
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 16 August 2023
Sec. Livestock Genomics
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1184764
This article is part of the Research TopicFrom Agriculture Genome to Phenome: Genome-Wide Association, Prediction and SelectionView all 11 articles
 Kai Zhang1
Kai Zhang1 Guozhi Wang2Lihuan Wang1Bin Wen1
Guozhi Wang2Lihuan Wang1Bin Wen1 Xiangchao Fu1Ning Liu1Zhiju Yu1Wensu Jian1Xiaolin Guo1Hanzhong Liu1*
Xiangchao Fu1Ning Liu1Zhiju Yu1Wensu Jian1Xiaolin Guo1Hanzhong Liu1* Shi-Yi Chen2*
Shi-Yi Chen2*Coat color is an important phenotypic characteristic of the domestic rabbit (Oryctolagus cuniculus) and has specific economic importance in the Rex rabbit industry. Coat color varies considerably among different populations of rabbits, and several causal genes for this variation have been thoroughly studied. Nevertheless, the candidate genes affecting coat color variation in Chinese Rex rabbits remained to be investigated. In this study, we collected blood samples from 250 Chinese Rex rabbits with six different coat colors. We performed genome sequencing using a restriction site-associated DNA sequencing approach. A total of 91,546 single nucleotide polymorphisms (SNPs), evenly distributed among 21 autosomes, were identified. Genome-wide association studies (GWAS) were performed using a mixed linear model, in which the individual polygenic effect was fitted as a random effect. We detected a total of 24 significant SNPs that were located within a genomic region on chromosome 4 (OCU4). After re-fitting the most significant SNP (OCU4:13,434,448, p = 1.31e-12) as a covariate, another near-significant SNP (OCU4:11,344,946, p = 7.03e-07) was still present. Hence, we conclude that the 2.1-Mb genomic region located between these two significant SNPs is significantly associated with coat color in Chinese Rex rabbits. The well-studied coat-color-associated agouti signaling protein (ASIP) gene is located within this region. Furthermore, low genetic differentiation was also observed among the six coat color varieties. In conclusion, our results confirmed that ASIP is a putative causal gene affecting coat color variation in Chinese Rex rabbits.
Among all farm animals, modern rabbits (Oryctolagus cuniculus) are among the most recently domesticated species, although the exact domestication date of the species remains controversial when examined on the basis of archeological records and genetic evidence (1, 2). However, it has been widely acknowledged that modern rabbits have a single domestication origin, resulting in lower genetic variation in comparison with other farm animals (3–5). More than 200 rabbit breeds have been officially registered in the Domestic Animal Diversity Information System (DAD-IS),1 and these show considerable morphological variation, such as in body size, coat color, and hair phenotype (6, 7). Among them, the Rex rabbit is well known for its short, dense, and smooth hair. This Rex rabbit phenotype is believed to have genetically originated from normal hair (8). Coat color is an important phenotypic trait in the fur industry, and at least 16 color varieties of Rex rabbits have been recognized by the American Rabbit Breeders Association (ARBA);2 however, the preferred coat color differs between different markets.
Coat color in mammals is determined by the relative amounts of eumelanin and phaeomelanin in melanocytes, and many studies have been conducted to identify coat-color-associated genes and causal mutations in domestic animals during the past two decades (9). In domestic rabbits, melanocortin 1 receptor (MC1R) is the first gene to have been thoroughly studied, and several causal mutations have been successfully identified as affecting coat color (10, 11). As a competitive ligand to MC1R in the melanin synthesis pathway (12), a premature stop mutation of the agouti signaling protein (ASIP) gene has been reported to be responsible for the non-agouti black coat color in rabbits (13). Additionally, a premature stop mutation of the tyrosinase-related protein 1 (TYRP1) gene is associated with brown coat color in rabbits (14). Based on the gene expression patterns observed in Rex rabbits of various colors, it has been suggested that the POU class 2 homeobox 1 gene (POU2F1) affects fur color formation in Rex rabbits (15). The variability of the tyrosinase (TYR) gene has been studied in domestic and wild European rabbits; this work has confirmed the effects of missense mutations on coat colors (16). The genetic polymorphisms of five candidate genes were genotyped to investigate their associations with different coat colors in rabbits (17). In addition to loci determining different coat colors, it was found that both eumelanic and pheomelanic pigmentations can be further diluted more or less under genetical control of the dilute locus: for example, black can be diluted to gray. Fontanesi et al. (18) successfully mapped the dilute locus of rabbits to the melanophilin (MLPH) gene and identified a frameshift mutation associated with the dilute coat color.
Due to the wide application of high-throughput sequencing technologies, large numbers of genome-wide variants can now be discovered and genotyped at an affordable cost (19). Among these technologies, restriction-site-associated DNA sequencing (RAD-seq) is a cost-efficient approach for investigating genome-wide variants, especially in non-model species (20); the approach was first proposed in 2008 and is characterized by sequencing of small genomic fragments that are randomly digested by restriction enzyme (s). The RAD-seq approach has been widely used for population genetics and genome-wide association studies (GWAS) [such as by (21–24)]. In rabbits, genetic diversity and population structure have been investigated using genome-wide single nucleotide polymorphisms (SNPs) that have been generated using the RAD-seq approach (25, 26). In this study, we similarly employed the RAD-seq approach to identify genome-wide SNPs, which we subsequently used for GWAS with six coat color varieties of Chinese Rex rabbits. The results could help us to better understand the underlying genetic basis of coat colors in Chinese Rex rabbits.
Venous blood was collected from the marginal ear veins of 250 Rex rabbits raised at the Research Farm of Sichuan Academy of Grassland Sciences. The rabbits consisted of six coat color varieties: 40 White Rex (WT), 42 Californian Rex (CL), 42 Black Rex (BL), 42 Chinchilla Rex (CC), 42 Dark Chinchilla Rex (DC), and 42 Light Chinchilla Rex (LC). Among these varieties, WT, CL, BL, and CC exhibit different coat colors, and there are two varieties of CC with a darker (DC) and lighter (LC) coat color, respectively (Figure 1). There was no genetic relationship within three generations among any of the sampled animals according to pedigree information. Genomic DNA was extracted using the Axy-Prep Genomic DNA Miniprep Kit (Axygen Bioscience, USA).
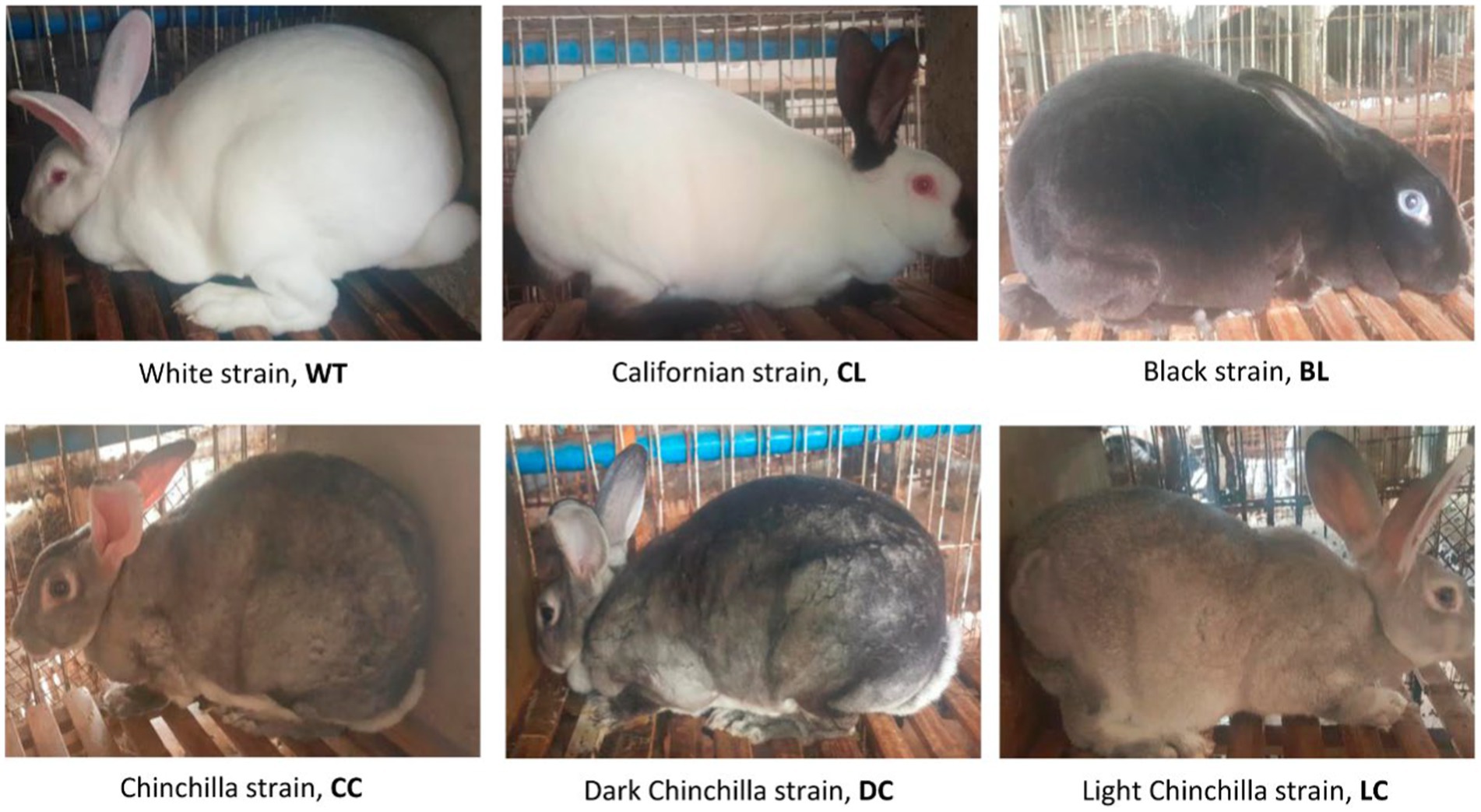
Figure 1. Phenotypes of the six coat color varieties of Rex rabbit included in this study.
Based on preliminary investigation on the reference genome sequences of rabbits, the restriction enzyme EcoRI (NEB, Beijing) was successfully used to digest genomic DNA (~1 μg per sample used). Sequencing libraries were constructed according to the recommended pipeline (20). In brief, P1 adapter sequence was first added to the digested fragments; this was followed by sequential steps of sample pooling, random shearing, and fragment size-based selection using agarose gel. Subsequently, DNA was ligated to a second adapter (P2) with divergent ends. DNA fragments of ~400 bp in length were selected to construct the sequencing libraries. Finally, the libraries were sequenced on an Illumina HiSeq platform and 150 bp paired-end reads were generated (Novogene Co. Ltd., Beijing).
After the initial sequencing images were converted into sequence files in the FASTQ format using a standard pipeline, we first investigated the Qphred value-based error rate. Using the fastp software package (27), low-quality reads were discarded according to three criteria (26): (1) reads containing adaptor sequences, (2) reads containing ambiguous bases for more than 10% of the total length, and (3) reads containing low-quality bases (Qphred value <5) for more than 50% of the total length. If either member of the paired reads was marked as low quality, both pairs were discarded. After these steps, we obtained clean reads and subjected them to the following analyses.
All clean reads were mapped to a rabbit reference genome (UM_NZW_1.0) using the BWA software with default parameters (28). Subsequently, we employed the GATK toolkit v3.8 (29) for discovery and genotyping of small variants (SNPs and InDels) among all samples according to GATK Best Practices recommendations (30, 31); in this process, the duplicate removal, realignment, and hard filtering steps were performed with default parameters. After exclusion of all InDels, a raw set of SNPs was obtained. SNPs were removed if they had a coverage depth < 3, calling rate < 90% for the genotypes or individuals, minor allele frequency (MAF) < 0.05, and extreme deviation from Hardy–Weinberg equilibrium (HWE, p < 10−8). Finally, we extracted biallelic SNPs and generated a clean set of SNPs. The missing genotypes were further imputed using the Beagle software package v5.4 with default parameters (32).
We investigated the genomic distribution of clean SNPs and the transition/transversion ratio using the ANNOVAR software package (33). Nucleotide diversity (π) for each locus was calculated using the vcftools software package (34). The PopSc toolkit (35) was used to calculate the polymorphism information content (PIC), inter-variety Wright’s FST, and intra-variety Wright’s FIS (36). The pairwise p-distances among all samples were calculated from all SNPs using the TreeBeST software package (TreeSoft) and then subjected to the construction of a phylogenetic tree according to the neighbor-joining method (37); this phylogenetic tree was visualized using the ggtree R package (38).
In GWAS, the six coat color varieties of Rex rabbits were arbitrarily coded using the ordinal values of WT = 1, LC = 2, CC = 3, DC = 4, CL = 5, and BL = 6. To avoid potential bias arising from the arbitrary coding of coat colors, the reverse order was employed in independent repeat performance of GWAS. The effect of each SNP was estimated using a mixed linear model implemented in the GCTA software package (39):
where is the vector of coat colors coded above; is the mean term is the fixed effect of the SNP tested for association; is a vector containing the genotype score for the tested SNP; is the vector of individual random polygenic effects with , where is the genomic relationship matrix and is the additive genetic variance; is the incidence matrix for ; and is a vector of random residual effects with , where is an identity matrix and is the residual variance. After estimation of the SNP effects, the most significant SNP was selected and further added as a covariate to the mixed linear model described above. A Bonferroni approach was used for correction of multiple comparisons in the GWAS results (40).
We obtained 208.51 Gb raw paired-end reads (approximately 1.5 billion reads) across all the sequenced samples, from which 208.48 Gb clean reads (0.83 Gb per sample) were generated after the quality control steps. On average, 98.9% of the clean reads were successfully aligned against the reference genome. A total of 5,162,522 raw SNPs were generated on 21 autosomes, and we finally obtained 91,546 high-quality biallelic SNPs according to our custom filtering process. These SNPs were distributed across the whole genome, and an average of 42.3 SNPs per Mb genomic region was comparably observed among all autosomes (Figure 2A). The mean MAF was ~0.25 (Figure 2B). There were 64,311 transitions and 27,235 transversions (transition/transversion ratio = 2.36). Using the reference annotation of the rabbit genome (UM_NZW_1.0), we inferred the locations of the SNPs. SNPs were distributed within exons (N = 1,648), introns (N = 34,037), 1 kb upstream/downstream regions of genes (N = 1,411), and intergenic regions (N = 54,450).
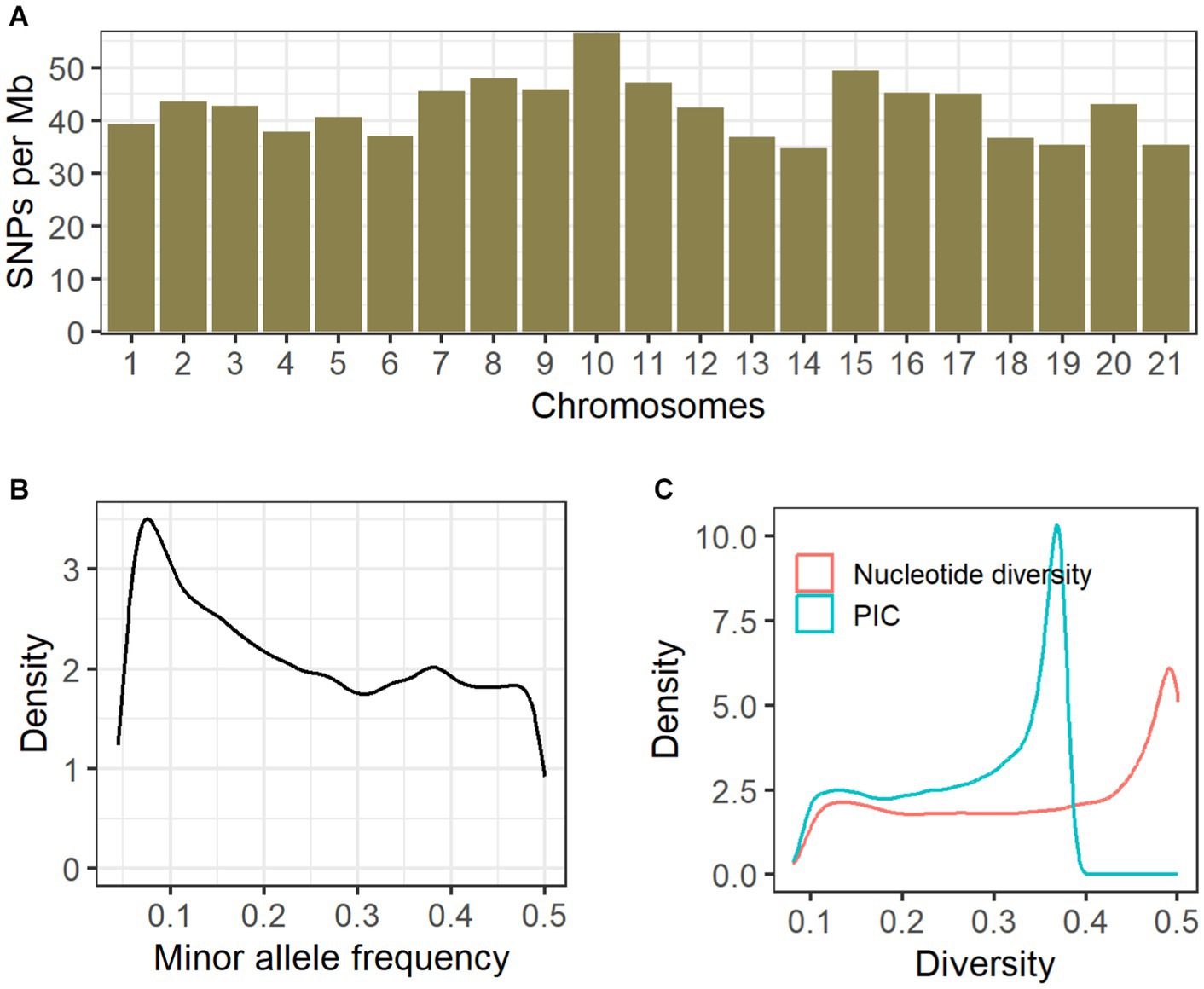
Figure 2. Genomic distribution and genetic diversity of SNPs. For all clean SNPs, we investigated the genomic distribution (A), minor allele frequencies (B), and the density distribution of nucleotide diversity and polymorphism information content (C).
Among all clean SNPs, the distribution density of nucleotide diversity exhibited a single peak close to 0.5, and a similar pattern was also observed for PIC (Figure 2C). The median and mean values for nucleotide diversity across the six coat color varieties were 0.3518 and 0.3185, respectively (Table 1); among these, the Black Rex showed the highest degree of nucleotide diversity, with a median of 0.3672 and a mean of 0.3340. The Black Rex and Californian Rex had the highest and lowest PIC, with mean values of 0.2649 and 0.2440, respectively. Furthermore, there were no obvious differences among the coat color varieties in relation to genetic diversity.
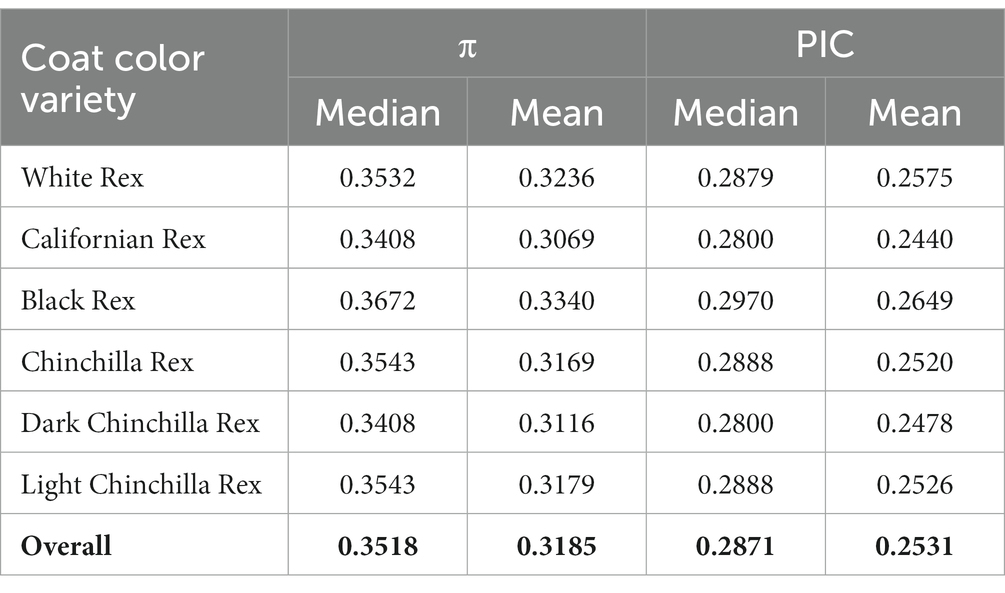
Table 1. Nucleotide diversity (π) and polymorphism information content (PIC) in different coat color varieties of Rex rabbit.
The highest and lowest degrees of inter-variety differentiation were observed between the Dark Chinchilla Rex and the Californian Rex (FST = 0.0962), and between the Chinchilla Rex and the Dark Chinchilla Rex (FST = −0.0002), respectively (Figure 3A). Intra-variety inbreeding coefficients (FIS) ranged from −0.1221 in the Californian Rex to −0.0522 in the Black Rex. According to the phylogenetic tree for all samples (Figure 3B), both the White Rex and the Californian Rex formed their own clusters and were separated from the other breeds. Next, most of the Black Rex rabbits were clustered together and were almost distinguishable. However, there was no obvious clustering pattern among the Chinchilla Rex and the other two breeds.
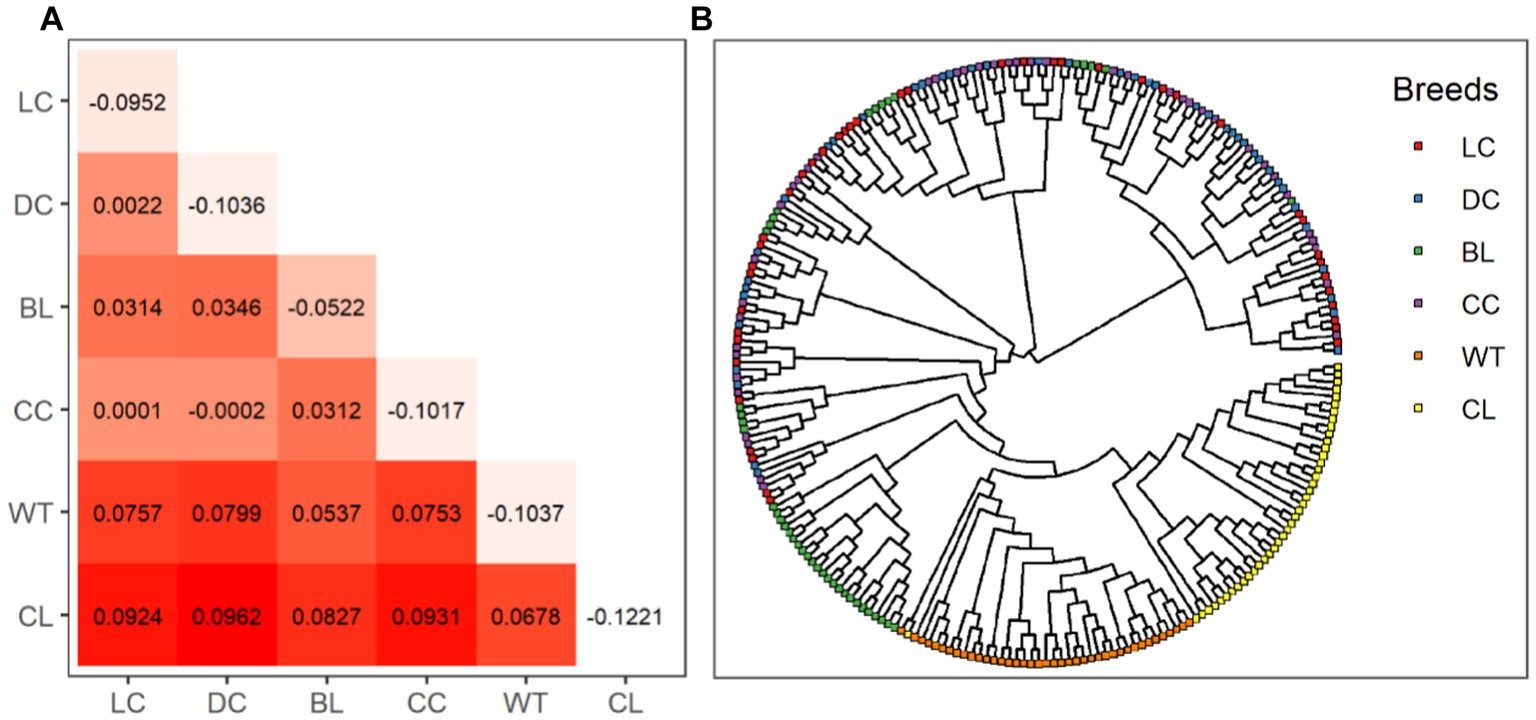
Figure 3. Genetic structures among the six coat color varieties of Rex rabbit. The matrix (A) shows pairwise Wright’s FST values in the lower triangle and FIS values in diagonal cells. The phylogenetic tree for all 250 animals is shown in (B). WT, White Rex; CL, Californian Rex; BL, Black Rex; CC, Chinchilla Rex; DC, Dark Chinchilla Rex; LC, Light Chinchilla Rex.
The association analysis results are shown in Figure 4. A total of 24 SNPs were detected as statistically significant; all of these were located within a 3.01-Mb genomic region on chromosome 4 (OCU4). After fitting the most significant SNP (OCU4:13,434,448; p = 1.31e-12) as a covariate, the association signal within this region noticeably decreased, but it still almost reached the threshold for significance (OCU4:11,344,946; p = 7.03e-07). The allelic frequencies of the two SNPs within each population are shown in Table 2; notably, OCU4:13,434,448 was completely fixed in the three non-Chinchilla populations. When both SNPs (OCU4:13,434,448 and OCU4:11,344,946) were simultaneously fitted as covariates, there was no longer any significant association signal within this region. Upon reverse-coding of the coat color, the association results did not change noticeably (Supplementary Figure S1).
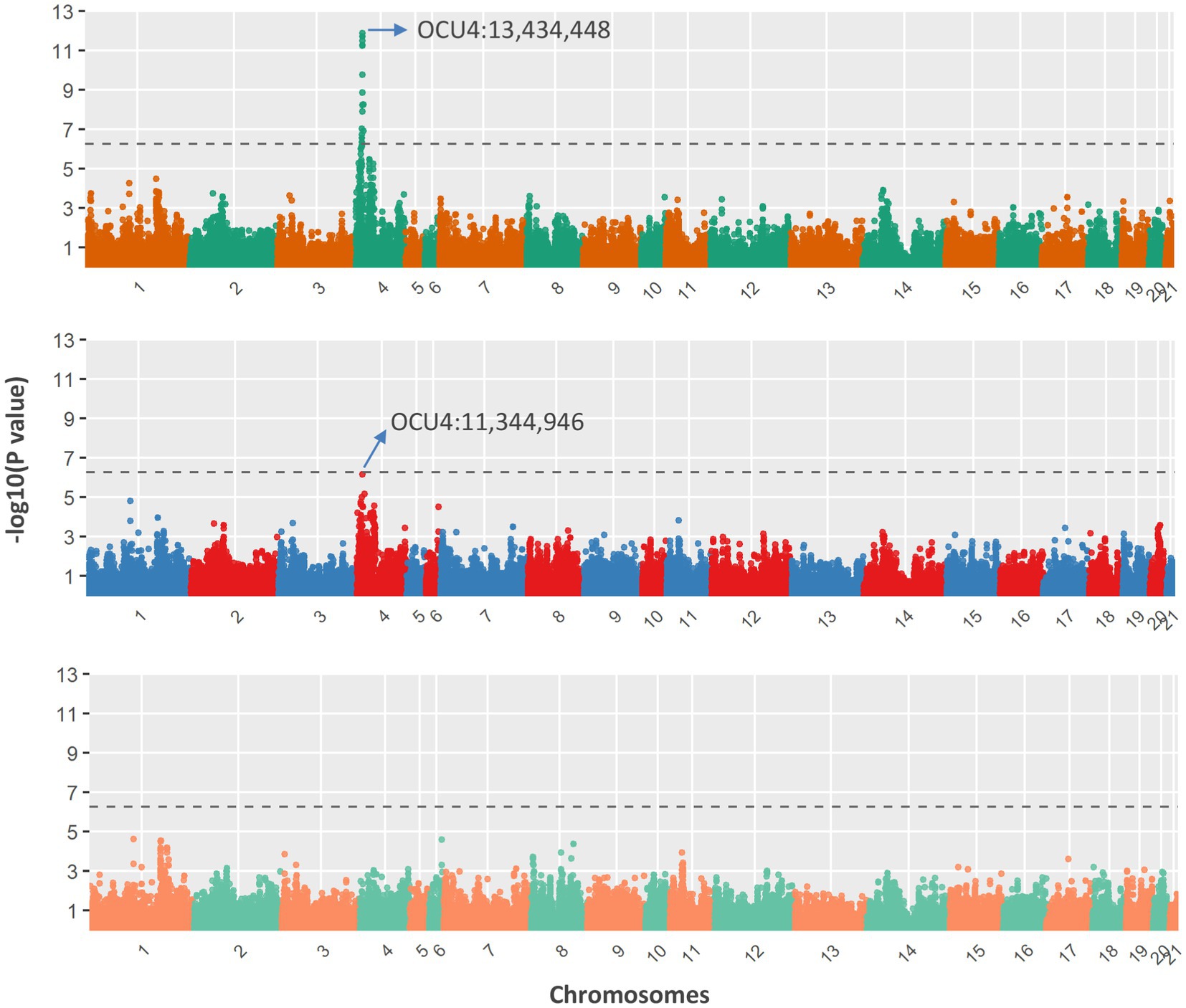
Figure 4. Genome-wide association with coat colors of Chinese Rex rabbits. After testing all SNP effects with a mixed linear model (top panel), the most significant SNP (OCU4:13,434,448) was fitted as a covariate for re-testing of SNP effects (middle panel). Both significant SNPs (OCU4:13,434,448 and OCU4:11,344,946) were simultaneously fitted as covariates for re-testing of SNP effects (bottom panel). The dashed line represents the genome-wide significance threshold.
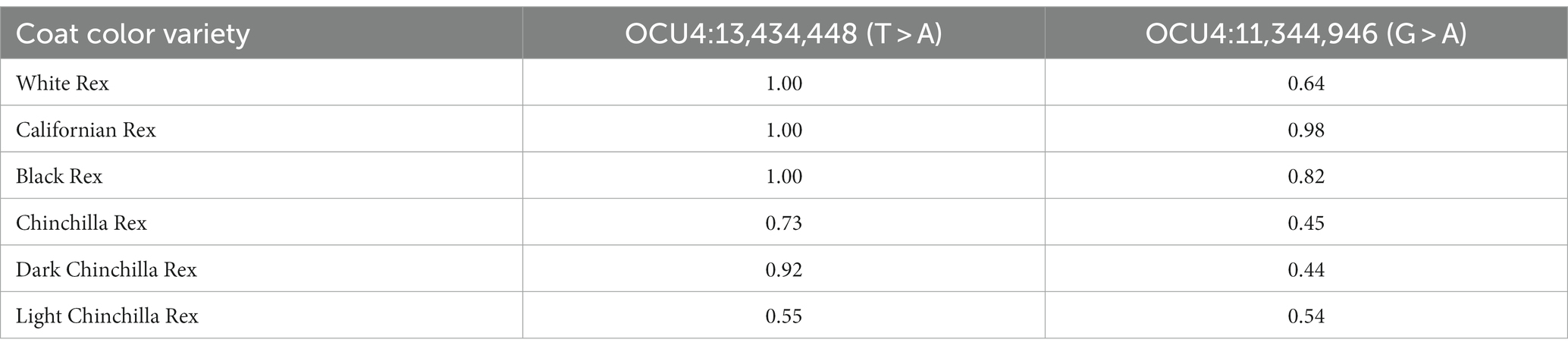
Table 2. Frequencies of reference alleles for the two (near-) significant SNPs.
We further investigated the annotated genes within this candidate genomic region (including 500 kb upstream of OCU4:11,344,946 and 500 kb downstream of OCU4:13,434,448), identifying 51 positional candidate genes in total. Among these genes, the well-studied ASIP gene, which is significantly associated with agouti coat color in rabbits, was located 150 kb upstream of the most significant SNP (OCU4:13,434,448). However, the second-most independently significant SNP (OCU4:11,344,946) was located at a large distance, 1.7 Mb upstream of the ASIP gene. Besides ASIP, no other known coat-color-associated gene was found within this candidate genomic region.
Coat color is an important phenotypic characteristic in domestic animals and has been directly subjected to artificial selection (41). It has also been proposed that hundreds of loci/genes play a role in affecting coat color, which (in combination with diverse selection preferences among humans) has ultimately resulted in considerable variation in a wide range of domestic animals (9). In addition to being farmed for the production of meat, wool, and fur, modern rabbits have been kept as a pet animal worldwide, with specific emphasis on the subjective selection of coat color. Therefore, rabbits could represent an ideal case for the identification of candidate genes and causal mutations affecting the expression of different coat colors. With the use of a cost-efficient method, genome-wide genetic variants could be discovered de novo through implementation high-throughput surveys, such as GWAS, for economically important traits and for the investigation of population genetic structures. In this study, we collected six coat color varieties of Rex rabbits raised in China and employed a high-throughput approach to successfully identify genome-wide and evenly distributed SNPs.
Coat color in mammals is generally considered to be a qualitative trait, although the phenotypic variations are genetically determined by polygenes. Therefore, the genome-wide scanning approach has been increasingly widely used to reveal coat-color-associated candidate genes and causal mutations. For example, Li et al. (42) genotyped ~50 k SNPs and employed a GWAS approach to identify three known pigmentation genes in sheep. In the Iranian Markhoz goat, a total of six genes have been identified as being associated with black, brown, and white coat colors using a GWAS approach (43). Based on the newly discovered SNPs in this study, we also conducted the first GWAS for coat color in Chinese Rex rabbits. Our results revealed that a 2.1-Mb genomic region (OCU4:11,344,946 – 13,434,448) containing ASIP, which has been shown in previous studies to be significantly associated with coat color (13), is also significantly associated with coat color in Rex rabbits. In a previous study of Rex rabbits with different coat colors, Yang et al. (44) found that ASIP had three alleles and was extensively expressed in all analyzed tissues. Recently, an 11-kb deletion spanning the promoter and first exon of ASIP has been suggested to be the most likely causal variant for the black-and-tan phenotype in rabbits (45). In the present study, we confirmed that ASIP is a putative causal gene affecting coat color in Chinese Rex rabbits. In the melanocytes of the hair follicle, ASIP encodes a paracrine signaling molecule that promotes the synthesis of pheomelanin (46). However, further studies are needed to explore whether the two candidate SNPs identified in this study are causal variants or not; although both of them are located more than 100 kb away from ASIP (upstream and downstream), possible roles for these SNPs in regulating gene expression cannot be excluded. Another possibility is that the two candidate SNPs are closely linked to the potential causal variant(s).
In addition to the discovery of coat-color-associated candidate genes, both genetic diversity and population structures among the six coat color varieties of Rex rabbits were investigated using the set of genome-wide SNPs generated in this study. Our results revealed the differential genetic diversity among these coat color varieties, with the highest genetic diversity observed in the Black Rex. This result is consistent with those presented in a previous report on genetic diversity patterns among 29 domestic and wild rabbit populations, examined using microsatellite markers (47). Liu et al. (25) also investigated population structure among eight Chinese rabbit breeds (not including the Rex rabbit), whose FST values were significantly higher than our estimates in this study; this may suggest that genetic differentiation among different populations of Rex rabbits is relatively low in comparison with other indigenous breeds. In accordance with this possibility, less inter-variety genetic differentiation was observed, with lower Fst values, than in former reports (47, 48). Meanwhile, our clustering analysis revealed that only individuals of the White and Californian Rex rabbit varieties could be clustered together and distinguished from individuals of other varieties. Overall, our results revealed using genome-wide SNP information that there is low genetic differentiation among different coat color varieties of Chinese Rex rabbits.
In this study, we discovered a genome-wide set of SNPs for Chinese Rex rabbits and used these to perform association analyses for the coat color phenotype. Our results revealed a single genomic region that is significantly associated with Rex coat color, and confirmed that the previously known coat-color-associated gene ASIP is a putative causal gene affecting coat color variation in Chinese Rex rabbits. Furthermore, low genetic differentiation was revealed among the six coat color varieties of Rex rabbit studied.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was approved by the Animal Care and Use Committee of Sichuan Academy of Grassland Sciences (YTS20B03). The study was conducted in accordance with the local legislation and institutional requirements.
KZ, HL, and S-YC: conceptualization. KZ and GW: formal analysis. LW, BW, XF, NL, ZY, WJ, and XG: resources. KZ: writing—original draft preparation. HL and S-YC: writing—review and editing. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Earmarked Fund for the China Agriculture Research System (CARS-43-A-3) and Science and Technology Department of Sichuan Province (2021YFYZ0033 and 2021YFYZ0009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1184764/full#supplementary-material
1. Carneiro, M, Afonso, S, Geraldes, A, Garreau, H, Bolet, G, Boucher, S, et al. The genetic structure of domestic rabbits. Mol Biol Evol. (2011) 28:1801–16. doi: 10.1093/molbev/msr003
2. Irving-Pease, EK, Frantz, LAF, Sykes, N, Callou, C, and Larson, G. Rabbits and the specious origins of domestication. Trends Ecol Evol. (2018) 33:149–52. doi: 10.1016/j.tree.2017.12.009
3. Frantz, LA, Bradley, DG, Larson, G, and Orlando, L. Animal domestication in the era of ancient genomics. Nat Rev Genet. (2020) 21:449–60. doi: 10.1038/s41576-020-0225-0
4. Lai, SJ, Liu, YP, Liu, YX, Li, XW, and Yao, YG. Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol Phylogenet Evol. (2006) 38:146–54. doi: 10.1016/j.ympev.2005.06.013
5. Liu, YP, Cao, SX, Chen, SY, Yao, YG, and Liu, TZ. Genetic diversity of Chinese domestic goat based on the mitochondrial DNA sequence variation. J Anim Breed Genet. (2009) 126:80–9. doi: 10.1111/j.1439-0388.2008.00737.x
6. Dorożyńska, K, and Maj, D. Rabbits-their domestication and molecular genetics of hair coat development and quality. Anim Genet. (2021) 52:10–20. doi: 10.1111/age.13024
7. Fontanesi, L. Rabbit genetic resources can provide several animal models to explain at the genetic level the diversity of morphological and physiological relevant traits. Appl Sci. (2021) 11:373. doi: 10.3390/app11010373
8. Diribarne, M, Mata, X, Chantry-Darmon, C, Vaiman, A, Auvinet, G, Bouet, S, et al. A deletion in exon 9 of the LIPH gene is responsible for the rex hair coat phenotype in rabbits (Oryctolagus cuniculus). PLoS One. (2011) 6:e19281. doi: 10.1371/journal.pone.0019281
9. Linderholm, A, and Larson, G. The role of humans in facilitating and sustaining coat colour variation in domestic animals. Semin Cell Dev Biol. (2013) 24:587–93. doi: 10.1016/j.semcdb.2013.03.015
10. Fontanesi, L, Scotti, E, Colombo, M, Beretti, F, Forestier, L, Dall’olio, S, et al. A composite six bp in-frame deletion in the melanocortin 1 receptor (MC1R) gene is associated with the Japanese brindling coat colour in rabbits (Oryctolagus cuniculus). BMC Genet. (2010) 11:59. doi: 10.1186/1471-2156-11-59
11. Fontanesi, L, Tazzoli, M, Beretti, F, and Russo, V. Mutations in the melanocortin 1 receptor (MC1R) gene are associated with coat colours in the domestic rabbit (Oryctolagus cuniculus). Anim Genet. (2006) 37:489–93. doi: 10.1111/j.1365-2052.2006.01494.x
12. Lu, D, Willard, D, Patel, IR, Kadwell, S, Overton, L, Kost, T, et al. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. (1994) 371:799–802. doi: 10.1038/371799a0
13. Fontanesi, L, Forestier, L, Allain, D, Scotti, E, Beretti, F, Deretz-Picoulet, S, et al. Characterization of the rabbit agouti signaling protein (ASIP) gene: transcripts and phylogenetic analyses and identification of the causative mutation of the nonagouti black coat colour. Genomics. (2010) 95:166–75. doi: 10.1016/j.ygeno.2009.11.003
14. Utzeri, VJ, Ribani, A, and Fontanesi, L. A premature stop codon in the TYRP 1 gene is associated with brown coat colour in the European rabbit (Oryctolagus cuniculus). Anim Genet. (2014) 45:600–3. doi: 10.1111/age.12171
15. Yang, N, Zhao, B, Hu, S, Bao, Z, Liu, M, Chen, Y, et al. Characterization of POU2F1 gene and its potential impact on the expression of genes involved in fur color formation in rex rabbit. Genes. (2020) 11:575. doi: 10.3390/genes11050575
16. Utzeri, VJ, Ribani, A, Schiavo, G, and Fontanesi, L. Describing variability in the tyrosinase (TYR) gene, the albino coat colour locus, in domestic and wild European rabbits. Ital J Anim Sci. (2021) 20:181–7. doi: 10.1080/1828051X.2021.1877574
17. Jia, X, Ding, P, Chen, S-Y, Zhao, S, Wang, J, and Lai, S-J. Analysis of MC1R, MITF, TYR, TYRP1, and MLPH genes polymorphism in four rabbit breeds with different coat colors. Animals. (2021) 11:81. doi: 10.3390/ani11010081
18. Fontanesi, L, Scotti, E, Allain, D, and Dall’olio, S. A frameshift mutation in the melanophilin gene causes the dilute coat colour in rabbit (Oryctolagus cuniculus) breeds. Anim Genet. (2014) 45:248–55. doi: 10.1111/age.12104
19. Davey, JW, Hohenlohe, PA, Etter, PD, Boone, JQ, Catchen, JM, and Blaxter, ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. (2011) 12:499–510. doi: 10.1038/nrg3012
20. Baird, NA, Etter, PD, Atwood, TS, Currey, MC, Shiver, AL, Lewis, ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. (2008) 3:e3376. doi: 10.1371/journal.pone.0003376
21. Li, Y, Li, B, Yang, M, Han, H, Chen, T, Wei, Q, et al. Genome-wide association study and fine mapping reveals candidate genes for birth weight of Yorkshire and Landrace pigs. Front Genet. (2020) 11:183. doi: 10.3389/fgene.2020.00183
22. Masharing, N, Sodhi, M, Chanda, D, Singh, I, Vivek, P, Tiwari, M, et al. ddRAD sequencing based genotyping of six indigenous dairy cattle breeds of India to infer existing genetic diversity and population structure. Sci Rep. (2023) 13:9379. doi: 10.1038/s41598-023-32418-6
23. Narum, SR, Buerkle, CA, Davey, JW, Miller, MR, and Hohenlohe, PA. Genotyping-by-sequencing in ecological and conservation genomics. Mol Ecol. (2013) 22:2841–7. doi: 10.1111/mec.12350
24. Xu, P, Xu, S, Wu, X, Tao, Y, Wang, B, Wang, S, et al. Population genomic analyses from low-coverage RAD-Seq data: a case study on the non-model cucurbit bottle gourd. Plant J. (2014) 77:430–42. doi: 10.1111/tpj.12370
25. Liu, C, Wang, S, Dong, X, Zhao, J, Ye, X, Gong, R, et al. Exploring the genomic resources and analysing the genetic diversity and population structure of Chinese indigenous rabbit breeds by RAD-seq. BMC Genomics. (2021) 22:573. doi: 10.1186/s12864-021-07833-6
26. Ren, A, Du, K, Jia, X, Yang, R, Wang, J, Chen, S-Y, et al. Genetic diversity and population structure of four Chinese rabbit breeds. PLoS One. (2019) 14:e0222503. doi: 10.1371/journal.pone.0222503
27. Chen, S, Zhou, Y, Chen, Y, and Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
28. Li, H, and Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
29. Mckenna, A, Hanna, M, Banks, E, Sivachenko, A, Cibulskis, K, Kernytsky, A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
30. Depristo, MA, Banks, E, Poplin, R, Garimella, KV, Maguire, JR, Hartl, C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
31. Van Der Auwera, GA, Carneiro, MO, Hartl, C, Poplin, R, Del Angel, G, Levy-Moonshine, A, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. (2013) 43:11.10.1. doi: 10.1002/0471250953.bi1110s43
32. Browning, BL, Zhou, Y, and Browning, SR. A one-penny imputed genome from next-generation reference panels. Am J Hum Genet. (2018) 103:338–48. doi: 10.1016/j.ajhg.2018.07.015
33. Wang, K, Li, M, and Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
34. Danecek, P, Auton, A, Abecasis, G, Albers, CA, Banks, E, Depristo, MA, et al. The variant call format and VCFtools. Bioinformatics. (2011) 27:2156–8. doi: 10.1093/bioinformatics/btr330
35. Chen, SY, Deng, F, Huang, Y, Li, C, Liu, L, Jia, X, et al. PopSc: computing toolkit for basic statistics of molecular population genetics simultaneously implemented in web-based calculator, Python and R. PLoS ONE. (2016) 11:e0165434. doi: 10.1371/journal.pone.0165434
36. Wright, S. The genetical structure of populations. Ann Eugenics. (1949) 15:323–54. doi: 10.1111/j.1469-1809.1949.tb02451.x
37. Kumar, S, Stecher, G, and Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
38. Yu, G, Smith, DK, Zhu, H, Guan, Y, and Lam, TTY. Ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. (2017) 8:28–36. doi: 10.1111/2041-210X.12628
39. Yang, J, Lee, SH, Goddard, ME, and Visscher, PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. (2011) 88:76–82. doi: 10.1016/j.ajhg.2010.11.011
40. Bland, JM, and Altman, DG. Multiple significance tests: the Bonferroni method. BMJ. (1995) 310:170. doi: 10.1136/bmj.310.6973.170
41. Wiener, P, and Wilkinson, S. Deciphering the genetic basis of animal domestication. Proc R Soc Lond B Biol Sci. (2011) 278:3161–70. doi: 10.1098/rspb.2011.1376
42. Li, MH, Tiirikka, T, and Kantanen, J. A genome-wide scan study identifies a single nucleotide substitution in ASIP associated with white versus non-white coat-colour variation in sheep (Ovis aries). Heredity. (2014) 112:122–31. doi: 10.1038/hdy.2013.83
43. Nazari-Ghadikolaei, A, Mehrabani-Yeganeh, H, Miarei-Aashtiani, SR, Staiger, EA, Rashidi, A, and Huson, HJ. Genome-wide association studies identify candidate genes for coat color and mohair traits in the Iranian Markhoz goat. Front Genet. (2018) 9:105. doi: 10.3389/fgene.2018.00105
44. Yang, C, Ge, J, Chen, S, Liu, Y, Chen, B, and Gu, Z. Sequence and gene expression analysis of the agouti signalling protein gene in rex rabbits with different coat colours. Ital J Anim Sci. (2015) 14:3810. doi: 10.4081/ijas.2015.3810
45. Letko, A, Ammann, B, Jagannathan, V, Henkel, J, Leuthard, F, Schelling, C, et al. A deletion spanning the promoter and first exon of the hair cycle-specific ASIP transcript isoform in black and tan rabbits. Anim Genet. (2020) 51:137–40. doi: 10.1111/age.12881
46. Del Bino, S, Duval, C, and Bernerd, F. Clinical and biological characterization of skin pigmentation diversity and its consequences on UV impact. Int J Mol Sci. (2018) 19:2668. doi: 10.3390/ijms19092668
47. Alves, JM, Carneiro, M, Afonso, S, Lopes, S, Garreau, H, Boucher, S, et al. Levels and patterns of genetic diversity and population structure in domestic rabbits. PLoS One. (2015) 10:e0144687. doi: 10.1371/journal.pone.0144687
Keywords: Rex rabbits, coat color, GWAS, RAD-seq, genome
Citation: Zhang K, Wang G, Wang L, Wen B, Fu X, Liu N, Yu Z, Jian W, Guo X, Liu H and Chen S-Y (2023) A genome-wide association study of coat color in Chinese Rex rabbits. Front. Vet. Sci. 10:1184764. doi: 10.3389/fvets.2023.1184764
Edited by:
Kefei Chen, Curtin University, AustraliaReviewed by:
Peipei Ma, Shanghai Jiao Tong University, ChinaCopyright © 2023 Zhang, Wang, Wang, Wen, Fu, Liu, Yu, Jian, Guo, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi-Yi Chen, c3ljaGVuc2F1QGdtYWlsLmNvbQ==; Y2hlbnN5QHNpY2F1LmVkdS5jbg==; Hanzhong Liu, aGFuemhvbmc2Njg4QHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.