the poultry flock from external sources, or alternatively the reversion to virulence of attenuated commercial SG9R vaccines.
Research approval for this study was obtained from the Department of Agricultural, Land Reform and Rural Development (DALRRD) permit no. 12/11/1/1/8 (1608 LH), and ethical approval was obtained from the Research and Animal Ethics Committees of the University of Pretoria, under project no. REC187-19.
The field isolates used in this study (n = 9) were obtained from the NOSA Pty (Ltd) veterinary laboratory, Centurion, South Africa, which originally cultured and identified SG from the splenic material of chickens submitted by veterinarians for routine diagnosis, from poultry farms with a case history and where SG infection was suspected (Table 1). Flocks were immunized according to the manufacturers' recommendations, i.e., the Cevac®-SG vaccine initial vaccination was at 4 weeks followed by a second vaccination at 6 to 8 weeks, and the Nobilis® SG9R vaccine was administered at 6 weeks followed by boosters at 12-week intervals. The two freeze-dried vaccine strains, Nobilis® SG9R (batch no. A155AJ01) from MSD Animal Health Pty (Ltd) and Cevac® S. Gallinarum 1000D (batch no. 044/21) from Ceva Animal Health Pty (Ltd), were purchased from the manufacturers and reconstituted by adding 10 ml of distilled water to each vial. Whole-genome sequencing data for two of the Nobilis® SG9R vaccines, SG strain SG9Ra (PRJNA206379), and SG9Rb (PRJNA206380), were available from the National Center for Biotechnology Information (NCBI) (RRID:SCR_006472) (22), but these sequences were generated from vaccine batches in 2001 and 2009, respectively; therefore, it was decided to sequence more recent and locally sourced batches of the vaccines.
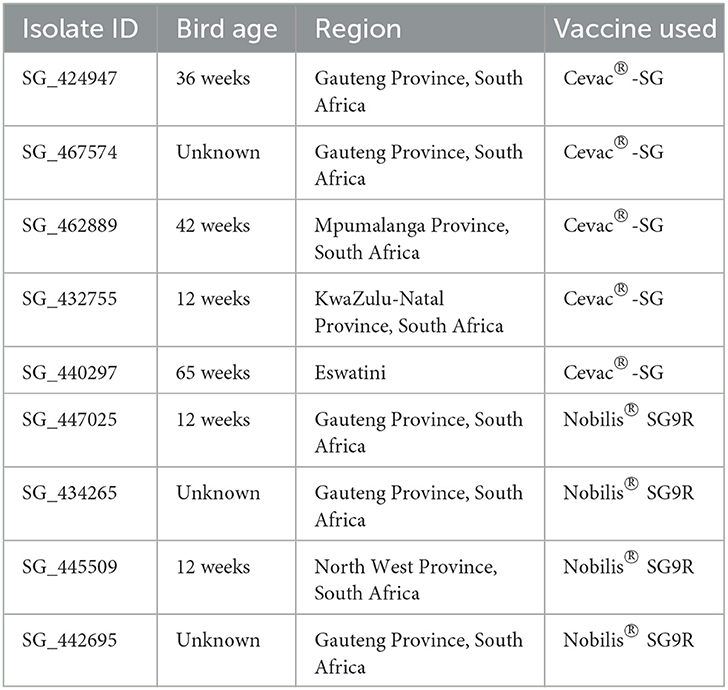
Table 1. Salmonella gallinarum field isolates analyzed in this study.
To verify purity, the field isolates and vaccine strains were cultured on Columbia blood agar with 5% horse serum and MacConkey agar without crystal violet for 24 h at 37°C at the Bacteriology laboratory of the Department of Veterinary Tropical Diseases, University of Pretoria. All isolates were confirmed to be pure and were subsequently inoculated into 100 ml of brain–heart infusion (BHI) broth and propagated for 24 h at 37°C. Cells were pelleted from 1.5 ml of cultures using centrifugation (10,000 x g at 4°C for 10 min). Then genomic DNA was isolated using the PureLink® Genomic DNA Kit (Invitrogen) according to the manufacturer's instructions.
The isolated genomic DNA was submitted to the Central Analytical Facility, University of Stellenbosch for Ion Torrent Sequencing on the Ion S5 sequencer at 200 x coverage. The quality of the read data was evaluated using FastQC (RRID:SCR_014583) (23), followed by read trimming using the trimming tool in CLC Genomics Workbench version 8.5.1 (CLC Bio-Qiagen, Aarhus, Denmark). The reads were de novo assembled in CLC Genomics Workbench using the default parameters with a 200 bp cutoff size for the contigs, and the resulting contigs were submitted to the public databases for molecular typing and microbial genome diversity (PubMLST) Species ID tool (RRID:SCR_012955) (24, 25). The quality of the whole-genome assemblies was assessed using the Quality Assessment Tool for Genome Assemblies (QUAST) (RRID:SCR_001228) (26, 27).
Ion Torrent sequence reads data produced in this study as well as Illumina sequencing reads data of the 2001 (SRR1045124) and 2009 (SRR1045125) Nobilis® SG9R vaccine were assembled to the reference SG strain 287/91 (NC_011274.1) using CLC Genomic Workbench version 8.5.1. All complete SG genomes available from the NCBI database (https://www.ncbi.nlm.nih.gov/data-hub/genome, accessed: 30 September 2022) were downloaded, namely S. Gallinarum strain 287/91, SG9 (NZ_CM001153), 07Q015 (NZ_CP077760), SCPM-O-B-4493 (NZ_CP088134), SCPM-O-B-4548 (NZ_CP088142), and ATCC9184 (NZ_CP019035) (18). Salmonella enterica subsp. enterica serovar Typhimurium strain LT2 (S. Typhimurium) (NC_003197) was used as the outgroup. The phylogenetic and molecular evolution (PhaME) analysis tool was used to extract the SNPs between the isolates, vaccine strains, and all available complete S. Gallinarum reference genomes (28). The PhaME pipeline also constructed a phylogeny using IQ-Tree (RRID:SCR_017254) with automatic model selection and ultrafast bootstrap with 1000 iterations. The consensus maximum likelihood tree was viewed in FigTree Version 1.4.4 (RRID:SCR_008515) (29–31).
The sequencing read data generated in this study and sequencing reads data of the 2001 Nobilis® SG9R vaccine (SRR1045124) were uploaded to the Galaxy web platform using the public server at usegalaxy.org (RRID:SCR_006281) (32). The rapid haploid variant calling and core genome alignment tool (Snippy) version 4.6.0 with default parameters was then used for variant analysis using SG strain 9 (NZ_CM001153) and its plasmid (CM001154) as the reference (33). Default parameters include a minimum coverage of 10 reads per position and a minimum of 90% of reads must differ from the reference to be considered a variant.
De novo assembled genomes were submitted to the RAST server and annotated (RRID:SCR_014606) (34). The Mauve Contig Mover (MCM) tool of the Multiple Genome Alignment (Mauve) program was used to align and sort the order of the contigs compared to the SG strain SG9 reference genome (RRID:SCR_012852) (35, 36). The reordered contigs were then used to perform a progressive Mauve alignment.
DNA from nine SG isolates and two vaccine strains was sequenced, and the complete consensus genomes were de novo assembled. Ion torrent whole-genome sequencing produced good quality reads between 230 and 372 x coverage and 50 to 51% GC content. Using the de novo assembled contigs in PubMLST, all the isolates were confirmed as SG. The isolates' consensus genomes covered between 99.12 and 99.74 % of the reference strain (Table 2). A reference genome coverage of 100% is ideal, but factors such as sequencing errors resulting in difficulties with repetitive homopolymer regions, AT-rich region, a common problem in Ion Torrent sequencing, as well as gene repeats, insertions, and deletions could affect this value. Another measure of the quality of an assembly is the benchmarking universal single-copy ortholog (BUSCO) score that considers the number of highly conserved genes present or absent. For both vaccines and seven of the isolates, > 95% of the core genes could be identified and were therefore considered good assemblies. The remaining two isolates, SG_432755 and SG_440297, were considered adequate assemblies with 94.59% and 93.92% of the core genes identified, respectively.
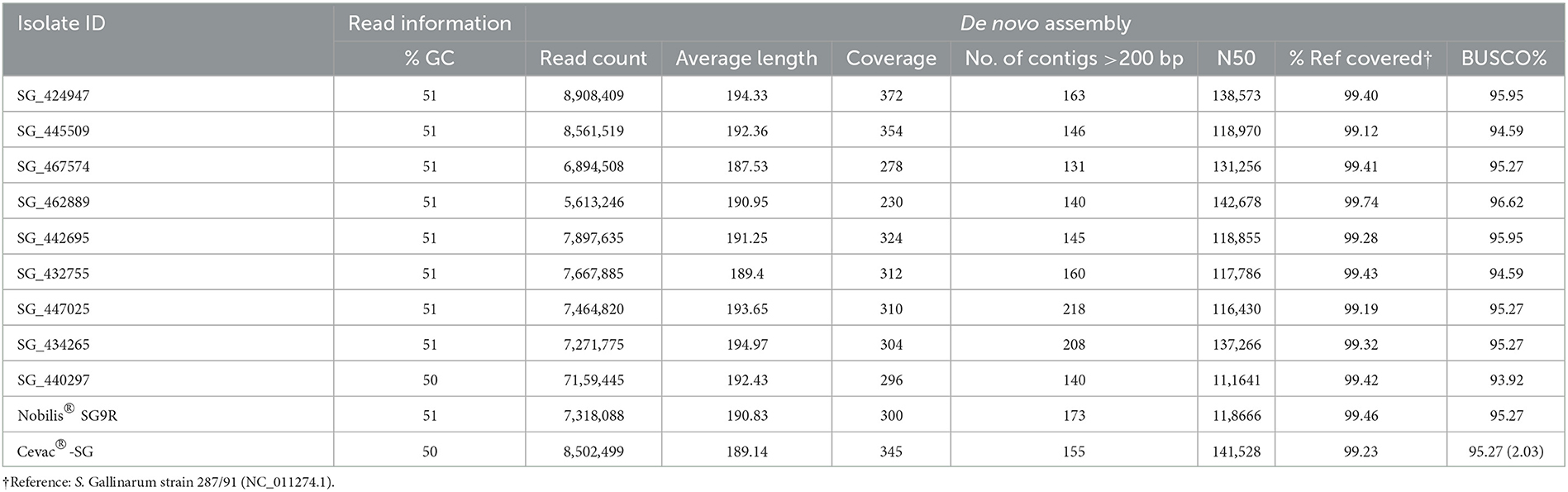
Table 2. Sequencing and de novo assembly results.
A maximum likelihood SNP phylogenetic tree was used to infer the relationship of the isolates to available complete SG reference genomes (Figure 1). As expected, the vaccines were closely related to the parental SG9 vaccine strain (100% bootstrap support), but all the isolates were also closely related to the SG9 wild-type strain (100% bootstrap support). The isolates also formed two distinct clades with 93% bootstrap support. Isolates SG_440297, SG_467575, SG_462889, SG_424947, and SG_432755 formed a clade with the Cevac®-SG vaccine; and SG_442695, SG_447025, SG_434265, and SG_445509 formed a clade with the Nobilis® SG9R vaccine, which is consistent with the vaccines used in those flocks. The 2001 and 2009 Nobilis® SG9R vaccines grouped more closely with the newly sequenced Nobilis®SG9R vaccine as expected, but a low bootstrap support (59%) was obtained. The low bootstrap value could be the result of improvements in sequencing technologies or more likely that some natural mutations occurred over time during the production of this vaccine, but no data for the production method were available in the public domain. The 2001 and 2009 Nobilis® SG9R strains were previously found to be identical (17); therefore, only the 2001 sequence was used for further analysis.
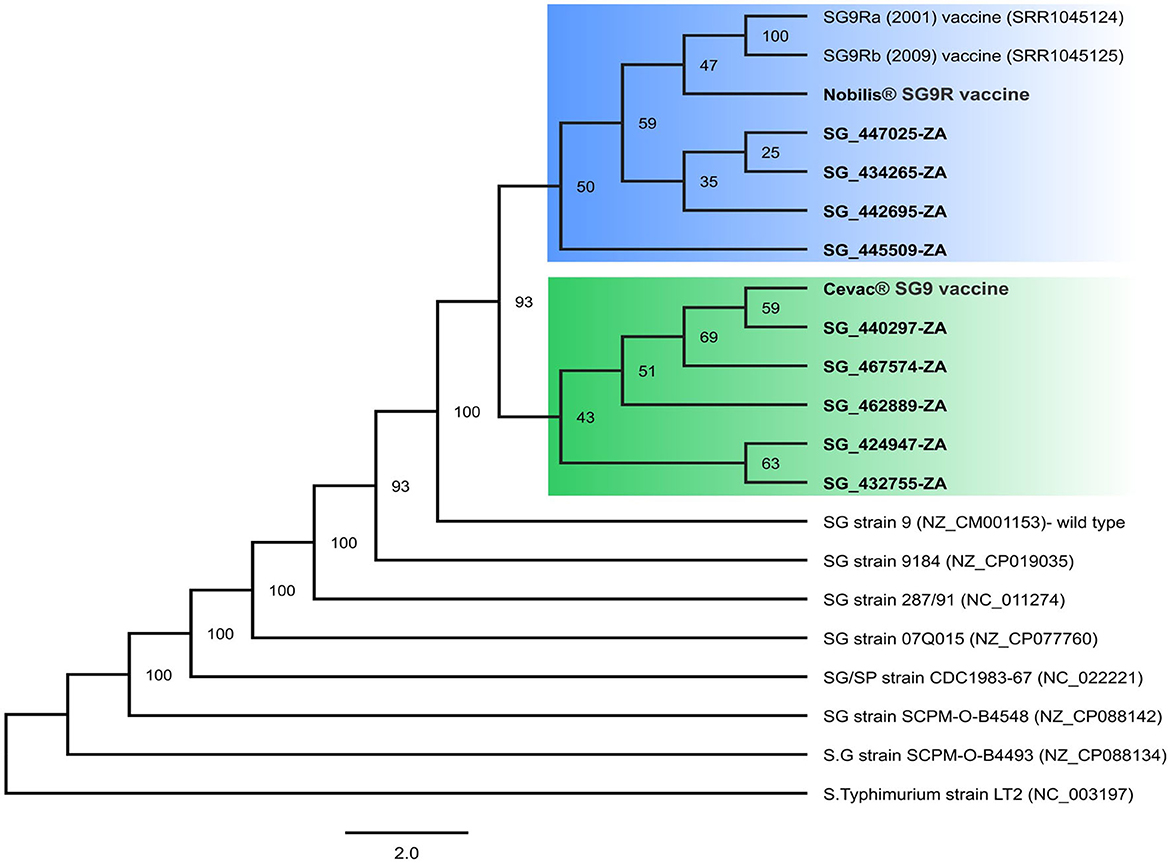
Figure 1. SNP-based maximum likelihood phylogeny of complete SG genomes. Thousand bootstrap trees were constructed with bootstrap support (%), shown at the nodes. Sequences generated in this study are highlighted in boldface. Two distinct clades related to the Nobilis®SG9R and Cevac®SG9 vaccines are highlighted in blue and green, respectively.
An SNP analysis using wild-type strain SG9 as a reference revealed that the Cevac® SG vaccine strain had 20 variations (6 deletions, 9 insertions, and 5 SNPs) in total. The SNPs included the previously described SNPs associated with attenuation, i.e., point mutations in the aceE gene resulting in an amino acid change and the rfaJ gene resulting in a premature stop codon (Table 3). Isolate SG__424947 with 21 variations (6 deletions, 8 insertion, and 7 SNPs), SG_467574 with 18 variations (6 deletions, 7 insertions, and 5 SNPs), SG_462889 with 22 variations (5 deletions, 8 insertions, and 9 SNPs), SG6_432755 with 18 variations (5 deletions, 8 insertions, and 5 SNPs), and SG_440297 with 17 variations (6 deletions, 7 insertions, and 4 SNPs) lacked the two characteristic point mutations in aceE and rfaJ, but shared other variations with the vaccine strains. The Cevac® SG vaccine shared three SNPs with these five isolates in the intergenic region (IGR) upstream from the golB gene and missense mutations in the yihX and mscM genes.
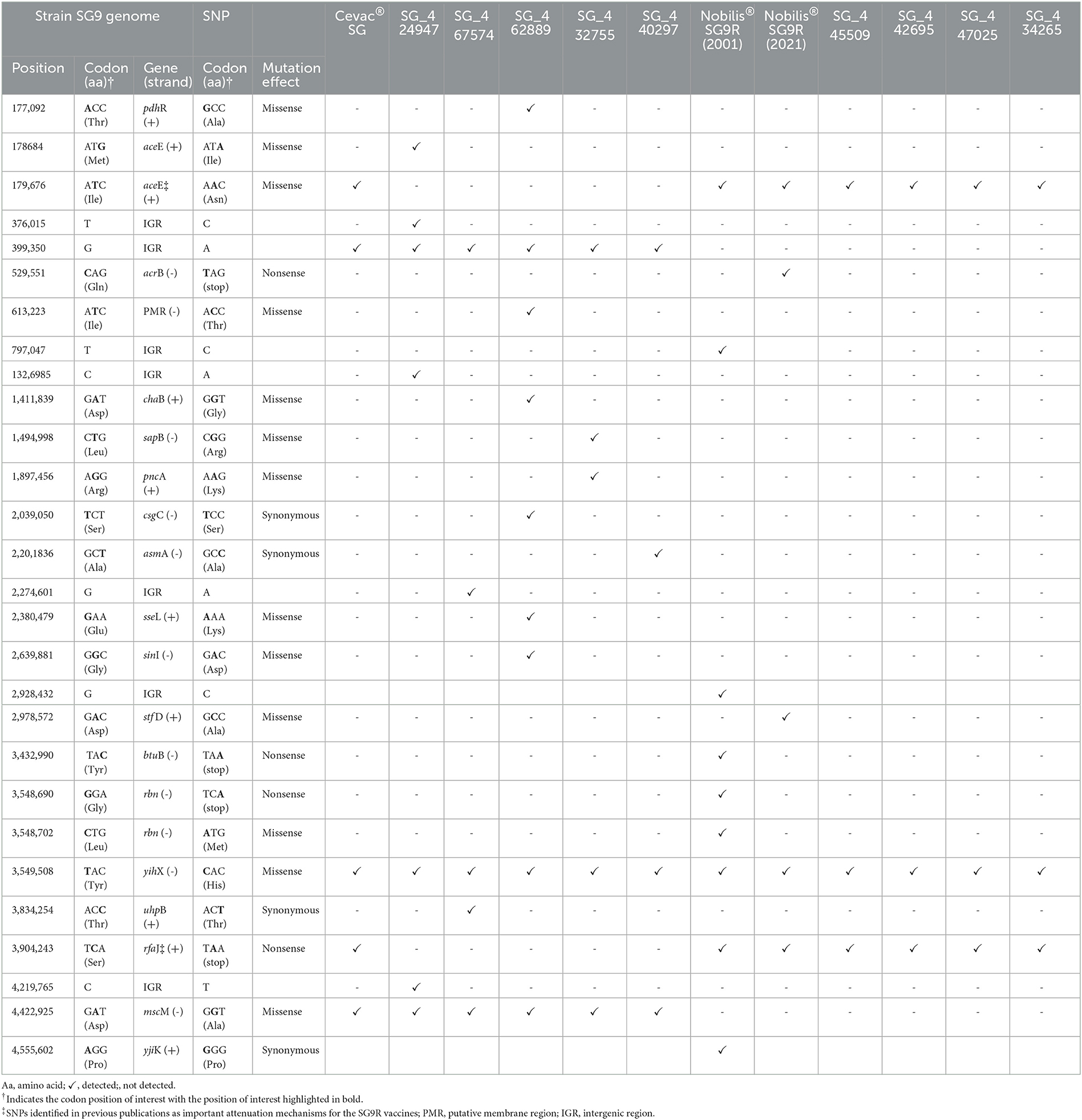
Table 3. Core SNP analysis of southern African isolates and the vaccines compared to S. Gallinarum strain SG9 (NZ_CM001153).
Vaccine strain Nobilis® SG9R (2001) and the newly sequenced Nobilis® SG9R had 14 (5, deletions and 9 SNPs) and 19 (6 deletions, 8 insertions, and 5 SNPs) sequence variations, respectively. Both vaccines contained the aforementioned characteristic point mutations in the aceE and rfaJ genes and only shared one other SNP in the yihX gene. SG_445509 with 16 variations (5 deletions, 8 insertions, and 3 SNPs), SG_442695 with 18 variations (6 deletions, 9 insertions, and 3 SNPs), SG_447025 with 17 variations (6 deletions, 8 insertions, and 3 SNPs), and SG_434265 with 17 variations (6 deletions, 8 insertions, and 3 SNPs) also contained both aforementioned point mutations in the aceE and rfaJ genes. All four isolates shared only one additional SNP with the vaccine strains in the yihX gene. Two other unique SNPs in the acrB and stf D genes were observed in the more recent Nobilis® SG9R vaccine.
The Cevac® SG vaccine had 11 insertions and deletions (indels) in common with the 5 isolates that were grouped in the same clade. Only three of the indels were in genes, i.e., a frameshift insertion in the ybjX, a disruptive insertion in the hydH gene, and a disruptive deletion in the sadA gene (Table 4). All three of these indels were also found in the isolates that formed part of the Nobilis vaccine clade. The new Nobilis® SG9R vaccine shared two additional deletions with the isolates that were in the same clade, i.e., a disruptive deletion in the kef C gene and a conservative deletion in the ftsK gene. The deletion in the kef C gene was also found in the other isolates, but not in the Cevac® SG vaccine, and the deletion in the ftsK gene was found in the Cevac® SG vaccine and only two other isolates (SG_467574 and SG_440297). The Nobilis® SG9R (2001) vaccine strain only had five deletions, of which only one was in the same location as all the isolates and other vaccine strains, and it is in an intergenic region (IGR).
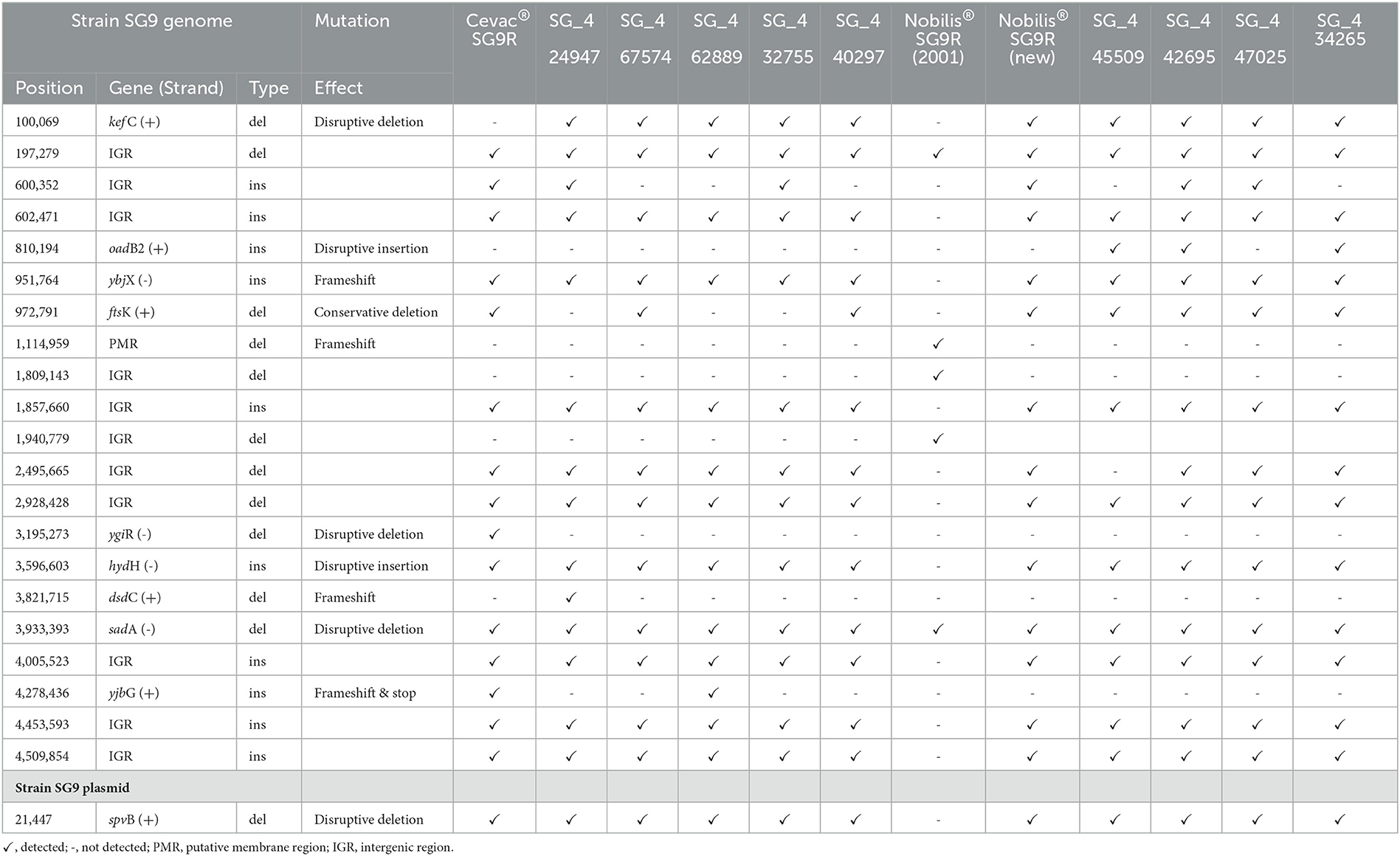
Table 4. Insertion and deletion variations in vaccine strains, southern African isolates, and the S. Gallinarum strain compared with SG9 reference strain (NZ_CM001153) and its plasmid (CM001154).
An SNP analysis of the plasmid revealed only a single disruptive deletion in the polyproline linker (PPL), a proline repeat region, of the spvB gene but it was present in all the vaccines and isolates. The plasmid sequence data for Nobilis® SG9R (2001) were not available (Table 4).
A comparative genome alignment progressive Mauve produced nine locally collinear blocks (LCBs) (Figure 2). Full synteny was observed between the isolates, vaccines, and SG strain 9, with small areas of genome rearrangement observed near the 5′end of the genome. The reference genome had a similar arrangement, but the contig overlapping this region was split and arranged to preserve the genome coordinates compared to the closely related reference SG strain 287/91 (37).

Figure 2. Alignment S. Gallinarum strain 9, SG9R vaccine strains, isolates, and S. Gallinarum strain 287/91. All the genomes were aligned with progressive Mauve using the default parameters. Similarly colored LCB blocks indicate homologous regions and lines linking these regions between the genomes.
The strain SG9R is the most widely used SG vaccine strain in the world and one of the only two strains registered for use as vaccines in South Africa to prevent fowl typhoid fever. Reversion of the vaccine strain has been studied previously with varying results. In this study, we used an approach similar to that of Van Immerseel et al. (17), combining comparative whole-genome analysis with SNP detection to investigate the possible reversion to the virulence of vaccines during outbreaks of fowl typhoid fever in South Africa and Eswatini in 2017. SNP-based maximum likelihood phylogeny analysis showed that all the local field isolates were more closely related to the vaccine and SG9 strain than any of the other available SG reference strain genomes.
The SNP analysis revealed the anticipated known markers of attenuation in the SG9R vaccine strains. A missense point mutation in the second position of a codon encoding an isoleucine (Ile) resulted in an amino acid change to asparagine (Asn) in the pyruvate dehydrogenase (aceE) gene and a nonsense mutation in the second codon in the rfaJ gene at position 3,904,243 from a serine (Ser) to a stop codon. However, whether the mutation in the aceE gene affects the function of the pyruvate dehydrogenase E1 component is unknown (13). The premature stop codon in the aceE gene which encodes a lipopolysaccharide (LPS) 1,2,-glucosyltransferase could result in a truncated LPS core and loss of the O-antigen side chain (15, 38). A missense point mutation in the yihX gene found in all vaccines resulted in a tyrosine (Tyr) to histidine (His) amino acid change. The yihX gene encodes glucose-1-phosphatase, which is a putative enzyme with an unknown function in SG, but possibly plays a role in metabolism and is upregulated by the PhoP/Q system, that controls the expression of numerous genes involved in the virulence and survival of Gram-negative bacteria (39).
Van Immerseel et al. (17) previously reported no changes between the 2001 and 2009 Nobilis®-SG9R vaccines and only identified four SNPs in total, in the aceE, rfaJ, btuB, and galT genes. Only the first three were located using the Snippy pipeline in this study, which could be attributed to the different technologies used. A comparison of the 2001 and newly sequenced Nobilis®-SG9R vaccines demonstrated that the genome of the vaccine has changed slightly over the past two decades, but the older and newer vaccines were still more closely related to each other than to the field isolates. The more recent Nobilis®-SG9R vaccine also contained substantially more insertions and deletions compared to the 2001 Nobilis®-SG9R vaccine, but most of these were in the IGR with one disruptive deletion in the sadA gene. The sadA gene encodes a trimeric autotransporter adhesin and plays a role in biofilm formation and adhesion in S. typhimurium. However, a loss in this gene does not cause a significant effect on infection (40). This deletion was also observed in the Cevac® SG vaccine, and all the isolates tested in this study and could be a potential marker to distinguish between vaccine- and wild-type strains.
The four isolates, SG_445509, SG_442695, SG_447025, and SG_434265, that were closely related to the Nobilis®-SG9R vaccine contained both known markers of attenuation in aceE and rfaJ genes, a point mutation in the yihX gene, plus intact spv, SPI-1 and SPI-2 gene clusters (data not shown), and as such these are revertant vaccines and most likely the cause of the outbreaks observed on the respective farms (15). The remaining five isolates were closely related to the Cevac® SG vaccine, but none of these isolates contained the known attenuation markers. However, these isolates were closely related to and formed a distinct clade with the Cevac® SG vaccine. Point mutations resulting in intact aceE and rfaJ genes along with intact spv, SPI-1, and SPI-2 gene clusters were most likely the reason for the virulence observed in these flocks. Van Immerseel et al. (17) similarly reported that these mutations were the likely cause of the reversion to virulence observed in Belgium. These isolates furthermore shared a point mutation in the yihX gene, and one additional missense mutation in the mscM gene was found in all the isolates and the Cevac® SG vaccine. The mscM gene encodes a miniconductance mechanosensitive channel and plays a role in regulating osmotic pressure (41). This protein is not well-characterized in Salmonella, and the effect of mutations remains unknown (42).
Analysis of the indels revealed two insertions and one deletion shared between all the isolates and vaccines sequenced in this study. A frameshift insertion was identified in the ybjX (also known as somA) gene, which encodes a virK homolog protein and plays a minor role in virulence in Salmonella enterica serovar Enteritidis (SE) by modulating membrane proteins, affecting bacterial motility, secretion, and altering the membrane proteins to evade host adaptive immune response (43, 44). Mckelvey et al. (43) also demonstrated that this gene plays a role in SE persistence in chickens. SG is a non-motile pathogen; therefore, its motility would not be affected, but mutations in this gene could affect the ability of SG to evade the host immune response as has been shown in SE (43). A disruptive insertion was found among all the isolates sequenced in this study in the hydH gene, which encodes the sensor kinase of the two-component zinc resistance-associated regulator (ZraS) and possibly plays a role in Salmonella infection, acting as a signal (45). Even though neither of these insertions was observed in the 2001 Nobilis®-SG9R vaccine, it is an interesting discovery that warrants further investigation to determine if these are markers of attenuation in the newer vaccine strain and to differentiate vaccine from wild-type strains.
A disruptive deletion in the kef C gene was observed in all the isolates and the new Nobilis®-SG9R vaccine, but not in the Cevac® SG vaccine. This gene encodes the glutathione-regulated potassium-efflux system protein KefC which controls the efflux of potassium and protects against electrophiles in Escherichia coli (46). Little information is available on this gene in Salmonella including any effects mutations may have. A conservative deletion in the ftsK gene was found in the Cevac® SG vaccine, and only two isolates were closely related to it, as well as in the Nobilis®-SG9R vaccine and all the isolates related to it. The ftsK encodes DNA translocase FtsK and plays a role in chromosome segregation, but Wang et al. (47) found that indels in this gene probably have no effect on the protein function.
The spvB virulence plasmid gene encodes Mono (ADP-ribosyl) transferase SpvB and inhibits phagocyte function when it is secreted into the macrophage cytoplasm (15). Both of the vaccines and all the isolates contained the expected lenght of nine prolines in the PPL. The length of the PPL is linked to the increased virulence, thus, even if the PPL is only nine proline residues long, it is still intact and the strain can still show diminished virulence, along with the intact SPI-1 and SPI-2 type III secretion system (48).
There are some limitations in this study that could be addressed in future research. Previous studies, like that of Van Immerseel et al. (17) and De Carli et al. (17) used genomic comparison with SNP detection and phylogenetics to show how field strains were related to the SG9R vaccine and this study also focused only on similar genomic methods. However, the virulence of the field strains was not studied in vivo and would be advantageous in future studies to confirm that observed SG symptoms were due to the reversion of the vaccine to virulence and not due to possible residual virulence of the vaccine.
In conclusion, all the field isolates from a recent spate of fowl typhoid outbreaks in South Africa and Eswatini were closely related to the SG9R vaccine strains in use, and since no wild-type strains were identified, reversion to virulence of the vaccine is the most likely cause of these outbreaks. Reversion to virulence in the Nobilis®-SG9R vaccine was associated with intact known virulence factors, spv, SPI-1, and SPI-2, whereas the virulence of Cevac®-SG vaccine-derived revertants was likely associated with point mutations resulting in intact aceE and rfaJ genes. The latter findings still require in vivo verification, as does the cause driving the consistent selection of these specific point mutations. Additional markers identified in this study, i.e., an SNP in the yihX gene, insertions in the ybjX and hydH genes, and a deletion in the sadA gene could, with further investigation, prove to be useful in distinguishing current SG9R vaccines from wild-type strains by targeted PCR assays.
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/genbank/), accession numbers CP118112–CP118133 and SRR23450481–SRR23450491.
Ethical approval was obtained from the Research and Animal Ethics Committees of the University of Pretoria, under project no. REC187-19.
AB and CA contributed to the conception and design of the study. AB organized the database, performed the bioinformatic analyses, and wrote the first draft of the manuscript. Both authors contributed to the manuscript revision, read, and approved the submitted version.
This study and AB's postdoctoral fellowship were funded by the National Research Foundation SARChI grant no. N00705/114612.
The authors are grateful to the veterinarians and NOSA Laboratory (Pty) Ltd. for providing the isolates used in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. World Organization for Animal Health. “Fowl typhoid and pullorum disease,” in Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2022, 8th Edn (Office international des epizooties). Available onlne at https://www.woah.org/en/what-we-do/standards/codes-and-manuals/terrestrial-manual-online-access/ (accessed June 21, 2023).
2. Zhou X, Kang X, Zhou K, Yue M, A. Global dataset for prevalence of Salmonella Gallinarum between 1945 and 2021. Scien Data. (2022) 9:495. doi: 10.1038/s41597-022-01605-x
3. Shivaprasad H, Barrow P. Pullorum Disease and Fowl Typhoid. Diseases of Poultry, 12th ed, eds YM Saif, AM Fadley, JR Glisson, LR McDougald, LK Nolan, and DE Swayne. Ames, IA: Blackwell Publishing, (2008), p. 620-34.
4. Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, Churcher C, et al. Comparative genome analysis of salmonella enteritidis Pt4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. (2008) 18:1624–37. doi: 10.1101/gr.077404.108
5. Barrow P, Simpson J, Lovell M, Binns M. Contribution of Salmonella-Gallinarum large plasmid toward virulence in fowl typhoid. Infect Immun. (1987) 55:388–92. doi: 10.1128/iai.55.2.388-392.1987
6. Rychlik I, Gregorova D, Hradecka H. Distribution and function of plasmids in Salmonella Enterica. Vet Microbiol. (2006) 112:1–10. doi: 10.1016/j.vetmic.2005.10.030
7. Purchase C, Picard J, McDonald R, Bisschop S, A. Comparison of the oral application and injection routes using the onderstepoort biological products fowl typhoid vaccine, its safety, efficacy and duration of protection in commercial laying hens. J South Af Vet Assoc-Tydskrif Van Die Suid-Afrikaanse Vet Veren. (2008) 79:39–43. doi: 10.4102/jsava.v79i1.239
8. Shivaprasad H. Fowl Typhoid and pullorum disease. Revue Scient Et Technique-Office International Des Epizoo. (2000) 19:405–24. doi: 10.20506/rst.19.2.1222
9. Revolledo L. Vaccines and vaccination against fowl typhoid and pullorum disease: an overview and approaches in developing countries. J Appl Poult Res. (2018) 27:279–91. doi: 10.3382/japr/pfx066
10. Jiang Z, Kang X, Song Y, Zhou X, Yue M. Identification and evaluation of novel antigen candidates against salmonella pullorum infection using reverse vaccinology. Vaccines. (2023) 11:865. doi: 10.3390/vaccines11040865
11. Desin TS, Köster W, Potter AA. Salmonella vaccines in poultry: past, present and future. Expert Rev Vaccines. (2013) 12:87–96. doi: 10.1586/erv.12.138
12. Lee Y, Mo I, Kang M. Protective efficacy of live Salmonella Gallinarum 9r vaccine in commercial layer flocks. Avian Pathol. (2007) 36:495–8. doi: 10.1080/03079450701691278
13. Kang M-S, Kwon Y-K, Kim H-R, Oh J-Y, Kim M-J, An B-K, et al. Differential identification of salmonella enterica serovar gallinarum biovars gallinarum and pullorum and the biovar gallinarum live vaccine strain 9r. Vet Microbiol. (2012) 160:491–5. doi: 10.1016/j.vetmic.2012.05.041
14. Kang M-S, Kwon Y-K, Jung B-Y, Kim A, Lee K-M, An B-K, et al. Differential identification of Salmonella Enterica Subsp. enterica serovar gallinarum biovars gallinarum and pullorum based on polymorphic regions of glgc and spec genes. Vet Microbiol. (2011) 147:181–5. doi: 10.1016/j.vetmic.2010.05.039
15. Kwon H, Cho S. Pathogenicity of Sg 9r, a rough vaccine strain against fowl typhoid. Vaccine. (2011) 29:1311–8. doi: 10.1016/j.vaccine.2010.11.067
16. Kang M-S, Kwon Y-K, Kim H-R, Oh J-Y, Kim M-J, An B-K, et al. Comparative proteome and transcriptome analyses of wild-type and live vaccine strains of Salmonella Enterica Serovar Gallinarum. Vaccine. (2012) 30:6368–75. doi: 10.1016/j.vaccine.2012.08.048
17. Van Immerseel F, Studholme D, Eeckhaut V, Heyndrickx M, Dewulf J, Dewaele I, et al. Salmonella Gallinarum field isolates from laying hens are related to the vaccine strain SG9R. Vaccine. (2013) 31:4940–5. doi: 10.1016/j.vaccine.2013.08.033
18. De Carli S, Graf T, Kipper D, Lehmann F, Zanetti N, Siqueira F, et al. Molecular and phylogenetic analyses of Salmonella Gallinarum trace the origin and diversification of recent outbreaks of fowl typhoid in poultry farms. Vet Microbiol. (2017) 212:80–6. doi: 10.1016/j.vetmic.2017.11.001
19. Kim NH, Ko DS, Ha EJ, Ahn S, Choi K-S, Kwon H-J. Optimized detoxification of a live attenuated vaccine strain. (Sg9r) to improve vaccine strategy against fowl. Typhoid Vacc. (2021) 9:122. doi: 10.3390/vaccines9020122
20. Silva E, Snoeyenbos G, Weinack OM, Smyser C. Studies on the use of 9r strain of Salmonella Gallinarum as a vaccine in chickens. Avian Dis. (1981) 3:38–52. doi: 10.2307/1589825
21. Bouzoubaa K, Nagaraja K, Kabbaj F, Newman J, Pomeroy B. Feasibility of using proteins from Salmonella Gallinarum vs. 9r Live vaccine for the prevention of fowl typhoid in chickens. Avian Dis. (1989) 33:385–91. doi: 10.2307/1591094
22. Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. (2022) 50:D20–d6. doi: 10.1093/nar/gkab1112
23. Andrews S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. (2010). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed February 15, 2023).
24. Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. (2012) 158(Pt 4):1005–15. doi: 10.1099/mic.0.055459-0
25. Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: bigsdb software, the pubmlstorg website and their applications. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
26. Gurevich A, Saveliev V, Vyahhi N, Tesler G. Quast: quality assessment tool for genome assemblies. Bioinformatics. (2013) 29:1072–5. doi: 10.1093/bioinformatics/btt086
27. Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with quast-Lg. Bioinformatics. (2018) 34:i142–i50. doi: 10.1093/bioinformatics/bty266
28. Shakya M, Ahmed SA, Davenport KW, Flynn MC, Lo C-C, Chain PSG. Standardized phylogenetic and molecular evolutionary analysis applied to species across the microbial tree of life. Sci Rep. (2020) 10:1723. doi: 10.1038/s41598-020-58356-1
29. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. Iq-tree 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
30. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. Modelfinder: fast model selection for accurate phylogenetic estimates. Nat Methods. (2017) 14:587–9. doi: 10.1038/nmeth.4285
31. Rambaut A. Figtree. Tree Figure Drawing Tool. (2009). Available online at: http://treebioedacuk/software/figtree/ (accessed February 15, 2023).
32. Community TG. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. (2022) 50:W345–W51. doi: 10.1093/nar/gkac610
33. Seemann T,. Snippy: Fast Bacterial Variant Calling from NGS Reads. (2015). Available at https://github.com/tseemann/snippy (accessed February 15, 2023).
34. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The rast server: rapid annotations using subsystems technology. BMC Genomics. (2008) 9:75. doi: 10.1186/1471-2164-9-75
35. Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. (2004) 14:1394–403. doi: 10.1101/gr.2289704
36. Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. Reordering contigs of draft genomes using the mauve aligner. Bioinformatics. (2009) 25:2071–3. doi: 10.1093/bioinformatics/btp356
37. Richardson EJ, Limaye B, Inamdar H, Datta A, Manjari KS, Pullinger GD, et al. Genome sequences of salmonella enterica serovar typhimurium, choleraesuis, dublin, and gallinarum strains of well- defined virulence in food-producing animals. J Bacteriol. (2011) 193:3162–3. doi: 10.1128/JB.00394-11
38. Lyman M, Steward J, Roantree R. Characterization of the virulence and antigenic structure of Salmonella typhimurium strains with lipopolysaccharide core defects. Infect Immun. (1976) 13:1539–42. doi: 10.1128/iai.13.6.1539-1542.1976
39. Charles RC, Harris JB, Chase MR, Lebrun LM, Sheikh A, LaRocque RC, et al. comparative proteomic analysis of the phop regulon in Salmonella Enterica serovar typhi vs. typhimurium. PLoS ONE. (2009) 4:e6994. doi: 10.1371/journal.pone.0006994
40. Raghunathan D, Wells TJ, Morris FC, Shaw RK, Bobat S, Peters SE, et al. Sada, a trimeric autotransporter from Salmonella Enterica Serovar typhimurium, can promote biofilm formation and provides limited protection against infection. Infect Immun. (2011) 79:4342–52. doi: 10.1128/IAI.05592-11
41. Compton EL, Mindell JA. Bacterial ion channels. EcoSal Plus. (2010) 4:2. doi: 10.1128/ecosalplus.3.3.2
42. Altendorf K, Booth IR, Gralla J, Greie J-C, Rosenthal AZ, Wood JM. Osmotic stress. EcoSal Plus. (2009) 3:5. doi: 10.1128/ecosal.5.4.5
43. McKelvey JA, Yang M, Jiang Y, Zhang S. Salmonella Enterica serovar enteritidis antimicrobial peptide resistance genes aid in defense against chicken innate immunity, fecal shedding, and egg deposition. Infect Immun. (2014) 82:5185–202. doi: 10.1128/IAI.02387-14
44. Detweiler CS, Monack DM, Brodsky IE, Mathew H, Falkow S. Virk, Soma and Rcsc Are important for systemic Salmonella enterica serovar typhimurium infection and cationic peptide resistance. Mol Microbiol. (2003) 48:385–400. doi: 10.1046/j.1365-2958.2003.03455.x
45. Huang Y, Leming CL, Suyemoto M, Altier C. Genome-wide screen of salmonella genes expressed during infection in pigs, using in vivo expression technology. Appl Environ Microbiol. (2007) 73:7522–30. doi: 10.1128/AEM.01481-07
46. Ferguson GP, Nikolaev Y, McLaggan D, Maclean M, Booth IR. Survival during exposure to the electrophilic reagent N-ethylmaleimide in Escherichia Coli: role of Kefb and Kefc potassium channels. J Bacteriol. (1997) 179:1007–12. doi: 10.1128/jb.179.4.1007-1012.1997
47. Wang Y, Jia B, Xu X, Zhang L, Wei C, Ou H, et al. Comparative genomic analysis and characterization of two salmonella enterica serovar enteritidis isolates from poultry with notably different survival abilities in egg whites. Front Microbiol. (2018) 9:111. doi: 10.3389/fmicb.2018.02111
Keywords: Salmonella gallinarum, fowl typhoid, reversion to virulence, SG9R, whole-genome comparison, vaccine
Citation: Beylefeld A and Abolnik C (2023) Salmonella gallinarum strains from outbreaks of fowl typhoid fever in Southern Africa closely related to SG9R vaccines. Front. Vet. Sci. 10:1191497. doi: 10.3389/fvets.2023.1191497
Received: 22 March 2023; Accepted: 05 June 2023;
Published: 05 July 2023.
Edited by:
Min Yue, Zhejiang University, ChinaReviewed by:
Paul Barrow, University of Surrey, United KingdomCopyright © 2023 Beylefeld and Abolnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda Beylefeld, YW1hbmRhLmJleWxlZmVsZEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.