 Huarun Sun†
Huarun Sun† Junkai Zhang
Junkai Zhang Gongzheng Hu
Gongzheng Hu- Department of Pharmacology and Toxicology, College of Veterinary Medicine, Henan Agricultural University, Zhengzhou, China
Integrative conjugative elements (ICEs) are important carriers for disseminating resistance genes. We have previously reported a novel element ICEHpa1 carrying seven antibiotic resistance genes, which could be self-transmissible relying on the novel T4SS. To identify novel ICEHpa1 variants from 211 strains and novel T4SS encoded in ICEHpa1, and to explore the relationships in these ICEs, four complete sequences of ICEs were identified by WGS analysis and antimicrobial susceptibility testing was determined by broth microdilution. In addition, a comparative analysis of these ICEs was conducted with bioinformatic tools, and the transfer abilities of these ICEs were confirmed by conjugation. Four ICEHpa1 variants ICEGpa1818, ICEGpa1808, ICEGpa1807, and ICEGpa1815 with different resistance gene profiles were characterized, and their hosts showed different resistance spectrums. All ICEs shared the same backbone and were inserted into the tRNALeu site, and all resistance regions were inserted into the same target site between the accessory and integration regions. This study analyzed complete sequences of ICEs from the ICEHpa1 family and identified novel T4SS and insertion element ISGpa2. Diverse resistance genes extensively exist in these ICEs, serving as a reservoir for resistance genes and facilitating their dissemination.
Introduction
Glaesserella parasuis is a gram-negative bacterium usually involved in respiratory tract infections, polyarthritis, fibrinous polyserositis and meningitis in swine (1). G. parasuis is a primary swine pathogen causing respiratory disease worldwide that can result in huge economic losses (2). β-lactam, tetracycline and aminoglycoside antibiotics are the drugs of choice for treating infections in animals and zoonotic diseases in humans. Plasmid-mediated β-lactam, tetracycline and aminoglycoside resistance genes have been reported in G. parasuis (3–6), but these plasmid-mediated resistance genes are not self-transmissible through conjugation for lacking conjugal transfer proteins. The conventional view holds that prokaryotic evolution occurs due to clonal divergence and periodic selection (7). Horizontal gene transfer is another driving force in microbial evolution that allows microbes to acquire new genes and phenotypes, contributing to the diversification and adaptation of micro-organisms (8). Integrative conjugative elements (ICEs) are important carriers for horizontal gene transfer in bacterial populations (9). Hitherto, 28 ICEs families have been identified (https://bioinfo-mml.sjtu.edu.cn/ICEberg2/) in various Gram-positive and Gram-negative bacteria. We recently identified two ICEs from G. parasuis that carry multiple resistance genes, ICEHpa1 and its variant ICEGpa1804, in the genomes from G. parasuis isolates (10). According to comparative sequence analysis, ICEHpa1 and ICEGpa1804 shared highly conserved ICE backbone, including replication, stabilization, type IV secretion system (T4SS) and integration without obvious structural homology with ICEs from reported families.
This study described four novel ICEHpa1 variants among 211 G. parasuis isolates that shared a common ICE backbone with ICEHpa1 and identified a novel T4SS. Moreover, we defined ICEHpa1 family and proposed a model for the emergence of ICEHpa1 elements that involve the continual recruitment of the different resistance gene modules. This work will shed light on the characteristics of the ICEs from ICEHpa1 family and puts forward innovative insights into the considerable diversity of genes and potential accessory functions encoded by the variable DNA in these ICEs.
Materials and methods
Bacterial strains and DNA sequence analysis
Two hundred and eleven strains were collected from pigs with respiratory diseases from pig farms and animal hospitals in Henan (n = 58), Hubei (n = 47), Hunan (n = 19), Anhui (n = 17), Jiangxi (n = 29), Shaanxi (n = 19), and Shanxi (n = 22) provinces of China from 2016 to 2019. The genomic DNA of the 211 strains was extracted using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) and then were sequenced via Illumina Hiseq platform and assembled by SPAdes (11). Resistance genes were determined via the CGE server (https://cge.cbs.dtu.dk/services/) (12). Subsequently, selected strains YHP1818, GHP1808, GHP1807 and YHP1815 with different antimicrobial resistance gene profiles were completely sequenced using a combined Illumina HiSeq and Nanopore sequencing approach. Initially, the quality of the raw sequence reads was checked using FastQC (Q30 > 85%). Adapter trimming of the Illumina reads was performed using Trimmomatic, and Nanopore reads shorter than 1,000 bp were removed. Sequencing reads as short-read and long-read data were assembled with Unicycler 0.4.4 with the hybrid assembly strategy (13, 14). Automated genome annotation was generated using the NCBI prokaryotic genome annotation pipeline (https://submit.ncbi.nlm.nih.gov/subs/genome/). Comparative analysis was conducted using the genome comparison visualizer Easyfig (15).
Serotyping and MLST
Serovars of the host strains were determined using the primers that Jia mentioned (16). Seven housekeeping genes (atpD, infB, mdh, rpoB, 6pgd, g3pd and frdB) were amplified and sequenced as had been described (17), following registration of sequences at https://pubmlst.org/organisms/glaesserella-parasuis in terms of allele numbers and STs assignment. These data were analyzed via software available on the website.
Susceptibility testing
Due to the unavailability of an approved method for G. parasuis, MICs of YHP1818, GHP1808, GHP1807 and YHP1815 were determined by the broth microdilution method (18) using cation-adjusted Mueller-Hinton broth containing 10% fetal bovine serum and 0.01% NAD, as suggested by a previous report (19). The tested antimicrobial agents were oxytetracycline, amoxicillin, enrofloxacin, streptomycin, gentamicin, tilmicosin, florfenicol, sulfamethoxazole/trimethoprim (19/1) and colistin. Actinobacillus pleuropneumoniae ATCC27090 and Escherichia coli ATCC 25922 were used as control strains.
Evolutionary analyses of the core T4SS genes
Evolutionary analyses were implemented by MEGA7 (20), involving reference sequences derived from the NCBI database. Phylogenetic trees were created automatically by applying Neighbor-Join and BioNJ algorithms for core genes based on their respective amino acid sequences to further explore the evolution of the novel T4SS. The traC/virB4, traD/virD4, and traI genes encoded the typical T4SS proteins (21) and were ideal for analyzing the evolution of the T4SS.
Conjugal transfer of the G. Parasuis ICEs
The circular intermediates of these ICEs were detected by PCR and sequencing with primers ICE-out-F and ICE-out-R as described previously (10). To investigate the conjugal transfer of these ICEs, this study adopted the YHP1818, GHP1808, GHP1807, and YHP1815 strains as the donors and G. parasuis V43 (rifampicin-resistance) as the recipient. As previously described (10), matings were performed with selection on TSA plates supplemented with 10% fetal bovine serum, 0.01% NAD, 8 mg/L oxytetracycline, and 100 mg/L rifampicin. Selected transconjugants were confirmed with PCR for the presence of the virB4 gene using the primers, susceptibility testing and MLST.
Nucleotide sequence accession numbers
The complete sequences of the four chromosomes carrying ICEGpa1818, ICEGpa1808, ICEGpa1807 and ICEGpa1815 have been submitted to GenBank with the following accession numbers: CP071487, CP071490, CP071491 and CP071489. The complete sequence of the plasmid p1807 in GHP1807 has been deposited in the GenBank database with accession number CP071492.
Results
General genetic features of host strains carrying ICEs and antimicrobial susceptibility
The whole-genome sequencing showed that 29 of 211 G. parasuis strains contain a conserved ICEHpa1-like genetic backbone. Twenty nine strains were distributed into 12 different STs. The highest prevalence was observed for ST280 (6/29), followed by ST428 (5/29) and ST430 (5/29). Moreover, these 29 strains were assigned to 7 serovars. Serovars 4 (11/29) and 1 (10/29) were the most prevalent serovars. Table 1 shows the genetic features of 29 host strains carrying ICEs. Host strains YHP1818 [tet(B)], GHP1807 [tet(B), strA, strB, aphA1, sul2, and floR], GHP1808 [tet(B), blaRob−1 and aac(6)'-Ie-aph(2')-Ia] and YHP1815 [tet(B), blaRob−1, strA, strB, aphA1, and sul2] have different resistance gene profiles. The chromosomes of four host strains range from 2,289,620 bp to 2,435,338 bp and the GC contents of four chromosomes (40.03, 39.98, 40.02, and 39.93%) are very close. WGS analysis indicated that the strain GHP1807 contain one chromosome and a 5,215 bp plasmid, while YHP1818, GHP1808, and YHP1815 all contain just one chromosome. In the strain GHP1807, the floR gene was located on the small plasmid p1807, but other genes tet(B), strA, strB, aphA1, and sul2 were located on the ICEHpa1 variant, i.e., ICEGpa1807. In the other three strains, all resistance genes were located on the new ICEHpa1 variants (ICEGpa1818, ICEGpa1808, and ICEGpa1815), and all ICEs shared extensive sequence homology and highly conserved gene order. ICEHpa1 is the first to be identified and characterized (10) and thus was selected as a reference to define the ICEHpa1 family. The GC contents of the four ICEs are all lower than the rest of the genomes, and all ICEs are inserted into the tRNALeu sites. Table 2 shows the genetic features of the ICEs from ICEHpa1 family.
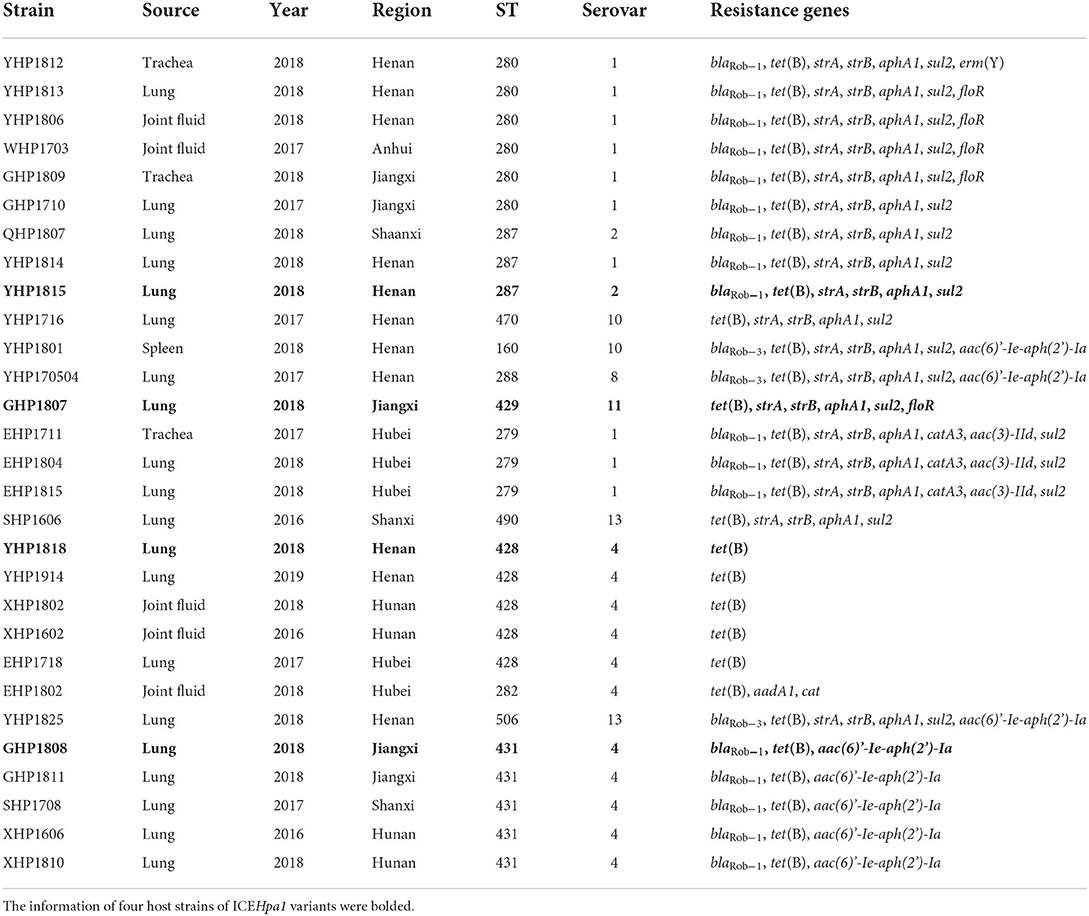
Table 1. General genetic features of 29 host strains carrying ICEs.
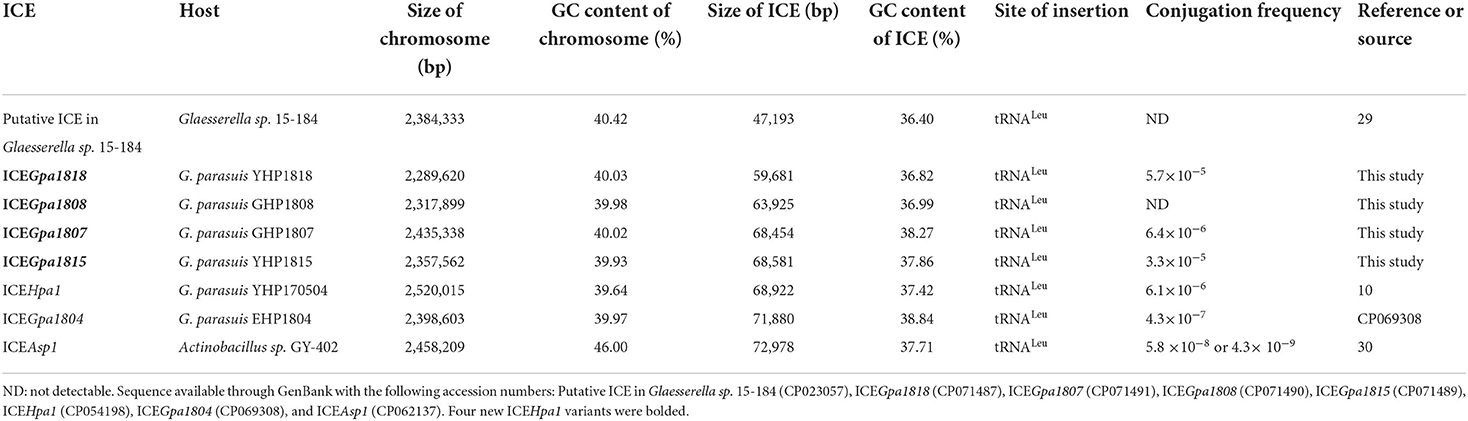
Table 2. General genetic features of ICEs from ICEHpa1 family and their host strains.
Strain YHP1818 exhibited high MIC of oxytetracycline (64 mg/L); GHP1808 showed high MICs of oxytetracycline (64 mg/L), amoxicillin (128 mg/L), streptomycin (128 mg/L), and gentamicin (128 mg/L); GHP1807 exhibited high MICs of oxytetracycline (64 mg/L), streptomycin (128 mg/L), sulfamethoxazole/trimethoprim (128 mg/L) and florfenicol (16 mg/L), and YHP1815 showed high MICs of oxytetracycline (64 mg/L), amoxicillin (16 mg/L), streptomycin (128 mg/L) and sulfamethoxazole/trimethoprim (128 mg/L) (Table 3).
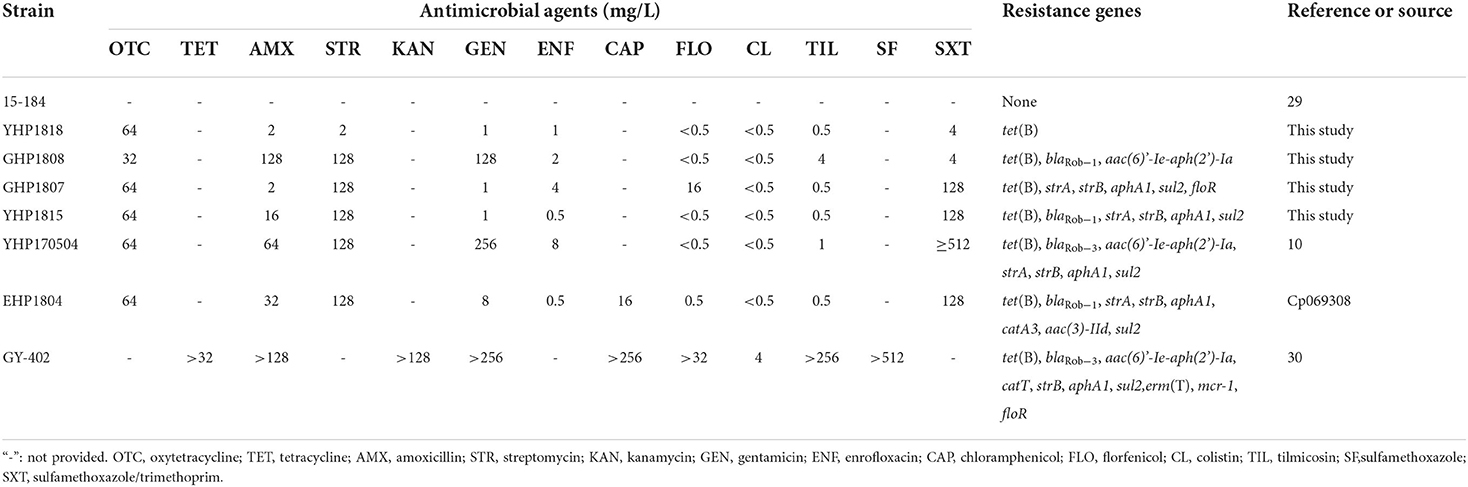
Table 3. MICs and resistance genes in host isolates.
Comparative sequence analysis of the T4SS
Comparative sequence analysis revealed that these four ICEs shared highly conserved ICE backbone, including replication, stabilization, T4SS and integration. Based on the bioinformatic analyses, 23 ORFs (tfc1 to tfc23) that followed the putative replication module of ICEHpa1 were considered to encode a novel lineage T4SS (Figure 1A). Nineteen of 23 genes exhibited homology to known T4SS components (Table 4), a highly conserved module of the ICEs from the ICEHpa1 family, with 95 to 100% DNA similarity. T4SS of ICEHpa1 shares low sequence homology with ICEHin1056 (most closely, 14% cover and 72.21% identity) and putative ICE in Gallibacterium anatis UMN179 (14% cover and 71.79% identity) (Figure 2). Table 4 shows the deduced amino acid sequence similarities of the putative T4SS components of ICEHpa1 with the T4SS genes of ICEHin1056 and putative ICE in Gallibacterium anatis UMN179. T4SSs, with multi-subunit cell envelope spanning structures, comprise some genes encoding the Secretion channel, Pilus and Surface filamen (21, 22). Five ORFs were transcribed in a forward orientation, while the rest 18 ORFs were transcribed in the opposite direction (Figure 1A). Seven of the 23 ORFs from the putative T4SS cluster of the ICEHpa1 have transmembrane helices, playing an important role in transfer (Figure 1A). Furthermore, sequence analysis with SignalP 3.0 Server (23) revealed that 3 of the 23 genes of this gene cluster contain signal peptide sequences that are typical for genes involved in T4SSs.
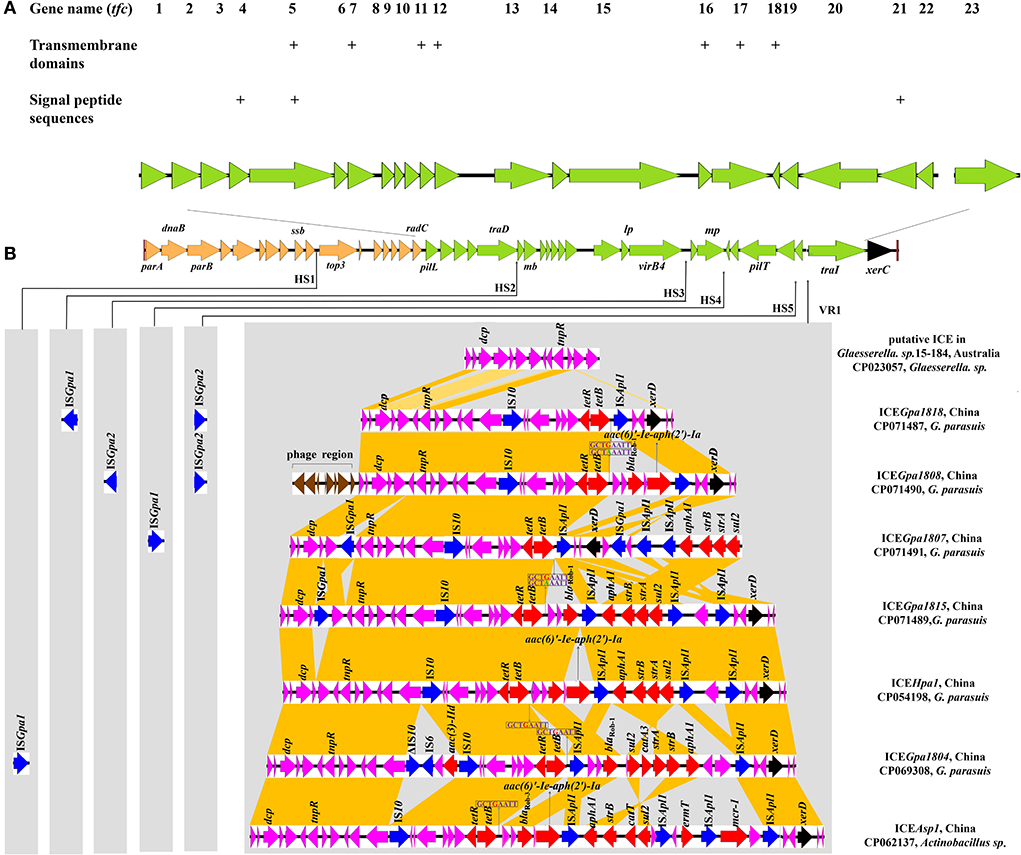
Figure 1. (A) Schematic representation of eight ICEHpa1 family ICEs and localization of the putative T4SS (green). This novel T4SS is encoded by 23 genes. Seven of the 23 ORFs from putative T4SS of the ICEHpa1 have transmembrane helices (indicated by plus signs) and 3 of the 23 genes are predicted to contain signal peptide sequences (indicated by plus signs). (B) Structural comparison between eight ICEs from ICEHpa1 family: the putative ICE in Glaesserella sp.15-184 (CP023057), ICEGpa1818 (CP071487), ICEGpa1807 (CP071491), ICEGpa1808 (CP071490), ICEGpa1815 (CP071489), ICEHpa1 (CP054198), ICEGpa1804 (CP069308), and ICEAsp1 (CP062137). Thin arrows indicate the sites of insertion for hotspots (HS1-HS5) and variable region (VR1). The different genes are shown in different colors among these ICEs. The putative replication modules, T4SS modules, integration genes, phage proteins, accessory genes, resistance genes and IS elements are in orange, green, black, brown, purple, red, and blue, respectively. Regions of >68% homology in VR1 are marked by dark orange shading.

Table 4. The deduced amino acid sequence similarities of the putative T4SS components of ICEHpa1 with the T4SS genes of ICEHin1056 and putative ICE in Gallibacterium anatis UMN179.
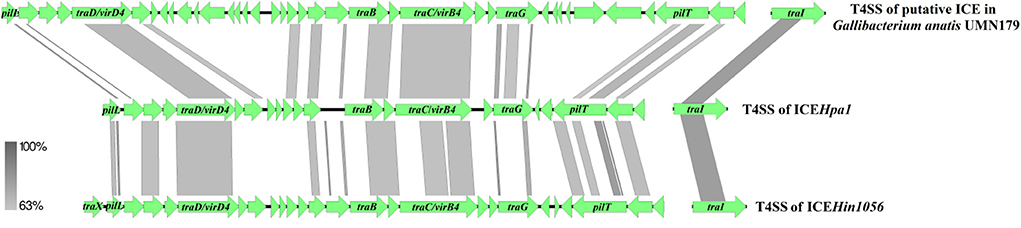
Figure 2. Compared the T4SS of ICEHpa1 (CP054198) with putative ICE in Gallibacterium anatis UMN179 (CP002667) and ICEHin1056 (AJ627386). Regions of >63% homology are marked by gray shading.
The bioinformatic analysis showed tfc5 exhibits homology with traD/virD4, a gene encoding T4SS conjugal transfer protein VirD4 in the DNA transport (24). tfc15 is homologous to traC/virB4 gene encoding an important T4SS component VirB4, which is required for the assembly of the system and substrate transfer (25). tfc13 and tfc17 are homologous to traB and traG, respectively, whose products are required for type IV secretion pilus assembly. Moreover, tfc23 has homology to traI gene encoding a relaxase to transfer N-terminally-fused Cre to target cells (26). tfc1 and tfc20 are homologous with pilL and pilT, respectively, and encode outer membrane lipoproteins responsible for thin pilus biosynthesis (27, 28).
Evolutionary analyses of the TraC/VirB4, TraD/VirD4 and trai proteins
The phylogenetic tree analysis indicate that the TraC/VirB4, TraD/VirD4 and TraI proteins are on a separate branch of the phylogenetic tree, respectively (Figure 3). Moreover, the trees for TraC/VirB4 and TraI proteins are extremely similar, of which the ICEHpa1 and putative ICE in Gallibacterium anatis UMN179 formed an evolutionarily separate group that differed from other ICEs and plasmids. Besides, TraD/VirD4 in ICEHpa1 exhibits a different branching pattern alongside ICEHin1056 members, putative ICE in Haemophilus somnus 2,336 and putative ICE in Gallibacterium anatis UMN179. From the silico bioinformatic analysis, it could be concluded that the gene cluster encoded structural components of a novel putative T4SS, which is evolutionarily different from previously described systems.
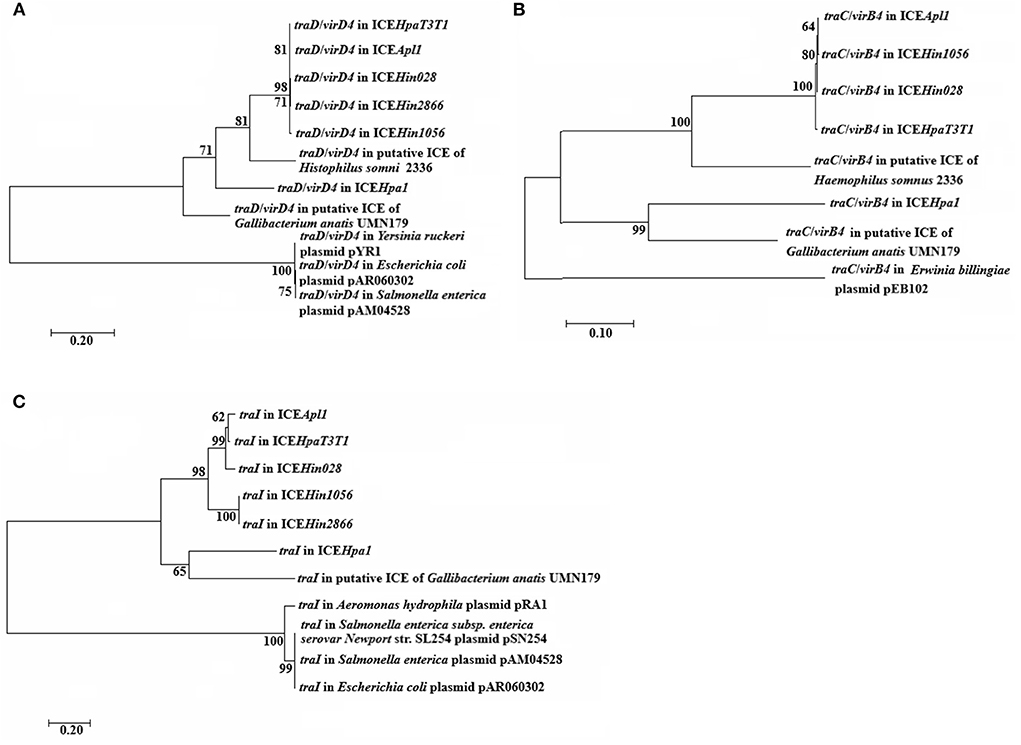
Figure 3. Phylogenetic analysis of (A) traD/virD4, (B) traC/virB4, and (C) traI genes. Amino acide sequences of the indicated these core genes were used to generate the phylogenetic trees shown. Bootstrap values are indicated at branch points. The individual scale bars represent genetic distances and reflect the number of substitutions per residue.
Analysis of VR1 regions in ICEHpa1 family ICEs
Apart from the core genes, five potential “hotspots” (HS1-HS5) and one “variable region”(VR1) were interspaced into the conserved backbone of ICEs. Two novel insertion elements, ISGpa1 and ISGpa2, were distributed between five hotspots (HS1-HS5) and the variable region (VR1). ISGpa1 and ISGpa2 with high host specificity, however, were only present in the ICEs from G. parasuis.
This study coded variable DNAs in VR1 for resistance to antibiotics and bacteriophage infection, which conferred adaptive functions to various environments. The VR1 mainly consisted of three segments in an accessory genes region, a multidrug-resistant region (MRR) and an integration region. Unlike other ICEs, upstream of three segments, a 5,137 bp cassette encoding eight proteins, was predicted as a prophage-related element in ICEGpa1808. The contents of the accessory genes region and the integration region in other resistance ICEs were almost identical except for ICEGpa1807 and ICEGpa1815. The integration region of VR1 in ICEGpa1807 was inserted into the MRR, which varied in the position and direction from other resistance ICEs in Figure 1B. An insertion element, ISGpa1, was found downstream of the zincin-like metallopeptidase domain-containing protein. Similarly, the ISGpa1 was also observed in the accessory genes region of ICEGpa1815, with a different direction from ICEGpa1807.
Compared with the putative ICE in the genome of Glaesserella sp.15-184 (28), except for ICEGpa1807, MRRs were inserted into the same site in the resistant ICEs between the segment tnpR-hp and integration gene xerD. The MRR of ICEGpa1818 was characterized by a truncated transposon Tn10, remaining two regulatory genes lysR and arsR, a sodium/glutamate symporter, an antibiotic biosynthesis monooxygenase, an amino acid-binding protein, a hypothetical protein, the repressor gene tetR, and the tetracycline resistance gene tet(B), as well as 83 bp of the downstream region. Correspondingly, the host strain harboring ICEGpa1818 showed a high MIC of oxytetracycline (64 mg/L). The truncated Tn10 was also found on the left of the MRR of ICEGpa1808. Moreover, a 2,804 bp segment carrying genes blaRob−1 and aac(6)'-Ie-aph(2')-Ia from G. parasuis pQY431 was observed immediately downstream of the truncated transposon Tn10. Correspondingly, the host strain harboring ICEGpa1808 showed high MICs of oxytetracycline (32 mg/L), amoxicillin (128 mg/L), streptomycin (128 mg/L) and gentamicin (128 mg/L). The resistance gene region of ICEGpa1807 comprised two parts with five different resistance genes and four complete insertion sequences (one IS10 copy and three ISApl1 copies). A truncated Tn10 and an ISApl1 were identified upstream of the integration region of VR1. Downstream of the integration region of VR1, two ISApl1 copies, the four resistance genes aphA1, strB, strA and sul2, were detected and all oriented in the same direction. The host strain harboring ICEGpa1807 showed high MICs of oxytetracycline (64 mg/L), streptomycin (128 mg/L), and sulfamethoxazole/trimethoprim (128 mg/L) accordingly. Immediately downstream of the truncated Tn10, a blaRob−1, an intact transposon Tn6742, and an additional ISApl1 element were found in the MRR of ICEGpa1815. The host strain harboring ICEGpa1815 showed high MICs of oxytetracycline (64 mg/L), amoxicillin (16 mg/L), streptomycin (128 mg/L) and sulfamethoxazole/trimethoprim (128 mg/L).
Apart from ICEs from G. parasuis, two additional putative ICEs from Glaesserella sp. (29) and Actinobacillus sp. (30) from Pasteurellaceae were observed to contain similar genetic backbone. According to comparative analysis of MRRs from ICEHpa1 family ICEs, antibiotic resistance genes tet(B), blaRob−1, blaRob−3, aac(6)'-Ie-aph(2')-Ia, aphA1, strA, strB, sul2, aac(3)-IId, catA3, erm(T) and mcr-1 were observed in the MRRs of ICEHpa1 family ICEs (Figure 4). Truncated Tn10 harboring the antibiotic resistance determinant tet(B) existed in all resistance ICEs without tetC, tetD and IS10-R. The truncated Tn10 in tested ICEs, however, did not abolish the tetracyclines resistance activity of the host strain. An 8 bp' GCTGAATT' or 'GCTAAATT' was found immediately downstream of the 6,687 bp truncated Tn10 in ICEs of this family (Figure 1B), indicating that the specific locus was recognized, and the Tn10 in these ICEs was truncated. Based on the acquired truncated Tn10, other acquired resistance genes [blaRob−1, blaRob−3, aac(6)'-Ie-aph(2')-Ia] were probably accumulated from small plasmids by homologous recombination, but aphA1, strA, strB, sul2, catA3, erm(T) and mcr-1 were acquired by a series of insertions that were meditated by ISApl1 (Figures 4, 5). The tandem multiplication of the ISApl1-flanked antibiotic resistance genes probably caused the expansion of the MRRs.
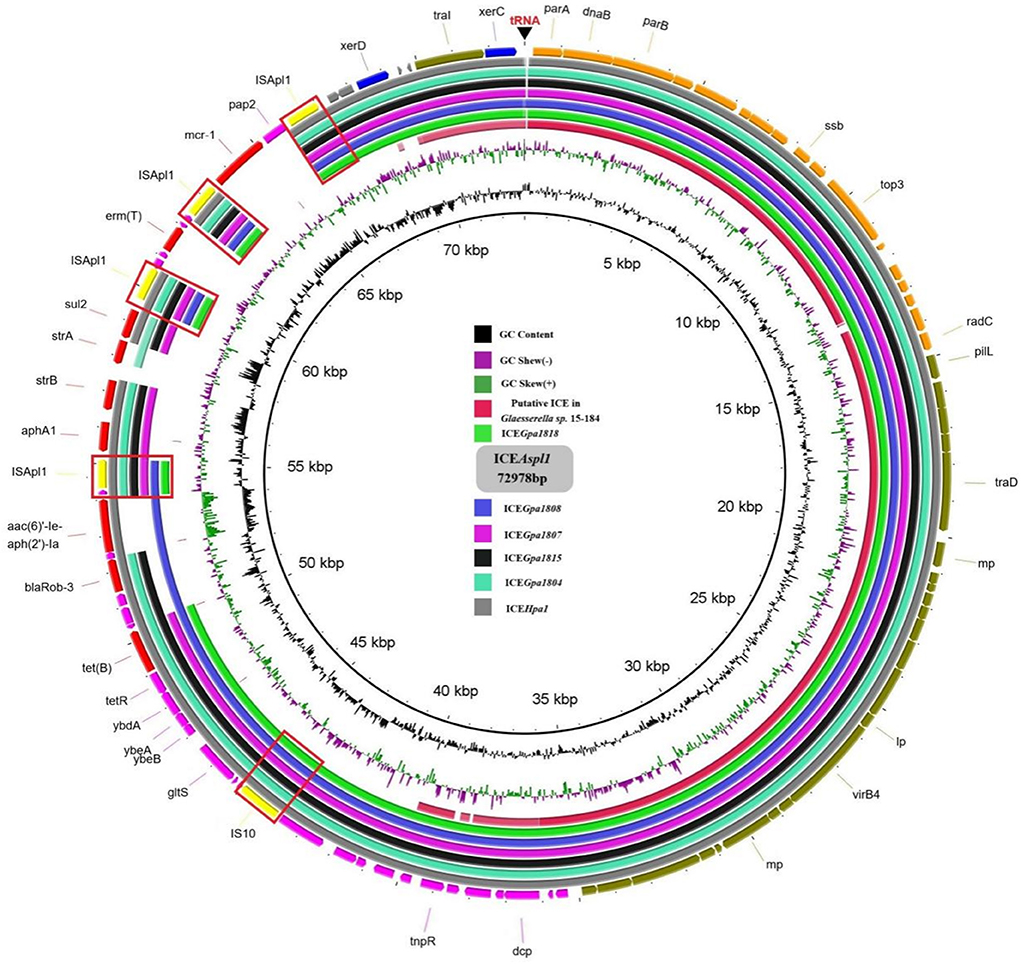
Figure 4. Comparison of the circular form of the ICEHpa1 family ICEs. The outer ring comprises the coding sequences (CDSs) of the ICEAsp1. Circular form of the ICEAsp1 was used as a reference to compare with the the putative ICE in Glaesserella sp.15-184 (CP023057), ICEGpa1818 (CP071487), ICEGpa1808 (CP071490), ICEGpa1807 (CP071491), ICEGpa1815 (CP071489), ICEGpa1804 (CP069308), and ICEHpa1 (CP054198). Key features of the ICEAsp1 are highlighted in different colors, replication genes and stability-associated genes are in orange, transfer-associated genes are in olive, IS elements are in yellow, ARGs are in red, other genes are in purple and integrase genes are in blue. IS elements were marked by the boxes.
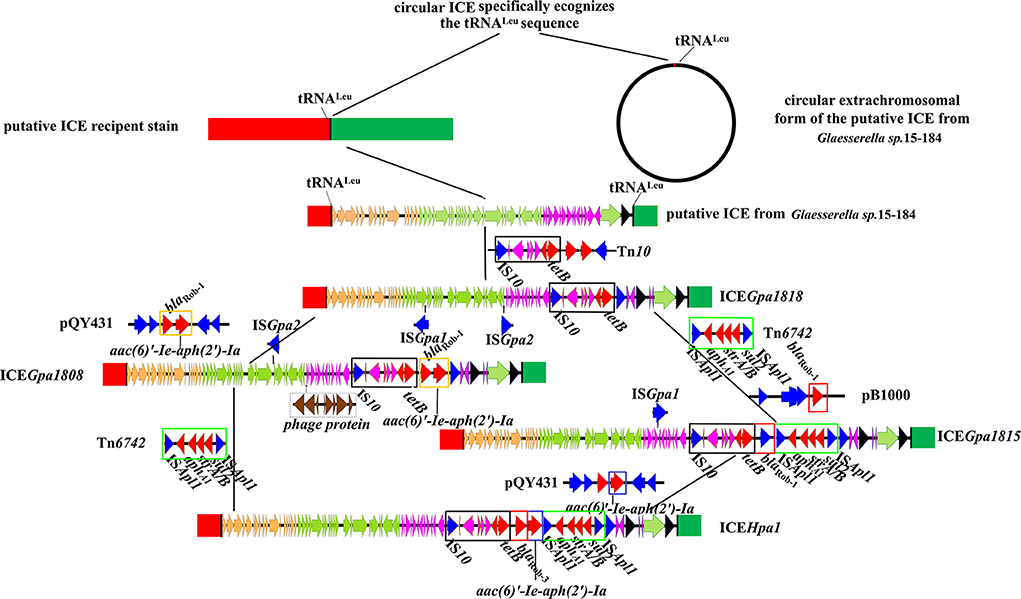
Figure 5. Schematic representation of the stepwise evolution process of ICEHpa1. Genes are presented as broad arrows, with the arrowhead indicating the direction of transcription. The reference sequences include the putative ICE in Glaesserella sp.15-184 (CP023057), ICEGpa1818 (CP071487), ICEGpa1808 (CP071490), ICEGpa1815 (CP071489), ICEHpa1 (CP054198), transposon Tn10 (AF162223), transposon Tn6742 (MN844034), plasmid pQY431 (KC405065), and plasmid pB1000 (DQ840517).
Two potential formation processes of MRR of ICEHpa1
In most cases, all ICEs exhibited extensive DNA sequence homologies and preserved gene order, but the main difference lay in their MRRs. High sequence similarities (some of 100%) were observed, especially in the putative replication and T4SS modules (Figures 4, 5). Based on the bioinformatic analyses of a series of ICEs of the ICEHpa1 family, we deduced two possible formation processes of MRR of ICEHpa1 (Figure 5).
Primitively, ICEHpa1-like circular extrachromosomal form with no resistance gene recognized specific tRNALeu site and then was integrated into the potential host chromosome. The putative ICE in the genome of Glaesserella sp.15-184 was regarded as the original structure of the ICEHpa1. ICEGpa1818 was formed as the putative ICE in the genome of Glaesserella sp.15-184 captured partial transposon Tn10 DNA sequences. Subsequently, episomal small transposon Tn6742 was inserted downstream of the tet (B) of ICEGpa1818, thus forming a new element. When the new element and small plasmid pB1000 collided under certain conditions, this element captured the blaRob−1 gene via homologous recombination to form a novel element ICEGpa1815. Ultimately, ICEGpa1815 got the bifunctional enzyme AAC(6')-Ie-APH(2")-Ia from small plasmid pQY43 via homologous recombination.
The other potential formation process of MRR of ICEHpa1 was observed via comparative sequence analysis. The putative ICE in the genome of Glaesserella sp.15-184 captured partial transposon Tn10 DNA sequences, and acquired the segment carrying the genes blaRob−1 and aac(6)'-Ie-aph(2')-Ia via homologous recombination from G. parasuis pQY43. Subsequently, the intact transposon Tn6742 was acquired via ISApl1-mediated insertion.
Conjugal transfer of four ICEs
Inverse PCR with primers were positive for four host strains carrying ICEs which indicated the presence of the intermediate circular form of ICEGpa1818, ICEGpa1807, ICEGpa1815, and ICEGpa1808. Transfers occurred with frequencies of 5.7 × 10−5, 6.4 × 10−6 and 3.3 × 10−5 transconjugants per donor cell (YHP1818, GHP1807, and YHP1815) for strain V43, respectively. However, conjugation experiments failed to obtain transconjugants of GHP1808, despite repeated attempts. Together, these results demonstrated that the ICEGpa1818, ICEGpa1807, and ICEGpa1815 were active and transferable ICE elements. The conjugation frequencies of the ICEs tested were recorded in Table 2.
Identification of a novel insertion element, ISGpa2
A novel DNA segment with a structural characteristic of insertion elements was identified in these novel ICEs with the BLAST analysis, and designated as ISGpa2 according to ISs nomenclature (https://www-is.biotoul.fr/). The nucleotide sequence of insertion sequence ISGpa2 was deposited in the ISFinder database.
ISGpa2 was 1,180-bp long with a GC content of 42.29%, containing a single 972 bp ORF. Comparisons of the sequence with the ISFinder and GenBank databases revealed that ISGpa2 belonged to the IS110 family. The ISGpa2 transposase exhibited extensive amino acid sequence homology with the putative transposase of ISNme5 from Neisseria meningitidis FAM18 (69.4% coverage with 56.6% identity) and ISPye59 from Paracoccus yeei TT13 (50.5% coverage with 33.0% identity). Like other members of the IS110 family, no DRs or IRs were found in ISGpa2. ISGpa2 also exists in the G. parasuis reference strains SH0104 (CP024412), SH0165 (CP001321), HPS412 (CP041334) and some other isolates.
Analysis of the FloR-carrying plasmid in GHP1807
In GHP1807, plasmid p1807 is 5,215 bp and consists of 3 ORFs encoding the florfenicol resistance protein FloR, transcriptional regulator LysR and a potential Rep protein involved in plasmid replication (Figure 6). Comparative sequence analysis revealed that p1807 almost shares 100% identity with the pHPSGC but misses 64 bp. The segment carrying floR and lysR also exists in pHPSF1 with different backbones from G. parasuis. In pHPSF1, the segment carrying floR and lysR was integrated to the plasmid backbone, which consists of mobilization genes mobA/L and mobC, and rep gene. It seems that the segment carrying floR and lysR originating from pHPSF1 was integrated to new plasmid backbone to generate a novel plasmid p1807.
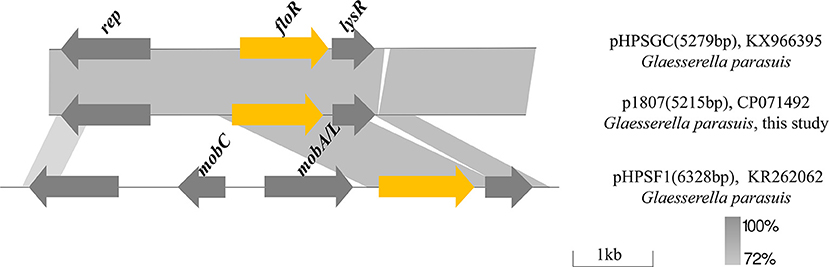
Figure 6. Comparison of plasmids p1807 (CP071492), pHPSGC (KX966395), and pHPSF1 (KR262062). Arrows indicate gene positions and transcriptional orientation. Regions of >72% homology are marked by gray shading.
Discussion
According to previous reports, plasmids are the main carriers to disseminate resistance genes among G. parasuis strains (3–6). In this study, 29 strains contain the conserved ICE genetic backbone among the 211 G. parasuis strains, indicating that ICEs are the predominant carriers for various resistance genes in G. parasuis. Strains YHP1818, GHP1807, GHP1808, YHP1815, YHP170504, and EHP1804 contained similar ICEs (ICEGpa1818, ICEGpa1808, ICEGpa1807, ICEGpa1815, ICEHpa1, and ICEGpa1804, respectively) that were isolated from various places (Henan, Jiangxi, Jiangxi, Henan, Henan and Hubei). Similar situations have been observed in the other species of bacteria. Strains contained highly similar genetic backbones but were isolated at divergent places and in different years (31, 32).
Host strains carrying ICEs were distributed into 7 different serovars (serovar 4, 1, 2, 10, 13, 8 and 11), which suggested a tendency for these ICEs from the ICEHpa1 family to be inherited by horizontal transfer rather than vertical transmission. In addition, these ICEs tended to have greater mobility among G. parasuis isolates. Meanwhile, 21 G. parasuis host strains were distributed with two serovars (serovar 4 and 1), indicating that these serovars clinical isolates were the main host strains for ICEs, and no restriction barriers hindered the transfer between these serovars. However, multiple conjugation experiments for GHP1808 failed, the reason for which is unknown. Non-transferability of ICEGpa1808 may be caused by the the insertion of the prophage-related element upstream the MRR or the transfer frequency in this strain is rather low.
ICEs from ICEHpa1 family are typically mosaic and modular and grouped on the element as they contained functional modules from different sources. None of the ICEs from ICEHpa1 family contained nucleotide sequences, indicating recent acquisition from other families than Pasteurellaceae. This observation proved that the ICEHpa1 family was evolving by descent within its host species without recombining with other species-specific families. Additionally, this ICEHpa1 family appeared to be shared only by strains associated with swine. The characteristics of traC/virB4, traD/virD4 and traI trees revealed that the great degree of recombination covered these core genes common evolutionary history or/and that these genes had evolved independently. Furthermore, different core genes co-existed in a host chromosome in tandem via recombination to form a novel T4SS.
ICEs are integrated into and excises from DNA via an ICE-encoded recombinase (9). Putative ICE in the genome of Glaesserella sp.15-184 carrying recombinase XerC was proved to integrate into, and excise from the host chromosome (29), and such was the case of ICEHpa1 harboring recombinase XerC and XerD. These findings suggested that ICEHpa1 family ICEs were probably integrated into and excised using an ICE-encoded XerC recombinase, while resistance regions were integrated at the specific attachment site by XerD recombinase. A range of resistance genes was located in the genome's ICEs (Table 3, Figure 1B), contributing to bacteria's diversification and adaptation. The resistance regions of these ICEs were likely assembled with different parts from other small plasmids from G. parasuis, and other transposons. These resistance genes conferred resistance to many antibacterial agents, including β-lactams, tetracyclines, aminoglycosides, sulfonamides, chloramphenicol, and macrolides colistin. In each case, the resistance genes are always carried on small transposons, with tetracycline resistance genes on Tn10, β-lactamase-encoding genes on Tn3, colistin resistance genes on Tn6330 and aminoglycoside and sulfonamide resistance genes on Tn6742 (10, 30). Tn10 was one of the most extensively studied transposable elements and an original case of small composite transposon where two same IS10 elements cooperated to mediate the transposition of antibiotic resistance genes (33). Consisting of indirectly repeating insertion sequences IS10-L and IS10-R flanking tet genes, Tn10 was involved in tetracyclines resistance (34, 35). Truncated Tn10 carrying the antibiotic resistance determinant tet (B) presented in all resistance ICEs that did not contain the tetC, tetD and IS10-R. Truncated Tn10 in tested ICEs, however, did not abolish the tetracyclines resistance activity of the host strain, which verified Richard's previous conclusion that the genes tetC and tetD were not involved in tetracycline resistance in the transposon Tn10 (34, 35). Most transposons contained drug-resistant cassettes in divergent ICEs, which increased the possibility for these resistance genes to be captured after acquiring ICEs by host strains. Furthermore, unlike other ICEs from the ICEHpa1 family, upstream of three segments and eight phage-related ORFs were found in ICEGpa1808, but the role of the phage cassette in the resistance ICEs transmission remained unclear.
A recent study confirmed that ICEAsp1 from Actinobacillus sp. is a new member of the ICEHpa1 family (30). ICEAsp1 is the first resistance ICE related to the ICEHpa1 family other than G. parasuis. Compared with the resistance gene region of ICEHpa1, the strA gene was replaced by a novel chloramphenicol resistance gene catT in the resistance gene region of ICEAsp1. An erm (T) gene and a putative transposon Tn6330 carrying colistin resistance gene mcr-1 were located immediately downstream of these modules. Colistin has been viewed as the last resort in treating Gram-negative bacterial infections (36). The emergence of ICEAsp1 carrying a colistin resistance gene mcr-1 signified that the ICEs from ICEHpa1 family could act as a reservoir for mcr-1 and accelerate the spread of colistin resistance.
Two potential formation processes of MRR of ICEHpa1 were deduced from the dataset, suggesting that the spread of ICEHpa1-mediated drug resistance was not strictly related to specific evolution processes. Various resistance genes were gradually captured by ICEHpa1. Conceivably, the ICEs from the ICEHpa1 family as the carriers should have a larger resistance gene load than small plasmids derived from Pasteurella (3–6). Despite limited knowledge of resistance genes-mobilizing vehicles, it could be deduced that resistance genes embedded in ICEHpa1 structures were capable of broader horizontal transmission with the benefit of abundant is elements.
The structural comparison showed that the formation of ICEHpa1 probably resulted from the abundance of ISApl1, ISGpa1 and ISGpa2 elements. Interestingly, ISApl1, originally identified in the A. pleuropneumoniae (37), was observed in the resistance gene region among all resistance ICEs from the ICEHpa1 family. ISApl1 promoted the adaptive evolution of bacteria and helped the host adapt to distinct environments by accumulating resistance genes. Multiple resistance genes aphA1, strB, catT, sul2, erm (T) and mcr-1 were observed in ICEAsp1, involving a series of insertion events meditated by ISApl1. Moreover, two newly identified IS elements with high host specificity, ISGpa1 and ISGpa2, only exist in the G. parasuis reference strains. Except for ICEHpa1, ISGpa1 and ISGpa2 were sporadically inserted in potential “hotspots” and “variable region” of the other five resistance ICEs from G. parasuis. Multiple copies ISGpa1 and ISGpa2 could serve as crossover points for homologous recombination events and play a vital role in genome flexibility, adaptation, and evolution of G. parasuis genomes. Moreover, the two novel insertion elements are critical to transferring ICEs among G. parasuis strains.
Conclusions
This study revealed that the diverse ICEs from the ICEHpa1 family recently emerged in the isolates of G. parasuis, Glaesserella sp. and Actinobacillus sp. from Pasteurellaceae. A novel T4SS from these ICEs was characterized, and potential “hotspots” (HS1-HS5) and one “variable region” (VR1) were defined in this study. In addition, the potential formation processes of MRR of ICEHpa1 were deduced, and ISGpa2, a novel insertion element, was identified. The ICEs from the ICEHpa1 family are the predominant carriers for various resistance genes. More in-depth research on ICEHpa1 family ICEs will be carried out in our laboratory.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
GH and HS conceived and designed the experiments. HS, JZ, QM, FY, and HW produced the data. HS, YZ, YP, LY, and HW analyzed the data. HS, HW, and GH wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was financed by the National Key Research and Development Program of China [2016YFD05101304].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Oliveira S, Pijoan C. Haemophilus parasuis: new trends on diagnosis epidemiology and control. Vet Microbiol. (2004) 99:1–12. doi: 10.1016/j.vetmic.2003.12.001
2. Oliveira S, Galina L, Pijoan C. Development of a PCR test to diagnose Haemophilus parasuis infections. J Vet Diagn Invest. (2001) 13:495–501. doi: 10.1177/104063870101300607
3. Yang SS, Sun J, Liao XP, Liu BT Li LL, Li L, et al. Co-location of the erm(T) gene and blaROB−1 gene on a small plasmid in Haemophilus parasuis of pig origin. J Antimicrob Chemother. (2013) 68:1930–2. doi: 10.1093/jac/dkt112
4. Moleres J, Santos-López A, Lázaro I, Labairu J, Prat C, Ardanuy C, et al. Novel blaROB−1-bearing plasmid conferring resistance to β-lactams in Haemophilus parasuis isolates from healthy weaning pigs. Appl Environ Microbiol. (2015) 81:3255–67. doi: 10.1128/AEM.03865-14
5. Lancashire JF, Terry TD, Blackall PJ, Jennings MP. Plasmid-encoded Tet B tetracycline resistance in Haemophilus parasuis. Antimicrob Agents Chemother. (2005) 49:1927–31. doi: 10.1128/AAC.49.5.1927-1931.2005
6. Chen P, Liu Y, Wang Y, Li WT, Bi DR, He QG. Plasmid mediated streptomycin and sulfonamide resistance in Haemophilus parasuis. J Animal Vet Adv. (2012) 11:1106–9. doi: 10.3923/javaa.2012.1106.1109
7. Peter GJ, Ford DW, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. (2002) 19:2226–38. doi: 10.1093/oxfordjournals.molbev.a004046
8. Juhas M, Power PM, Harding RM, Ferguson DJP, Dimopoulou ID, Elamin ARE, et al. Sequence and functional analyses of Haemophilus spp. genomic islands. Genome Biol. (2007) 8:R237. doi: 10.1186/gb-2007-8-11-r237
9. Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. (2010) 8:552–63. doi: 10.1038/nrmicro2382
10. Sun HR, Cui XD, Liu XK, Li SH, Yi KF, Pan YS, et al. Molecular characterization of a novel integrative conjugative element ICEHpa1 in Haemophilus parasuis. Front Microbiol. (2020) 11:1884. doi: 10.3389/fmicb.2020.01884
11. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
12. He DD, Wang LL, Zhao SY, Liu LP, Liu JH, Hu GZ, et al. A novel tigecycline resistance gene, tet(X6), on an SXT/R391 integrative and conjugative element in a Proteus genomospecies 6 isolate of retail meat origin. J Antimicrob Chemother. (2020) 75:1159–64. doi: 10.1093/jac/dkaa012
13. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
14. Li R, Xie M, Dong N, Lin D, Yang X, Wong MHY, et al. Efficient generation of complete sequences of MDR-encoding plasmids by rapid assembly of MinION barcoding sequencing data. Gigascience. (2018) 7:1–9. doi: 10.1093/gigascience/gix132
15. Sullivan M, Petty N, Beatson S. Easyfig: a genome comparison visualizer. Bioinformatics. (2011) 27:1009–10. doi: 10.1093/bioinformatics/btr039
16. Jia A, Zhou R, Fan H, Yang K, Ming L. Development of serotype-specific PCR assays for typing of Haemophilus parasuis circulating in southern China. J Clin Microbiol. (2017) 55:3249–57. doi: 10.1128/JCM.00688-17
17. Mullins MA, Register KB, Brunelle BW, Aragon V, Galofré-Mila N, Baylese D, et al. A curated public database for multilocus sequence typing (MLST) and analysis of Haemophilus parasuis based on an optimized typing scheme. Vet Microbiol. (2013) 162:899–906. doi: 10.1016/j.vetmic.2012.11.019
18. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated From Animals, 4th ed. Wayne, PA: CLSI supplement VET08 CLSI (2018).
19. Prüller S, Turni C, Blackall PJ, Beyerbach M, Klein G, Kreienbrock L, et al. Towards a Standardized Method for Broth Microdilution Susceptibility Testing of Haemophilus parasuis J Clin Microbiol (2017) 55:264–73. doi: 10.1128/JCM.01403-16
20. Kumar S, Stecher G. Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 70 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
21. Juhas M, Crook DW, Hood DW. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol. (2010) 10:2377–86. doi: 10.1111/j.1462-5822.2008.01187.x
22. Juhas M, Crook DW, Dimopoulou ID, Lunter G, Harding RM, Ferguson DJP, et al. Novel type IV secretion system involved in propagation of genomic islands. J Bacteriol. (2007) 189:761–71. doi: 10.1128/JB.01327-06
23. Bendtsen JD, Nielsen H, Heijne G. Brunak SS. Improved prediction of signal peptides: SignalP 30. J Mol Biol. (2004) 340:783–95. doi: 10.1016/j.jmb.2004.05.028
24. Christie PJ. Type IV secretion: intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol Microbiol. (2001) 40:294–305. doi: 10.1046/j.1365-2958.2001.02302.x
25. Rabel C, Grahn AM, Lurz R, Lanka E. The VirB4 family of proposed traffic nucleoside triphosphatases: common motifs in plasmid RP4 trbE are essential for conjugation and phage adsorption. J Bacteriol. (2003) 185:1045–58. doi: 10.1128/JB.185.3.1045-1058.2003
26. Christie PJ, Whitaker N, González-Rivera C. BBA review revised mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta. (2014) 1843:1578–91. doi: 10.1016/j.bbamcr.2013.12.019
27. Sakai D, Komano T. The pilL and pilN genes of Incl1 plasmids R64 and Collb-P9 encode outer membrane lipoproteins responsible for thin pilus biosynthesis. Plasmid. (2000) 43:149–52. doi: 10.1006/plas.1999.1434
28. Sakai D, Komano T. Genes required for plasmid R64 thin-pilus biogenesis: identification and localization of products of the pilK, pilM, pilO, pilP, pilR, and pilT genes. J Bacteriol. (2002) 184:444–51. doi: 10.1128/JB.184.2.444-451.2002
29. Watt AE, Browning GF, Legione AR, Bushell RN, Stent A, Cutler RS, et al. A novel Glaesserella sp. Isolated from severe respiratory infections in pigs has a mosaic genome with virulence factors putatively acquired by horizontal transfer. Appl Environ Microbiol. (2018) 17:84. doi: 10.1128/AEM.00092-18
30. Gao Y, Xia L, Pan R, Xuan HY, Guo HD, Song QQ, et al. Identification of mcr-1 and a novel chloramphenicol resistance gene catT on an integrative and conjugative element in an Actinobacillus strain of swine origin. Vet Microbiol. (2021) 254:108983. doi: 10.1016/j.vetmic.2021.108983
31. Wozniak RA, Fouts DE, Spagnoletti M, Colombo MM, Ceccarelli D, Garriss G, et al. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. (2009) 5:e1000786. doi: 10.1371/journal.pgen.1000786
32. Luo P, He XY, Wang YH. Liu, QT, Hu CQ. Comparative genomic analysis of six new-found integrative conjugative elements (ICEs) in Vibrio alginolyticus. BMC Microbiol. (2016) 16:79. doi: 10.1186/s12866-016-0692-9
33. Halling SM, Simons RW, Way JC, Walsh RB, Kleckner N. DNA sequence organization of IS10-right of Tn10 and comparison with IS10-left. Proc Natl Acad Sci U S A. (1982) 798:2608–12. doi: 10.1073/pnas.79.8.2608
34. Jorgensen RA, Reznikoff WS. Organization of structural and regulatory genes that mediate tetracycline resistance in transposon Tn10. J Bacteriol. (1979) 138:705–14. doi: 10.1128/jb.138.3.705-714.1979
35. Chalmers R, Sewitz S, Lipkow K, Crellin P. Complete nucleotide sequence of Tn10. J Bacteriol. (2000) 182:2970–2. doi: 10.1128/JB.182.10.2970-2972.2000
36. Li XS, Liu BG, Dong P, Li FL, Yuan L, Hu GZ. The prevalence of mcr-1 and resistance characteristics of Escherichia coli isolates from diseased and healthy pigs. Diagn Microbiol Infect Dis. (2017) 91:63–5. doi: 10.1016/j.diagmicrobio.2017.12.014
Keywords: multiresistance, G. parasuis, ICE, T4SS, ICEHpa1 family
Citation: Sun H, Zhang J, Miao Q, Zhai Y, Pan Y, Yuan L, Yan F, Wu H and Hu G (2022) Genomic insight into the integrative conjugative elements from ICEHpa1 family. Front. Vet. Sci. 9:986824. doi: 10.3389/fvets.2022.986824
Received: 05 July 2022; Accepted: 02 August 2022;
Published: 19 August 2022.
Edited by:
Xu Wang, Huazxhong Agricultural University, ChinaReviewed by:
Dexi Li, Henan Agricultural University, ChinaJohn Dustin Loy, University of Nebraska-Lincoln, United States
Copyright © 2022 Sun, Zhang, Miao, Zhai, Pan, Yuan, Yan, Wu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Wu, aHVhdG9uZ3poaTY2QDE2My5jb20=; Gongzheng Hu, eWFvbGlsYWJAMTYzLmNvbQ==
†These authors have contributed equally to this work