 Iryna V. Goraichuk1,2
Iryna V. Goraichuk1,2 Anton Gerilovych1
Anton Gerilovych1 Vitaliy Bolotin1Olexii Solodiankin1
Vitaliy Bolotin1Olexii Solodiankin1 Kiril M. Dimitrov2Oleksandr Rula1Nataliia Muzyka1Oleksandr Mezinov3Borys Stegniy1Olena Kolesnyk1Mary J. Pantin-Jackwood2
Kiril M. Dimitrov2Oleksandr Rula1Nataliia Muzyka1Oleksandr Mezinov3Borys Stegniy1Olena Kolesnyk1Mary J. Pantin-Jackwood2 Patti J. Miller2
Patti J. Miller2 Claudio L. Afonso2
Claudio L. Afonso2 Denys Muzyka1,4*
Denys Muzyka1,4*- 1National Scientific Centre, Institute of Experimental and Clinical Veterinary Medicine, Kharkiv, Ukraine
- 2Exotic and Emerging Avian Viral Diseases Research Unit, Southeast Poultry Research Laboratory, US National Poultry Research Center, Agricultural Research Service, USDA, Athens, GA, United States
- 3The F.E. Falz-Fein Biosphere Reserve “Askania Nova”, National Academy of Agrarian Sciences of Ukraine, Askania-Nova, Kherson Oblast, Ukraine
- 4Department of Zoology, H.S. Skovoroda Kharkiv National Pedagogical University, Kharkiv, Ukraine
Newcastle disease virus (NDV) infects a wide range of bird species worldwide and is of importance to the poultry industry. Although certain virus genotypes are clearly associated with wild bird species, the role of those species in the movement of viruses and the migratory routes they follow is still unclear. In this study, we performed a phylogenetic analysis of nineteen NDV sequences that were identified among 21,924 samples collected from wild and synanthropic birds from different regions of Ukraine from 2006 to 2015 and compared them with isolates from other continents. In synanthropic birds, NDV strains of genotype II, VI, VII, and XXI of class II were detected. The fusion gene sequences of these strains were similar to strains detected in birds from different geographical regions of Europe and Asia. However, it is noteworthy to mention the isolation of vaccine viruses from synanthropic birds, suggesting the possibility of their role in viral transmission from vaccinated poultry to wild birds, which may lead to the further spreading of vaccine viruses into other regions during wild bird migration. Moreover, here we present the first publicly available complete NDV F gene from a crow (genus Corvus). Additionally, our phylogenetic results indicated a possible connection of Ukrainian NDV isolates with genotype XXI strains circulating in Kazakhstan. Among strains from wild birds, NDVs of genotype 1 of class I and genotype I of class II were detected. The phylogenetic analysis highlighted the possible exchange of these NDV strains between wild waterfowl from the Azov-Black Sea region of Ukraine and waterfowl from different continents, including Europe, Asia, and Africa.
1. Introduction
Newcastle disease virus (NDV), or Avian orthoavulavirus 1, is a member of the recently separated subfamily Avulavirinae in the Paramyxoviridae family (1). NDV is capable of infecting a wide range of bird species and is of great economic importance to both large poultry enterprises and private farms. Since the discovery of the first NDV in the 1920s, an array of data has accumulated illustrating the genetic and pathogenic diversity of the viral strains (2). According to the Terrestrial Manual (World Organization for Animal Health, formerly the Office International des Epizooties), (WOAH, founded as OIE) Terrestrial Manual, NDV strains are divided into three main pathogenicity groups: velo-, meso-, and lentogenic (3, 4). Velogenic strains cause hemorrhages, reduced egg production, intestinal lesions, and neurological symptoms with high mortality in infected chickens. Mesogenic strains cause disease with respiratory or neurological symptoms but little mortality. Lentogenic strains can cause subclinical respiratory or intestinal infections and are considered low-virulent. Avian orthoavulavirus 1 strains are divided into two classes based on the nucleotide sequence of the fusion (F) protein gene. Class I consists mainly of lentogenic strains isolated from wild birds worldwide and includes only one genotype (5). Class II contains 21 genotypes with velo-, meso- and lentogenic strains that have been detected in a wide variety of host species around the world (6).
The fusion protein is an important determinant of NDV pathogenicity (7, 8). It is synthesized as an inactive precursor (F0), which is proteolytically cleaved by host proteases into two polypeptides (F1 and F2) for the virus particles to be infectious. The efficiency of proteolytic cleavage is dependent on the host cell and the virus strain (9–11). F proteins of velogenic and mesogenic NDV virus strains are characterized by the presence of multiple basic amino acids at the F0 cleavage sequence which are recognized by ubiquitous host cell proteases (12–14). In contrast, the F0 protein of lentogenic strains has a monobasic cleavage site, which is cleavable by a restricted number of certain host proteases. These differences in the F gene sequences which correlate with different virulence phenotypes are prime targets for the development of molecular biological approaches to identify and characterize NDV isolates. However, the viral population may acquire mutations and adaptive changes in response to different pressures by the host's immune system (15, 16). Together with large genetic diversity it can affect real-time PCR specificity for NDV detection, particularly virulence determination (17, 18) and vaccine efficacy (19, 20) since both are typically based on the genetic sequence of the F protein, most often affected by vaccine pressure.
Although a wealth of information about this virus has accumulated to date, the role of wild birds in the global circulation and epidemiology of certain NDV genotypes remains unclear. The expansion of the surveillance program for Avian orthoavulavirus 1 will improve our understanding of the global epidemiological picture, as well as help effectively prevent Newcastle Disease (ND). A large-scale study of the potential host range of NDV among wild birds in Africa showed the year-round presence of viruses, as well as their phylogenetic relationship with strains of domestic birds in the study area (21). Currently, in the territory of Eurasia, few studies of the potential host range of Avian orthoavulavirus 1 have been carried out, and the seasonality of infection has not been established. Even though there are many reports of NDV isolated from wild birds in this region, more than 30 countries remain unstudied or with limited data (22–29). Therefore, studies performed at the major stopping point locations for migratory birds can be of great contribution toward gaining more knowledge on the potential host range of NDV therein.
Ukraine occupies a unique geographical location in central and Eastern Europe, where the West Asia-East Africa flyways of wild migratory birds cross the Black Sea-Mediterranean, and East Atlantic flyways (30, 31). The natural conditions such as climate and the abundance of wetlands with an area of more than 590,000 ha in Ukraine contribute to the year-round presence of a large number of wild birds. This is especially evident during the period of seasonal migrations and wintering, when numerous wild bird species pass from North Asia and Europe to the Mediterranean, Africa, and Southwest Asia, and also cross from the Baltic and Caspian Seas to the Black and Mediterranean Seas, and from western Siberia and Kazakhstan to Western Europe and North Africa (32). The Azov-Black Sea region is one of the densest territories of Eastern Europe from an ornithological point of view. This region is historically an area of nesting, flight, migratory stops, and wintering for many bird species. Therefore, a large number of waterfowl and waterbirds from the Central part of Eurasia winter in the Azov-Black Sea region of Ukraine or stop there during migrations. As a result, wild bird populations from Ukraine and West Europe can interact with birds from Asia and Africa (33).
Domestic poultry also plays a role in the circulation of Avian orthoavulavirus 1. According to data from the United Nations Food and Agriculture Organization (FAO), the total poultry presence in the world (chickens, ducks, turkeys, geese, and guinea fowl) was ~35 billion birds in 2020 (34). Chickens accounted for 94% of the world's poultry population with about 46% of these located in the territory of Asia. To help keep ND under control in developed countries, industrial poultry farms are recommended to vaccinate their birds (mainly chickens and turkeys) and isolate them from the external environment to avoid contact with wild and synanthropic bird species (35, 36). But cases of possible transmission of the NDV vaccine strain from poultry to wild birds have been described (23, 37, 38). There are also reports of velogenic strains in wild birds (39). Currently, it is not well studied if lentogenic strains of NDV from wild birds may cause respiratory infection in poorly vaccinated poultry birds following exposure. However, a few cases of the change in the virus virulence were well-documented. Retrospective studies showed that cumulative mutations at the fusion protein cleavage site acquired through natural transmission at the poultry farms lead to change in the virulence of originally lentogenic virus during NDV outbreak of genotype I in Australia (40–42). Another study demonstrated an initially non-pathogenic strain from a wild duck acquiring pathogenicity through passaging in chickens (43).
Currently, the issue of the introduction and circulation of lentogenic NDV vaccine strains into the wild bird population is under consideration and discussion, thus it is important to assess the presence of vaccine spillovers to wild and synanthropic birds as well (23). Therefore, this article presents surveillance data of Avian orthoavulavirus 1 circulation in a natural reservoir from eight different regions in the South and East of Ukraine, including migratory bird's stopping points located in the Azov-Black Sea region. Part of these results was previously reported (5, 23, 27, 29, 44).
2. Materials and methods
2.1. Sample collection
Samples from wild and synanthropic birds were collected in Ukraine between 2006 and 2015 during active and passive surveillance (29, 45). Wild birds were defined as those occurring in a natural habitat other than poultry and synanthropic birds were defined as undomesticated birds that live in close association with people. A total of 21,924 samples were collected, among them, 21,854 samples were collected from 103 species of wild birds during active surveillance and 70 samples from synanthropic birds during passive surveillance. The sampling sites were located in the Azov-Black Sea region [Kherson, Mykolaiv, Odesa, Zaporizhzhya, and the Autonomous Republic (AR) of Crimea regions] and the East of Ukraine (Donetsk, Dnipro, and Kharkiv regions) (Figure 1). The samples' background data, including bird species and location, were recorded (Supplementary Tables S1, S2).
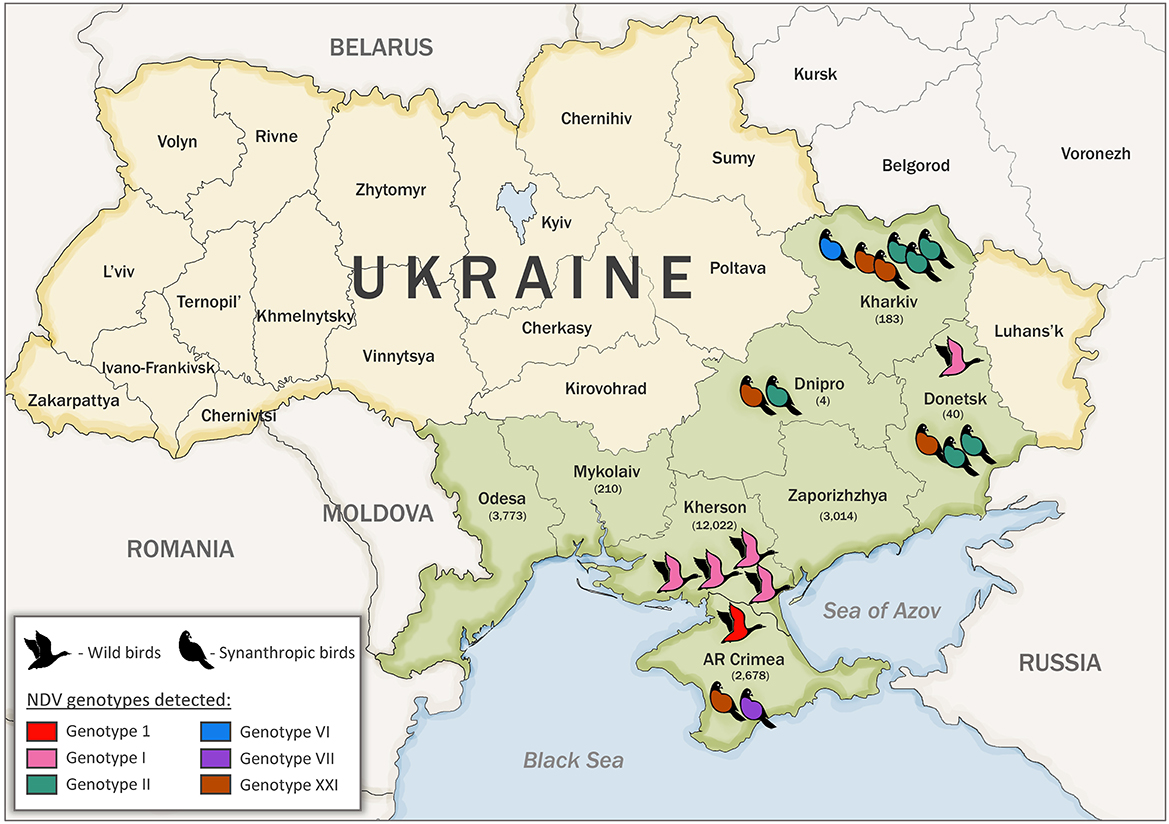
Figure 1. Sample collection number, bird type, and location in Ukraine. Sampled regions are indicated in green color. The number of collected samples are indicated in parenthesis under the name of each region. Genotypes detected in this study are shown in red (genotype 1, class I), pink (genotype I, class II), teal (genotype II, class II), blue (genotype VI, class II), purple (genotype VII, class II), and brown (genotype XXI, class II).
Sampling from wild birds was carried out in cooperation with ornithologists, who helped identify the bird species. Cloacal swabs were collected from apparently healthy live-trapped and hunted wild birds. Fresh feces were collected from places of mass bird accumulation. Feces were collected only if the origin and type of bird had been established. Immediately after sampling, samples of the biological material from wild birds were placed in tubes with a transport medium (Hank's balanced salt solution containing 0.5% lactalbumin, 10% glycerol, 200 U penicillin, 0.200 mg streptomycin, 100 U polymyxin, 0.250 mg gentamicin, and 50 U nystatin per ml) then stored and transported on ice to the laboratory and further stored in liquid nitrogen.
Samples of spleen, brain, liver, and intestines were collected from dead synanthropic birds (pigeons and gray crows) in Ukraine between 2006 and 2015. All dead synanthropic birds were found in locations of their natural habitat within cities in Kharkiv, Dnipro, Donetsk, Odesa, and AR Crimea regions. Samples were chilled at 4°C, transported to the lab, and stored at −80°C. Before analysis, samples were thawed and suspended in transport media (10% w/v).
2.2. Virus isolation and identification
Each fecal/cloacal swab medium or tissue suspension supernatant was inoculated (0.2 ml) into five 9- to 11-day-old specific-pathogen-free (SPF) embryonated chicken eggs (ECEs) using standard methods as described previously (46, 47). Allantoic fluids from all inoculated ECEs were harvested and tested for hemagglutination activity with chicken red blood cells using a hemagglutination assay (HA) (48). All HA-positive samples were analyzed in the hemagglutination inhibition (HI) assay using reference antisera to influenza A virus subtypes H1–H16 and avian paramyxoviruses (APMV-1–APMV-9) according to previous recommendations (47, 49, 50). The identification of avian influenza viruses and APMV-4, APMV-6, APMV-7, and APMV-13 have been previously described (29, 32, 51, 52).
Pathogenicity evaluation was performed on eleven selected NDV isolates (KF851268–KF851270, KJ914671, KJ914672, KU133362–KU133365, KY042127, KY042128, MZ101338) using the intracerebral pathogenicity index (ICPI) assay on 1-day-old SPF chickens following established procedures at the Southeast Poultry Research Laboratory (SEPRL), U.S. Department of Agriculture (USDA), Athens, GA, USA (3).
2.3. RNA extraction and sequencing
All HI-identified NDV isolates were subject to sequencing to determine their genotype and virulence. Viral RNA was extracted from infected allantoic fluids using TRIzol LS (Invitrogen, USA) following the manufacturer's instructions. The nucleotide sequences of the complete coding region of the F protein gene were determined by utilizing the RT-PCR/sequencing approach (53). The amplification reaction was performed using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Life Technologies, USA) with overlapped primer pairs for sequencing the complete F gene of different NDV genotypes from class I and II as described previously by Miller et al. (24). All RT-PCR products were subjected to electrophoresis in a 1% agarose gel (0.5X TBE). The appropriately sized DNA bands were excised from the gel and purified using the QuickClean II Gel Extraction Kit (GenScript, USA) and subjected to DNA sequencing. Nucleotide sequencing was performed for 19 samples on an ABI Sanger sequencer (Applied Biosystems, USA) with fluorescent dideoxy-nucleotide terminators at SEPRL, USDA, Athens, GA, USA. Sequence editing and assembly were performed using the SeqMan software of the LaserGene package (DNASTAR, USA).
2.4. Phylogenetic analysis
Genotype and sub-genotype identification were based on the phylogenetic topology and evolutionary distances between different taxonomic groups using a pilot dataset as described by Dimitrov et al. (5). Multiple sequence alignments of the NDV complete F gene sequences were produced using the MAFFT version 7 software (54). The pilot tree of class I and II NDV isolates (n = 98) was constructed using the Maximum-likelihood method based on the General Time-Reversible (GTR) model with a discrete gamma distribution (+G) and allowing for invariant sites (+I) with statistical analysis based on 1,000 bootstrap replicates, as implemented in MEGA7 (55). The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. For all analyses, the codon positions included were 1st, 2nd, 3rd, and non-coding, and all positions containing gaps and missing data were eliminated. A total of 1,661 positions were included in the pilot analysis of the complete F gene dataset.
For each genotype detected (1 of class I and I, II, VI, VII, and XXI of class II), more detailed phylogenetic trees were constructed using the most closely related sequences detected in BLAST. The Roman numerals presented in the name of each sequence in the phylogenetic tree represent the respective sub-genotype, followed by the GenBank accession number, host name, country of isolation, strain designation, and year of isolation (if available).
The complete F gene data set used for the phylogenetic analysis was also used to estimate the average evolutionary distances comparing Ukrainian NDV isolates to other relative strains. Pair-wise analysis was conducted using the maximum composite likelihood model using MEGA7 software (56). The rate variation among sites was modeled with a gamma distribution (shape parameter = 4).
3. Results
3.1. Geographic distribution of the viruses sequenced
The serological examination of biological material collected from 21,854 wild and 70 synanthropic birds belonging to 105 species and 11 different orders was conducted during 2006–2015. The largest number of samples was collected from birds of the order Anseriformes (15,013 samples), followed by Charadriiformes (4,737 samples), and Passeriformes (1,562 samples). The main sampling sites (~99% of the samples) for wild birds were located in the Azov Black Sea region. This region is the meeting point of the transcontinental migration routes of different wild birds from Siberia, Africa, Europe, and Asia. The rest of the biological samples were collected in the eastern region of Ukraine (Figure 1). NDV was identified in 31 (0.14%) samples from asymptomatic wild birds and 24 (34.29%) dead synanthropic birds by the HI test, part of which was previously reported (22, 29). All three overlapping regions of the complete F gene were successfully amplified by RT-PCR for 18 isolates, previously classified as NDV based on the serological HI test (Table 1). Two complete F genes of different NDV genotypes were amplified and sequenced for one isolate, which brought the total number of obtained sequences to 19. Some of the viruses were sequenced and previously published (5, 23, 27, 29, 44). Sequenced NDV isolates were obtained from dead synanthropic birds and asymptomatic wild waterfowl in different regions of Ukraine (Table 1). Twelve NDV isolates were attained from synanthropic birds (pigeons and crows). Isolates from synanthropic birds were obtained from four different regions: three originated from the Kharkiv region and one each from the Dnipro region, the Donetsk region, and the Autonomous Republic of Crimea (Figure 1). The six isolates from wild birds all came from the Kherson region in the south of Ukraine.
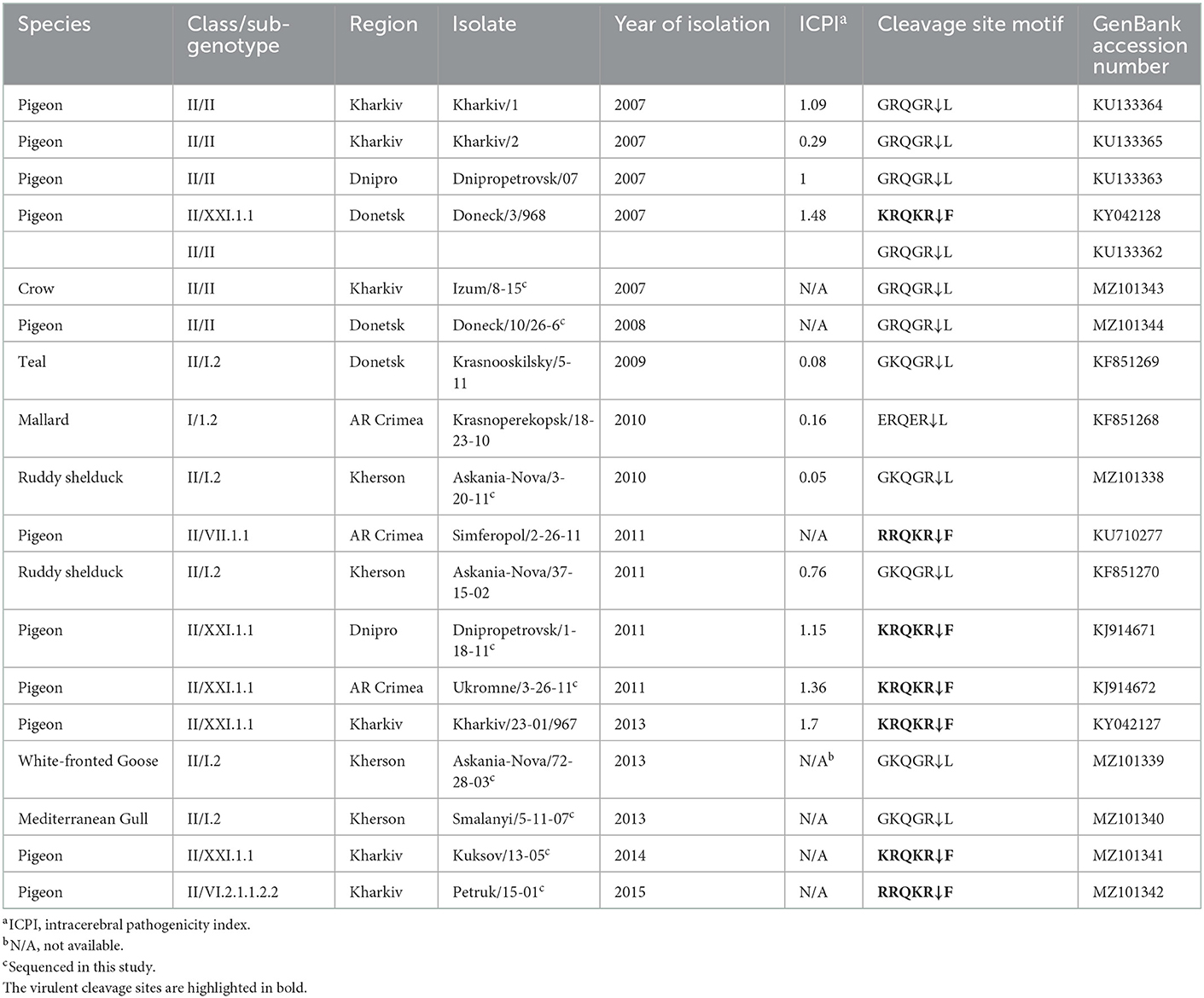
Table 1. Background information data for NDV isolates recovered in Ukraine between 2006 and 2015.
3.2. Pathotype characterization
NDV pathogenicity markers were examined for all 19 NDV sequences. The deduced amino acid sequences of the fusion protein cleavage site revealed that sequences of seven Ukrainian isolates from this study presented four basic amino acids at the C-terminus of the F2 protein from residues 112–116 and phenylalanine at residue 117 (Table 1, highlighted in bold), and based on the WOAH definition of vNDV molecular pathotyping (3) they are considered as virulent strains. The cleavage site motifs of these Ukrainian isolates were 112KRQKR↓117 (n = 5) and 112RRQKR↓F117 (n = 2). All seven viruses were isolated from synanthropic pigeons in the Dnipro, Donetsk, Kharkiv, and AR Crimea regions of Ukraine. The rest of the cleavage site motifs were represented by sequences 112GRQGR↓L117 (n = 6), 112GKQGR↓L117 (n = 5), and 112ERQGR↓L117 (n = 1) that are typical for viruses of low virulence.
Pathogenicity evaluation was performed for eleven Ukrainian viruses using ICPI. The ICPI values ranged from 0.05 to 1.7 (Table 1). Four viruses had an ICPI value below 0.7, which characterizes them as lentogenic (avirulent) which is in agreement with the predictions based on deduced amino acid cleavage site sequences. ICPI values equal to or above 0.7 indicate a virulent strain. Virulent strains with ICPI values below 1.5 are mesogenic (moderate virulence) and those with values above 1.5 are velogenic NDV strains (3, 55, 57). Thus, six viruses were classified as mesogenic and one (pigeon/Ukraine/Kharkiv/23-01-/967/2013) as velogenic (3, 55).
3.3. Genotypic characterization
A dataset of 79 complete F gene coding sequences of class I and II NDV isolates retrieved from GenBank was added to the 19 sequences from this study and used to construct a pilot tree for the preliminary identification of NDV genotypes as previously described (5). Based on the pilot tree, one NDV isolate belonged to class I and 18 to class II (Figure 2). Among class II isolates, five were of genotype I, six sequences were of genotype II, one was of genotype VI, one was of genotype VII, and five were of genotype XXI (Figure 2).

Figure 2. Phylogenetic analysis of NDV class I and II isolates based on the complete fusion gene sequences constructed with the maximum likelihood method, based on the general time-reversible model in MEGA v. 7.0.26. The percentage of trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The analysis involved 98 nucleotide sequences, 79 represent all class I and II sub-genotypes described by Dimitrov et al. (5) and 19 were collected from wild and synanthropic birds in Ukraine. All positions containing gaps and missing data were eliminated. There were a total of 1,661 positions in the final data set. The isolates used in this study are shown in colors. The Roman numerals presented in the taxa names in the phylogenetic tree represent the respective sub-genotype for each isolate, followed by the GenBank identification number, host name, country of isolation, strain designation, and year of isolation (if available).
3.4. Genotype 1 isolates
The single NDV isolate of class I was collected from a mallard in AR Crimea in 2010 (KF851268) and belongs to sub-genotype 1.2 of class I (Figure 3), previously known as sub-genotype 1c (5, 58). The highest homology of this isolate is with the two strains—pochard in Finland from 2006 (EU493454) and duck in China from 2007 (JF893453), sharing 98.81% nucleotide identity.

Figure 3. Phylogenetic analysis of NDV class I isolates based on the complete fusion gene sequences constructed with the maximum likelihood method with 1,000 bootstrap replicates. The analysis involved 44 nucleotide sequences. The tree was rooted to the oldest class I NDV isolate EF564833/Canada goose/USA/OH/78/1987. All positions containing gaps and missing data were eliminated. There were a total of 1,662 positions in the final data set. The isolates used in this study are shown in red. The Roman numerals on the right of the phylogenetic tree represent the respective sub-genotype for each isolate in accordance with the current classification (5) and the former nomenclature (58) is listed in parenthesis for reference.
3.5. Genotype I isolates
All five isolates of class II genotype I were classified as sub-genotype I.2 (Figure 4). These strains were collected from wild birds in the Kherson and Donetsk regions and shared between 98.50 and 99.82% nucleotide identity. Among them, the isolate collected from a white-fronted goose (MZ101339) had the highest nucleotide identity of 99.4% with the isolate collected from a Mediterranean gull (MZ101340) in 2013, which had an even higher nucleotide identity of 99.82% with another Ukrainian strain collected from ruddy shelduck (genus Tadorna) in 2011 (KF851270) (29). All these viruses were collected in the Kherson region. The fourth strain (MZ101338) from the Kherson region, isolated from ruddy shelduck in 2010, was most similar to an isolate originating from central Eurasia (Novosibirsk region) in 2010 (KX352836) (25) and shared 99.88% nucleotide identity. The only virus from the Donetsk region was collected from teal in 2009 (KF851269) and had the highest nucleotide homology with the strain isolated from mallard in Luxembourg in 2008 (HE972213) (59).

Figure 4. Phylogenetic analysis of NDV genotype I and II class II isolates based on the complete fusion gene sequences constructed with the maximum likelihood method with 1,000 bootstrap replicates. The analysis involved 65 nucleotide sequences (a sequence from genotype IV AY741404/fowl/UK/Herts/33/1933 was included as an outgroup). All positions containing gaps and missing data were eliminated. There were a total of 1,661 positions in the final data set. The isolates used in this study are shown in pink (genotype I) and teal (genotype II). The Roman numerals on the right of the phylogenetic tree represent the respective sub-genotype for each isolate in accordance with the current classification (5) and the former nomenclature (58) is listed in parenthesis for reference.
3.6. Genotype II isolates
All six NDV genotype II isolates (KU133362–KU133365, MZ101343, and MZ101344) were 100% identical (Figure 4). These isolates were collected from dead pigeons in Dnipro, Donetsk, and Kharkiv regions and a dead crow (genus Corvus) in the Kharkiv region in 2007 (23). These viruses were identical to the LaSota vaccine strain (MH392212) (60) and other vaccine strains isolated from different species of birds across different continents (37).
3.7. Genotype VI isolates
The single NDV genotype VI isolate was derived from a pigeon located in the Kharkiv region of Ukraine (MZ101342) and belongs to sub-genotype VI.2.1.1.2.2, formerly classified as sub-genotype VIk (Figure 5) (5, 58). The highest homology of this isolate is to a strain isolated from a pigeon in Belgium in 2011 (JX901124) (61), sharing 99.16% nucleotide identity.
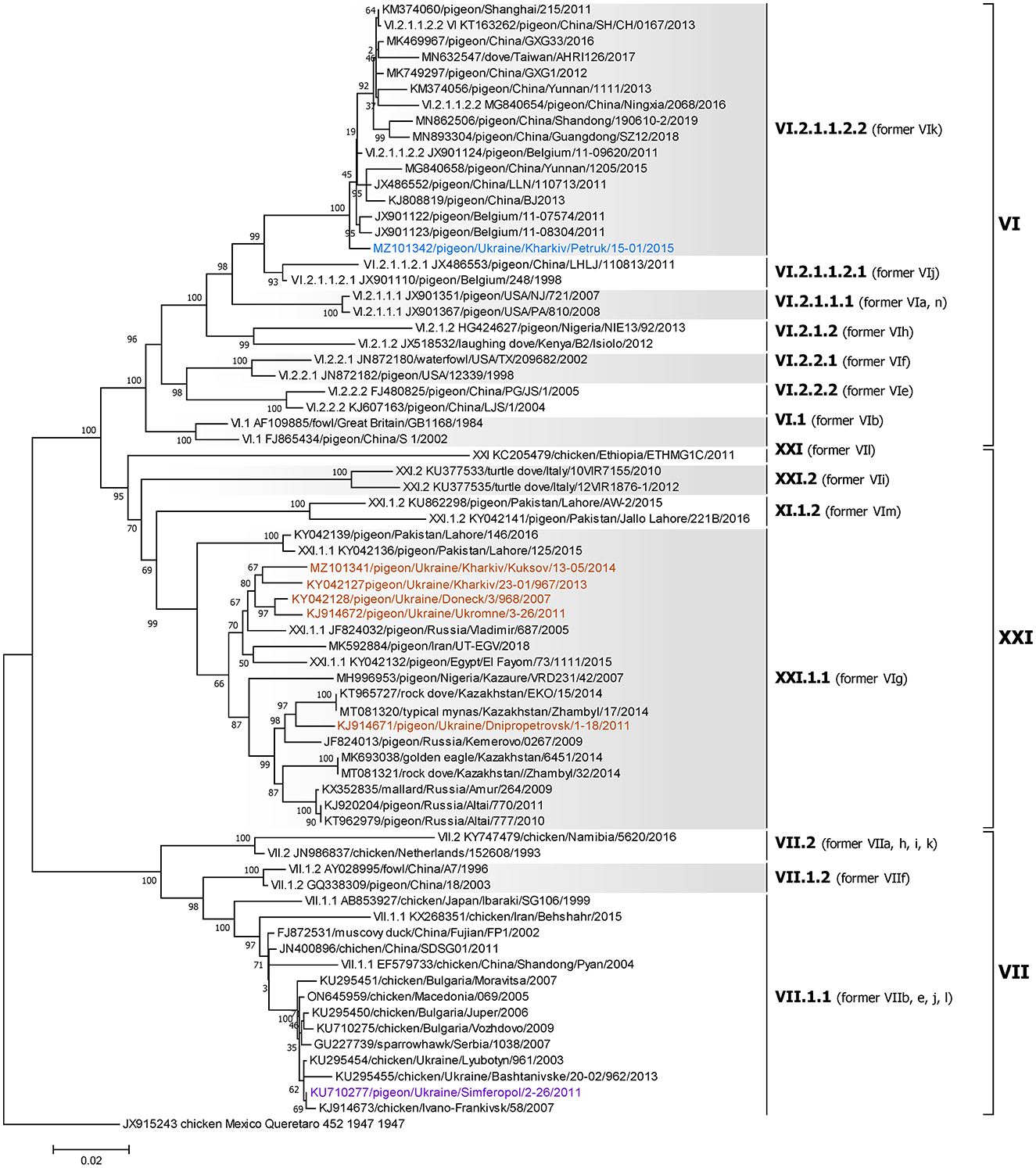
Figure 5. Phylogenetic analysis of NDV genotype VI, VII, and XXI class II isolates based on the complete fusion gene sequences constructed with the maximum likelihood method with 1,000 bootstrap replicates. The analysis involved 71 nucleotide sequences (a sequence from genotype XVI JX915243/chicken/Mexico/Queretaro/452/1947 was included as an outgroup). All positions containing gaps and missing data were eliminated. There were a total of 1,660 positions in the final data set. The isolates used in this study are shown in blue (genotype VI), purple (genotype VII), and brown (genotype XXI). The Roman numerals on the right of the phylogenetic tree represent the respective sub-genotype for each isolate in accordance with the current classification (5) and the former nomenclature (58) is listed in parenthesis for reference.
3.8. Genotype VII isolates
The single NDV genotype VII isolate was derived from a pigeon located in Simferopol, AR Crimea (KU710277) and belongs to sub-genotype VII.1.1, formerly classified as sub-genotype VIId (Figure 5) (5, 58). The highest homology of this isolate is to a strain isolated from a chicken in the Kharkiv region of Ukraine in 2003 (KU295454) (44), sharing 99.88% nucleotide identity.
3.9. Genotype XXI isolates
All five genotype XXI isolates were detected in Ukrainian pigeons and belong to sub-genotype XXI.1.1, previously known as VIg (Figure 5) (5, 58). These strains were collected in Donetsk, Dnipro, Kharkiv, and AR Crimea regions and shared between 95.91 and 98.98% nucleotide identity. Of these, three (KJ914672, KY042127, and MZ101341), that were collected in Kharkiv and AR Crimea, had the highest homology to another Ukrainian strain (KY042128) isolated from a pigeon in the Donetsk region in 2007 (27), sharing 98.98, 98.13, and 98.07% nucleotide identity, respectively (27, 28). Another Ukrainian sub-genotype XXI.1.1 sequence from the Dnipro region (KJ914671) shared 98.01% homologies with two strains isolated from pigeons in Kazakhstan in 2014 (KT965727 and MT081320) (62).
4. Discussion
Our previous studies, as well as this one, provide essential information on the epidemiology of the NDV isolates from synanthropic and wild birds in Ukraine. In this study, we used Sanger sequencing to obtain complete F gene sequences of NDV in Ukraine from 2006 to 2015. Our study reports repeated NDV detection of sub-genotype I.2 in wild birds collected at the stopping points of migratory birds in Ukraine, as well as the first occurrence of sub-genotype VI.2.1.1.2 and the continuous presence of sub-genotypes II and XXI.1.1 in Ukrainian synanthropic birds. Additionally, we present the first publicly available complete NDV F gene from a crow (genus Corvus).
The Azov-Black Sea region of Ukraine is part of three transcontinental wild bird migration routes: the West Asia-East Africa, East Atlantic, and Black Sea-Mediterranean flyways (30, 31, 45). This region is comprised of areas for transit, stops during migration, and nesting for many bird species, which makes it one of the highly important regions in Eurasia for monitoring and studying the global circulation of NDV and predicting the emergence of new strains possibly transmitted by wild birds.
The single class I NDV isolate was detected from wild waterfowl at a major stopping point location in AR Crimea and belonged to the 1.2 sub-genotype, formerly known as 1c. This virus clustered together with strains also isolated from wild waterfowl of the order Anseriformes (family Anatidae) in China and Finland. The high identity between these isolates from Europe and Asia supports our hypothesis of intercontinental viral transmission by migratory birds.
All detected class II NDV isolates from wild waterfowl and shorebirds collected at the major stopping point locations of migratory routes in Ukraine, belonged to genotype I, which is consistent with our previous study (29, 63) and confirms the continuous threat of virus introduction from wild birds. All viruses were obtained from members of the orders Anseriformes (family Anatidae) and Charadriiformes (family Laridae). Of the five genotype I strains analyzed, all were classified as sub-genotype I.2. These isolates grouped together with isolates previously detected in different hosts at various locations, including (25, 29). Luxembourg, the North Caucasian, Siberian, and Far East Federal Districts of Russia, China, Taiwan, and Nigeria which confirms the intercontinental spread of the virus by migratory birds. Similar findings were reported in our previous studies where epidemiological connections of Avian Paramyxoviruses between Europe and Africa were shown (29). The high identity between isolates from wild birds in Europe and Asia, and their close phylogenetic relationship with strains from Africa, support our hypothesis of virus exchange along the Black Sea-Mediterranean and Asian-East African migratory flyways and highlight the possibility of intercontinental viral transmission.
Special attention should be given to the data obtained from the phylogenetic analysis of NDVs isolated from synanthropic birds in Ukraine. Six of these isolates, which were isolated from pigeons and a crow in three Eastern and Southern regions of Ukraine in 2007 and 2008, were classified as genotype II. To the best of our knowledge, there was no complete NDV F gene of any sub-genotype from crows in public databases prior to this study. However, partial F gene sequences of viruses of genotypes II isolated from crows in India in 2002 (AY339400) and Pakistan in 2017 (MN728799–MN728801) were available in the GenBank database (64). Interestingly, Ukrainian viruses were identical to the vaccine strain LaSota, which is widely used in Ukraine and around the world as a live vaccine against NDV (37). Industrial and backyard poultry farming is very developed in these Eastern regions of Ukraine. Furthermore, one of the last NDV outbreaks in Ukraine was previously recorded in one of those regions (Kharkiv region) in 2006 (3, 29). Because of that outbreak, a number of anti-epizootic measures were implemented, involving vaccination against NDV in industrial and backyard farms (including those where poultry had direct contact with synanthropic birds). We supposed the presence of a vaccine virus found in the synanthropic birds could be a result of contact with vaccinated poultry. In recent decades, the number of genotypes has increased. It is likely that the spread of a vaccine strain could contribute to an increase in the genetic diversity of NDV (32). Therefore, special attention must be paid to the distribution of vaccine strains in wild birds in order to understand the consequences of global vaccination.
NDV isolates of XXI genotype were identified in five dead pigeons with clinical signs of disease, which not only confirmed that sub-genotype XXI.1.1 is seemingly maintained in pigeons in the East of Ukraine (Donetsk and Kharkiv regions) from 2007 to 2014 (27) but also was detected for the first time in the South of Ukraine (Dnipro and Simferopol regions). Even though the isolates collected in Donetsk, Kharkiv, and AR Crimea regions grouped together, the isolate collected in the Dnipro region in 2011 was highly similar to isolates from pigeons and wild birds from genus Acridotheres (KT965727 and MT081320) from Kazakhstan in 2014 (62). This further highlights the possibility of continuous intercontinental viral spread. This may also indicate an additional link in the distribution of genotype XXI strains or its association with a host preference for pigeons. Interestingly, two different NDV sub-genotypes (II and XXI.1.1) were detected in a pigeon from Donetsk collected in 2007. This was possible due to the utilization of two different sets of primers to amplify the complete F gene of different NDV genotypes. However, this is rare, because in some cases we were not able to amplify a complete F gene even though APMV-1 was confirmed in these isolates by serological methods. We are speculating that it was due to acquired mutations in the NDV F gene which resulted in the primer's mismatch. Also, we can't exclude the possibility of the cross-reactivity of different serotypes by serology, which we previously observed and reported in an isolate collected from a white-fronted goose in the Kherson region in 2011 (51, 52). This isolate weakly cross-reacted with APMV-1 and APMV-7 antisera in serology, but we were unable to amplify NDV's F gene by utilizing the set of primers for routine NDV detection. However, random whole-genome next-generation sequencing allowed us to assemble the complete genome and discover a new serotype of APMV (named APMV-13), which explained the previous inability to amplify the complete F gene using primers specific for NDV. This highlights the need for implementing random next-generation sequencing as a routine diagnostic tool in order to better perceive the complete epidemiological situation.
To the best of our knowledge, only viruses of sub-genotypes II, VII.1.1, and XXI.1.1 have been previously reported to circulate in pigeons in Ukraine (23, 27, 44, 65). In this study, we report the first identification of genotype VI NDV in Ukraine. This virulent NDV isolate, classified as sub-genotype VI.2.1.1.2, was obtained from a deceased pigeon in the Kharkiv region in 2015.
To obtain a complete picture of the distribution of NDV along the migratory flyways and to determine all circulating genotypes among wild waterfowl, it is necessary to continue the annual monitoring of NDV in the Azov-Black Sea region, as one of the major stopping points for migratory birds. Additional data will help to assess the degree of involvement of wild birds in the spread of virulent strains, that can be especially dangerous for poultry production. Further monitoring of NDV in synanthropic bird species will provide useful data for the study of vaccine strains widespread globally.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MZ101338, MZ101339, MZ101340, MZ101341, MZ101342, MZ101343, MZ101344, KF851268, KF851270, KJ914671, KJ914672, KU710277, KF851269, KU133362, KU133363, KU133364, KU133365, KY042127, and KY042128.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the National Scientific Center Institute of Experimental and Clinical Veterinary Medicine.
Author contributions
Conceptualization: IG, AG, DM, and CA. Funding acquisition: CA and BS. Project administration and coordination: CA, AG, and DM. Methodology: IG. Sample preparation: IG, VB, OS, OR, NM, OM, and OK. Data curation and formal analyses: IG, KD, PM, and CA. Writing—original draft: IG and DM. Writing—review and editing: PM, CA, and MP-J. All authors have read and agreed to the published version of the manuscript.
Funding
The work with Ukrainian isolates was supported by the U.S. Defense Threat Reduction Agency and by the USDA, ARS CRIS Project 6040-32000-064. Part of the research was funded by USDA Project P444, through the Ukrainian Science and Technology Center. Part of the research was done in the frame of the Joint Ukrainian-Austrian R&D Project (contract #M7-2021, 11.11.2021 and #M37-2022, 24.05.2022).
Acknowledgments
We would like to acknowledge Timothy Olivier and Dawn Williams-Coplin for their technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1026296/full#supplementary-material
Supplementary Table S1. Number of samples of biological material collected from wild birds of different species in different regions of Ukraine from 2006 to 2015.
Supplementary Table S2. Number of samples of biological material collected from synanthropic birds of different species in different regions of Ukraine from 2006 to 2015.
References
1. Rima B, Balkema-Buschmann A, Dundon WG, Duprex P, Easton A, Fouchier R, et al. ICTV virus taxonomy profile: paramyxoviridae. J Gen Virol. (2019) 100:1593–4. doi: 10.1099/jgv.0.001328
2. Kaleta EF, Baldauf C. Newcastle Disease in Free-Living and Pet Birds. Newcastle: Springer (1988), 197–246.
3. OIE. Newcastle Disease (Infection with Newcastle Disease Virus). Manual of Diagnostic Tests and Vaccinesfor Terrestrial Animals. Geneva: World Organization for Animal Health (2021).
5. Dimitrov KM, Abolnik C, Afonso CL, Albina E, Bahl J, Berg M, et al. Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infect Genet Evol. (2019) 74:103917. doi: 10.1016/j.meegid.2019.103917
6. Dimitrov KM, Ramey AM, Qiu X, Bahl J, Afonso CL. Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect Genet Evol. (2016) 39:22–34. doi: 10.1016/j.meegid.2016.01.008
7. Oberdorfer A, Werner O. Newcastle disease virus: detection and characterization by PCR of recent German isolates differing in pathogenicity. Avian Pathol. (1998) 27:237–43. doi: 10.1080/03079459808419330
8. Russell PH, Samson ACR, Alexander DJ. Newcastle disease virus variations. Appl Virol Res. (1990) 2:177–95.
9. Nagai Y, Klenk HD, Rott R. Proteolytic cleavage of the viral glycoproteins and its significance for the virulence of Newcastle disease virus. Virology. (1976) 72:494–508. doi: 10.1016/0042-6822(76)90178-1
10. Nagai Y, Shimokata K, Yoshida T, Hamaguchi M, Iinuma M, Maeno K, et al. The spread of a pathogenic and an apathogenic strain of Newcastle disease virus in the chick embryo as depending on the protease sensitivity of the virus glycoproteins. J Gen Virol. (1979) 45:263–72. doi: 10.1099/0022-1317-45-2-263
11. Wang Y, Bi Y, Yu W, Wei N, Wang W, Wei Q, et al. Two mutations in the HR2 region of Newcastle disease virus fusion protein with a cleavage motif “RRQRRL” are critical for fusogenic activity. Virol J. (2017) 14:185. doi: 10.1186/s12985-017-0851-0
12. Toyoda T, Sakaguchi T, Imai K, Inocencio NM, Gotoh B, Hamaguchi M, et al. Structural comparison of the cleavage-activation site of the fusion glycoprotein between virulent and avirulent strains of Newcastle disease virus. Virology. (1987) 158:242–7. doi: 10.1016/0042-6822(87)90261-3
13. Pritzer E, Kuroda K, Garten W, Nagai Y, Klenk HD. A host range mutant of Newcastle disease virus with an altered cleavage site for proteolytic activation of the F protein. Virus Res. (1990) 15:237–42. doi: 10.1016/0168-1702(90)90031-6
14. Panda A, Huang Z, Elankumaran S, Rockemann DD, Samal SK. Role of fusion protein cleavage site in the virulence of Newcastle disease virus. Microb Pathog. (2004) 36:1–10. doi: 10.1016/j.micpath.2003.07.003
15. Miller PJ, Kim LM, Ip HS, Afonso CL. Evolutionary dynamics of Newcastle disease virus. Virology. (2009) 391:64–72. doi: 10.1016/j.virol.2009.05.033
16. Kim SH, Wanasen N, Paldurai A, Xiao S, Collins PL, Samal SK. Newcastle disease virus fusion protein is the major contributor to protective immunity of genotype-matched vaccine. PLoS ONE. (2013) 8:e74022. doi: 10.1371/journal.pone.0074022
17. Kim LM, Afonso CL, Suarez DL. Effect of probe-site mismatches on detection of virulent Newcastle disease viruses using a fusion-gene real-time reverse transcription polymerase chain reaction test. J Vet Diagn Invest. (2006) 18:519–28. doi: 10.1177/104063870601800601
18. Rue CA, Susta L, Brown CC, Pasick JM, Swafford SR, Wolf PC, et al. Evolutionary changes affecting rapid identification of 2008 Newcastle disease viruses isolated from double-crested cormorants. J Clin Microbiol. (2010) 48:2440–8. doi: 10.1128/JCM.02213-09
19. Miller PJ, King DJ, Afonso CL, Suarez DL. Antigenic differences among Newcastle disease virus strains of different genotypes used in vaccine formulation affect viral shedding after a virulent challenge. Vaccine. (2007) 25:7238–46. doi: 10.1016/j.vaccine.2007.07.017
20. Cardenas-Garcia S, Diel DG, Susta L, Lucio-Decanini E, Yu Q, Brown CC, et al. Development of an improved vaccine evaluation protocol to compare the efficacy of Newcastle disease vaccines. Biologicals. (2015) 43:136–45. doi: 10.1016/j.biologicals.2014.11.003
21. Cappelle J, Caron A, Servan De Almeida R, Gil P, Pedrono M, Mundava J, et al. Empirical analysis suggests continuous and homogeneous circulation of Newcastle disease virus in a wide range of wild bird species in Africa. Epidemiol Infect. (2015) 143:1292–303. doi: 10.1017/S095026881400185X
22. Muzyka D, Pantin-Jackwood M, Stegniy B, Afonso C, editors. Avian Paramyxovirus Serotypes Circulating in Wild Bird Populations of the Azov-Black Sea Region of Ukraine in 2006–2011. Meeting Abstract (2013).
23. Ayala AJ, Dimitrov KM, Becker CR, Goraichuk IV, Arns CW, Bolotin VI, et al. Presence of vaccine-derived newcastle disease viruses in wild birds. PLoS ONE. (2016) 11:e0162484. doi: 10.1371/journal.pone.0162484
24. Miller PJ, Dimitrov KM, Williams-Coplin D, Peterson MP, Pantin-Jackwood MJ, Swayne DE, et al. International biological engagement programs facilitate newcastle disease epidemiological studies. Front Public Health. (2015) 3:235. doi: 10.3389/fpubh.2015.00235
25. Yurchenko KS, Zhou P, Kovner AV, Zavjalov EL, Shestopalova LV, Shestopalov AM. Oncolytic effect of wild-type Newcastle disease virus isolates in cancer cell lines in vitro and in vivo on xenograft model. PLoS ONE. (2018) 13:e0195425. doi: 10.1371/journal.pone.0195425
26. Glushchenko AV, Alikina TY, Yurchenko KS, Shekunov EV, Gulyaeva MA, Matsuno K, et al. Nearly complete genome sequence of a Newcastle disease virus strain isolated from a wild garganey. Microbiol Resour Announc. (2019) 8:19. doi: 10.1128/MRA.01072-19
27. Sabra M, Dimitrov KM, Goraichuk IV, Wajid A, Sharma P, Williams-Coplin D, et al. Phylogenetic assessment reveals continuous evolution and circulation of pigeon-derived virulent avian avulaviruses 1 in Eastern Europe, Asia, and Africa. BMC Vet Res. (2017) 13:291. doi: 10.1186/s12917-017-1211-4
28. Pchelkina IP, Manin TB, Kolosov SN, Starov SK, Andriyasov AV, Chvala IA, et al. Characteristics of pigeon paramyxovirus serotype-1 isolates (PPMV-1) from the Russian Federation from 2001 to 2009. Avian Dis. (2013) 57:2–7. doi: 10.1637/10246-051112-Reg.1
29. Muzyka D, Pantin-Jackwood M, Stegniy B, Rula O, Bolotin V, Stegniy A, et al. Wild bird surveillance for avian paramyxoviruses in the Azov-Black Sea regions of Ukraine (2006–2011) reveals epidemiological connections with Europe and Africa. Appl Environ Microbiol. (2014) 80:5427–38. doi: 10.1128/AEM.00733-14
30. Diadicheva E, Matsievskaya N. Migration routes of waders using stopover sites in the Azov-Black Sea region, Ukraine. Vogelwarte. (2000) 40:161–78. Available online at: https://www.zobodat.at/pdf/Vogelwarte_40_1999_0161-0178.pdf
31. Kulak MV, Ilinykh FA, Zaykovskaya AV, Epanchinzeva AV, Evstaphiev IL, Tovtunec NN, et al. Surveillance and identification of influenza A viruses in wild aquatic birds in the Crimea, Ukraine (2006–2008). Avian Dis. (2010) 54:1086–90. doi: 10.1637/9272-020510-ResNote.1
32. Muzyka D, Pantin-Jackwood MJ, Spackman E, Smith DM, Rula O, Muzyka N, et al. Isolation and genetic characterization of avian influenza viruses isolated from wild birds in the Azov-Black Sea region of Ukraine (2001–2012). Avian Dis. (2016) 60:365–77. doi: 10.1637/11114-050115-Reg
33. Boere GC, Galbraith CA, Stroud DA. Waterbirds Around the World: A Global Overview of the Conservation, Management and Research of the World's Waterbird Flyways. London: Stationery Office (2006).
34. FAO. Gateway to Poultry Production and Products. New York, NY: FAO (2020). Available online at: https://www.fao.org/poultry-production-products/production/poultry-species/en/ (accessed August 3, 2022).
35. Miller PJ, Koch G. Newcastle disease. In:Swayne DE, Glisson JR, McDougald LR, Nolan LK, Suarez DL, Nair VL, , editor. Diseases of Poultry. 13 ed. Ames, IA: Wiley-Blackwell in Partnership with the American Association of Avian Pathologists (2013). p. 89–138.
36. Dimitrov KM, Afonso CL, Yu Q, Miller PJ. Newcastle disease vaccines: a solved problem or a continuous challenge? Vet Microbiol. (2017) 206:126–36. doi: 10.1016/j.vetmic.2016.12.019
37. Welch CN, Shittu I, Abolnik C, Solomon P, Dimitrov KM, Taylor TL, et al. Genomic comparison of Newcastle disease viruses isolated in Nigeria between 2002 and 2015 reveals circulation of highly diverse genotypes and spillover into wild birds. Arch Virol. (2019) 164:2031–47. doi: 10.1007/s00705-019-04288-9
38. Cardenas Garcia S, Navarro Lopez R, Morales R, Olvera MA, Marquez MA, Merino R, et al. Molecular epidemiology of Newcastle disease in Mexico and the potential spillover of viruses from poultry into wild bird species. Appl Environ Microbiol. (2013) 79:4985–92. doi: 10.1128/AEM.00993-13
39. Vidanović D, Sekler M, Asanin R, Milić N, Nisavić J, Petrović T, et al. Characterization of velogenic Newcastle disease viruses isolated from dead wild birds in Serbia during 2007. J Wildl Dis. (2011) 47:433–41. doi: 10.7589/0090-3558-47.2.433
40. Afonso CL. Virulence during Newcastle disease viruses cross species adaptation. Viruses. (2021) 13:115. doi: 10.3390/v13010110
42. Kirkland PD. Virulent Newcastle disease virus in Australia: in through the 'back door'. Aust Vet J. (2000) 78:331–3. doi: 10.1111/j.1751-0813.2000.tb11786.x
43. Shengqing Y, Kishida N, Ito H, Kida H, Otsuki K, Kawaoka Y, et al. Generation of velogenic Newcastle disease viruses from a nonpathogenic waterfowl isolate by passaging in chickens. Virology. (2002) 301:206–11. doi: 10.1006/viro.2002.1539
44. Dimitrov KM, Bolotin V, Muzyka D, Goraichuk IV, Solodiankin O, Gerilovych A, et al. Repeated isolation of virulent Newcastle disease viruses of sub-genotype VIId from backyard chickens in Bulgaria and Ukraine between 2002 and 2013. Arch Virol. (2016) 161:3345–53. doi: 10.1007/s00705-016-3033-2
45. Muzyka D, Pantin-Jackwood M, Spackman E, Stegniy B, Rula O, Shutchenko P. Avian influenza virus wild bird surveillance in the Azov and Black Sea regions of Ukraine (2010–2011). Avian Dis. (2012) 56:1010–6. doi: 10.1637/10157-040912-ResNote.1
46. Alexander DJ. Newcastle Disease Virus and Other Avian Paramyxoviruses. A Laboratory Manual for the Isolation and Identification of Avian Pathogens. 4 ed. Kennett Square, PA: The American Association of Avian Pathologists (1998). p. 156-63.
47. OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 8th ed. Paris: OIE (2018).
48. American Association of Avian Pathologists. A Laboratory Manual for the Isolation, Identification and Characterization of Avian Pathogens. 5th ed. Athens, GA: American Association of Avian Pathologists (2008).
49. Capua I, Alexander DJ. Avian Influenza and Newcastle Disease: A Field and Laboratory Manual. 1st ed. Milan: Springer (2009). p. 186. doi: 10.1007/978-88-470-0826-7_1
50. Spackman. Animal Influenza Virus. New York, NY: Humana Press (2014). doi: 10.1007/978-1-4939-0758-8
51. Goraichuk I, Sharma P, Stegniy B, Muzyka D, Pantin-Jackwood MJ, Gerilovych A, et al. Complete genome sequence of an avian paramyxovirus representative of putative new serotype 13. Genome Announc. (2016) 4:16. doi: 10.1128/genomeA.00729-16
52. Goraichuk I, Poonam S, Dimitrov K, Stegniy B, Muzyka D, Pantin-Jackwood M, et al. Phylogenetic analysis of the complete genome of the APMV-13 isolate from Ukraine. In: Proceedings of the 17th International Congress on Infectious Disease 7th International Congress on Infectious Diseases: Journal of Infectious Disease. International Journal of Infectious Diseases (2016). p. 459. doi: 10.1016/j.ijid.2016.02.972
53. Diel DG, Miller PJ, Wolf PC, Mickley RM, Musante AR, Emanueli DC, et al. Characterization of Newcastle disease viruses isolated from cormorant and gull species in the United States in 2010. Avian Dis. (2012) 56:128–33. doi: 10.1637/9886-081111-Reg.1
54. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
55. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 70 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
56. Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. (2004) 101:11030–5. doi: 10.1073/pnas.0404206101
57. Alexander DJ. Newcastle disease and other avian paramyxoviruses. Rev Sci Tech. (2000) 19:443–62. doi: 10.20506/rst.19.2.1231
58. Diel DG, da Silva LH, Liu H, Wang Z, Miller PJ, Afonso CL. Genetic diversity of avian paramyxovirus type 1: proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infect Genet Evol. (2012) 12:1770–9. doi: 10.1016/j.meegid.2012.07.012
59. Snoeck CJ, Marinelli M, Charpentier E, Sausy A, Conzemius T, Losch S, et al. Characterization of newcastle disease viruses in wild and domestic birds in Luxembourg from 2006 to 2008. Appl Environ Microbiol. (2013) 79:639–45. doi: 10.1128/AEM.02437-12
60. Butt SL, Taylor TL, Volkening JD, Dimitrov KM, Williams-Coplin D, Lahmers KK, et al. Rapid virulence prediction and identification of Newcastle disease virus genotypes using third-generation sequencing. Virol J. (2018) 15:179. doi: 10.1186/s12985-018-1077-5
61. Van Borm S, Rosseel T, Steensels M, van den Berg T, Lambrecht B. What's in a strain? Viral metagenomics identifies genetic variation and contaminating circoviruses in laboratory isolates of pigeon paramyxovirus type 1. Virus Res. (2013) 171:186–93. doi: 10.1016/j.virusres.2012.11.017
62. Orynbayev MB, Fereidouni S, Sansyzbai AR, Seidakhmetova BA, Strochkov VM, Nametov AM, et al. Genetic diversity of avian avulavirus 1 (Newcastle disease virus genotypes VIg and VIIb) circulating in wild birds in Kazakhstan. Arch Virol. (2018) 163:1949–54. doi: 10.1007/s00705-018-3815-9
63. Gerilovych A, Stegniy B, Potkonjak A, Bolotin V, Solodyankin O. Genotyping of Newcastle disease virus strains, allocated in Ukraine in 1967–2007 (Genotypes 1, 2 and 4). Contemp Agric. (2009) 58:58–67. Available online at: http://polj.uns.ac.rs/wp-content/uploads/arhiva-savremena-poljoprivreda/2009Savremenapoljoprivreda34.pdf
64. Tirumurugaan KG, Vinupriya MK, Vijayarani K, Kumanan K. Analysis of the fusion protein cleavage site of Newcastle disease virus isolates from India reveals preliminary evidence for the existence of II, VI, and VII genotypes. Indian J Virol. (2011) 22:131–7. doi: 10.1007/s13337-011-0044-1
65. Gerilovych A, Potkonjak A. Molecular evolution of newcastle disease virus in Ukraine. Contemp Agric. (2009) 58:46–55. Available online at: http://polj.uns.ac.rs/wp-content/uploads/arhiva-savremena-poljoprivreda/2009Savremenapoljoprivreda12.pdf
Keywords: NDV, Avian orthoavulavirus 1, surveillance, sequencing, bird migration, synanthropic, pigeon, Ukraine
Citation: Goraichuk IV, Gerilovych A, Bolotin V, Solodiankin O, Dimitrov KM, Rula O, Muzyka N, Mezinov O, Stegniy B, Kolesnyk O, Pantin-Jackwood MJ, Miller PJ, Afonso CL and Muzyka D (2023) Genetic diversity of Newcastle disease viruses circulating in wild and synanthropic birds in Ukraine between 2006 and 2015. Front. Vet. Sci. 10:1026296. doi: 10.3389/fvets.2023.1026296
Received: 23 August 2022; Accepted: 02 January 2023;
Published: 19 January 2023.
Edited by:
Kimberly VanderWaal, University of Minnesota Twin Cities, United StatesReviewed by:
Chantal Snoeck, Luxembourg Institute of Health, LuxembourgUrsula Höfle, University of Castilla-La Mancha, Spain
Copyright © 2023 Goraichuk, Gerilovych, Bolotin, Solodiankin, Dimitrov, Rula, Muzyka, Mezinov, Stegniy, Kolesnyk, Pantin-Jackwood, Miller, Afonso and Muzyka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denys Muzyka,  ZG11enlrYTc3QGdtYWlsLmNvbQ==
ZG11enlrYTc3QGdtYWlsLmNvbQ==