
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 09 March 2023
Sec. Veterinary Infectious Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1140655
This article is part of the Research TopicGenomics And Proteomics Approaches for The Improved Biologics Against Infectious AgentsView all 6 articles
 Muhammad Nafees Ur Rehman1
Muhammad Nafees Ur Rehman1 Farman Ullah Dawar2
Farman Ullah Dawar2 Jifeng Zeng1,3Lixia Fan1Wei Feng1Mengqi Wang1Nuo Yang1Guiying Guo1
Jifeng Zeng1,3Lixia Fan1Wei Feng1Mengqi Wang1Nuo Yang1Guiying Guo1 Jiping Zheng1,3*
Jiping Zheng1,3*Edwardsiella tarda is a Gram-negative, facultative anaerobic rod-shaped bacterium and the causative agent of the systemic disease “Edwardsiellosis”. It is commonly prevalent in aquatic organisms with subsequent economic loss and hence has attracted increasing attention from researchers. In this study, we investigated the complete genome sequence of a highly virulent isolate Edwardsiella tarda SC002 isolated from hatchlings of the Siamese crocodile. The genome of SC002 consisted of one circular chromosome of length 3,662,469 bp with a 57.29% G+C content and four novel plasmids. A total of 3,734 protein-coding genes, 12 genomic islands (GIs), 7 prophages, 48 interspersed repeat sequences, 248 tandem repeat sequences, a CRISPR component with a total length of 175 bp, and 171 ncRNAs (tRNA = 106, sRNA = 37, and rRNA = 28) were predicted. In addition, the coding genes of assembled genome were successfully annotated against eight general databases (NR = 3,618/3,734, COG = 2,947/3,734, KEGG = 3,485/3,734, SWISS-PROT = 2,787/3,734, GO = 2,648/3,734, Pfam = 2,648/3,734, CAZy = 130/3,734, and TCDB = 637/3,734) and four pathogenicity-related databases (ARDB = 11/3,734, CARD = 142/3,734, PHI = 538/3,734, and VFDB = 315/3,734). Pan-genome and comparative genome analyses of the complete sequenced genomes confirmed their evolutionary relationships. The present study confirmed that E. tarda SC002 is a potential pathogen bearing a bulk amount of antibiotic resistance, virulence, and pathogenic genes and its open pan-genome may enhance its host range in the future.
Edwardsiella tarda is a versatile bacterium that infects a broad range of organisms such as fish, amphibians, reptiles, birds, and mammals (1–3). Edwardsiella tarda infections show a broad spectrum of clinical manifestations in various organisms. For instance, Miyazaki and Kaige (4) reported necrotic lesions on multiple organs of Tilapia (Tilapia nilotica), Koeboelkuti et al. (5) reported multiple subcutaneous abscesses on grass snakes, while White et al. (6) reported hemorrhagic enteritis of pelicans. Some studies also observed septicemia and bacteremia in farmed hatchling crocodiles (7, 8), and meningitis, gastroenteritis, and soft tissue infections in humans (9, 10). Particularly, E. tarda is one of those pathogens infecting both cultured and wild aquatic species (3, 11). Hence, aquatic life is usually prone to acquiring E. tarda infections, and this fact has been extensively focused on to protect the aquaculture industry.
Studies on phenotyping, genotyping, and whole-genome sequencing reveal E. tarda's diversity and pathogenicity (12, 13). The three genetically different taxa of E. tarda possess various degrees of pathogenicity in different hosts. Nearly all the fish pathogenic E. tarda isolates were acknowledged as Edwardsiella anguillarum and Edwardsiella piscicida (14, 15). Currently, the genus Edwardsiella of gamma Proteobacteria (16) consists of five species of Edwardsiella, namely, tarda, hoshinae, ictaluri, anguillarum, and piscicida. Particularly, E. tarda has the ability to attack epithelial cells (17, 18) and macrophages (19) where it multiplies intracellularly. It is one of the important steps of its pathogenesis by sabotaging the fish immunity and triggering systemic hemorrhagic septicemia (20). In addition, E. tarda uses numerous virulence factors such as secretion systems (T3SS and T6SS), adhesin, fimbrial adhesin-like protein (FimA), invasin (Inv), ferric uptake regulator (Fur), hemolysin (Hly), protease (PR), catalase (CAT), peroxidase (POD), and superoxide dismutase (SOD), siderophore, outer membrane proteins (OMPs), lipopolysaccharide (LPS), and two-component systems (TCSs) (3, 21).
From the economic point of view, the Siamese crocodile (Crocodylus siamensis) is a widely bred species in the South China region. A previous examination showed that immature Siamese crocodiles are vulnerable to genetic abnormalities, pilling up and asphyxia, mouth sores, skin diseases, gastropathy, and fungal and bacterial infections. Among bacterial infections, E. tarda was considered the main Edwardsiella species of septicemia among hatchlings and was controlled by the antibiotic “oxytetracycline” (7). However, later a new outbreak of Edwardsiellosis was reported and its subsequent investigation showed tetracycline-resistant E. tarda isolates (8). In the present study, from the aforementioned outbreak, a lethal isolate “E. tarda SC002” was subjected to whole-genome sequencing (8) and their genetic properties, virulence factors, invasive properties, and drug resistance were explored. The present study provides genomic insights into E. tarda, which will be indirectly helpful for candidate vaccine preparation against outbreaks of Edwardsiellosis in the aquaculture industry.
Edwardsiella tarda SC002 was previously isolated from hatchlings of a diseased Siamese crocodile (8) and grown in BHI broth at 28 C overnight. After further sub-culturing up to the 40th generation, the bacterial genomic DNA from the exponential growth phase was extracted by the SDS technique. The extracted DNA was then subjected to 1% agarose gel electrophoresis for detection purposes and measured by a Qubit® 2.0 Fluorometer (Thermo Scientific).
The whole genome was sequenced using the Nanopore PromethION platform and the Illumina NovaSeq PE150 platform (Novogene Beijing, China). For sample preparation, 1 μg of DNA per sample was used as input material. According to the manufacturer's guidelines, sequencing libraries were prepared using NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA). In brief, samples were sliced up to 350 bp by the sonication technique. The fragments were then end-polished, poly A-tailed, and ligated to a full-length adaptor for Illumina sequencing with further PCR amplification. Eventually, the amplified products were purified (AMPure XP system) and analyzed via Agilent2100 Bioanalyzer and quantified by real-time PCR. A hybrid assembly was generated using Unicycler software on short-read Illumina PE150 sequencing and long-read Nanopore sequencing data. The distribution of sequencing depth was counted, and the assembled sequence was specified as a chromosomal or a plasmid sequence according to size and alignment. The assembled sequence of whether it is a circular or a linear genome was also checked.
Various important components of the genome, including (CDSs) coding DNA sequences, repetitive sequences, non-coding RNAs (ncRNAs), genomic islands (GIs), transposons, prophages, and clustered regularly interspaced short palindromic repeat (CRISPR) elements, were predicted. The GeneMarkS package was used for retrieval of the associated coding gene.1 The RepeatMasker2 for the prediction of interspersed repeat sequences and the Tandem repeat finder for the detection of tandem repeats (22) were applied. tRNAscan-SE and rRNAmmer were used for the prediction of transfer RNA (tRNA) and ribosomal RNA (rRNA) genes, respectively (23, 24). The conserved small RNAs (snRNAs) were predicted by BLAST against the Rfam database (25). The IslandPath-DIOMB and the transposonPSI programs were applied to predict GIs, and transposons based on BLAST sequences, respectively (26). Furthermore, PHAST and CRISPRFinder were used for the identification of prophages and CRISPR elements, respectively.3
Some important databases for gene functional annotation, such as Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), Non-Redundant Protein Database (NR), Transporter Classification Database (TCDB), and SWISS-PROT databases, were used. A whole-genome BLAST with parameters such as E-value < 1e-5 and minimal identity > 40% was performed against the aforementioned seven databases. The secretory proteins and membrane proteins were predicted by the SignalP database and TMHMM Server, respectively.4 5 In addition, type secretory proteins (types I–VII) were predicted by the EffectiveT3 tool.6 Gene clusters for secondary metabolism were evaluated by the antiSMASH tool (28). For the assessment of pathogenicity, the Pathogen–Host Interactions (PHI), the Virulence Factors Database (VFDB), and the Antibiotic Resistance Genes Database (ARDB) were used. Carbohydrate-active enzymes were identified by the Carbohydrate-Active enZYmes Database (CAZy).
A genomewide comparison of the genus Edwardsiella members and species from related genera in the family Enterobacteriaceae, including the Escherichia coli (E. coli) strain 97-3250 (NZ_CP027599) and the Salmonella enterica strain FDAARGOS_878 (NZ_CP065718), was performed by Mauve genome alignment using the MegAlign_17 tool. A phylogenetic tree was constructed using the maximum-likelihood (ML) technique with the statistical support of 1,000 bootstrap replicates. Further analysis for carrying out comparative genomics was based on average nucleotide identity (ANI) values. The ANI values between the two genomes were calculated by the ANI calculator of Kostas lab (29) and visualized by a heatmap generated by the package ggplot2 in the statistical software R (version 2021-09.1+372). PGAweb, an online server, was used to estimate E. tarda pan-genome; however, incomplete genomes were excluded from the analysis (30).
Edwardsiella tarda SC002 is comprised of a circular chromosome of 3,662,469 bp with 57.29% of G+C content (Figure 1). The predicted genetic components are 3,734 genes, 12 genomic islands (GIs), a CRISPR component with a total length of 175 bp, and 106, 37, and 28 tRNAs, sRNAs, and rRNAs, respectively. Furthermore, 7 prophages (4 on chromosome and 3 on different plasmids), 48 interspersed repeat sequences (including LTR and one unknown repeat), and 248 tandem repeat sequences (including TRF, minisatellite, and microsatellite DNA) were predicted (Table 1). In addition, the predicted 3,734 coding DNA sequences (CDSs) have an average length of 916 bp, representing 82.83% of the total genome.
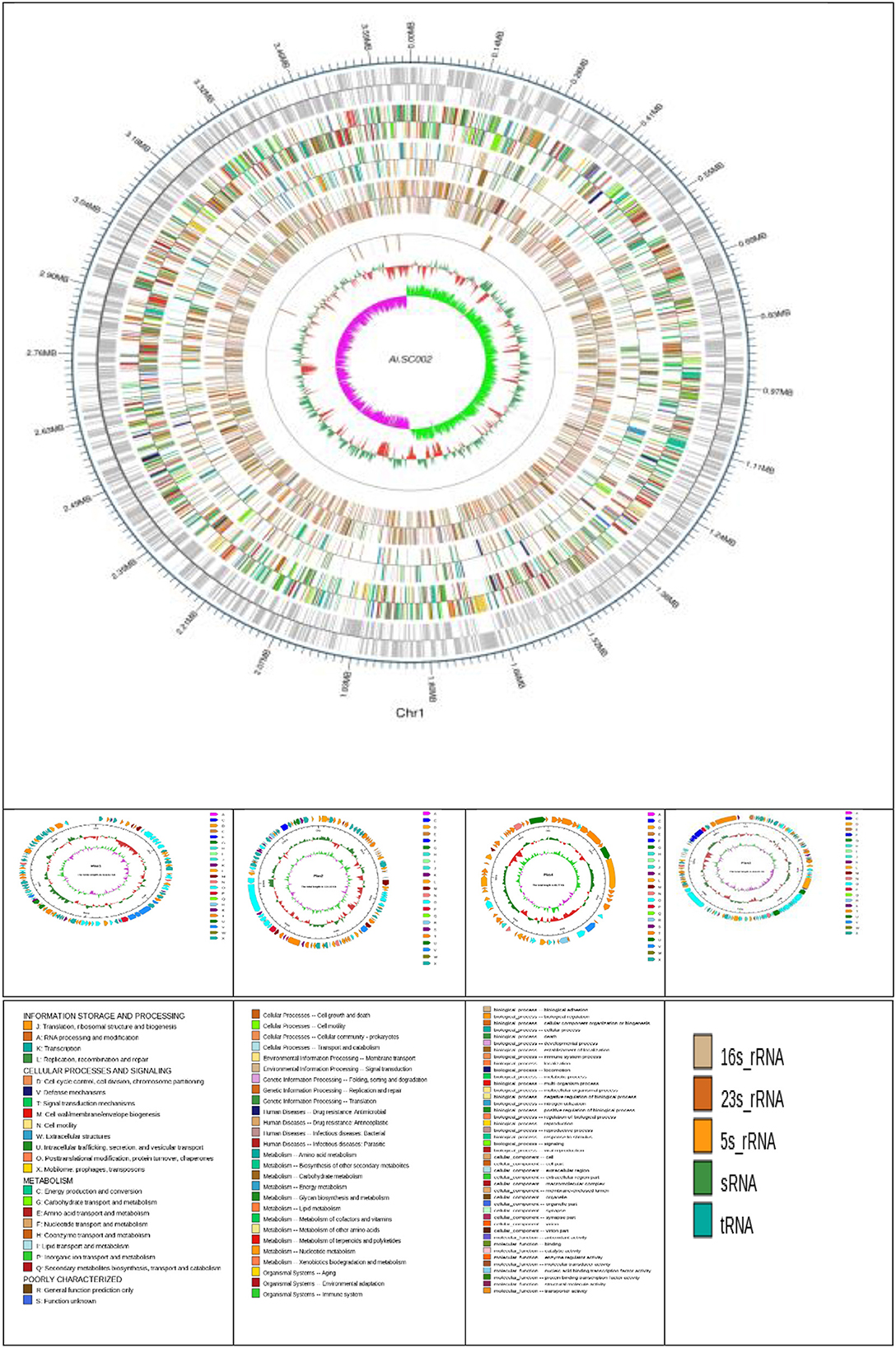
Figure 1. Atlas of the chromosome and four plasmids of Edwardsiella tarda SC002. From the outside to the center: genome sequence coordinates, genes encoded on forward and reverse strands, followed by COG annotation, KEGG annotation, GO annotation, ncRNA, GC content, and GC skew (G–C/G+C) GC/G+C.
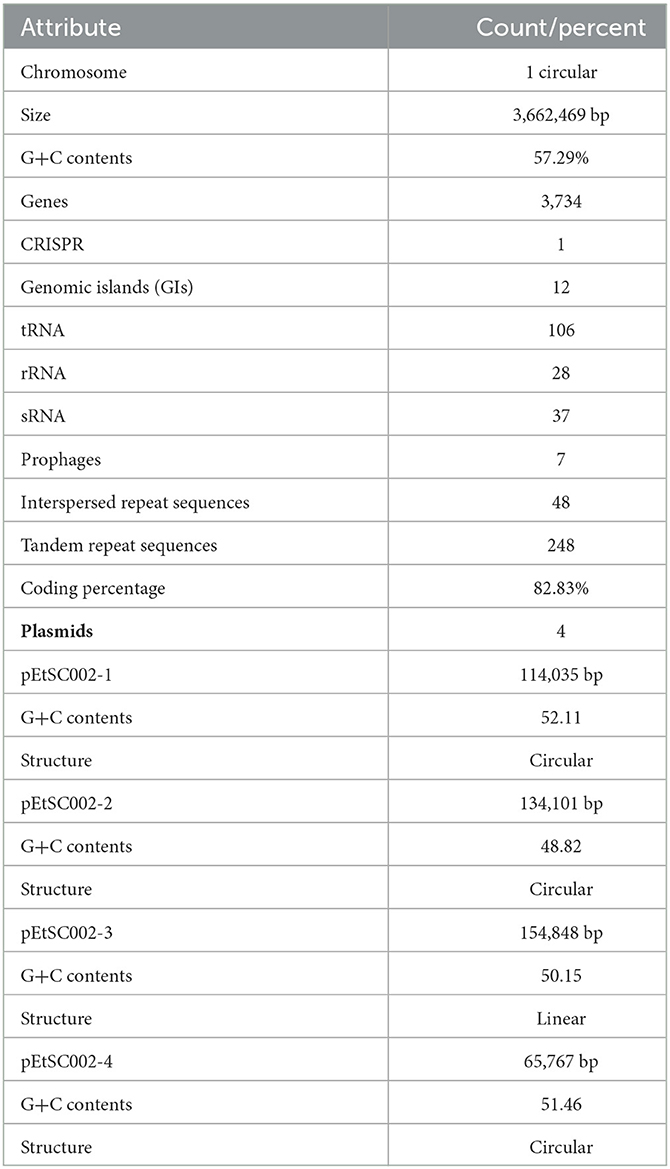
Table 1. Overall genomic features of the Edwardsiella tarda SC002.
Four novel plasmids, namely pEtSC002-1, pEtSC002-2, pEtSC002-3, and pEtSC002-4, were observed, in which pEtSC002-1, pEtSC002-2, and pEtSC002-4 plasmids were circular with the sizes of 114,035 bp, 134,101 bp, and 65,767 bp, respectively, and pEtSC002-3 plasmid was linear with the size of 154,848 bp (Figure 1).
pEtSC002-1 encodes various Tra proteins (M, X, and Y domains), replication initiation protein DnaC, and plasmid partition proteins (ParA and ParM), which show its resemblance to IncP plasmid. Additionally, the most abundant antibiotic resistance genes (blaTEM, qnrS1, strB, sul2, aph3, sul1, cml, and tetA) were found in the pEtSC002-1 plasmid.
pEtSC002-2 encodes a few Tra proteins (M, T, I, and Y domains), which indicate that it is a conjugative plasmid. However, plasmid partition proteins, such as SopA, SopB, and ParB, including plasmid replication initiator protein IncFII RepA, were encoded, indicating that it is an IncFII plasmid. There were no antibiotic resistance genes found in this plasmid; however, bacteriocin and colicin V were encoded which can act as efflux pumps conferring antibiotic resistance.
pEtSC002-3 was more unique than other plasmids observed in this study, a non-circular plasmid and one that encodes the maximum abundant number of Tra proteins (A, B, C, D, E, F, G, H, I, K, L, M, N, Q, T, U, V, W, X, and Y), indicating that it has a strong conjugation potential. Furthermore, plasmid partition proteins, such as ParA, ParB, and ParM, and a plasmid replication protein, DnaC of the IncFII RepA family, were encoded, indicating that it is also an IncFII plasmid.
pEtSC002-4 found in this study was a circular conjugative plasmid with various Tra (A, B, C, D, E, F, G, H, I, K, L, N, Q, T, U, and V) and Trb (B, C) proteins. The plasmid replication IncFII RepA protein family and ParA protein were found, indicating that it is an IncFII plasmid.
The G+C content shows high variability in the E. tarda SC002 genome (Figure 1). A significant part (213,631 bp; 5.17%) of the genome (4,131,220 bp) is comprised of genomic islands (GIs, n = 12), representing a complex structure of the genome. In addition, an adequate amount (n = 48) of interspersed repeat sequences (0.138% of 4,131,220 bp) and a variable number of tandem repeats (VNTRs) (n = 151, 0.435% of 4,131,220 bp) were detected. The GIs usually encoded hypothetical proteins, followed by the transposase, the integrase, the transcription regulator, transporter proteins, plasmid partition proteins, the terminase, phage proteins, and others (Table 2). The seven prophages (5.68% of 4,131,220 bp) were discerned, of which four were on a chromosome (average GC% = 53.19) and three were on two plasmids (average GC% = 50.56). Furthermore, prophages 4, 5, and 6 overlapped on several CDS regions of GIs 1, 4, and 5, respectively, on the chromosome. A valuable amount of ncRNA (1.3% of 4,131,220 bp) was observed as tRNA (n = 106), rRNA (n = 28), and sRNA (n = 37). A CRISPR component of 175 bp was found on plus strand of chromosome with 41 bp spacer and 31 bp direct repeat sequences (Table 1).
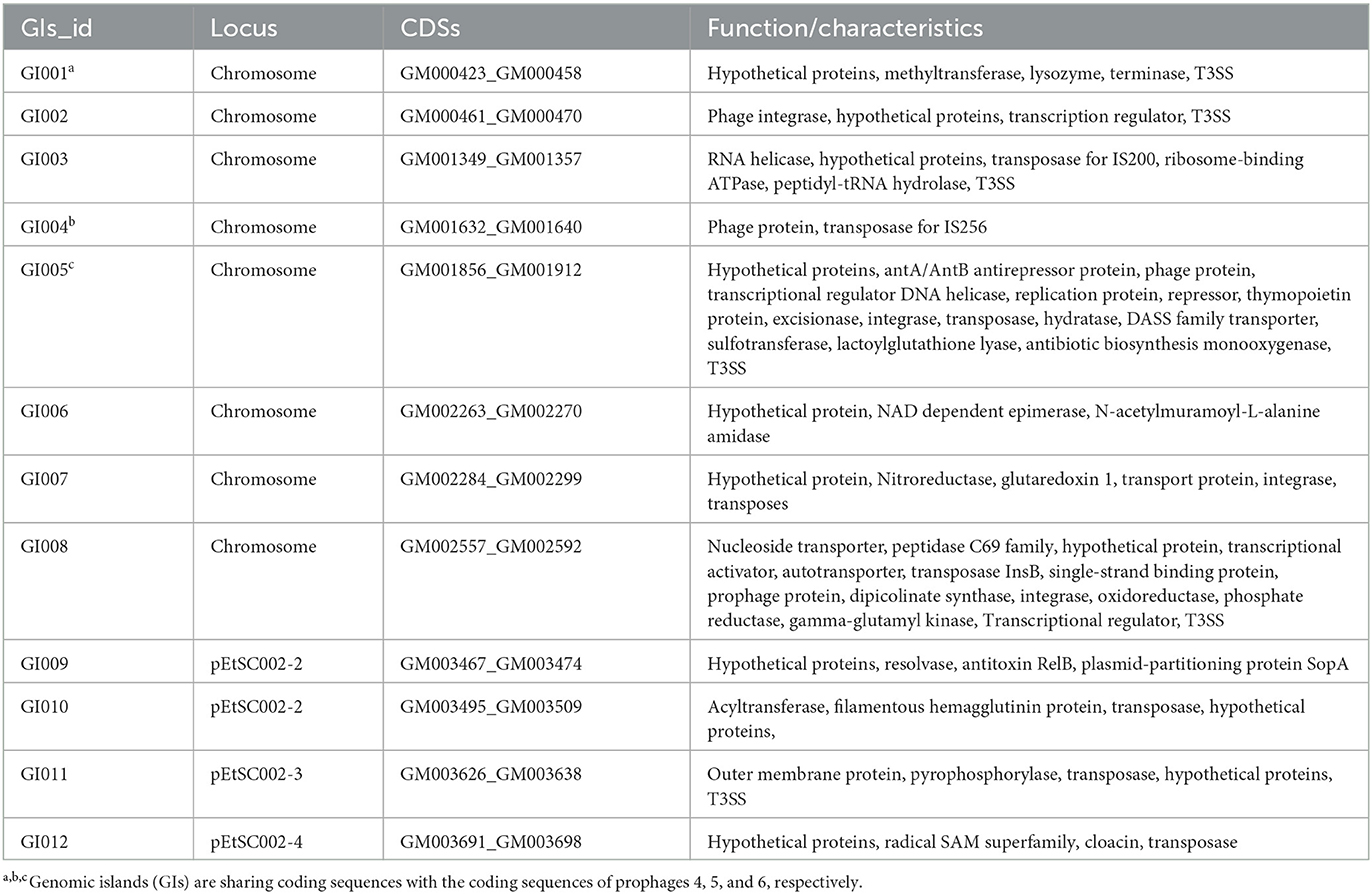
Table 2. Overview of the genomic islands in the Edwardsiella tarda SC002.
Gene functions were predicted by various databases such as COG, KEGG, GO, NR, Pfam, CAZy, and TCDB. The most abundant gene functions were predicted by NR (n = 3,618/3,734), followed by KEGG (n = 3,485/3,734), COG (n = 2,947/3,734), GO (2,648/3,734), and Pfam (2,648/3,734), TCDB (637/3,734), and CAZy (130/3,734).
Based on the orthology examination, the COG database predicted 2,947 genes (78.92%), and these genes were distributed into 23 functional categories (Figure 2A). According to the COG categorization, the five most rich annotated functions were amino acid transport and metabolism (284 genes), carbohydrate transport and metabolism (248 genes), energy production and conversion (232 genes), transcription (226 genes), and translation, and ribosomal structure and biogenesis (225 genes). Furthermore, 166 hypothetical genes were discerned, which may need to be explored by further studies. In addition, 16 genes for extracellular structures, 166 genes each for transposons and prophages, and one gene were found for RNA processing and modification (Supplementary Tables S1, S2; Figure 2A).

Figure 2. Gene functional annotation of the Edwardsiella tarda genome. (A) COG annotation distribution. (B) KEGG annotation distribution. (C) GO annotation distribution. (D) NR annotation distribution. (E) CAZy annotation distribution. (F) TCDB annotation distribution. (G) TCDB annotation distribution. (H) PHI annotation distribution.
The KEGG database annotation represents a total of 2,510 genes. Followed by the classification of the KEGG orthology (KO) database, the annotated genes were distributed into six categories: metabolism (1,663), environmental information processing (317), genetic information processing (190), cellular processes (175), human diseases (126), and organismal systems (39) (Figure 2B). According to the orthology results, the most populated class was represented by metabolic pathways, global and overview maps from the “Metabolism” category, with 587 genes. The second abundant class was the biosynthesis of secondary metabolites from the “Metabolism” category (267 genes), followed by microbial metabolism in diverse environments (184 genes) and biosynthesis of antibiotics (175 genes) from the “Metabolism” category. In addition, two-component systems (TCSs) and ABC transporters were discerned with 138 and 123 genes, respectively, from the “Environmental Information Processing” category (Supplementary Tables S1, S2).
According to GO analysis, a total of 2,648 protein-encoding genes were annotated (Figure 2C). The annotated genes were mainly categorized into 43 subfunctional items. These were further distributed into three major categories: biological process (22 subfunctions), cellular component (12 subfunctions), and molecular function (9 subfunctions). However, a total of 1,525 different genes, followed by 1,500, and 532 belonging to the cellular process, metabolic process, and localization, respectively, of the biological process category were annotated (Supplementary Tables S1, S2).
According to the NR database, a total of 3,618 genes were annotated within 80 different bacterial species. The top 20 species in which E. tarda have 3,087 genes were followed by Edwardsiella species, in which Edwardsiella has 110 genes, Escherichia coli (NZ_CP027599) has 52 genes, Edwardsiella hoshinae (E. hoshinae) (NZ_CP065626) has 48 genes, and Salmonella enterica (CP006631) has 47 genes, respectively (Supplementary Tables S1, S2; Figure 2D).
The Pfam database analysis annotated a total of 2,648 genes, which are grouped into 294 clans. The five most abundant gene containing clans were cl0023.33 followed by cl0063.24, clan0123.17, cl0344.3, and cl0015.19 with 1,041, 675, 514, 361, and 92 genes, respectively (Supplementary Tables S1, S2).
The CAZy analysis shows that 130 genes are matching with 59 CAZy-family genes. The CAZy families GT2, GH23, and CMB50 have the most abundant number of genes, which were 14, 13, and 12, respectively. However, the CAZy class annotation shows that GH, GT, and CBM have 55, 50, and 22 matching genes (Supplementary Tables S1, S2; Figure 2E).
Transporter Classification Database (TCDB) annotated a total of 637 genes and categorized them into 7 classes and 18 subclasses. Class 3 has the highest number of genes followed by class 2 and class 1 with 209, 202, and 89 genes, respectively (Figure 2F). The subclassification revealed that the highest number of genes belong to subclass 2.A, followed by 3.A, 1.B, 9.B, and 3.D with 198, 167, 50, 50, and 42 genes, respectively (Supplementary Tables S1, S2; Figure 2G).
To evaluate the pathogenicity of SC002, databases, such as ARDB, CARD, PHI, and VFDB, were searched. According to Antibiotic Resistance Genes Database (ARDB) analysis, 11 genes were annotated, of which two each of tetracycline, streptomycin, and sulfonamide resistance and 1 each of aminoglycosides, bacitracin, penicillin, fluoroquinolone, and chloramphenicol resistance were found (Supplementary Table S3). The Comprehensive Antibiotic Resistance Database (CARD) analysis annotated a total of 142 genes, in which macB was the most prevalent, with eight genes followed by five evgS and four each of gadX and TaeA genes (Supplementary Tables S1, S3). The pathogen–host interactions database (PHI) annotates a total of 538 genes categorized into six classes, of which the most populated one was “reduced virulence”, followed by “unaffected pathogenicity” and “hypervirulence” with 303, 137, and 36 genes, respectively (Figure 2H). The Virulence Factor Database (VFDB) detected a total of 315 genes, and the most common genes were related to virulence factors such as “flagellar protein,” “LOS,” and “capsule” (Supplementary Tables S1, S3).
A gene cluster is a pool of genes on DNA encoding similar proteins. These proteins collectively perform a generalized function. Gene cluster size is inconsistent, from a few genes to some 100 genes (31). For secondary metabolite evaluation, there are five clusters encoded by the genome. Cluster 1 was identified as homoserine lactone that consists of 17 genes, Cluster 2 was betalactone with 18 genes, Cluster 3 was bacteriocin with 11 genes, Cluster 4 was thiopeptide with 21 genes, and Cluster 5 was siderophore with 11 genes (Figure 3). Furthermore, Clusters 1–4 were encoded by chromosome, while Cluster 5 was present on pEtSC002-4 (Table 3). All of the aforementioned clusters have 100% homology with reference species, except Cluster 4, which has 33% homology. Upon secretory protein analysis, 209 gene-encoding proteins were predicted as signaling proteins. Furthermore, 129 genes were predicted for encoding T3SS proteins, of which 16 genes were encoded by all the plasmids.
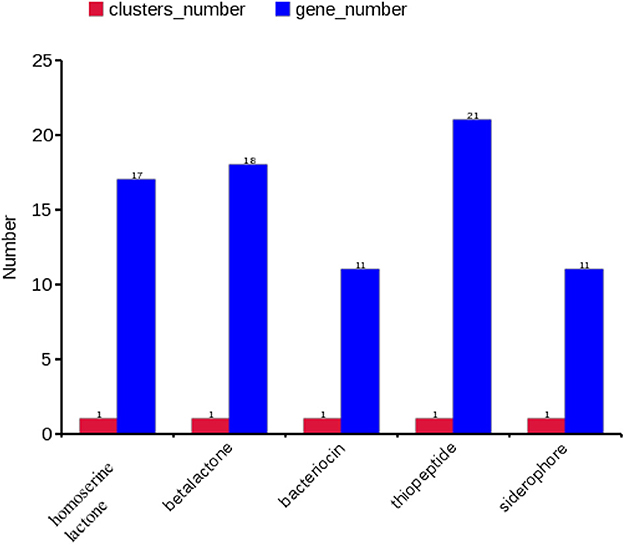
Figure 3. Secretory protein annotation of an SC002 genome. Five clusters with their respective numbers of genes.
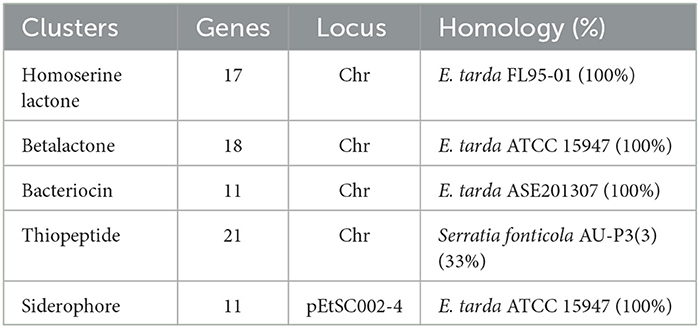
Table 3. Effector/secretory protein profiling encoded by the genome.
For evolutionary relationships, SC002 and 10 strains of the genus Edwardsiella including two outgroup organisms were compared. A phylogenetic tree was constructed based on conserved aligned blocks, which shows a clearly divergent lineage. Four E. tarda strains were clustered together and one strain of E. tarda was branched with E. piscicida species (previously known as E. tarda) (Figure 4A). To further explore the evolutionary relationships, ANI values were discerned. The ANI values were compared to estimate genomic differences and relatedness between two genomes. According to our results (Figure 4B), the genomes of four E. tarda strains shared ANI values ranging from 99.34 to 99.45%. These values are above the threshold of 94–96% identity, which is usually considered a speciation edge (32). However, the strain ET_1 shared ANI values of 83% with E. tarda strains and 99.85% with E. piscicida strains (formerly known as E. tarda), which clearly shows that it is a distinct species currently classified as E. tarda.
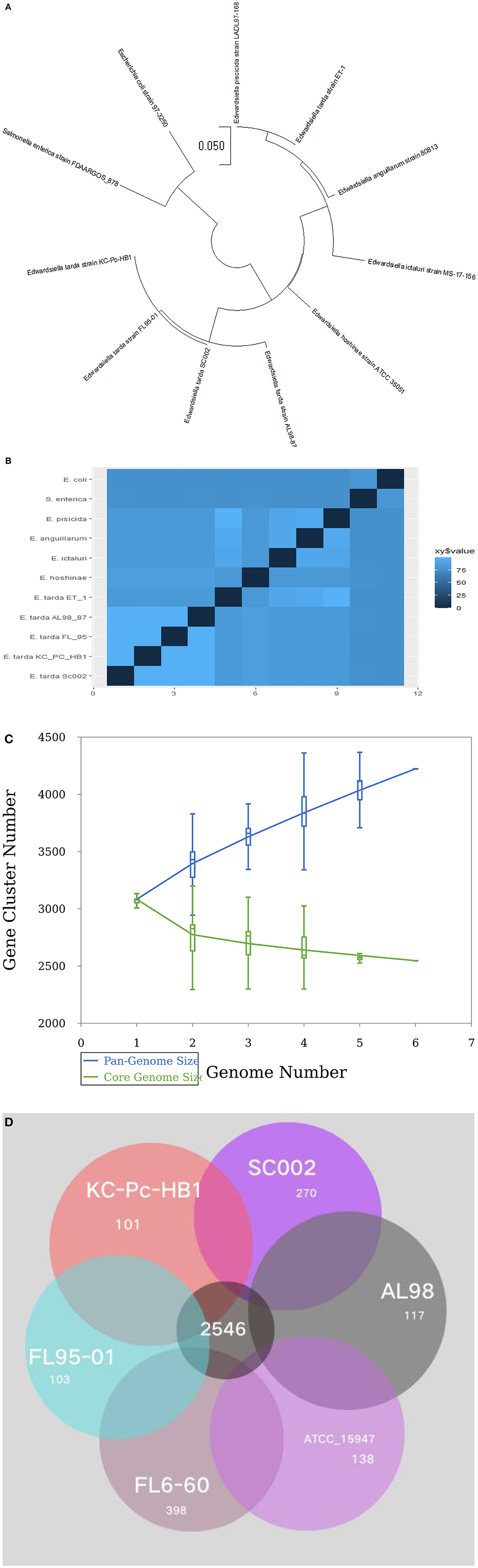
Figure 4. Phylogenetic and comparative genomics analysis: (A) A total of 11 genomes of Edwardsiella species, including two outgroup organisms—Salmonella enterica and Escherichia coli, were analyzed by the Mauve algorithm and a phylogenetic tree was constructed based on conserved aligned blocks of complete genomes. (B) A heatmap of 11 genomes based on the average nucleotide identity (ANI) values between two species. (C) A line chart represents an open pan-genome of six Edwardsiella tarda strains. (D) Venn diagram of core and unique genes shared by a comparison of six known E. tarda strains.
Pan-genome analysis for six E. tarda strains (Figure 4C), shows that the species have an open pan-genome. It is comprised of 4,222 cluster genes, distributed into 2,546 core cluster genes (60%), 558 accessory genes (13.2%), and 1,118 unique genes (26.5%). A number of variable genes ranging from 101–398 were recognized in the unique genes. The maximum and minimum numbers of unique genes were observed in the E. tarda strains of FL6-60 (NC_017309) and KC_Pc_HB1 (NZ_CP023706), respectively (Figure 4D). Notably, our strain SC002 contained 270 unique genes (Figure 4D) and shares a higher number of core (conserved) genes with strains ATCC_15947 (2639), FL95-01 (2637), FL6-60 (2634), AL98 (2633), and KC_PC_HB1 (2631). Collectively, these results indicated that the genome structure of the six E. tarda strains has high conservation and diversity.
Edwardsiellosis is a global issue for the aquaculture industry in the current era. Therefore, an understanding regarding the genetic makeup of Edwardsiella is needed that may be helpful in finding ways to overcome their outbreak in the aquatic species. Accordingly, this study isolated E. tarda SC002, a virulent strain from Siamese crocodile hatchlings in the Hainan Province of China, and their whole genome was sequenced to determine its pathogenicity, virulence, and niche-related properties. The SC002 genome has relatively higher coding and non-coding genes (tRNA, rRNA, and sRNA) than the other sequenced Edwardsiella species (such as KC_Pc_HB1, C07-087, and FL95-01) and is relevant to the rapid growth of the bacterium (33). The 5S rRNA gene was found to double in our study, which is similar to that mentioned in a previous study (34). Previously, the isolate SC002 was clustered with ATCC 15945 (8), indicating that it is a genotype of mammalian origin (13, 35). The G+C content of SC002 was 57.29%, which is similar to a previous study where the G+C content of Edwardsiella genomes ranges from 56.8 to 59.80% (36).
Unlike the published Edwardsiella complete genomes having one or two plasmids, the SC002 genome contained four novel plasmids (34, 37–39). Subsequently, BLAST analysis shows that the pEtSC002-1 was identical to the S. enterica plasmid (NZ_CP037959.1), while pEtSC002-2, pEtSC002-3, and pEtSC002-4 were identical to the E. tarda KC_PC_HB1 plasmid (NZ_CP023707.1) (40, 41). The antibiotic resistance determinants were only observed on pEtSC002-1 where the resistance gene patterns (sul1, sul2, cml, tetA, aph33, and aph6) were similar to those of EIB202 plasmid resistome (33). The β-lactam and fluoroquinolone resistance genes were additional to pEtSC002-1, indicating that this plasmid is more virulent than the latter plasmid. Three T3SS genes were annotated on pEtSC002-1, whereas plasmid pEIB202 encodes an incomplete set of T4SS genes which may play a role in horizontal gene transfer (HGT) (34). However, the detection of two prophages on pEtSC002-1 may enhance its capability for producing genetic exchange among bacteria. Furthermore, all plasmids share conjugative plasmid features and encode for Tra, replication initiation, and plasmid partition proteins, which shows that these plasmids might belong to the IncP plasmid that was capable of undergoing replication and stable inheritance in a wide variety of Gram-negative bacteria (42).
According to our study, 12 genomic islands (GIs) were detected in SC002, whereas 11 in E. tarda FL6-60, 24 in E. piscicida, and 31 in E. ictaluri, which shows that the taxawise GIs are frequent as E. tarda < E. piscicida < E. ictaluri (34). However, 5 out of 12 GIs of SC002 encode for T3SS proteins, whereas in EIB202, two GIs: GI7 and GI17 encode for T3SS and T6SS, respectively (34), which shows that these GIs contribute to the pathogenicity of SC002.
The analysis of the SC002 genome suggests that it has the ability to deal with environmental changes due to the presence of gene regulation systems, such as two-component systems (TCSs) and quorum sensing systems. In the genome of SC002, a total of 138 TCS genes and 50 quorum-sensing genes were detected, which were more than those of the previously reported E. piscicida (34). Usually, TCS genes are adjacent; however, similar to a previous study (34), the barA/UvrY, arcA/B, and cheA/B/Y, TCSs in the present study are unusually organized. The presence of some well-known TCSs explored previously in E. tarda, such as PhoP/Q (43) and QseB/C (44), might have a role in the pathogenicity and virulence of SC002.
Edwardsiella tarda is capable of surviving and persisting intracellularly in phagocytes followed by systemic infection (45). To resist phagocyte-mediated killing, various essential strategies, such as enzyme production including CAT, POD, and SOD, have been implicated by E. tarda to neutralize reactive oxygen species (ROS), while TTSS and T6SS contribute to invasion and subversion of the host cells (46). In the present study, 129 T3SS genes were predicted; however, in a previously reported E. tarda KC-Pc-HB1, no T3SS/T6SS genes were detected (41), indicating that SC002 is a more virulent strain.
A previous study revealed the mechanism of E. tarda host adaptation and interspecies discrepancies within the species of the genus Edwardsiella (35). Phylogenomics and pairwise ANI comparison show that the E. tarda strain ET_1 was clustered to E. piscicida, calling for reconsideration of the genus Edwardsiella. Horizontal gene transfer (HGT) in E. tarda was responsible for the acquisition of the locus of enterocyte effacement (LEE) (47). Host specificity in Staphylococcus aureus was associated with host-adaptive gene transformation via mobile genetic elements (MGEs) (48). According to the present study, E. tarda contains an open pan-genome, which is congruent with a previous study (49). Consequently, the pathogen has a chance to expend its host niches in the future. Further study is needed to experimentally prove the function of unique genes in host specificity and evolution. E. tarda is a well-known aquatic pathogen affecting the aquaculture industry worldwide. Its target is not only to attack high-value fish species such as turbot but also to inflict injuries on birds, reptiles, and mammals. Therefore, strict measures to control the pathogen and proper vaccine candidate development are needed.
The whole-genome sequence of SC002 was characterized as highly virulent and multidrug resistant (MDR). A thorough examination of the genome sequence shows that the pathogen has an array of drug-resistant genes on pEtSC002-1. The conjugative and prophage determinants of the observed plasmids further considered that the contents of the genome are partly structured in various aquatic ecological niches during its life cycle. An adequate amount of TCSs' quorum sensing and T3SS genes that showed the diverse nature of the pathogen were confirmed. By exploring the understanding of the pathogenesis, virulence, and host specificity of the organism and by adopting the approach of “reverse vaccinology”, this study lays the foundation for candidate vaccine development.
The complete genome sequence of the Edwardsiella tarda SC002 chromosome and the plasmids pEtSC002-1, pEtSC002-2, pEtSC002-3, and pEtSC002-4 have been deposited to the GenBank under accession numbers CP116675, CP116676, CP116677, CP116678, and CP116679 respectively.
The animal study was reviewed and approved by Hainan University Laboratory Animal Care Committee (HULACA180703). Written informed consent was obtained from the owners for the participation of their animals in this study.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study received funding from the National Natural Science Foundation of China (Grants 32060788 to JZe and 32060131 to JZh) and the Hainan Provincial Natural Science Foundation of China (Grants 321MS007 to GG, 821MS029 to NY, 821RC1052 to LF, and 2019RC084 to JZh).
We acknowledge the kind support of our laboratory members for performing E. tarda genome sequencing data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1140655/full#supplementary-material
2. ^http://www.repeatmasker.org/
3. ^http://phast.wishartlab.com (27).
4. ^https://services.healthtech.dtu.dk/service.php?SignalP-5.0
5. ^https://services.healthtech.dtu.dk/service.php?TMHMM-2.0
1. Michael J, Abbott SL. Infections associated with the genus Edwardsiella: the role of Edwardsiella tarda in human disease. Clin Infect Dis. (1993) 17:742–8. doi: 10.1093/clinids/17.4.742
2. Yousuf R, How S, Amran M, Hla K, Shah A, Francis A. Edwardsiella tarda septicemia with underlying multiple liver abscesses. Malays J Pathol. (2006) 28:49–53.
3. Park SB, Aoki T, Jung TS. Pathogenesis of and strategies for preventing Edwardsiella tarda infection in fish. Vet Res. (2012) 43:1–11. doi: 10.1186/1297-9716-43-67
4. Miyazaki T, Kaige N. Comparative histopathology of edwardsiellosis in fishes. Fish Pathol. (1985) 20:219–27. doi: 10.3147/jsfp.20.219
5. Koeboelkuti LB, Czirjak GA, Miklos T. Edwardsiella tarda associated subcutaneous abscesses in a captive grass snake (Natrix natrix, Squamata: Colubridae). Kafkas Univ Vet Fak Derg. (2013) 19:1061–3.
6. White FH, Simpson CF, Williams Jr, LE, Isolation Isolation of Edwardsiella tarda from aquatic animal species and surface waters in Florida. J Wildl Dis. (1973) 9:204–8.
7. Guo G, Jiang J, Yang N, Wang P, Zhang L, Wang Y, et al. An investigation of sudden death in farmed infant Siamese crocodiles during winter and spring in Hainan, China. Indian J Anim Res. (2018) 52:1058–62. doi: 10.18805/ijar.v0iOF.6999
8. Rehman MN, Wang Y, Pan J, Han Y, Yang N, Wang X, et al. Histological and molecular characterization of Edwardsiella tarda infection in Siamese crocodile (Crocodylus siamensis) hatchlings. Aquaculture. (2021) 535:736367. doi: 10.1016/j.aquaculture.2021.736367
9. Slaven EM, Lopez FA, Hart SM, Sanders CV. Myonecrosis caused by Edwardsiella tarda: a case report and case series of extraintestinal E. tarda infections. Clin Infect Dis. (2001) 32:1430–3. doi: 10.1086/320152
10. Nelson J, Nelson C, Carter J. Extraintestinal manifestations of Edwardsiella tarda infection: a 10-year retrospective review. J La State Med Soc. (2009) 161:103–6.
11. Miniero Davies Y, Xavier de Oliveira MG, Paulo Vieira Cunha M, Soares Franco L, Pulecio Santos SL, Zanolli Moreno L, et al. Edwardsiella tarda outbreak affecting fishes and aquatic birds in Brazil. Vet Q. (2018) 38:99–105. doi: 10.1080/01652176.2018.1540070
12. Buján N, Mohammed H, Balboa S, Romalde JL, Toranzo AE, Arias CR, et al. Genetic studies to re-affiliate Edwardsiella tarda fish isolates to Edwardsiella piscicida and Edwardsiella anguillarum species. Syst Appl Microbiol. (2017) 41:30–7. doi: 10.1016/j.syapm.2017.09.004
13. Griffin MJ, Quiniou SM, Cody T, Tabuchi M, Ware C, Cipriano RC, et al. Comparative analysis of Edwardsiella isolates from fish in the eastern United States identifies two distinct genetic taxa amongst organisms phenotypically classified as E.tarda. Vet Microbiol. (2013) 165:358–72. doi: 10.1016/j.vetmic.2013.03.027
14. Zhang Y, Arias CR. Identification and characterization of an intervening sequence within the 23S ribosomal RNA genes of Edwardsiella ictaluri. Syst Appl Microbiol. (2007) 30:93–101. doi: 10.1016/j.syapm.2006.04.004
15. Du M, Chen J, Zhang X, Li A, Li Y, Wang Y. Retention of virulence in a viable but nonculturable Edwardsiella tarda isolate. Appl Environ Microbiol. (2007) 73:1349–54. doi: 10.1128/AEM.02243-06
16. Abbott SL, Janda JM. The genus Edwardsiella. Prokaryotes. (2006) 6:72–89. doi: 10.1007/0-387-30746-X_4
17. Ling SHM, Wang XH, Xie L, Lim TM, Leung KY. Use of green fluorescent protein (GFP) to study the invasion pathways of Edwardsiella tarda in in vivo and in vitro fish models. Microbiology. (2000) 146:7–19. doi: 10.1099/00221287-146-1-7
18. Phillips AD, Trabulsi LR, Dougan G, Frankel G. Edwardsiella tarda induces plasma membrane ruffles on infection of HEp-2 cells. FEMS Microbiol Lett. (1998) 161:317–23. doi: 10.1111/j.1574-6968.1998.tb12963.x
19. Srinivasa RPS, Yamada Y, Leung KY. A major catalase (KatB) that is required for resistance to H2O2 and phagocyte-mediated killing in Edwardsiella tarda. Microbiology. (2003) 149:2635–44. doi: 10.1099/mic.0.26478-0
20. Sahoo P, Swain P, Sahoo SK, Mukherjee SC, Sahu AK. Pathology caused by the bacterium Edwardsiella tarda in Anabas testudineus (Bloch). Asian Fish Sci. (2000) 13:357–62. doi: 10.33997/j.afs.2000.13.4.007
21. Leung KY, Siame BA, Tenkink BJ, Noort RJ, Mok YK. Edwardsiella tarda-virulence mechanisms of an emerging gastroenteritis pathogen. Microbes Infect. (2012) 14:26–34. doi: 10.1016/j.micinf.2011.08.005
22. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. (1999) 27:573–80. doi: 10.1093/nar/27.2.573
23. Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. BioRxiv. (2021) 46:614032. doi: 10.1093/nar/gkab688
24. Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. (2007) 35:3100–8. doi: 10.1093/nar/gkm160
25. Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, et al. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinf. (2018) 62:e51. doi: 10.1002/cpbi.51
26. Bertelli C, Brinkman FS. Improved genomic island predictions with IslandPath-DIMOB. Bioinf. (2018) 34:2161–7. doi: 10.1093/bioinformatics/bty095
27. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. (2007) 35:W52–7. doi: 10.1093/nar/gkm360
28. Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. (2017) 45:W36–41. doi: 10.1093/nar/gkx319
29. Rodriguez RLM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints. (2016) 4:e1900ve1901. doi: 10.7287/peerj.preprints.1900v1
30. Chen X, Zhang Y, Zhang Z, Zhao Y, Sun C, Yang M, et al. PGAweb: a web server for bacterial pan-genome analysis. Front Microbiol. (2018) 9:1910. doi: 10.3389/fmicb.2018.01910
31. Gangman Y, Sing-Hoi S, Thon MR. Identifying clusters of functionally related genes in genomes. Bioinformatics. (2007) 23:1053–60. doi: 10.1093/bioinformatics/btl673
32. Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. (2009) 106:19126–31. doi: 10.1073/pnas.0906412106
33. Xiao J, Wang Q, Liu Q, Wang X, Liu H, Zhang Y. Isolation and identification of fish pathogen Edwardsiella tarda from mariculture in China. Aquacult Res. (2008) 40:13–7. doi: 10.1111/j.1365-2109.2008.02101.x
34. Wang Q, Yang M, Xiao J, Wu H, Wang X, Lv Y, et al. Genome sequence of the versatile fish pathogen Edwardsiella tarda provides insights into its adaptation to broad host ranges and intracellular niches. PLoS ONE. (2009) 4:e7646. doi: 10.1371/journal.pone.0007646
35. Yang M, Lv Y, Xiao J, Wu H, Zheng H, Liu Q, et al. Edwardsiella comparative phylogenomics reveal the new intra/inter-species taxonomic relationships, virulence evolution and niche adaptation mechanisms. PLoS ONE. (2012) 7:e36987. doi: 10.1371/journal.pone.0036987
36. Tekedar HC, Blom J, Kalindamar S, Nho S, Karsi A, Lawrence ML. Comparative genomics of the fish pathogens Edwardsiella ictaluri 93-146 and Edwardsiella piscicida C07-087. Microb Genomics. (2020) 6:322. doi: 10.1099/mgen.0.000322
37. Wang R, Tekedar HC, Lawrence ML, Chouljenko VN, Kim J, Kim N, et al. Draft genome sequences of Edwardsiella ictaluri strains LADL11-100 and LADL11-194 isolated from zebrafish Danio rerio. Genome Announc. (2015) 3:e01449–15. doi: 10.1128/genomeA.01449-15
38. Shao S, Qiliang L, Qin L, Haizhen W, Jingfan X, Zongze S, et al. Phylogenomics characterization of a highly virulent Edwardsiella strain ET080813T encoding two distinct T3SS and three T6SS gene clusters: propose a novel species as Edwardsiella anguillarum sp. nov. Syst Appl Microbiol. (2015) 38:36–47. doi: 10.1016/j.syapm.2014.10.008
39. Williams ML, Gillaspy AF, Dyer DW, Thune RL, Waldbieser GC, Schuster SC, et al. Genome sequence of Edwardsiella ictaluri 93-146, a strain associated with a natural channel catfish outbreak of enteric septicemia of catfish. J Bacteriol. (2012) 194:740–1. doi: 10.1128/JB.06522-11
40. Liao YS, Chen BH, Hong YP, Teng RH, Wang YW, Liang SY, et al. Emergence of multidrug-resistant Salmonella enterica serovar Goldcoast strains in Taiwan and international spread of the ST358 clone. Antimicrob Agents Chemother. (2019) 63:e01122–19. doi: 10.1128/AAC.01122-19
41. Lee K, Kim HK, Park SK, Sohn H, Cho Y, Choi YM, et al. First report of the occurrence and whole-genome characterization of Edwardsiella tarda in the false killer whale (Pseudorca crassidens). J Vet Med Sci. (2018) 80:1041–6. doi: 10.1292/jvms.17-0590
42. Siddique A, Figurski DH. The active partition gene incC of IncP plasmids is required for stable maintenance in a broad range of hosts. J Bacteriol. (2002) 184:1788–93. doi: 10.1128/JB.184.6.1788-1793.2002
43. Lv Y, Xiao J, Liu Q, Wu H, Zhang Y, Wang Q. Systematic mutation analysis of two-component signal transduction systems reveals EsrA-EsrB and PhoP-PhoQ as the major virulence regulators in Edwardsiella tarda. Vet Microbiol. (2012) 157:190–9. doi: 10.1016/j.vetmic.2011.12.018
44. Wang X, Wang Q, Yang M, Xiao J, Liu Q, Wu H, et al. QseBC controls flagellar motility, fimbrial hemagglutination and intracellular virulence in fish pathogen Edwardsiella tarda. Fish Shellfish Immunol. (2011) 30:944–53. doi: 10.1016/j.fsi.2011.01.019
45. Rao PSS, Lim TM, Leung KY. Opsonized virulent Edwardsiella tarda strains are able to adhere to and survive and replicate within fish phagocytes but fail to stimulate reactive oxygen intermediates. Infect. Immun. (2001) 69:5689–97. doi: 10.1128/IAI.69.9.5689-5697.2001
46. Tan YP, Zheng J, Tung SL, Rosenshine I, Leung KY. Role of type III secretion in Edwardsiella tarda virulence. Microbiology. (2005) 151:2301–13. doi: 10.1099/mic.0.28005-0
47. Nakamura Y, Takano T, Yasuike M, Sakai T, Matsuyama T, Sano M. Comparative genomics reveals that a fish pathogenic bacterium Edwardsiella tarda has acquired the locus of enterocyte effacement (LEE) through horizontal gene transfer. BMC Genomics. (2013) 14:642. doi: 10.1186/1471-2164-14-642
48. Matuszewska M, Murray GG, Harrison EM, Holmes MA, Weinert LA. The evolutionary genomics of host specificity in Staphylococcus aureus. Trends Microbiol. (2020) 28:465–77. doi: 10.1016/j.tim.2019.12.007
Keywords: Edwardsiella tarda SC002, genome annotation, pan-genome, genome sequencing, comparative genomics
Citation: Rehman MNU, Dawar FU, Zeng J, Fan L, Feng W, Wang M, Yang N, Guo G and Zheng J (2023) Complete genome sequence analysis of Edwardsiella tarda SC002 from hatchlings of Siamese crocodile. Front. Vet. Sci. 10:1140655. doi: 10.3389/fvets.2023.1140655
Received: 09 January 2023; Accepted: 08 February 2023;
Published: 09 March 2023.
Edited by:
Farhan Anwar Khan, University of Agriculture, Peshawar, PakistanReviewed by:
Ahmad Ud Din, Sichuan University, ChinaCopyright © 2023 Rehman, Dawar, Zeng, Fan, Feng, Wang, Yang, Guo and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiping Zheng, amlwaW5nLnpoZW5nQGhhaW5hbnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.