
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 19 August 2022
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.978243
 Zhi-ying Xu1,2,3,4†
Zhi-ying Xu1,2,3,4† Han Gao1,2,3,4†
Han Gao1,2,3,4† Qi-yuan Kuang1,2,3,4
Qi-yuan Kuang1,2,3,4 Jia-bao Xing1,2,3,4Zhi-yuan Wang1,2,3,4Xin-yu Cao1,2,3,4Si-jia Xu1,2,3,4Jing Liu1,2,3,4Zhao Huang1,2,3,4
Jia-bao Xing1,2,3,4Zhi-yuan Wang1,2,3,4Xin-yu Cao1,2,3,4Si-jia Xu1,2,3,4Jing Liu1,2,3,4Zhao Huang1,2,3,4 Ze-zhong Zheng1,2,3,4
Ze-zhong Zheng1,2,3,4 Lang Gong1,2,3,4
Lang Gong1,2,3,4 Heng Wang1,2,3,4
Heng Wang1,2,3,4 Mang Shi5*
Mang Shi5* Gui-hong Zhang1,2,3,4*Yan-kuo Sun1,2,3,4*
Gui-hong Zhang1,2,3,4*Yan-kuo Sun1,2,3,4*African swine fever (ASF) outbreak have caused tremendous economic loss to the pig industry in China since its emergence in August 2018. Previous studies revealed that many published sequences are not suitable for detailed analyses due to the lack of data regarding quality parameters and methodology, and outdated annotations. Thus, high-quality genomes of highly pathogenic strains that can be used as references for early Chinese ASF outbreaks are still lacking, and little is known about the features of intra-host variants of ASF virus (ASFV). In this study, a full genome sequencing of clinical samples from the first ASF outbreak in Guangdong in 2018 was performed using MGI (MGI Tech Co., Ltd., Shenzhen, China) and Nanopore sequencing platforms, followed by Sanger sequencing to verify the variations. With 22 sequencing corrections, we obtained a high-quality genome of one of the earliest virulent isolates, GZ201801_2. After proofreading, we improved (add or modify) the annotations of this isolate using the whole genome alignment with Georgia 2007/1. Based on the complete genome sequence, we constructed the methylation profiles of early ASFV strains in China and predicted the potential 5mC and 6mA methylation sites, which are likely involved in metabolism, transcription, and replication. Additionally, the intra-host single nucleotide variant distribution and mutant allele frequency in the clinical samples of early strain were determined for the first time and found a strong preference for A and T substitution mutation, non-synonymous mutations, and mutations that resulted in amino acid substitutions into Lysine. In conclusion, this study provides a high-quality genome sequence, updated genome annotation, methylation profile, and mutation spectrum of early ASFV strains in China, thereby providing a reference basis for further studies on the evolution, transmission, and virulence of ASFV.
African swine fever (ASF) is a highly contagious viral disease in swine which exhibit a high mortality rate approaching 100% (1–3). ASF virus (ASFV), the causative agent of ASF, belongs to the order Asfuvirales, family Asfarviridae, and genus Asfivirus (4). ASFV was first introduced in northeast China in August 2018. Subsequently, the virus was isolated and analyzed (5, 6). Since then, ASFV has been causing series of outbreaks which resulted in significant economic loss nationwide (7). Recently, the emergence of ASFV strains with more genomic diversity and lower virulence in domestic pigs has been reported in China (8, 9).
To perform variant, transcriptome, and proteome analysis of ASFV, a high-quality genome is necessary (10). Several early strains, from the outbreaks in China, were isolated and sequenced using either Sanger sequencing or next-generation sequencing (NGS). However, these technologies could not overcome the limitations associated with sequencing large genomes, as the ASFV genome (170~193 kb) contains a series of homopolymers and repeat regions, including inverted terminal repeats of variable length (11, 12). Using data from three sequencing technologies, Nanopore, Illumina, and Sanger sequencing, 71 sequencing errors and several regional mistakes were corrected in the high-quality genome of the Georgia 2007/1 strain (13). Thus, there is still a need for a high-quality genome sequence for the ASFV strain that caused early outbreaks in China to be used as a reference for further research.
Intra-host single nucleotide variants (iSNVs), also known as quasispecies, are generated during virus evolutionary processes that occur within a host. Previous studies have reported that intra-host diversity of viral genomes was correlated with the virus virulence, immunogenicity, and infectivity (14). While consensus-level sequencing of ASFV is relatively commonplace in molecular epidemiology, few studies have explored the genetic characteristics of iSNVs and the mutational pool of ASFV. Currently, the characteristic of iSNVs of circulating ASFVs were found to be diverse unveiled by our lab (unpublished data). Especially, the characteristics of iSNVs of early strain are important and unavailable. It can serve as a reference for further studies on tracing the route, evolutionary patterns, and virulence of ASFV.
Herein, we used the MGI (MGI Tech Co., Ltd., Shenzhen, China) platform, Nanopore long-read sequencing, and Sanger sequencing to obtain a high-quality genome of the first virulent isolates of the ASFV in Guangdong. The genomic characteristics were also examined, including updated genomic annotations and predicted methylation modifications. Additionally, the iSNVs of this strain were explored. The results of this study provide in-depth knowledge of the early strains from the outbreak in China.
Field samples were collected from the first ASF outbreak in Zhuhai, Guangdong Province, China in December 2018. Infected pigs showed 100% mortality. The spleens, lymph nodes, and other tissue specimens were collected from a single pig in one ASFV-positive herd. The virus was subsequently isolated from lymph nodes using porcine bone macrophages that were extracted from 4-week-old specific pathogen-free pigs (15). This virus was designated the name ASFV_GZ201801_2.
To avoid the accumulation of single nucleotide variants during the passage process, viral DNA for MGI platform sequencing was extracted directly from specific tissues. The first-generation culture of porcine bone macrophages was used to prepare extracellular virions for Nanopore sequencing. The QIAamp DNA Mini Kit (51304, Qiagen, Germany) was used for extraction, and the extracted viral DNA was purified using the AMPure XP Purification Kit (A63880, Beckman Coulter, USA). The quality and concentration were evaluated using Nanodrop Lite (ND-LITE-PR, ThermoFisher, USA) and Qubit 4.0 Fluorometer (Q33238, Invitrogen, USA). All these procedures were performed according to the manufacturer's instructions.
The purified viral DNA from clinical sample was processed using the MGI platform for genome sequencing to generate sequence data, whereas the DNA from the first-generation porcine bone macrophage culture viral supernatant was used for Nanopore sequencing, which was performed according to the manufacturer's instructions. Library preparation was carried out using the Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies, UK) and Native Barcoding Expansion 1–12 (EXP-NBD104, Oxford Nanopore Technologies, UK). Subsequently, the library was loaded onto the R9.4.1 MinION flow cell chip (FLO-MIN106, Oxford Nanopore Technologies, UK) and sequenced. Base calling was performed using Guppy v2.1.3 (Oxford Nanopore Technologies, UK).
The data generated from the MGI platform were processed by removing adapter contamination and filtering using Trimmomatic v0.39 (16). Nanopore sequence data that had a quality value lower than Q20 were filtered using Fastp v0.20.1 (17). All filtered reads were hybrid-assembled using SPAdes v3.13.0 (18). The consensus sequence was subsequently generated from contigs, using iVar v1.3.1 (19). Subsequently, single nucleotide polymorphisms and indels were verified using bcftools v1.10.2 (20), and variants were validated by Sanger sequencing using clinical sample. Thereafter, genomic annotation was conducted using the Genome Annotation Transfer Utility (GATU) software; ASFV Georgia 2007/1 was used as the reference genome sequence (GenBank accession number: FR682468.2) (21).
To explore the genomic characteristics, ASFV references for genotype II, from GenBank, were downloaded and aligned using Mafft v7.471 (Table 1) (22). Multiple sequence alignment was performed and visualized using the R packages, RColorBrewer, and ggplot2 (23, 24). Nanopore fast5 files were used to screen potentially methylated nucleotides using Tombo v1.5.1 (25). Andefault model of Tombo was automatically selected to detect 5mC and 6mA methylation modifications in the ASFV_GZ201801_2 genome, and it subsequently computed the corresponding scores.
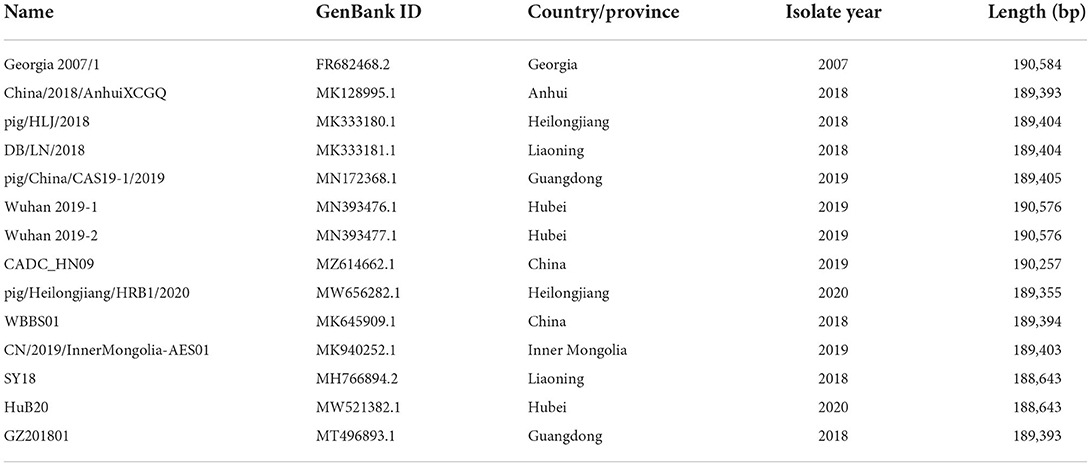
Table 1. Reference strains selected in this study.
The genome sequence that was validated by Sanger sequencing was used for iSNV analysis. Clean reads with high-quality bases (Q>20) were mapped to the validated genome sequence using BWA v0.7.17 (26) and then reformatted using SAMtools v1.10 (27). Subsequently, iVar v1.3.1, mentioned above, was then used to call iSNVs. Several criteria were used to screen for high-quality iSNVs: (1) total sequencing depth at iSNV site ≥100, (2) minor allele frequency ≥1%, and (3) depth of the minor allele ≥5. iSNVs that met the criteria were counted. To analyze the characteristics of the iSNVs, we calculated the preference of mutation types, the position of mutation, and the effect of mutation in the corresponding amino acid or ORF (Open Reading Frame).
The MGI/Nanopore sequencing data was processed, and the variants were identified using Sanger sequencing. Combining the advantage of MGI (High throughput), Nanopore (Long-read) and Sanger sequencing (High accuracy), we obtained a high-quality genome, named ASFV_GZ201801_2 (Genbank ID: ON263123). The genome sequence of this strain had 189,401 base pairs (bp) (Figure 1), with a high depth (MGI mean depth: 6656×, Nanopore mean depth: 74×) and total coverage (100%). Data from Nanopore platform were distributed evenly in the whole genome, and hybrid of sequencing data from two platforms led to a good resolution of ASFV genome. Compared our genome with early ASFV representative strains in China (ASFV Pig/HLJ/18: MK333180.1, ASFV China/2018/AnhuiXCGQ: MK128995.1), similar genome sizes found. ASFV_GZ201801_2 was annotated using ASFV Georgia 2007/1 as the reference genome. Furthermore, the novel annotations of this strain were explored.
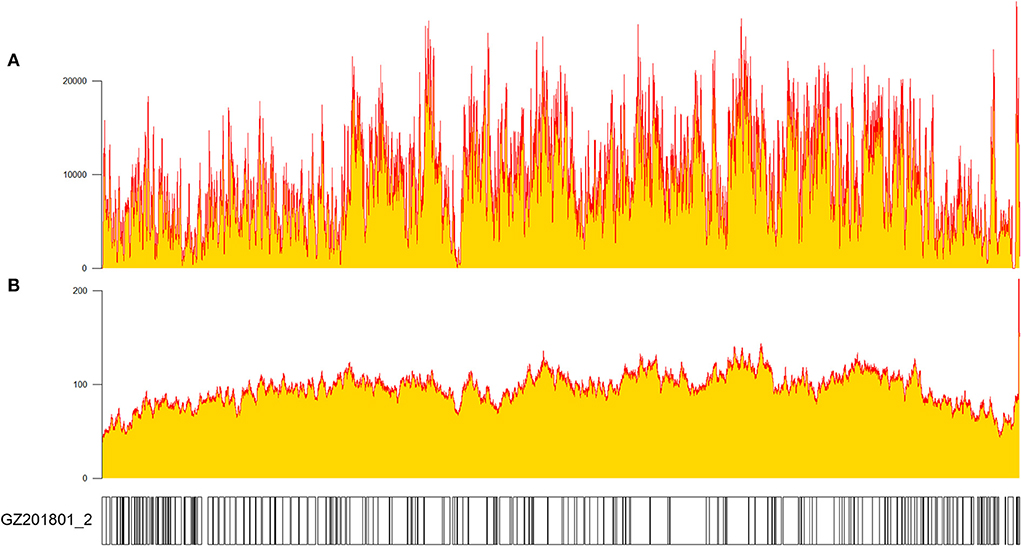
Figure 1. Depth and coverage of GZ201801_2 measured using SAMtools v1.10 after sequencing (27). (A) MGI platform sequencing and (B) Nanopore sequencing.
The annotations were manually examined and modified to fit any changes in ORFs, such as splits, fusions, or truncations. A total of 194 annotations were identified, eight of which were modified to accommodate changes caused by indels and nucleotide substitutions in the ORFs of ASFV_GZ201801_2. Modified annotations included the following ORFs: MGF 110-1L, ASFV G ACD 00190, MGF 110-11L, MGF 110-14L, MGF 360-10L, MGF 505-9R, NP419L, and I267L. The verified ASFV_GZ201801_2, comparing with ASFV_GZ201801 (179 ORFs), had clarified 14 novel annotations, which were 285L, ASFV_G_ACD_00120, ASFV_G_ACD_00350, ASFV_G_ACD_01980, C717R, hypothetical, hypothetical1, hypothetical2, MGF_100-3L, MGF_300-2R, MGF_360-16R, MGF_360-1Lb, MGF_360-19Ra, and MGF_360-19Rb (Figure 2). Notably, a series of genes in ASFV Georgia 2007/1 were annotated without the ATG initiation codon: DP60R, ASFV_G_ACD_00190, ASFV_G_ACD_00270, ASFV_G_ACD_00350, MGF_360-15R, NP1450L, and ASFV_G_ACD_01980, which started with leucine (UUA/UUG), while MGF_110-8L initiated with valine (GTG).
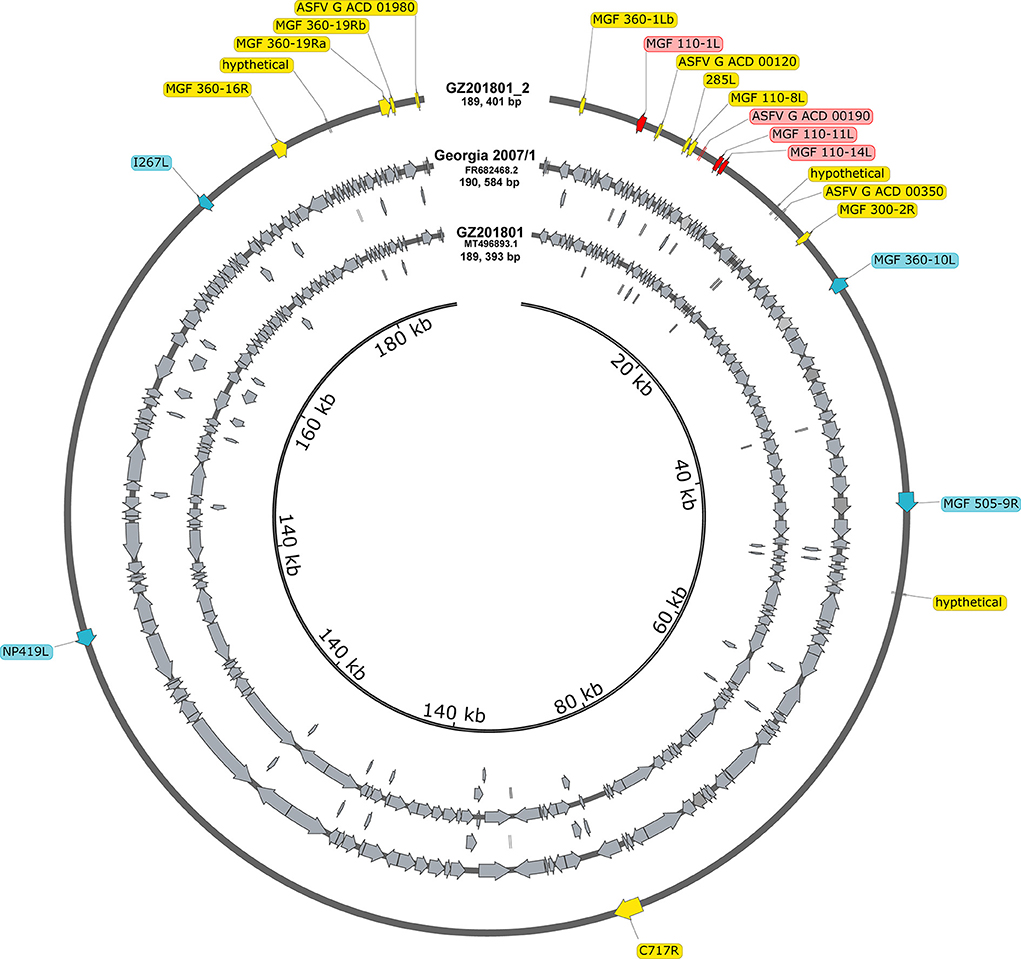
Figure 2. All annotations in the entire genome of Georgia 2007/1 and GZ201801. Compared to GZ201801, the genes marked with yellow represent the updated annotation of the GZ201801_2. The genes in blue and red represent different annotations GZ201801_2 in Georgia 2007/1, where blue represents different annotations due to site mutations, and red represents different annotations that appear owing to deletion or early termination.
To explore the genomic characteristics of ASFV_GZ201801_2, we aligned all the available ASFV whole genome sequences in China and ASFV Georgia 2007/1 that circulated in Eastern Europe, for more than a decade ago, with the improved ASFV_GZ201801_2. ASFV GZ201801_2, ASFV Georgia 2007/1, and other early strains shared several indels and substitutions, which led to high overall nucleotide identification (from 98.3% to approximately 100%). The 5′-end of the genome of ASFV_GZ201801_2 exhibited characteristics similar to those of ASFV Georgia 2007/1 (FR682468.2); however, several isolates from China demonstrated homogeneous 5′-end deletions, except for three strains (MN393476.1, MN393477.1, and MZ614662.1). We further compared our verified sequence to ASFV Pig/HLJ/2018 and found 5 indels and 6 substitutions located in homopolymer regions, which may be associated with sequencing errors. Similar findings were also observed in several other early strains from China. Furthermore, we demonstrated that the recent strains, in China, had undergone more variations and showed more diversity than earlier strains (Figure 3).
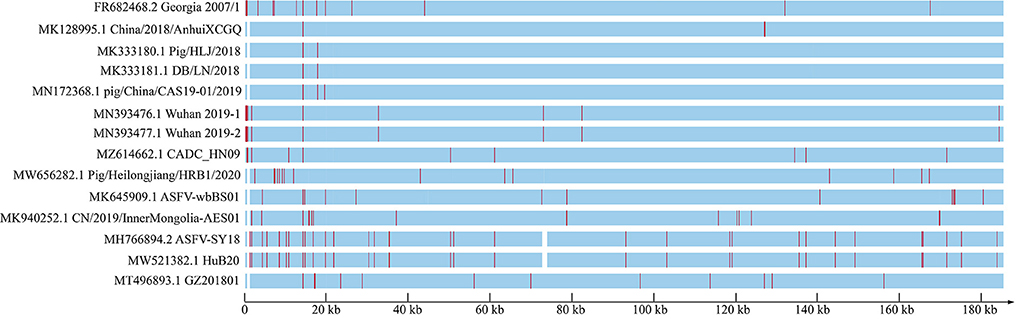
Figure 3. ASFV genome from China and Georgia 2007/1, which has been considered as the original strain circulating around Eurasia since 2007, showing the entire genome frame map of all genome-wide sequences compared to GZ201801_2. Each red line represents a site mutation, and the vacant positions represent deletion regions.
Potential 5mC and 6mA methylation were analyzed using Nanopore data. The methylation sites were predicted to be evenly distributed along the genome without remarkable regional preferences. In addition, a similar methylation state on the forward and reverse strands was noted, and the 6mA methylation scores at various sites on the entire genome were generally greater than those of 5mC methylation (Figures 4A–C). We predicted ten methylation sites, including their adjacent regions, in ASFV_GZ201801_2 that were most likely to have 5mC and 6mA modifications (Figures 4D,E). The genes that showed significant 5mC methylation included F778R, EP1242L, M448R, C257L, C475L, C962R, B646L, NP1450L, D1133L, and H359L, while the genes that showed significant 6mA methylation were MGF_100-1R, MGF_360-8L, A859L, F778R, F1055L, K205R, C962R, B318L, B354L, and CP2475L. The genes, including the potential 5mC and 6mA methylation sites, were primarily involved in metabolism, transcription, and replication (Tables 2, 3) (28).
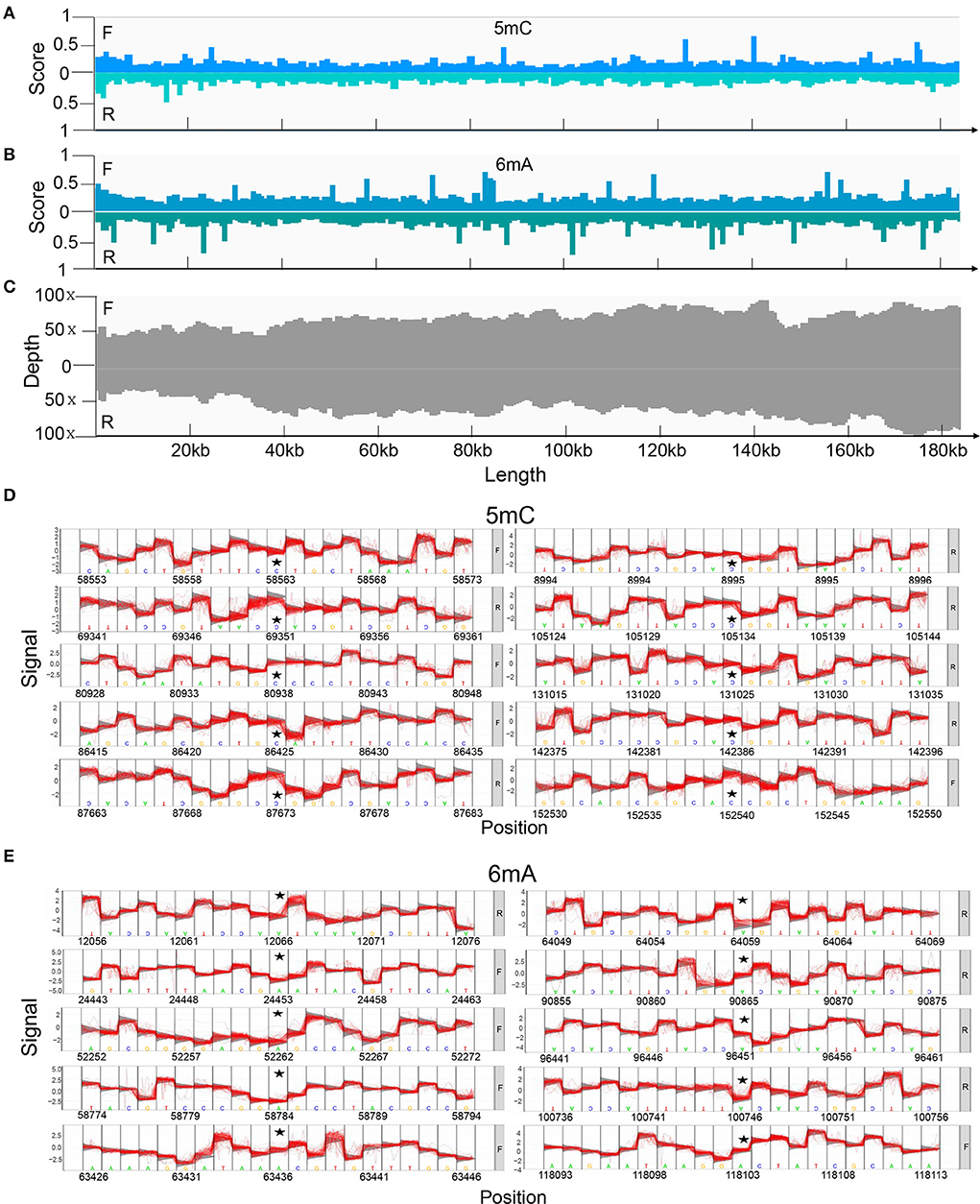
Figure 4. Methylation of positive and reverse strands in the genome predicted using Tombo and its corresponding depth. (A) The score of 5mC and (B) 6mA modification of positive and reverse strands. We compared the raw signal levels between sequenced samples and alternative models to obtain the statistical results of the corresponding sites, Tombo generated the correlation scores to evaluate the possibility of methylation on certain sites in the full-length viral genome. A higher score indicated that the site was more likely to undergo methylation modification. (C) Depth of Nanopore sequencing of positive and reverse strands. The top 10 methylation sites and their adjacent regions in the ASFV_GZ201801_2 that are most likely to be modified by (D) 5mC and (E) 6mA methylation also predicted using Tombo.
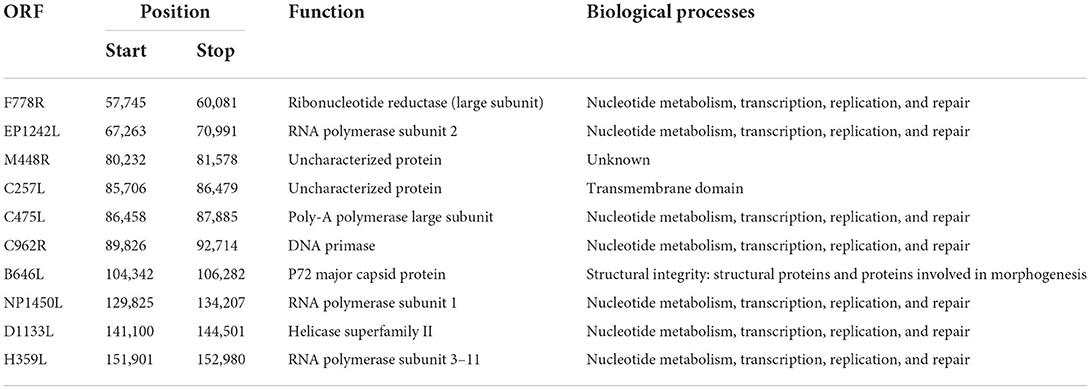
Table 2. Gene position, coded protein, and biological processes of the top 10 sites where 5mC methylation modification was predicted (28).

Table 3. Gene position, encoded protein, and biological process of the top 10 sites where the 6mA methylation was predicted (28–30).
Through the deep MGI sequencing of the genomic DNA, extracted from the clinical specimens, we achieved a high sequencing depth for all sites across the genome, with a mean depth of 6656 (coverage > 99%). Minor iSNVs were called by setting the threshold for minor allele frequency at 1% to exclude potential sequencing errors and minimize false discovery rates. The viral populations revealed a low frequency (<25%) cloud of iSNVs detectable across the entire genome. The minor iSNVs were distributed evenly on the genome, and marginally higher mutation frequencies were observed at the 5'-end than in the rest of the genome. Moreover, mutation preference across the genome was explored. To be specific, no obvious mutation preference could be observed when we calculated the mutations from a certain base to the other three bases (Figure 5A). On the contrary, when we calculated the mutations from the other three bases to a certain base, strong preference was found in Adenine (A) and Thymine (T) (Figure 5B). We further calculated the types of mutations in all the bases and their frequency and found that substitutions involving A and T occupied the largest proportion, reaching 28.044 and 17.343%, respectively. In addition, other mutations substituting Cytosine (C) with T or A occupied 10% above, whereas the rest of the mutations showed low mutation frequencies (Figure 5C). We further investigated the regions where the mutations were located, and 113 of the 190 iSNVs were found in the coding region (Figure 5D). Among all the genes, iSNVs were found slightly enrich in MGF 360-13La, MGF 505-7R, I96L, and MGF 360-21R. Moreover, the mutations were evenly distributed in the three positions of the codons which encode an amio acid, and the proportion of non-synonymous mutations was 76.11%, which was three times that of synonymous mutations (Figures 5E,F). More than half of the non-synonymous mutations showed a preference for lysine. Notably, 4 iSNVs of the non-synonymous mutations preferred to occur in the stop codon, which caused a frameshift mutation in the entire ORF; the four iSNVs were located in ASFV G ACD 00120, MGF 505-4R, K421R, and EP153R (Figure 5G).
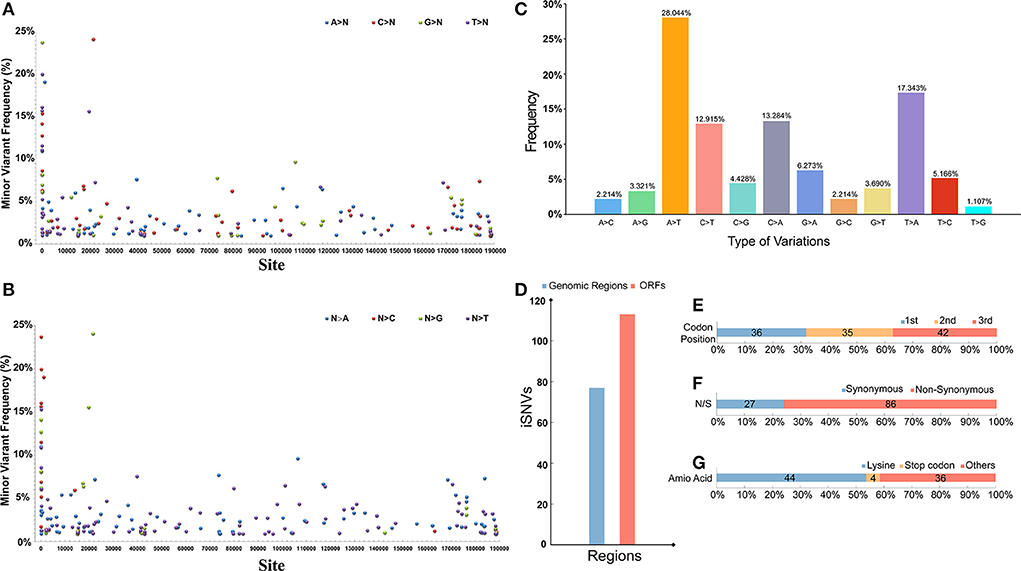
Figure 5. (A) The distribution and frequency of theiSNVs) that a certain base (A, T, C, G) mutate into other three bases. (B) The distribution and frequency of the iSNVs that the other three bases mutate into a certain base. (C) The percentage of different iSNVs mutation types. (D) Number of iSNVs in coding and non-coding regions. (E) Proportion of iSNVs occurring in different positions in codons across the genome. (F) Proportion of iSNVs causing non-synonymous and synonymous mutations. (G) Proportion of iSNVs that mutate into different type of amino acid. The number of iSNVs in each figure is indicated on the corresponding bar.
ASFV has been circulating in China for 5 years. Recently, low virulent strains with mutations and indels in the genomes have been detected; these strains have varying biological characteristics (8). The identification of mutations and deletions requires a high-quality genome. However, in China, there is still a lack of high-quality genomes of the early strains to use as references when studying the long-term effects of deletions and mutations. The complete genome of ASFV is typically obtained using either Sanger sequencing or NGS (11, 31). However, genome sequences generated from either one of these technologies do not reflect the actual genomic characteristics of ASFV, which has a large and intricate genome (13). The co-employment of Nanopore sequencing and NGS is an emerging strategy that is used to acquire complex microbial genomes, which combine the advantage of long sequencing reads from Nanopore and high accuracy from NGS (32, 33). Here, we combined the data generated from the MGI sequencing platform, Nanopore platforms, and Sanger sequencing to verify and obtain a high-quality genome of the lethal ASFV isolate, ASFV_GZ201801_2, which we collected during the early phase of the ASF pandemic in China.
Because of its complex genome, many genomic characteristics of ASFV remain unknown; thus, an accurate genome sequence was urgently needed. We compared ASFV_GZ201801_2 and ASFV Georgia 2007/1(FR682468.2) and detected a large number of site mutations, which had accumulated over the past decade. Furthermore, the genome sequence that we acquired, using samples from the first case of a high virulence ASF outbreak in China, could reflect the real epidemic strains from the early ASFV outbreak. In addition, we compared ASFV_GZ201801_2, ASFV Pig/HLJ/2018, and ASFV China/2018/AnhuiXCGQ and found that the strains from the early outbreaks in China shared a few same mutations. To conduct molecular epidemiology investigations of ASFV in China, a high-quality genome, isolated from the early strains from the first outbreak, is needed for reference. Many new site mutations have appeared in the recent Chinese strains, indicating the growing diversity of the ASFV strains in China. However, current circulating strains have shared several doubtful mutations when compared with early reference genome sequences, which may be caused by sequencing errors. Lacking a preferred reference would hinder further epidemiological studies of ASFV in China. Our verified genome sequence could be utilized as a reference for the early strains to trace the transmission route and evolution of ASFV.
Globally, the annotation of ASFV is constantly being updated; however, annotations of Chinese ASFV isolates still need to be confirmed. In our study, we found outdated and ambiguous annotations. We refined our annotations using the well-known sequence, ASFV Georgia 2007/1; several annotations were modified to accommodate changes in our genome. However, owing to the lack of transcriptome and proteome studies, the expression data of these ORFs was insufficient. There is a need to validate ASFV genome annotations reported by different studies worldwide.
Methylation of specific viral genes is involved in the transition from lytic to latent infection. Viral DNA methylation plays a crucial role in the life cycle of Epstein-Barr virus, adenovirus, and hepatitis B virus (34–38). Whether ASFV genome has DNA methylation and epigenetic regulation is to be discerned. There was research claimed that there is no ASFV methylation in infected Vero cells, while previous studies of the ASFV BA71V strain revealed the methylation of its 5′ cap (39, 40). With the development of technology, the methylation ASFV Wuhan 2019/1 was predicted using Nanopore which could detect methylation in high accuracy (41). Based on several recent clinical reports of ASF outbreaks in China, the period of onset of ASF in ASFV-positive pigs had extended, which might be a consequence of distinct ASFV DNA methylation. The mechanism related to epigenetic modifications underlying this phenomenon remains unclear and requires further exploration. Furthermore, previous studies have reported that 5mC and 6mA modifications potentially exist in the ASFV genome; these may be essential for virus-host interactions (41). In summary, our research contributes to the elucidation of the methylation profile of early AFSV strains in China using Nanopore sequencing technology, which provides a foundation for further research on the ASFV life cycle and immune regulation.
The iSNVs detected in early strains can be treated as the advantage mutation pool of ASFV, from which gain (or lose)-of-function mutations may be acquired by current or future epidemic strains; these iSNVs provide valuable information for epidemiology investigation and viral evolution studies. Eighty-five percent of the amino acid variations of the current SARS-CoV-2 genome correspond with iSNVs found in the early strains, which suggested that in-depth studies of the mutation pool of early strains were necessary. To characterize the landscape of within-host variants of early ASFV strains, we employed the deep MGI sequencing of viral DNA, extracted from ASFV-positive samples, to reveal the mutation spectrum, which may be used as a reference for current and future studies. The frequency of iSNVs in the ASFV genome was low, below 25%, which indicated a slow mutation rate. The frequency of substitution mutations, substituting with A or T, was substantially higher than the others. This indicated that the replication of ASFV showed preference, and the mechanism and effects of the replication process need to be further studied. The mutations were more concentrated in the coding region than in the non-coding region. Considering that the length of the coding region in ASFV is approximately 8.5 times the length of the non-coding region, the probability of mutation in the non-coding region is considerably higher than that of the coding region, which may be related to the regulation of ASFV. The regulatory role of the non-coding region between genes was previously reported (42), which also suggests that future research on ASFV should focus on the non-coding region. In addition, mutations have a similar probability of occurring at each location in the codons with no significant bias, which indicates that the choice of mutation does not favor a certain base in the codon. The probability of non-synonymous mutations is considerably higher than that of synonymous mutations. Non-synonymous mutations typically disappear owing to the effects of population selection, but their accumulation can lead to dramatic changes in the virulence, immunogenicity, and infectivity of the virus (43, 44), and afford an opportunity for viral adaptation (45). More than half of these non-synonymous mutations resulted in substitution with lysine, which is encoded by AAA. This mutation is commonly found near consecutive bases, which are widely distributed in the ASFV genome, but the mechanism underlying the preference for this type of mutation requires further investigation. To our knowledge, this is the first time that the mutation spectrum of early ASFV strains was determined. Our findings provide a reference to use in further investigations on molecular epidemiology, evolution, host adaptation, and pathogenicity of ASFV.
To our knowledge, this is the first time that the high-quality proofread genome sequence of GZ201801_2 and the mutant spectrum of early strains of ASFV were revealed. Our findings would provide a reference to pave the way for further investigations on molecular epidemiologic investigations, evolution, host adaptation, and pathogenicity of ASFV.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, ON263123.
Z-yX: writing original draft and software. HG: writing review and editing. Q-yK: data curation and validation. J-bX: visualization and validation. Z-yW: data curation. X-yC: validation. S-jX: formal analysis. JL: investigation. ZH and MS: methodology. Z-zZ: resources. LG: funding acquisition. HW: supervision. G-hZ and Y-kS: resources and project administration. All authors contributed to the article and approved the submitted version.
This study is funded by the National Key Research and Development Program of China (Grant Number 2021YFD1800101), the National Natural Science Foundation of China (Grant Number 32102704 and 31941004), the Key-Area Research and Development Program of Guangdong Province (Grant Number 2019B020211003), and the China Agriculture Research System of MOF and MARA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wang G, Xie M, Wu W, Chen Z. Structures and functional diversities of ASFV proteins. Viruses. (2021) 13:2124. doi: 10.3390/v13112124
2. Gaudreault NN, Madden DW, Wilson WC, Trujillo JD, Richt JA. African swine fever virus: an emerging DNA arbovirus. Front Vet Sci. (2020) 7:215. doi: 10.3389/fvets.2020.00215
3. Olesen AS, Lohse L, Boklund A, Halasa T, Gallardo C, Pejsak Z, et al. Transmission of African swine fever virus from infected pigs by direct contact and aerosol routes. Vet Microbiol. (2017) 211:92–102. doi: 10.1016/j.vetmic.2017.10.004
4. Guo F, Shi Y, Yang M, Guo Y, Shen Z, Li M, et al. The structural basis of African swine fever virus core shell protein p15 binding to DNA. FASEB J. (2021) 35:e21350. doi: 10.1096/fj.202002145R
5. Wen X, He X, Zhang X, Zhang X, Liu L, Guan Y, et al. Genome sequences derived from pig and dried blood pig feed samples provide important insights into the transmission of African swine fever virus in China in 2018. Emerg Microbes Infect. (2019) 8:303–6. doi: 10.1080/22221751.2019.1565915
6. Zhou X, Li N, Luo Y, Liu Y, Miao F, Chen T, et al. Emergence of African swine fever in China, 2018. Transbound Emerg Dis. (2018) 65:1482–4. doi: 10.1111/tbed.12989
7. Zhao D, Liu R, Zhang X, Li F, Wang J, Zhang J, et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg Microbes Infect. (2019) 8:438–47. doi: 10.1080/22221751.2019.1590128
8. Sun E, Huang L, Zhang X, Zhang J, Shen D, Zhang Z, et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg Microbes Infect. (2021) 10:2183–93. doi: 10.1080/22221751.2021.1999779
9. Sun E, Zhang Z, Wang Z, He X, Zhang X, Wang L, et al. Emergence and prevalence of naturally occurring lower virulent African swine fever viruses in domestic pigs in China in 2020. Sci China Life Sci. (2021) 64:752–65. doi: 10.1007/s11427-021-1904-4
10. Forth JH, Forth LF, Blome S, Hoper D, Beer M. African swine fever whole-genome sequencing-quantity wanted but quality needed. PLoS Pathog. (2020) 16:e1008779. doi: 10.1371/journal.ppat.1008779
11. Bao J, Wang Q, Lin P, Liu C, Li L, Wu X, et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 genotype II strains. Transbound Emerg Dis. (2019) 66:1167–76. doi: 10.1111/tbed.13124
12. Dixon LK, Sun H, Roberts H. African swine fever. Antiviral Res. (2019) 165:34–41. doi: 10.1016/j.antiviral.2019.02.018
13. Forth JH, Forth LF, King J, Groza O, Hubner A, Olesen AS, et al. A deep-sequencing workflow for the fast and efficient generation of high-quality African swine fever virus whole-genome sequences. Viruses. (2019) 11:846. doi: 10.3390/v11090846
14. Andersen KG, Shapiro BJ, Matranga CB, Sealfon R, Lin AE, Moses LM, et al. Clinical sequencing uncovers origins and evolution of lassa virus. Cell. (2015) 162:738–50. doi: 10.1016/j.cell.2015.07.020
15. Carrillo C, Borca MV, Afonso CL, Onisk DV, Rock DL. Long-term persistent infection of swine monocytes/macrophages with African swine fever virus. J Virol. (1994) 68:580–3. doi: 10.1128/jvi.68.1.580-583.1994
16. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
17. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
18. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
19. Castellano S, Cestari F, Faglioni G, Tenedini E, Marino M, Artuso L, et al. iVar, an interpretation-oriented tool to manage the update and revision of variant annotation and classification. Genes. (2021) 12:384. doi: 10.3390/genes12030384
20. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
21. Tcherepanov V, Ehlers A, Upton C. Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Genomics. (2006) 7:150. doi: 10.1186/1471-2164-7-150
22. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. (2019) 20:1160–6. doi: 10.1093/bib/bbx108
23. Neuwirth E. RColorBrewer: ColorBrewer Palettes. (2014). Available online at: https://rdrr.io/cran/RColorBrewer/man/ColorBrewer.html
25. Stoiber M, Quick J, Egan R, Eun Lee J, Celniker S, Neely RK, et al. De novo identification of DNA modifications enabled by genome-guided nanopore signal processing. bioRxiv. (2017). doi: 10.1101/094672
26. Jo H, Koh G. Faster single-end alignment generation utilizing multi-thread for BWA. Biomed Mater Eng. (2015) 26:S1791–6. doi: 10.3233/BME-151480
27. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
28. Dixon LK, Chapman DA, Netherton CL, Upton C. African swine fever virus replication and genomics. Virus Res. (2013) 173:3–14. doi: 10.1016/j.virusres.2012.10.020
29. Wang Q, Zhou L, Wang J, Su D, Li D, Du Y, et al. African swine fever virus K205R induces ER stress and consequently activates autophagy and the NF-kappaB signaling pathway. Viruses. (2022) 14:394. doi: 10.3390/v14020394
30. Ramirez-Medina E, Vuono EA, Pruitt S, Rai A, Espinoza N, Velazquez-Salinas L, et al. Evaluation of an ASFV RNA helicase gene A859L for virus replication and swine virulence. Viruses. (2021) 14:10. doi: 10.3390/v14010010
31. Wang X, Wang X, Zhang S, He Y, Chen X, Liu X, et al. Genetic characterization and variation of African swine fever virus China/GD/2019 strain in domestic pigs. Pathogens. (2022) 11:97. doi: 10.3390/pathogens11010097
32. McNaughton AL, Roberts HE, Bonsall D, de Cesare M, Mokaya J, Lumley SF, et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci Rep. (2019) 9:7081. doi: 10.1038/s41598-019-43524-9
33. Kafetzopoulou LE, Efthymiadis K, Lewandowski K, Crook A, Carter D, Osborne J, et al. Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro Surveill. (2018) 23:1800228. doi: 10.2807/1560-7917.ES.2018.23.50.1800228
34. Watanabe Y, Yamamoto H, Oikawa R, Toyota M, Yamamoto M, Kokudo N, et al. DNA methylation at hepatitis B viral integrants is associated with methylation at flanking human genomic sequences. Genome Res. (2015) 25:328–37. doi: 10.1101/gr.175240.114
35. Koumbi L, Karayiannis P. The epigenetic control of Hepatitis B virus modulates the outcome of infection. Front Microbiol. (2015) 6:1491. doi: 10.3389/fmicb.2015.01491
36. Hoelzer K, Shackelton LA, Parrish CR. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. (2008) 36:2825–37. doi: 10.1093/nar/gkn121
37. Tao Q, Robertson KD. Stealth technology: how Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clin Immunol. (2003) 109:53–63. doi: 10.1016/S1521-6616(03)00198-0
38. Ambinder RF, Robertson KD, Tao Q. DNA methylation and the Epstein-Barr virus. Semin Cancer Biol. (1999) 9:369–75. doi: 10.1006/scbi.1999.0137
39. Salas ML, Kuznar J, Vinuela E. Polyadenylation, methylation, and capping of the RNA synthesized in vitro by African swine fever virus. Virology. (1981) 113:484–91. doi: 10.1016/0042-6822(81)90176-8
40. Weber S, Hakobyan A, Zakaryan H, Doerfler W. Intracellular African swine fever virus DNA remains unmethylated in infected Vero cells. Epigenomics. (2018) 10:289–99. doi: 10.2217/epi-2017-0131
41. Jia L, Chen J, Liu H, Fan W, Wang D, Li J, et al. Potential m6A and m5C methylations within the genome of a Chinese African swine fever virus strain. Virol Sin. (2021) 36:321–4. doi: 10.1007/s12250-020-00217-2
42. Dunn LEM, Ivens A, Netherton CL, Chapman DAG, Beard PM. Identification of a functional small noncoding RNA of African swine fever virus. J Virol. (2020) 94:e01515–20. doi: 10.1128/JVI.01515-20
43. Ladner JT, Wiley MR, Mate S, Dudas G, Prieto K, Lovett S, et al. Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell Host Microbe. (2015) 18:659–69. doi: 10.1016/j.chom.2015.11.008
44. Illingworth CJ, Fischer A, Mustonen V. Identifying selection in the within-host evolution of influenza using viral sequence data. PLoS Comput Biol. (2014) 10:e1003755. doi: 10.1371/journal.pcbi.1003755
Keywords: African swine fever, annotation, methylation, mutation spectrum, viral genome
Citation: Xu Z-y, Gao H, Kuang Q-y, Xing J-b, Wang Z-y, Cao X-y, Xu S-j, Liu J, Huang Z, Zheng Z-z, Gong L, Wang H, Shi M, Zhang G-h and Sun Y-k (2022) Clinical sequencing uncovers the genomic characteristics and mutation spectrum of the 2018 African swine fever virus in Guangdong, China. Front. Vet. Sci. 9:978243. doi: 10.3389/fvets.2022.978243
Received: 25 June 2022; Accepted: 02 August 2022;
Published: 19 August 2022.
Edited by:
Suresh Kumar Tikoo, University of Saskatchewan, CanadaReviewed by:
Lauro Velazquez-Salinas, United States Department of Agriculture, United StatesCopyright © 2022 Xu, Gao, Kuang, Xing, Wang, Cao, Xu, Liu, Huang, Zheng, Gong, Wang, Shi, Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mang Shi, c2hpbTIzQG1haWwuc3lzdS5lZHUuY24=; Gui-hong Zhang, Z3VpaG9uZ3poQHNjYXUuZWR1LmNu; Yan-kuo Sun, eWFua3Vvc3VuQHNjYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.