
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 16 February 2023
Sec. Veterinary Infectious Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1092179
This article is part of the Research Topic Zoonotic Bacteria: Genomic Evolution, Antimicrobial Resistance, Pathogenicity, and Prevention Strategies View all 6 articles
 Hosny El-Adawy1,2*
Hosny El-Adawy1,2* Helmut Hotzel1
Helmut Hotzel1 Silvia García-Soto1
Silvia García-Soto1 Herbert Tomaso1
Herbert Tomaso1 Hafez M. Hafez3
Hafez M. Hafez3 Stefan Schwarz4,5
Stefan Schwarz4,5 Heinrich Neubauer1
Heinrich Neubauer1 Jörg Linde1
Jörg Linde1Campylobacter (C.) jejuni is a zoonotic bacterium of public health significance. The present investigation was designed to assess the epidemiology and genetic heterogeneity of C. jejuni recovered from commercial turkey farms in Germany using whole-genome sequencing. The Illumina MiSeq® technology was used to sequence 66 C. jejuni isolates obtained between 2010 and 2011 from commercial meat turkey flocks located in ten German federal states. Phenotypic antimicrobial resistance was determined. Phylogeny, resistome, plasmidome and virulome profiles were analyzed using whole-genome sequencing data. Genetic resistance markers were identified with bioinformatics tools (AMRFinder, ResFinder, NCBI and ABRicate) and compared with the phenotypic antimicrobial resistance. The isolates were assigned to 28 different sequence types and 11 clonal complexes. The average pairwise single nucleotide-polymorphisms distance of 14,585 SNPs (range: 0–26,540 SNPs) revealed a high genetic distinction between the isolates. Thirteen virulence-associated genes were identified in C. jejuni isolates. Most of the isolates harbored the genes flaA (83.3%) and flaB (78.8%). The wlaN gene associated with the Guillain–Barré syndrome was detected in nine (13.6%) isolates. The genes for resistance to ampicillin (blaOXA), tetracycline [tet(O)], neomycin [aph(3')-IIIa], streptomycin (aadE) and streptothricin (sat4) were detected in isolated C. jejuni using WGS. A gene cluster comprising the genes sat4, aph(3′)-IIIa and aadE was present in six isolates. The single point mutation T86I in the housekeeping gene gyrA conferring resistance to quinolones was retrieved in 93.6% of phenotypically fluoroquinolone-resistant isolates. Five phenotypically erythromycin-susceptible isolates carried the mutation A103V in the gene for the ribosomal protein L22 inferring macrolide resistance. An assortment of 13 β-lactam resistance genes (blaOXA variants) was detected in 58 C. jejuni isolates. Out of 66 sequenced isolates, 28 (42.4%) carried plasmid-borne contigs. Six isolates harbored a pTet-like plasmid-borne contig which carries the tet(O) gene. This study emphasized the potential of whole-genome sequencing to ameliorate the routine surveillance of C. jejuni. Whole-genome sequencing can predict antimicrobial resistance with a high degree of accuracy. However, resistance gene databases need curation and updates to revoke inaccuracy when using WGS-based analysis pipelines for AMR detection.
Campylobacter (C.) is recognized as the leading cause of bacterial gastroenteritis in humans and several animal species (poultry, cattle, pigs, sheep and goats) worldwide. C. jejuni is a commensal bacterium of animal species and, therefore, exposed to antimicrobial agents that are administered to animals for various reasons. Moreover, the environment can be contaminated with Campylobacter by litter and/or soil at farm premises (1). Poultry and their products are considered the most significant source of human campylobacteriosis (2, 3). As a consequence, antimicrobial resistance in C. jejuni is a growing problem (4, 5). Some clones of C. jejuni endure genetically stable over long periods of time, but C. jejuni can adapt to different environmental conditions by means of variation in the isolate's virulence (6).
In Germany, the prevalence of Campylobacter in poultry meat and chickens from 2001 through 2010 ranged from 14 to 34% and 6 to 64% per year, respectively (7). A voluntary monitoring program was conducted between 2004 and 2007 in broiler farms and the reported human incidence in Germany in order to identify the prevalence patterns of thermotolerant Campylobacter spp. showed that the peak in human campylobacteriosis preceded the peak in broiler prevalence in Lower Saxony (8). C. jejuni was isolated from different poultry species in Germany and showed a higher prevalence than C. coli (9).
It is necessary to understand virulence factors and molecular mechanisms contributing to pathogenesis of Campylobacter. Whole-genome sequences can be used for high-resolution genotyping and automatized detection of genetic markers for virulence, antimicrobial resistance and mobile genetic elements (10–12). Since the costs for WGS are decreasing, it has replaced traditional typing methods, such as pulsed-field gel electrophoresis (PFGE), multi-locus sequence typing (MLST) and serotyping for surveillance of bacterial infectious diseases by public health authorities (11–19). Consequently, WGS was also demonstrated for the investigation of virulence, clonality and antimicrobial resistance in Campylobacter isolated from poultry farms (20–23).
The WGS led to the creation of the core genome multi-locus sequence typing (cgMLST), a typing method encompassing hundreds of loci from the traditional seven loci of MLST (24). Additionally, studies using single nucleotide polymorphism (SNP) allow the establishment of the best phylogenetic relationship among different pathogens (25). The WGS is used for various purposes including novel antibiotic and diagnostic test development, studying the emergence of antibiotic resistance, disease surveillance, and direct infection control measures in both clinical settings and communities (26). The next-generation sequencing (NGS) systems available include Illumina Genome Analyzer (HiSeq, MiSeq), Life Technologies Ion Torrent, and the PacBio RX system (27).
The used WGS data revealed a high genetic diversity amongst C. jejuni isolated from broilers and definite types and virulence genes are implicated with the development of more severe human illness (28).
In Europe, the antimicrobial resistance of C. jejuni isolated from chickens and turkeys had to be reported every 2 years based on European Union Commission Implementing Decision 2013/652/EU (29). In Germany, the antimicrobial resistance of Campylobacter spp. isolated from broilers and turkeys was highest to ciprofloxacin, nalidixic acid and tetracycline whereas C. coli were more often resistant than C. jejuni and resistance was observed more frequently in turkeys than in broilers (30). The emergence of a high antimicrobial resistance and multidrug resistance was identified in C. jejuni isolated from commercial turkey farms in Germany (31).
The objective of this study was to analyze C. jejuni isolated from turkey flocks using WGS for high-resolution genotyping and to investigate their complete genomic potential concerning resistance to antimicrobial agents, plasmids and virulence-associated factors.
Sixty-six C. jejuni were isolated from 66 turkey flocks reared in different turkey farms in ten federal states in Germany, namely Baden-Wuerttemberg, Bavaria, Brandenburg, Mecklenburg-Western Pomerania, Lower-Saxony, North Rhine-Westphalia, Rhineland-Palatinate, Saxony, Saxony-Anhalt and Thuringia. The samples were collected from apparently healthy turkey flocks aged between the 12th and the 18th weeks (Table S1). The isolation was carried out according to ISO 10272 (32). All isolates were identified using MALDI-TOF MS and multiplex PCR assay as described previously by El-Adawy et al. (31).
The broth microdilution test was performed using commercially available microtitre plates TREK® Sensititre NLDMV2 (Trek Diagnostic Systems, Ltd., East Grinstead, UK) for the determination of the antimicrobial susceptibility of the 66 C. jejuni isolates to gentamicin, chloramphenicol, streptomycin, erythromycin, neomycin, amoxicillin, tetracycline, nalidixic acid, ciprofloxacin and metronidazole. The susceptibility test was performed according to CLSI recommendations and the plates were incubated under microaerophilic condition (CampyGen™, Oxoid Deutschland GmbH, Schwerte, Germany) at 37°C for 48 h (33). The results were read either visually or photometrically (Tecan Deutschland GmbH, Crailsheim, Germany) using the computer program easyWIN fitting (version V6.1, 2000; Tecan Deutschland GmbH, Crailsheim, Germany). C. jejuni ATCC 33560 (American Type Culture Collection, LGC Standards GmbH, Wesel, Germany) was used as reference strain for quality control in each batch of the broth microdilution tests. The resistance breakpoints for gentamicin, chloramphenicol, erythromycin, amoxicillin, tetracycline, nalidixic acid and ciprofloxacin were those recommended by the Clinical and Laboratory Standards Institute (33, 34) and in previously published literature (31, 35). The resistance breakpoint used for streptomycin was ≥ 64 μg/ml, as described previously (35). Since there were no CLSI breakpoints for neomycin, we used a tentative breakpoint for Escherichia coli of 32 μg/mL (36, 37). C. jejuni isolates were tested for resistance to metronidazole and the breakpoint for resistance at 16 mg/ml (38) was used.
Genomic DNA was extracted and purified from a 48 h bacterial culture on Mueller-Hinton blood agar plates (Oxoid Deutschland GmbH, Wesel, Germany) using QIAGEN® Genomic-tip 20/G Kit (QIAGEN®, Hilden, Germany) according to the manufacturer's instructions. The DNA was eluted in 200 μl elution buffer. DNA was quantified spectrophotometrically using a Nanodrop® ND-1000 (Fisher Scientific GmbH, Schwerte, Germany). The quality of the DNA was determined using the Qubit dsDNA BR Assay Kit (Invitrogen, Carlsbad CA, USA).
Sequencing libraries were created using the Nextera XT DNA Library Preparation Kit (Illumina Inc., San Diego, CA). Paired-end sequencing was performed on an Illumina MiSeq instrument according to the manufacturer's instructions (Illumina Inc., San Diego, CA) producing 300 bp long reads. Raw sequencing data were deposited by the European Nucleotide Archive (ENA) under the BioProject PRJEB55640. The bioinformatic analysis started with quality control of the raw paired-end reads from Illumina. The Linux-based bioinformatics pipeline WGSBAC v. 2.0.0 (https://gitlab.com/FLI_Bioinfo/WGSBAC) uses FastQC v. 0.11.7 (39) and calculates coverage of raw reads. Second, WGSBAC performs assembly using Shovill v. 1.0.4 (https://github.com/tseemann/shovill). To check the quality of the assembled genomes, WGSBAC uses QUAST v. 5.0.2 (40) and to identify potential contamination on both reads and assemblies, the pipeline uses Kraken 2 v. 1.1 (41) and the database Kraken2DB. For the investigation of antimicrobial resistance genes and virulence determinants, WGSBAC uses the software ABRicate (v. 0.8.10) (https://github.com/tseemann/abricate) and the databases: ResFinder (42), NCBI (43) and Virulence Factor Database (VFDB) (44). In addition, WGSBAC uses AMRFinderPlus (v. 3.6.10) (45) for the detection of chromosomal point mutations leading to AMR and organism-specific acquired resistance genes. For plasmid detection, Platon was used (46). BLAST search was performed with plasmid-borne contigs against NCBI's nucleotide database and hits were compared to recently published pTet-like plasmids (47, 48).
Annotation of the assembled genomes was performed by the software Bakta v. 1.6.1 (https://github.com/oschwengers/bakta) (49). Annotated features of interest were visualized using the software Geneious Prime v. 2021.0.1 (https://www.geneious.com).
For genotyping, WGSBAC uses classical multilocus sequence typing (MLST) on assembled genomes using the software mlst v. 2.16.1 that incorporates the PubMLST database for the seven gene C. jejuni/coli MLST scheme (https://pubmlst.org/organisms/campylobacter-jejunicoli). Core genome multilocus sequence typing was performed using the external software Ridom Seqsphere+ v. 5.1.0 with default settings and the specific core genome scheme (cgMLST v2). In addition, WGSBAC performs mapping-based genotyping using core-genome single nucleotide polymorphisms (cgSNPs) identified by Snippy v. 4.3.6 (https://github.com/tseemann/snippy) with standard settings. As reference the genome of C. jejuni NCTC 11168 (accession NC_002163.1) was used. For phylogenetic tree construction based on cgSNP analysis, WGSBAC uses the SNPs alignment matrix generated by Snippy and reconstructs the tree using RAxML (Randomized Axelerated Maximum Likelihood) v. 8 (50). The tree was rooted to the reference genome and visualized using the interactive Tree of Life (iTOL) v. 4 web tool (https://itol.embl.de/login.cgi).
Sequencing of the 66 C. jejuni isolates, yielded an average of 807,061 total reads (range: 156,702–2,263,714) per sample, with an average read length of 219 bp (Table S1) leading to an average read-coverage of 104-fold (range: 37–139). The assembled genomes consisted on average of 29 contigs (range: 20–256). The GC content was 30.43% and the genome size of the isolates was 1,700,350 bp on average (range: 1,410,342–1,883,846 bp).
The phylogentic and genotyping analysis displaying relatedness between the 66 C. jejuni isolates using cgMSLT based on cgSNP distances rooted to the reference genome revealed nine different groups according to how closely related the isolates were (Figure 1).
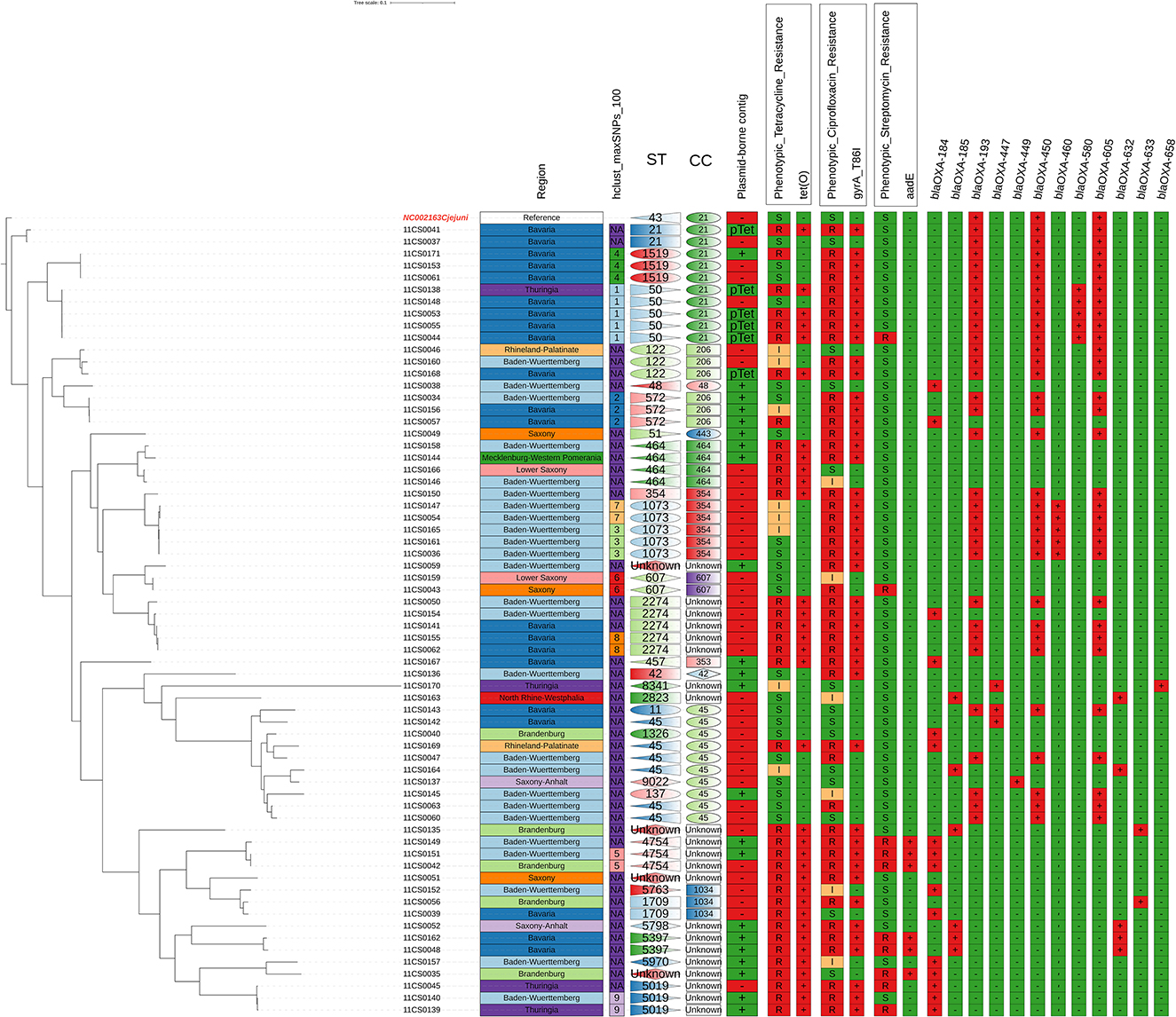
Figure 1. Phylogentic and genotyping analysis using cgMSLT (Phylogenetic trees based on cgSNP distances) rooted by the most distant isolate. Comparison between phenotypic and genotypic antimicrobial resistance of the tested 66 C. jejuni isolates with regard to the place of isolation and sequence types: correlation of susceptibility phenotypes and genotypes.
The origin and distribution of the 66 C. jejuni and their colonal complexes in 11 federal states in Germany were shown in Figure 2.

Figure 2. Origin of the 66 C. jejuni and their clonal complexes in 11 federal states in Germany.
The MLST analysis based on WGS revealed a high genetic diversity with 28 different sequence types (STs). For four isolates, a so far unknown MLST type was detected. The most prevalent STs found were ST 45 (9%), ST 50 (8%), ST 1073 (8%), ST 2274 (8%), and ST 464 (6%) (Table S1, Figure 1). The high genetic diversity was further indicated by an average core-gene Single Nucleotide-Polymorphisms distance of 14,585 cgSNPs (range 0–26,540 cgSNPs). Hierarchical clustering with a cut-off of 100 cgSNPs grouped 29 isolates into nine clusters, while the majority of isolates did not cluster. Finally, cgMLST analysis supported the finding of high genetic diversity, as 11 different clonal complexes (CCs) were detected among the 66 isolates (Figure 1, Table S1). For four isolates, an unknown clonal complex was identified. Both the SNP-based phylogeny and the cgMLST analysis indicated the genetic relatedness of isolates partly following the geographical origins from where they were isolated (Figures 1, 2). While the genetic diversity was generally high, some isolates were closely related (Figure 1, Table S1). For example, isolates 11CS0055 and 11CS0044 from Bavaria were indistinguishable (0 cgSNPs).
The two isolates 11CS0036 and 11CS0161 from samples obtained in Baden-Wuerttemberg were closely related and revealed only one cgSNP (Table S1), while other isolates (11CS0043 and 11CS0159) collected from two different federal states (Saxony and Lower Saxony) had two SNPs difference. Three SNPs difference was found in two isolates (11CS0042 and 11CS0151) from Baden-Wuerttemberg. Four isolates from Bavaria (11CS0061, 11CS0171, 11CS0062, and 11CS0155) seemed to be closely related with six SNPs difference (Table S1).
The genomic analysis of 66 C. jejuni isolates revealed in total 30 virulence-associated genes related to motility, chemotaxis, adhesion and invasion (Table 1, Table S1). Most of the isolates harbored the genes flaA (83.3%) and flaB (78.8%) coding for flagellin protein A and B, respectively. Among other virulence determinants, the gene cj1135 coding for the putative two-domain glucosyltransferase was present in 56% of the isolates. The gene rfbC coding for dTDP-4-dehydrorhamnose 35-epimerase was detected in 42.4% and the motility accessory factor PseD/Maf2 in 28.8% of the C. jejuni isolates. Genes for sialic acid synthase (neuB1) and UDP-N-acetylglucosamine 2-epimerase (neuC1) were detected in 16.6% of the isolates. In minor proportion, the combination of cj1136-138, neuA and cstIII genes was found in 15.2% of the C. jejuni isolates. Out of the 66 C. jejuni isolates, eight (12.1%) isolates harbored type IV secretion system genes (virB10, virB11, virB4, virB8, virB9, virD4 and cjp54). Seven isolates (10.6%) harbored cj421c, cj426c, cj1432c, cj1435c, cj1436c, cj1437c, cj1440c, virB10, virB11, virB4, virB8, virB9, virD4, and cjp54 genes (Table 1). The wlaN gene associated with the Guillain–Barré syndrome was identified in nine (13.6%) isolates.
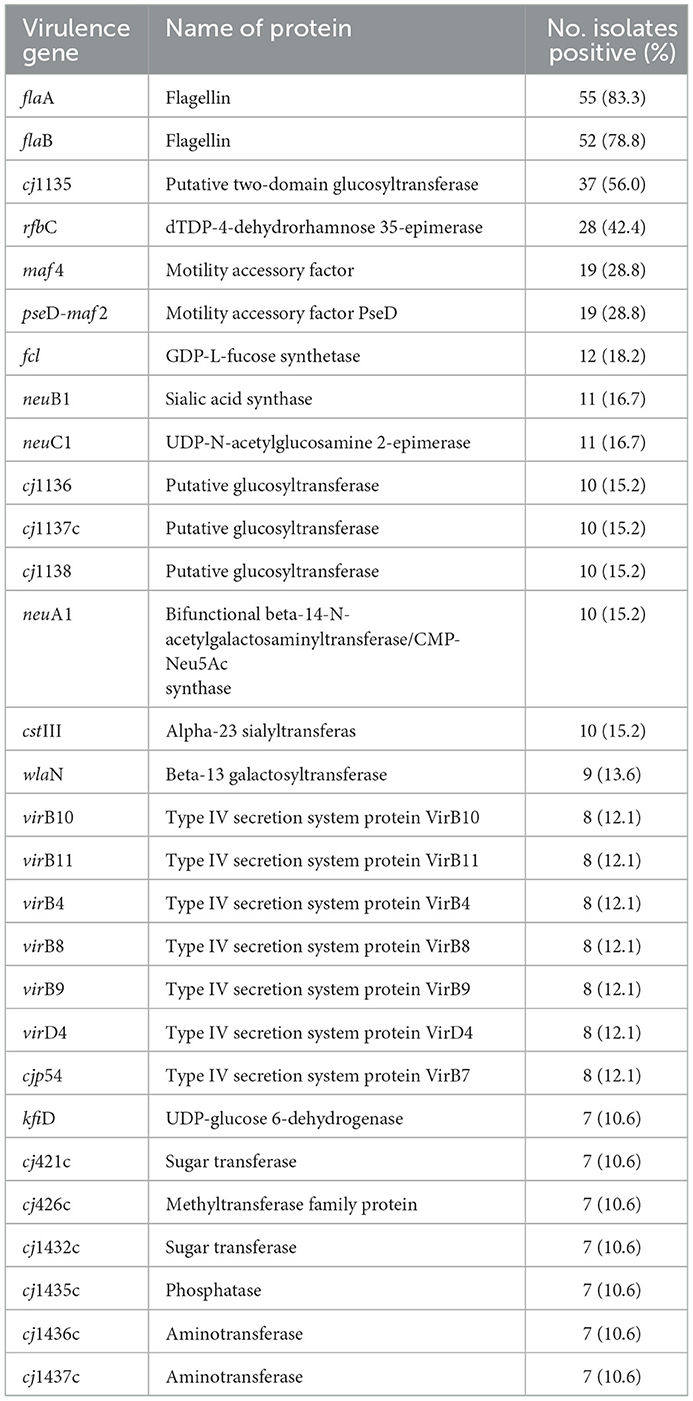
Table 1. Frequency of most significant virulence-associated genes found in in 66 Campylobacter jejuni isolates.
The results of antimicrobial susceptibility testing showed that all isolates were susceptible to gentamicin, erythromycin and chloramphenicol. Resistance to streptomycin, neomycin, tetracycline, nalidixic acid, ciprofloxacin and metronidazole was detected in 10 (15.2%), 18 (27.3%), 36 (54.5%), 44 (66.7%), 47 (71.2%), and 49 (74.2%) isolates, respectively (Table S1).
WGS analyses identified 18 acquired AMR genes that code for resistance to antimicrobials representing three different classes (tetracyclines, aminoglycosides and β-lactams) and point mutations in the gyrA gene coding for resistance to (fluoro) quinolones. The mutation associated with macrolide resistance was located in the gene for the ribosomal protein L22 (A103V) (Table 2, Table S1). The antimicrobial resistance gene associated with tetracycline resistance tet(O) was identified in 34 of the 66 C. jejuni isolates (51.5%) (Table 2, Table S1). Genes coding for aminoglycoside-modifying enzymes of two distinct families, aminoglycoside phosphotransferases (APHs) and aminoglycoside nucleotidyl transferases (ANTs) were detected. streptomycin resistance is encoded by aadE gene which was found in six out of ten (60.0%) phenotypically streptomycin-resistant C. jejuni. Out of 18 C. jejuni isolates resistant to neomycin, the aph(3′)-IIIa gene was detected in six (33.3%) isolates (Table 2). The sat4 gene encoding streptothricin resistance was detected in six (9.1%) C. jejuni isolated in this study. A gene cluster comprising the genes sat4, aph(3′)-IIIa and aadE was present in six isolates (Figure 3). Upstream the gene cluster, all the isolates have two ORFs, one codes for the HTH domain-containing protein and the other for a hypothetical protein. They are followed by the IS6-like element IS1216 family transposase. Downstream, variations within the isolates were detected: isolate 11CS0035 has the gene moeA1 that codifies for the product molybdopterin molybdenumtransferase MoeA and an unnamed gene that codes for the AraC family transcriptional regulator. Downstream of the gene cluster, in isolates 11CS0042, 11CS0149 and 11CS0151, an unnamed gene was found that codes for the putative motility protein and the gene nhaA2 that codes for the Na+/H+ antiporter NhaA. Isolates 11CS0162 and 11CS0048 have downstream an unnamed gene that codes for an ABC transporter substrate-binding protein and the gene skfB for the radical SAM protein.
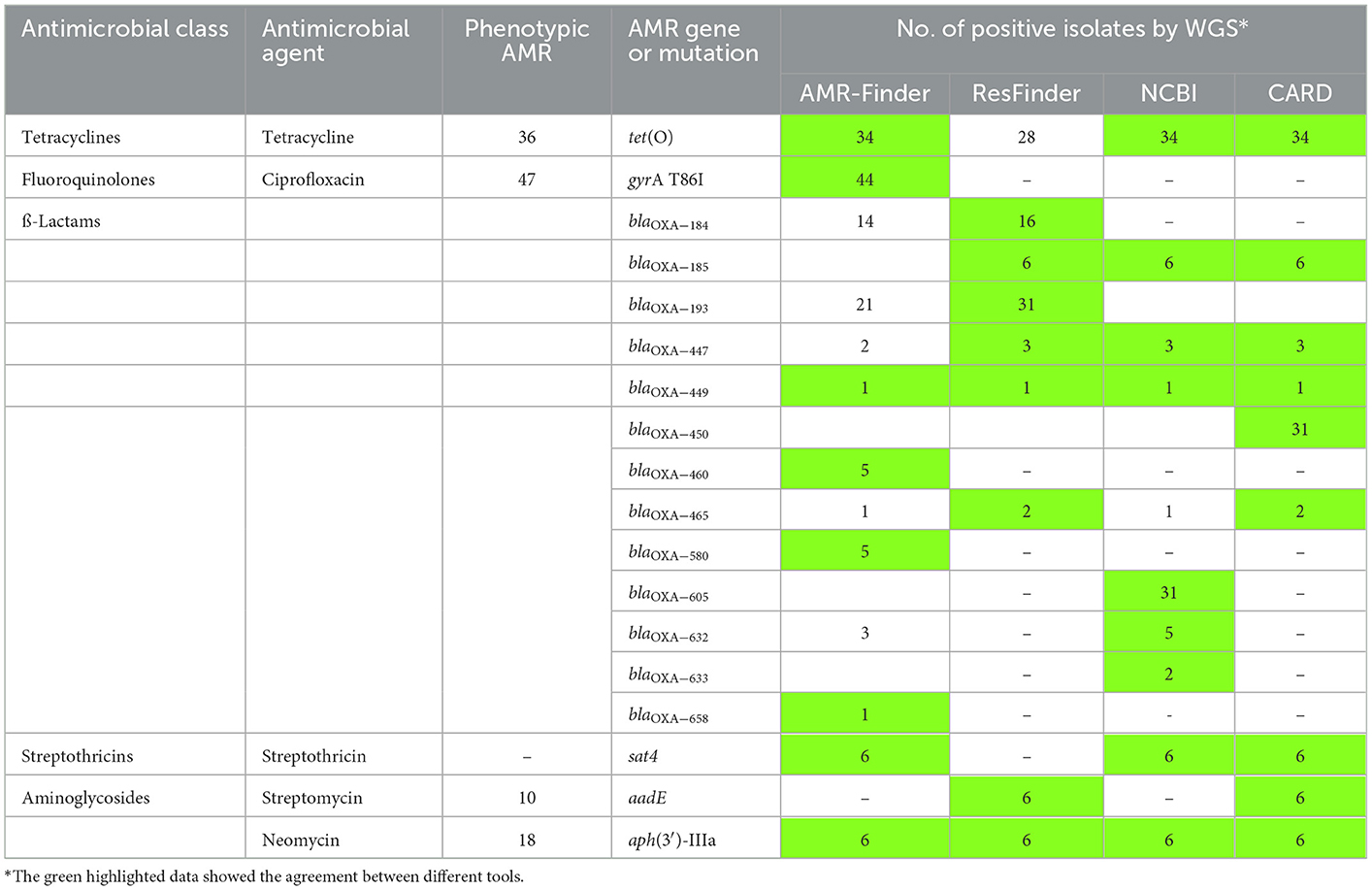
Table 2. Distribution of antimicrobial resistance genes/mutations in the tested 66 Campylobacter jejuni isolated using different tools of analysis and compatibility with phenotypic resistance.
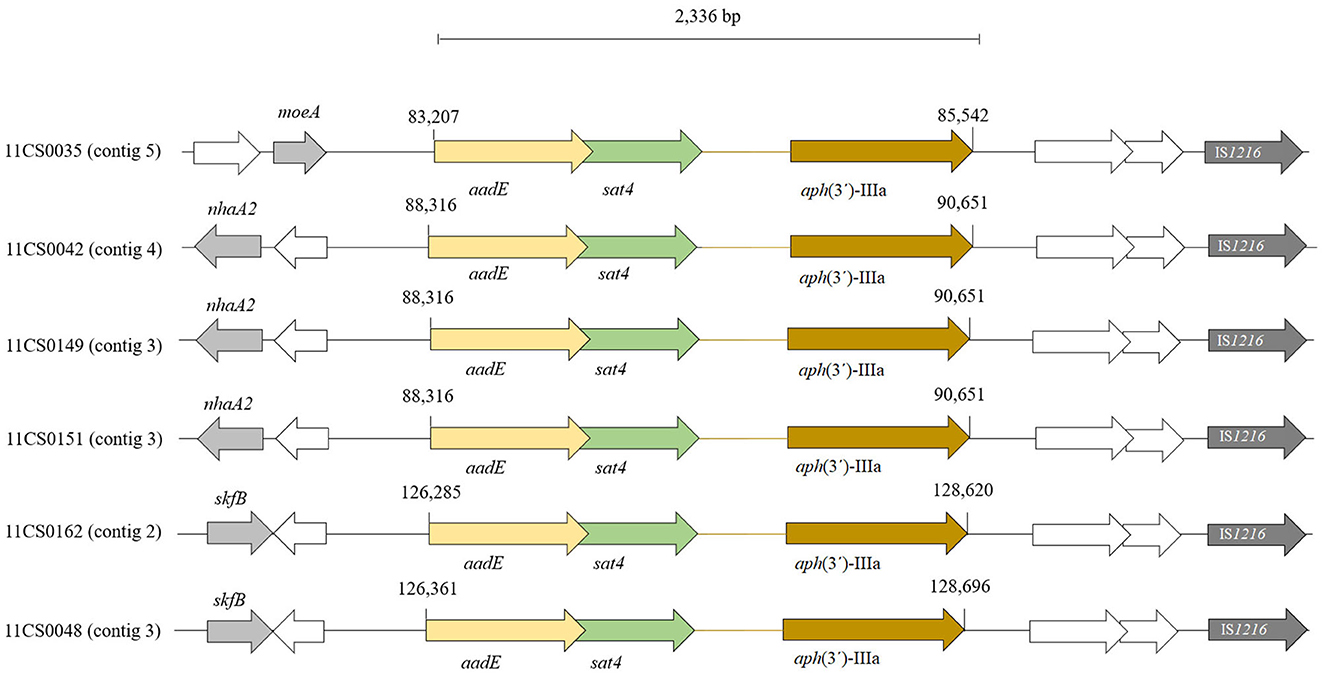
Figure 3. Distribution of aadE, sat4 and aph(3')-IIIa gene cluster found in six Campylobacter jejuni isolates. ORFs in white indicate unnamed genes.
Twelve known variants of blaOXA genes involved in β-lactam resistance were detected in this study using the results of different bioinformatic tools (AMRFinder, ResFinder, NCBI and ABRicate) (Table 2). Fifty-eight isolates harbored blaOXA genes (52 resistant, five susceptible and one intermediate against ampicillin) (Table S2). Out of 61 phenotypically ampicillin-resistant C. jejuni isolates, 57 (93.4%) carried at least one gene coding for β-lactamases of the OXA-like family.
The distribution of detected 13 blaOXA genes in 66 C. jejuni isolates is shown in Table S2 and Figure 1. In brief, blaOXA−184, blaOXA−185, blaOXA−193, blaOXA−447, blaOXA−449, blaOXA−450, blaOXA−460, blaOXA−465, blaOXA−580, blaOXA−605, blaOXA−632, blaOXA−633 and blaOXA−658-like genes were detected in 16 (24.2%), 6 (9.1%), 31 (47.0%), 3 (4.5%), 1 (1.5%), 31 (47.0%), 5 (7.6%), 2 (3.0%), 5 (7.6%), 31 (47.0%), 5 (7.6%), 2 (3.0%), 2 (3.0%), and 1 (1.5%) of 66 C. jejuni isolates, respectively (Table S2, Figure 1). Nineteen isolates harbored one gene coding for β-lactams of the OXA-like family (16 blaOXA−184, 1 blaOXA−465, 1 blaOXA−447 and 1 blaOXA−449. Twenty isolates carried three blaOXA genes together (blaOXA−193, blaOXA−450 and blaOXA−605) while seven and 11 isolates harbored two and four blaOXA genes, respectively (Table S2).
The accordance between phenotypic and genotypic antimicrobial resistance is shown in Table 2, Figure 1, and Table S1. Thirty-four (94.4%) out of 36 phenotypically tetracycline-resistant isolates harbored the tet(O) gene which confers tetracycline resistance (Table 2). Forty-seven (71.2%) C. jejuni isolates were phenotypically resistant to ciprofloxacin, 44 (93.6%) of them contained a chromosomal single point mutation in the gyrA gene which resulted in the amino acid substitutions C257T or 786I. All isolates carrying this mutation were resistant to (fluoro) quinolones (ciprofloxacin and nalidixic acid). Six isolates that were phenotypically intermediate to ciprofloxacin did not have any point mutation in the gyrA gene (Table 2). The cmeABCR multidrug efflux complex was present in all isolates.
Twenty-eight out of the 66 (42.4%) C. jejuni isolates contained at least one contig classified as plasmid-borne (Table S1). While the majority of those isolates (n = 18) contained one contig classified as plasmid-borne, isolates 11CS0052 and 11CS0059 contained five and four plasmid contigs, respectively, and another six isolates harbored two plasmids. The sizes of the plasmid-borne contigs detected by Platon ranged from 1,600 bp to 44,793 bp. Six isolates harbored a pTet-like plasmid-borne contig which carried the tet(O) gene and were phenotypically tetracycline-resistant. In the assembled genomes from six phenotypically tetracycline-resistant isolates, Platon identified a pTet-like plasmid-borne contig within a contig in which the tet(O) gene was also identified. BLAST search of the plasmid sequences in GenBank revealed that similar plasmids have been previously found in Campylobacter spp. Thirty-eight plasmid-borne contigs had a BLAST hit in C. jejuni, while for six, a hit in C. coli was found (Table S1).
The origin of the isolates, their phylogenetic relatedness, STs and association with antimicrobial resistance patterns are depicted in Figure 1. There is little correlation between genotypes and the numbers of AMR genes in the investigated isolates. Isolates assigned to STs 1073, 2274, 122, 5019, 4754, and 50 showed highly similar AMR gene profiles.
Whole-genome sequencing is a promising tool in public health as it is able to identify sources and routes of infections, to investigate outbreaks with the highest resolution and thus to improve surveillance of C. jejuni (51). This investigation presents an application of WGS for assessing the epidemiology of C. jejuni, isolated from turkey farms in Germany.
In the current investigation, 30 virulence-associated genes were identified in C. jejuni isolates. Interestingly, nine (13.6%) of the isolates carried the wlaN gene implicated in the Guillain–Barré syndrome (GBS), a polyneuropathic disorder damaging the peripheral nervous system and causing muscle weakness (52). In north-eastern Spain wlaN was detected in two (16.6%) C. jejuni isolates from broilers (20). The gene wlaN was also detected in 10.7% of the tested C. jejuni isolated from slaughterhouses for broilers in Southern Brazil (53) and in 4.7% and 23.8% of isolates from broiler feces and poultry meat in Japan, respectively (54). Isolates harboring the wlaN gene may have a higher pathogenic potential and can induce autoimmune disease in their hosts (53). A previous study showed that the presence of the wlaN gene increased the capacity of cell invasion (in vitro and in vivo) (55).
This study revealed on the one hand a high genetic diversity of the analyzed 66 C. jejuni isolates indicated by 28 STs, 11 CCs and an average pairwise cgSNP distance of 14,585. On the other hand, closely related isolates were found. In fact, six pairs of isolates with a cgSNP distance below 10 were found suggesting both regional persistence and spread of clones. In a recent study, high genetic diversity was found in C. jejuni collected during processing of caeca and neck skin samples of broilers. These isolates were assigned to ten sequence types, which belonged to seven clonal complexes, based on MLST. ST 257 was prevalent with 58 isolates assigned to it, followed by ST 51 with 25 isolates, ST 10089 with 16 isolates, ST 48 with 13 isolates and ST 50 with 12 isolates (56).
Campylobacter jejuni classified by the WHO as a “high priority pathogen” which gives it great concerns due to the emergence of antimicrobial resistance to multiple drugs including fluoroquinolones, macrolides and other clinically relevant classes which limits the alternative treatment for human campylobacteriosis (57).
WGS was used in this study to characterize and predict AMR in this collection of C. jejuni isolated of turkey farms from different federal states in Germany. The predicted antimicrobial resistance based on WGS data was concordant with the phenotypic resistance profiles in most cases.
Tetracycline resistance in Campylobacter is associated specifically with genes encoding ribosome protection proteins (RPPs) (58). In Campylobacter, tetracycline resistance genes can be located both in the chromosomal DNA and on plasmids (59). A clear trend toward an increase in the occurrence of tetracycline and (fluoro)quinolone resistance determinants among C. jejuni, linked to the spread of the co-occurring blaOXA−61 and tet(O)-tet(O/W/O) genes and the gyrA SNP, resulting in the amino acid substitution T86I, was found in the time span from 2001 to date in Europe (60).
The OXA-type β-lactamases confer resistance to the penicillins, although some are also able to cause resistance to cephalosporins and carbapenems. In 2007 European Food Safety Authority (EFSA) considered β-lactams as optional for monitoring at the European union (EU) level (61). A large proportion of C. jejuni produce β-lactamases. However, the β-lactamase of C. jejuni seems to play a role only in resistance to amoxicillin, ampicillin and ticarcillin (62). In the present investigation, a variety of 13 known β-lactam resistance genes (all blaOXA variants) were detected in 58 (87.9%) of the C. jejuni isolates, the most prevalent being blaOXA−193, blaOXA−450 and blaOXA−605 (n = 31; 47% of each) followed by blaOXA−184 (n = 16; 24.2%). These results were in accordance with a previous study in which two major β-lactamase genes, designated blaOXA−61 and blaOXA−184, were prevalent at 62.93 and 82.08% in C. jejuni from the poultry and other bird groups, respectively (63).
The finding of this study highlighted that C. jejuni is a reservoir for β-lactamase genes that might be transferred to other clinical or environmental bacteria. Thus, screening of C. jejuni for such genes may contribute to AMR surveillance in general.
Although point mutations at multiple positions in the gyrA gene associated with the resistance to fluoroquinolones in Campylobacter have been described (64), the gyrA mutation, that results in the amino acid substitution T86I, has been reported as the most prevalent mechanism in Campylobacter isolated from animals and humans (64–67). In the present investigation, 93.6% of C. jejuni isolates which were phenotypically resistant to both, ciprofloxacin and nalidixic acid, carried this point mutation in gyrA. This agrees with previous studies in which this mutation was identified in fluoroquinolone-resistant C. jejuni isolated from ruminants and poultry in Spain and Germany by SNP-PCR (68–70). These studies showed that this mutation is present for a long time in C. jejuni and still poses concern in isolates from farm animals.
The monitoring of the antimicrobial use in broilers in Germany between 2010 and 2016 showed the highest usage for aminoglycosides followed by fluoroquinolones and a substantial decrease for macrolides and tetracyclines. In turkey flocks, fluoroquinolones were used most frequently, followed by tetracyclines and macrolides. However, in contrast to broilers, the use of aminoglycosides was low in turkeys (30). Mechanisms of aminoglycoside resistance in Campylobacter were attributed to enzymatic drug modification (71) and mutations at the ribosomal binding sites (72). Aminoglycoside phosphotransferases (APHs) in Campylobacter are mainly encoded by the aph(3′)-III, which confers resistance to neomycin and amikacin whereas the aph(2″)-Ic gene confers resistance to gentamicin. Aminoglycoside O-nucleotidyltransferases (ANTs) in Campylobacter include ANT 6 and ANT 9, which confer resistance to streptomycin and spectinomycin, respectively (71, 73). The ant6-I genes encoding aminoglycoside O-nucleotidyl-transferases are widely spread among streptomycin-resistant C. jejuni (74). In this study, genes coding for aminoglycoside-modifying enzymes (APHs and ANTs) which confer resistance to amikacin, neomycin, gentamicin, streptomycin and spectinomycin were identified.
PCR was used previously for the determination of streptomycin resistance genes and to recognize ant[6]-Ia, ant[6]-Ib and other ant-like genes (74). Out of 10 phenotypically streptomycin-resistant C. jejuni, a gene cluster comprising the genes sat4, aph(3′)-IIIa and aadE was present in six (60%) connected with aminoglycoside resistance using WGS analysis.
In this study all isolates were phenotypically susceptible to erythromycin despite that five isolates carried a mutation in the gene for the ribosomal protein L22 that resulted in the amino acid substitution A103V, associated with macrolide resistance. A recent study conducted to investigate the genetic basis of antimicrobial resistance in C. coli and C. jejuni isolated from food animals, poultry processing and retail meat showed that the 23S rRNA (A2075G) mutation was identified only in C. coli isolates, while C. jejuni were more likely to harbor the aforementioned mutation in the gene for the L22 protein (23).
As Campylobacter are commensal bacteria that are exposed to various antimicrobial agents used in veterinary medicine, additional resistance mechanisms evolved in Campylobacter (5). The genes coding for aminoglycosides resistance are usually plasmid-borne (5). A mutation in the rpsL gene encoding the ribosomal protein S12 associated with streptomycin resistance was reported only in C. coli (72). The contribution of efflux to aminoglycoside resistance in Campylobacter is not completely proved, but is likely to be less important than the plasmid-borne genes coding for drug-modifying enzymes (5). Lack of knowledge may explain why resistance-associated genes have not been detected by WGS in four phenotypically streptomycin-resistant C. jejuni in the current investigation.
Here, the detection of genetic factors for AMR was performed using different tools and databases (AMRFinder, ResFinder, NCBI) in order not to miss any loci, as there is no single method that might be sufficient for the purpose alone (Table 2). Feldgarden et al. (45) found that AMRFinder appears to be a highly accurate AMR gene detection system (45).
In agreement with the results of this study, other previously published WGS-based studies demonstrated an overall very good concordance between genomic prediction and phenotypic determination of AMR (59, 67, 75, 76). Comparable results were reported by Feldgarden et al. for C. jejuni, using the NCBI AMRFinder tool, e.g., with a 98.9% correlation rate (45). A high correlation rate of 97.5% was also found in a recent study from England and Wales (76). There are multiple explanations for possible discrepancies between genotype and phenotype. There might be technical issues, such as low assembly quality (77–79). In this study, high quality of Illumina sequencing was achieved while long read sequencing might improve assembly contiguity (77) which may be helpful especially for plasmid detection and analysis (80). Another technical factor might be incomplete databases, which was counteracted by utilizing several tools. Microbiological factors influencing AMR genotype-phenotype correlation include transcriptional regulation, over-expression and under-expression of genes (e.g., efflux pumps), protein activation or modification or as well as novel resistance genes and mutations may be missed by the currently available databases and search tools (45, 77, 79).
Plasmids played an essential role in the ability of pathogenic bacteria to particularly overcome a new environment and are frequently associated with their virulence. Knowledge of plasmid genetics is significant for the understanding of the evolution and the origin of drug resistance genes (81). In the current investigation, 28 plasmid-borne contigs were detected in sequenced isolates using the Illumina MiSeq. The pTet-like plasmid-borne contig, which carries a tet(O) gene, was detected in six phenotypically resistant isolates. It has been reported previously that the tetracycline resistance is not always associated with the presence of pTet-like and in some isolates the gene is located on the chromosome (82). High prevalence of tetracycline resistant Campylobacter in chicken was explained in a previous study showed that horizontal transfer of tet(O) occurs rapidly and spontaneously without antimicrobial selection pressure between C. jejuni isolates in their intestine (83).
The results of this study emphasize the impact of WGS for in-depth genotyping, screening of virulence, clonality and antimicrobial resistance determinants in C. jejuni. In the present study, the antimicrobial resistance genes were mostly identified on the bacterial chromosome, while pTet-like plasmid-borne contigs that harbored the tet(O) gene were identified in six C. jejuni isolates from different regions (Bavaria and Thuringia) and ST types (122 and 50), suggesting intra-species dissemination of these types of plasmids. Combination of AMR databases are helpful for improving AMR detection in the absence of phenotypic data. Despite the high degree of correlation between phenotypic resistance and genotypes, the phenotypic susceptibility testing is still necessary.
This study revealed a relatively high genetic diversity of C. jejuni isolated from turkeys in German flocks while also genetically highly similar isolates were detected. This indicates persistence as well as spread of some C. jejuni clones. This finding has to be explored in the future in more detail.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: European Nucleotide Archive (ENA); PRJEB55640.
Ethics approval was not required for the study on animals because the bacterial isolates were taken from the authors' reference laboratory for Campylobacteriosis.
Conceived and designed the experiments: HE-A, HH, HN, JL, and HMH. Performed the experiments: HE-A, HH, and JL. Analyzed the data: HE-A, HH, HMH, SG-S, and JL. Contributed to the reagents/materials/analysis tools: HE-A, HH, HMH, HT, HN, SS, and JL. Wrote and corrected the manuscript: HE-A, HH, JL, SS, and HMH. Participated in the WGS that were used in the study: HE-A, SG-S, and JL. All authors read and approved the final manuscript.
We thank A. Hackbart, K. Cerncic, B. Hofmann and P. Methner at Friedrich-Loeffler-Institut, Jena, Germany for their excellent technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1092179/full#supplementary-material
1. Schets FM, Jacobs-Reitsma WF, van der Plaats RQJ, Heer LK, van Hoek AHAM, Hamidjaja RA, et al. Prevalence and types of Campylobacter on poultry farms and in their direct environment. J Water Health. (2017) 15:849–62. doi: 10.2166/wh.2017.119
2. Kaakoush NO, Castaño-Rodríguez N, Mitchell HM, Man SM. Global epidemiology of Campylobacter infection. Clin Microbiol Rev. (2015) 28:687–720. doi: 10.1128/CMR.00006-15
3. French NP Ji Z, Glen PC, Anne CM, Patrick JB, Kristin D, et al. Genomic analysis of fluoroquinolone- and tetracycline-resistant Campylobacter jejuni sequence type 6964 in humans and poultry, New Zealand, 2014–2016. Emerg Infect Dis. (2019) 25:2226. doi: 10.3201/eid2512.190267
4. Igwaran A, Okoh A. Human campylobacteriosis: a public health concern of global importance. Heliyon. (2019) 5:e02814-e. doi: 10.1016/j.heliyon.2019.e02814
5. Iovine NM. Resistance mechanisms in Campylobacter jejuni. Virulence. (2013) 4:230–40. doi: 10.4161/viru.23753
6. Manning G, Duim B, Wassenaar T, Wagenaar JA, Ridley A, Newell DG. Evidence for a genetically stable strain of Campylobacter jejuni. App Environ Microbiol. (2001) 67:1185–9. doi: 10.1128/AEM.67.3.1185-1189.2001
7. Schielke A, Rosner BM, Stark K. Epidemiology of campylobacteriosis in Germany – insights from 10 years of surveillance. BMC Infect Dis. (2014) 14:30. doi: 10.1186/1471-2334-14-30
8. Hartnack S, Doherr MG, Alter T, Toutounian-Mashad K, Greiner M. Campylobacter monitoring in German broiler flocks: an explorative time series analysis. Zoonoses Public Health. (2009) 56:117–28. doi: 10.1111/j.1863-2378.2008.01184.x
9. Atanassova V, Ring C. Prevalence of Campylobacter spp. in poultry and poultry meat in Germany. Int J Food Microbiol. (1999) 51:187–90. doi: 10.1016/S0168-1605(99)00120-8
10. Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. (2012) 13:601–12. doi: 10.1038/nrg3226
11. Biggs PJ, Fearnhead P, Hotter G, Mohan V, Collins-Emerson J, Kwan E, et al. Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage. PLoS ONE. (2011) 6:e27121. doi: 10.1371/journal.pone.0027121
12. Carrillo CD, Kruczkiewicz P, Mutschall S, Tudor A, Clark C, Taboada EN. A framework for assessing the concordance of molecular typing methods and the true strain phylogeny of Campylobacter jejuni and C. coli using draft genome sequence data. Front Cell Infect Microbiol. (2012) 2:57. doi: 10.3389/fcimb.2012.00057
13. Dingle KE, Colles FM, Wareing DR, Ure R, Fox AJ, Bolton FE, et al. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. (2001) 39:14–23. doi: 10.1128/JCM.39.1.14-23.2001
14. Magnússon SH, Guð*mundsdóttir S, Reynisson E, Rúnarsson ÁR, Harð*ardóttir H, Gunnarson E, et al. Comparison of Campylobacter jejuni isolates from human, food, veterinary and environmental sources in Iceland using PFGE, MLST and fla-SVR sequencing. J Appl Microbiol. (2011) 111:971–81. doi: 10.1111/j.1365-2672.2011.05100.x
15. Revez J, Llarena A, Schott T, Kuusi M, Hakkinen M, Kivistö R, et al. Genome analysis of Campylobacter jejuni strains isolated from a waterborne outbreak. BMC Genomics. (2014) 15:768. doi: 10.1186/1471-2164-15-768
16. Llarena A, Zhang J, Vehkala M, Välimäki N, Hakkinen M, Hänninen M, et al. Monomorphic genotypes within a generalist lineage of Campylobacter jejuni show signs of global dispersion. Microb genom. (2016) 2:e000088. doi: 10.1099/mgen.0.000088
17. Köser CU, Ellington MJ, Cartwright EJP, Gillespie SH, Brown NM, Farrington M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. (2012) 8:e1002824. doi: 10.1371/journal.ppat.1002824
18. Redondo N, Carroll A, McNamara E. Molecular characterization of Campylobacter causing human clinical infection using whole-genome sequencing: virulence, antimicrobial resistance and phylogeny in Ireland. PLoS ONE. (2019) 14:e0219088. doi: 10.1371/journal.pone.0219088
19. Katrine GJ, Kristoffer K, Mette RG, Birgitte N, Jørgen E, Hanne MH, et al. Whole-Genome Sequencing to detect numerous Campylobacter jejuni outbreaks and match patient isolates to sources, Denmark, 2015-2017. Emerg Infect Dis. (2020) 26:523. doi: 10.3201/eid2603.190947
20. Cantero G, Correa-Fiz F, Ronco T, Strube M, Cerdà-Cuéllar M, Pedersen K. Characterization of Campylobacter jejuni and Campylobacter coli broiler isolates by whole-genome sequencing. Foodborne Pathog Dis. (2018) 15:145–52. doi: 10.1089/fpd.2017.2325
21. Aksomaitiene J, Ramonaite S, Novoslavskij A, Malakauskas M, Kudirkiene E. Draft genome sequence of ciprofloxacin and ceftriaxone resistant Campylobacter jejuni MM26-781 assigned to novel ST isolated from common pigeon in Lithuania. Evolut Bioinform. (2019) 15:1176934319868469. doi: 10.1177/1176934319868469
22. Weis AM, Storey DB, Taff CC, Townsend AK, Huang BC, Kong NT, et al. Genomic comparison of Campylobacter spp. and their potential for zoonotic transmission between birds, primates, and livestock. Appl Environ Microbiol. (2016) 82:7165–75. doi: 10.1128/AEM.01746-16
23. Hull DM, Harrell E, van Vliet AHM, Correa M, Thakur S. Antimicrobial resistance and interspecies gene transfer in Campylobacter coli and Campylobacter jejuni isolated from food animals, poultry processing, and retail meat in North Carolina, 2018–2019. PLoS ONE. (2021) 16:e0246571. doi: 10.1371/journal.pone.0246571
24. Maiden MCJ, van Rensburg MJJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. (2013) 11:728–36. doi: 10.1038/nrmicro3093
25. Bogaerts B, Winand R, Fu Q, Van Braekel J, Ceyssens P, Mattheus W, et al. Validation of a bioinformatics workflow for routine analysis of whole-genome sequencing data and related challenges for pathogen typing in a European National Reference Center: Neisseria meningitidis as a Proof-of-Concept. Front Microbiol. (2019) 10. doi: 10.3389/fmicb.2019.00362
26. Köser CU, Ellington MJ, Peacock SJ. Whole-genome sequencing to control antimicrobial resistance. Trends Genetics. (2014) 30:401–7. doi: 10.1016/j.tig.2014.07.003
27. Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. (2012) 13:341. doi: 10.1186/1471-2164-13-341
28. Prendergast DM, Lynch H, Whyte P, Golden O, Murphy D, Gutierrez M, et al. Genomic diversity, virulence and source of Campylobacter jejuni contamination in Irish poultry slaughterhouses by whole genome sequencing. J Appl Microbiol. (2022) 133:3150–60. doi: 10.1111/jam.15753
29. Commission. E. Commission implementing decision 2013/652/EU of 12 November 2013 on the monitoring and reporting of antimicrobial resistance in zoonotic and commensal bacteria. Off J Eur Union L. (2013) 303:26-39.
30. Tenhagen BA, Flor M, Alt K, Knüver M, Buhler C, Käsbohrer A, et al. Association of antimicrobial resistance in Campylobacter spp. in broilers and turkeys with antimicrobial use. Antibiotics. (2021) 10:673. doi: 10.3390/antibiotics10060673
31. El-Adawy H, Hotzel H, Düpre S, Tomaso H, Neubauer H, Hafez HM. Determination of antimicrobial sensitivities of Campylobacter jejuni isolated from commercial turkey farms in Germany. Avian Dis. (2012) 56:685–92. doi: 10.1637/10135-031912-Reg.1
32. ISO. Microbiology of Food and Animal Feeding Stuffs – Horizontal Method for Detection and Enumeration of Campylobacter spp. – Part 1: Detection Method. Geneva: International Standards Organization (2017).
33. CLSI. Methods for Antimicrobial Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria Isolated From Animals, 1st Edition, CLSI supplement VET06. Wayne, PA (2017).
34. CLSI. Performance Standards for Antimicrobial Susceptibility Testing: 32nd Informational Supplement M100. Wayne, PA: CLSI (2022).
35. Gu W, Siletzky RM, Wright S, Islam M, Kathariou S. Antimicrobial susceptibility profiles and strain type diversity of Campylobacter jejuni isolates from turkeys in eastern North Carolina. Appl Environ Microbiol. (2009) 75:474–82. doi: 10.1128/AEM.02012-08
36. Vali L, Hamouda A, Hoyle DV, Pearce MC, Whitaker LHR, Jenkins C, et al. Antibiotic resistance and molecular epidemiology of Escherichia coli O26, O103 and O145 shed by two cohorts of Scottish beef cattle. J Antimicrob Chemother. (2007) 59:403–10. doi: 10.1093/jac/dkl491
37. Koga T, Aoki W, Mizuno T, Wakazono K, Ohno J, Nakai T, et al. Antimicrobial resistance in Campylobacter coli and Campylobacter jejuni in cynomolgus monkeys (Macaca fascicularis) and eradication regimens. J Microbiol Immunol Infect. (2017) 50:75–82. doi: 10.1016/j.jmii.2014.12.006
38. Dobbin G, Hariharan H, Daoust PY, Hariharan S, Heaney S, Coles M, et al. Bacterial flora of free-living double-crested cormorant (Phalacrocorax auritus) chicks on Prince Edward Island, Canada, with reference to enteric bacteria and antibiotic resistance. Comp Immunol Microbiol Infect Dis. (2005) 28:71–82. doi: 10.1016/S0147-9571(04)00059-1
39. Andrews S,. A Quality Control Tool for High Throughput Sequence Data UK. (2018). Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed August 26, 2022).
40. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
41. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Gen Biol. (2019) 20:257. doi: 10.1186/s13059-019-1891-0
42. Florensa AF, Kaas RS, Clausen P, Aytan-Aktug D, Aarestrup FM. ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb Genom. (2022) 8:748. doi: 10.1099/mgen.0.000748
43. Sayers EW, Beck J, Brister JR, Bolton EE, Canese K, Comeau DC, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. (2020) 48:D9–d16. doi: 10.1093/nar/gkz899
44. Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. (2005) 33:D325–D8. doi: 10.1093/nar/gki008
45. Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. (2019) 63:19. doi: 10.1128/AAC.00483-19
46. Schwengers O, Barth P, Falgenhauer L, Hain T, Chakraborty T, Goesmann A. Platon: identification and characterization of bacterial plasmid contigs in short-read draft assemblies exploiting protein sequence-based replicon distribution scores. Microb Genom. (2020) 6:398. doi: 10.1099/mgen.0.000398
47. Karlyshev AV. Complete genome sequencing of Campylobacter jejuni strain X reveals the presence of pVir- and pTet-like plasmids. Microbiol Resour Announc. (2022) 11:e0042122. doi: 10.1128/mra.00421-22
48. Marasini D, Karki AB, Buchheim MA, Fakhr MK. Phylogenetic relatedness among plasmids harbored by Campylobacter jejuni and Campylobacter coli isolated from retail meats. Front Microbiol. (2018) 9:2167. doi: 10.3389/fmicb.2018.02167
49. Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom. (2021) 7:1–13. doi: 10.1099/mgen.0.000685
50. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
51. Llarena A, Taboada E, Rossi M. Whole-Genome Sequencing in epidemiology of Campylobacter jejuni infections. J Clin Microbiol. (2017) 55:1269–75. doi: 10.1128/JCM.00017-17
52. Guirado P, Paytubi S, Miró E, Iglesias-Torrens Y, Navarro F, Cerdà-Cuéllar M, et al. Differential distribution of the wlaN and cgtB genes, associated with Guillain-Barré Syndrome, in Campylobacter jejuni isolates from humans, broiler chickens, and wild birds. Microorganisms. (2020) 8:325. doi: 10.3390/microorganisms8030325
53. Sierra-Arguello YM, Perdoncini G, Rodrigues LB. Ruschel dos Santos L, Apellanis Borges K, Quedi Furian T, et al. Identification of pathogenic genes in Campylobacter jejuni isolated from broiler carcasses and broiler slaughterhouses. Sci Rep. (2021) 11:4588. doi: 10.1038/s41598-021-84149-1
54. Datta S, Niwa H, Itoh K. Prevalence of 11 pathogenic genes of Campylobacter jejuni by PCR in strains isolated from humans, poultry meat and broiler and bovine faeces. J Med Microbiol. (2003) 52:345–8. doi: 10.1099/jmm.0.05056-0
55. Müller J, Schulze F, Müller W, Hänel I, PCR. detection of virulence-associated genes in Campylobacter jejuni strains with differential ability to invade Caco-2 cells and to colonize the chick gut. Vet Microbiol. (2006) 113:123–9. doi: 10.1016/j.vetmic.2005.10.029
56. Emanowicz M, Meade J, Burgess C, Bolton D, Egan J, Lynch H, et al. Antimicrobial resistance and genomic diversity of Campylobacter jejuni isolates from broiler caeca and neck skin samples collected at key stages during processing. Food Control. (2022) 135:108664. doi: 10.1016/j.foodcont.2021.108664
57. Rossi DA, Dumont CF, Santos ACS, Vaz MEL, Prado RR, Monteiro GP, et al. Antibiotic resistance in the alternative lifestyles of Campylobacter jejuni. Front Cell Infect Microbiol. (2021) 11:535757. doi: 10.3389/fcimb.2021.535757
58. Warburton PJ, Amodeo N, Roberts AP. Mosaic tetracycline resistance genes encoding ribosomal protection proteins. J Antimicrob Chemother. (2016) 71:3333–9. doi: 10.1093/jac/dkw304
59. Ocejo M, Oporto B, Lavín JL, Hurtado A. Whole genome-based characterisation of antimicrobial resistance and genetic diversity in Campylobacter jejuni and Campylobacter coli from ruminants. Sci Rep. (2021) 11:8998. doi: 10.1038/s41598-021-88318-0
60. Cobo-Díaz JF, González Del Río P, Álvarez-Ordóñez A. Whole Resistome analysis in Campylobacter jejuni and C. coli genomes available in public repositories. Front Microbiol. (2021) 12:662144. doi: 10.3389/fmicb.2021.662144
61. Authority. EFS. Report of the task force on zoonoses data collection including a proposal for a harmonized monitoring scheme of antimicrobial resistance in Salmonella in fowl (Gallus gallus), turkeys and pigs and Campylobacter jejuni and C. coli in broilers. EFSA J. (2007) 5:96r. doi: 10.2903/j.efsa.2007.96r
62. Aarestrup FM, Engberg J. Antimicrobial resistance of thermophilic Campylobacter. Vet Res. (2001) 32:311–21. doi: 10.1051/vetres:2001127
63. Marotta F, Garofolo G, di Marcantonio L, Di Serafino G, Neri D, Romantini R, et al. Antimicrobial resistance genotypes and phenotypes of Campylobacter jejuni isolated in Italy from humans, birds from wild and urban habitats, and poultry. PLoS ONE. (2019) 14:e0223804. doi: 10.1371/journal.pone.0223804
64. Luangtongkum T, Jeon B, Han J, Plummer P, Logue CM, Zhang Q. Antibiotic resistance in Campylobacter: emergence, transmission and persistence. Future Microbiol. (2009) 4:189–200. doi: 10.2217/17460913.4.2.189
65. Dahl LG, Joensen KG, Østerlund MT, Kiil K, Nielsen EM. Prediction of antimicrobial resistance in clinical Campylobacter jejuni isolates from whole-genome sequencing data. Eur J Clin Microbiol Infect Dis. (2021) 40:673–82. doi: 10.1007/s10096-020-04043-y
66. Meistere I, Kibilds J, Eglite L, Alksne L, Avsejenko J, Cibrovska A, et al. Campylobacter species prevalence, characterisation of antimicrobial resistance and analysis of whole-genome sequence of isolates from livestock and humans, Latvia, 2008 to 2016. Euro Surveill. (2019) 24:1800357. doi: 10.2807/1560-7917.ES.2019.24.31.1800357
67. Zhao S, Tyson GH, Chen Y, Li C, Mukherjee S, Young S, et al. Whole-genome sequencing analysis accurately predicts antimicrobial resistance phenotypes in Campylobacter spp. App Environ Microbiol. (2016) 82:459–66. doi: 10.1128/AEM.02873-15
68. Ocejo M, Oporto B, Hurtado A. Occurrence of Campylobacter jejuni and Campylobacter coli in cattle and sheep in Northern Spain and changes in antimicrobial resistance in two studies 10-years apart. Pathogens. (2019) 8: 8030098. doi: 10.3390/pathogens8030098
69. Oporto B, Juste RA, Hurtado A. Phenotypic and genotypic antimicrobial resistance profiles of Campylobacter jejuni Isolated from cattle, sheep, and free-range poultry faeces. Int J Microbiol. (2009) 2009:456573. doi: 10.1155/2009/456573
70. El-Adawy H, Ahmed MF, Hotzel H, Tomaso H, Tenhagen BA, Hartung J, et al. Antimicrobial susceptibilities of Campylobacter jejuni and Campylobacter coli recovered from organic turkey farms in Germany. Poult Sci. (2015) 94:2831–7. doi: 10.3382/ps/pev259
71. Fabre A, Oleastro M, Nunes A, Santos A, Sifré E, Ducournau A, et al. Whole-genome sequence analysis of multidrug-resistant Campylobacter isolates: a Focus on aminoglycoside resistance determinants. J Clin Microbiol. (2018) 56:e00390–18. doi: 10.1128/JCM.00390-18
72. Olkkola S, Juntunen P, Heiska H, Hyytiäinen H, Hänninen M. Mutations in the rpsL gene are involved in Streptomycin resistance in Campylobacter coli. Microb Drug Resist. (2010) 16:105–10. doi: 10.1089/mdr.2009.0128
73. Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug Resist Updat. (2010) 13:151–71. doi: 10.1016/j.drup.2010.08.003
74. Hormeño L, Ugarte-Ruiz M, Palomo G, Borge C, Florez-Cuadrado D, Vadillo S, et al. ant-I genes encoding aminoglycoside O-nucleotidyltransferases are widely spread among streptomycin resistant strains of Campylobacter jejuni and Campylobacter coli. Front Microbiol. (2018) 9:2515. doi: 10.3389/fmicb.2018.02515
75. Rokney A, Valinsky L, Vranckx K, Feldman N, Agmon V, Moran-Gilad J, et al. WGS-based prediction and analysis of antimicrobial resistance in Campylobacter jejuni isolates from Israel. Front Cell Infect Microbiol. (2020) 10:365. doi: 10.3389/fcimb.2020.00365
76. Painset A, Day M, Doumith M, Rigby J, Jenkins C, Grant K, et al. Comparison of phenotypic and WGS-derived antimicrobial resistance profiles of Campylobacter jejuni and Campylobacter coli isolated from cases of diarrhoeal disease in England and Wales, 2015–16. J Antimicrob Chemoth. (2020) 75:883–9. doi: 10.1093/jac/dkz539
77. Chen CY, Clark CG, Langner S, Boyd DA, Bharat A, McCorrister SJ, et al. Detection of antimicrobial resistance using proteomics and the comprehensive antibiotic resistance database: a case study. Proteomics Clin Appl. (2020) 14:e1800182. doi: 10.1002/prca.201800182
78. Doyle RM, O'Sullivan DM, Aller SD, Bruchmann S, Clark T, Coello Pelegrin A, et al. Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study. Microb Genom. (2020) 6:335. doi: 10.1099/mgen.0.000335
79. Zankari E, Allesøe R, Joensen KG, Cavaco LM, Lund O, Aarestrup FM. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemother. (2017) 72:2764–8. doi: 10.1093/jac/dkx217
80. Collineau L, Boerlin P, Carson CA, Chapman B, Fazil A, Hetman B, et al. Integrating whole-genome sequencing data into quantitative risk assessment of foodborne antimicrobial resistance: a review of opportunities and challenges. Front Microbiol. (2019) 10:1107. doi: 10.3389/fmicb.2019.01107
81. Batchelor RA, Pearson BM, Friis LM, Guerry P, Wells JM. Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiol. (2004) 150:3507–17. doi: 10.1099/mic.0.27112-0
82. Pratt A, Korolik V. Tetracycline resistance of Australian Campylobacter jejuni and Campylobacter coli isolates. J Antimicrob Chemother. (2005) 55:452–60. doi: 10.1093/jac/dki040
Keywords: Campylobacter jejuni, turkeys, genetic diversity, WGS, MLST, resistome, virulome
Citation: El-Adawy H, Hotzel H, García-Soto S, Tomaso H, Hafez HM, Schwarz S, Neubauer H and Linde J (2023) Genomic insight into Campylobacter jejuni isolated from commercial turkey flocks in Germany using whole-genome sequencing analysis. Front. Vet. Sci. 10:1092179. doi: 10.3389/fvets.2023.1092179
Received: 07 November 2022; Accepted: 23 January 2023;
Published: 16 February 2023.
Edited by:
Filippo Giarratana, University of Messina, ItalyCopyright © 2023 El-Adawy, Hotzel, García-Soto, Tomaso, Hafez, Schwarz, Neubauer and Linde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hosny El-Adawy,  aG9zbnkuZWxhZGF3eUBmbGkuZGU=
aG9zbnkuZWxhZGF3eUBmbGkuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.