 Lei Zhang
Lei Zhang Linlin Chen
Linlin Chen Hongtao Zhang
Hongtao Zhang Hongbin Si
Hongbin Si Xianyong Liu
Xianyong Liu Xun Suo
Xun Suo Dandan Hu
Dandan Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 22 July 2022
Sec. Parasitology
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.954725
This article is part of the Research TopicHost response to Veterinary Infectious Diseases: Role of Coding and Non-coding RNAs as Biomarkers and Disease ModulatorsView all 7 articles
Apicomplexan parasites have divergent biogenesis machinery for small RNA generation. Analysis has shown that parasites in Plasmodium and Cryptosporidium as well as many species in Leishmania or Trypanosoma do not have a complete machinery in small RNA biogenesis. Recently, the miRNA-generating system of Toxoplasma has been identified as plant/fungal-like and its miRNAome has been elucidated. However, the microRNA (miRNA) expression profiles and their potential regulatory functions in different stages of Eimeria tenella remain largely unknown. In this study, we characterized the RNA silencing machinery of E. tenella and investigated the miRNA population distribution at different life stages by high-throughput sequencing. We characterized the expression of miRNAs in the unsporulated oocyst, sporulated oocyst and schizogony stages, obtaining a total of 392 miRNAs. We identified 58 differentially expressed miRNAs between USO (unsporulated oocysts) and SO (sporulated oocysts) that were significantly enriched for their potential target genes in the regulation of gene expression and chromatin binding, suggesting an epigenetic modulation of sporulating by these miRNAs. In comparing miRNA expression at endogenous and exogenous developmental stages, twenty-four miRNAs were identified differently expressed. Those were mainly associated with the regulation of genes with protein kinase activity, suggesting control of protein phosphorylation. This is the first study about the evolution of miRNA biogenesis system and miRNA control of gene expression in Eimeria species. Our data may lead to functional insights into of the regulation of gene expression during parasite life cycle in apicomplexan parasites.
Coccidiosis in chickens caused by apicomplexan parasite Eimeria is a widespread intestinal disease (1). According to the latest estimates, the global cost of coccidiosis to the poultry industry is about £10 billion annually (2). Eimeria parasites have a complex developmental life cycle, with an exogenous phase in the environment, where oocysts excreted from chickens undergo differentiation (sporulation) and become infective, and an endogenous phase in the intestinal epithelial cells consisting of 3–5 rounds of schizogony, resulting in successive generations of schizonts (containing several merozoites), followed by sexual development and shedding of unsporulated oocysts (3).
MiRNAs are a class of non-coding RNA (ncRNA) that interact with messenger RNAs (mRNAs), leading to mRNA degradation or translation inhibition. It plays critical roles in a variety of biological processes, including development, metabolism, and apoptosis (4–6). In the biogenesis of miRNAs, primary miRNAs (pri-miRNAs) are transcribed by RNA polymerase II and undergo nuclear and cytoplasmic processing events, as carried out by the endoribonucleases Drosha and Dicer, respectively (7, 8). The mature miRNA, associated with the Argonaute (Ago) protein, is loaded onto the RNA-induced silencing complex (RISC) to interact with target mRNAs and regulate gene expression (9, 10). A single miRNA can silence multiple genes, while a single gene can be targeted by multiple miRNAs (11).
Apicomplexan parasites have divergent biogenesis machinery for small RNA generation. Argonaute and Dicer homologs were not found in Cryptosporidium and Plasmodium by analysis of their genome sequences, speculating that they do not have a mechanism for miRNA biosynthesis (12–14). This speculation is also supported by previous study (15). Examination of the available Trypanosoma genome sequences revealed the non-existence of identifiable remnants of DCL1, DCL2, or AGO1 homologs in T. cruzi. It was also found that there was no Dicer homolog in the Leishmania species (L. major, L. donovani), but a severely impaired pseudogene at the AGO1 locus was presented (16, 17). However, TbAGO1, a member of the Argonaute protein family, and two Dicer-like homologs, TbDCL1 and TbDCL2, were found in T. brucei. Meanwhile, endogenous small interfering RNAs or siRNAs were also identified (18). These findings also confirm the absence of a complete machinery underlying the generation of small RNA for many species of Leishmania or Trypanosome. In the study of Toxoplasma, the miRNA-generating system was identified as plant/fungal-like, and its miRNAome was elucidated (19). However, nothing is known about Eimeria miRNAs.
In this study, we characterized the RNA silencing machinery in E. tenella and investigated the distribution of miRNA population at different time periods by high-throughput sequencing. Different miRNAs and their potential target genes were analyzed between unsporulated oocysts, sporulated oocysts, and merozoites. A miRNA-mRNA interaction network was also constructed. This is the first study on Eimeria miRNAs, and our data will provide functional insights into parasite's lifecycle progression, and also basic knowledge for future studies on RNAi-dependent regulatory mechanisms in other apicomplexan parasites.
The use of animals in this study was approved by the Administration Committee of Laboratory Animals in Guangxi University and was performed in accordance with the Institutional Animal Care and Use Committee guidelines (Approval Number: Gxu-2021-013).
The E. tenella Houghton (ETH) strain was used throughout this work. Parasites were maintained and propagated by oral infection in 1-week-old broilers (Sanhuang chicken). Four-week-old AA broilers (Arbor Acres Poultry Breeding, Beijing, China) were used for the preparation of merozoites. Chickens were raised in a coccidia-free environment with ad libitum supply of filtered water and anticoccidial- and antibiotic-free feed. Procedures for parasite collection, purification and sporulation were carried out as described previously (20).
Four distinct developmental stages of the parasites were incorporated in this study: Unsporulated oocysts (USO), Sporulated oocysts (SO), merozoites at 108 h post-infection (Mer108) and merozoites at 120 h post-infection (Mer120). Purified unsporulated oocysts were collected from intestinal contents at 7 days post-infection (d.p.i.) from three groups of chicken (3 birds/group). Samples of sporulated oocysts were collected from the feces of three cages of chicken 9 d.p.i, and sporulated in 2.5% K2CrO4 (21). Caeca were collected from six groups of chicken at 108 and 120 h after infection, respectively. The merozoites for each sample were collected separately as reported by Schwarz et al. (22) with modifications. Briefly, sheared cecum was digested (0.50% sodium taurodeoxycholate hydrate and 0.25% trypsin in PBS) at 42°C for 30 min, filtered through gauze and centrifuged to obtain a precipitate containing dirty merozoites, which was further filtered to obtain clean merozoites. RNAs from all samples were extracted separately with Trizol regent, and genomic DNA was digested with DNase I (Qiagen, Hilden, Germany). Total RNAs were used for RNA-seq, and small RNA molecules were purified and used for miRNA-seq. RNA integrity was assessed using a 1.0% agarose gel. Thereafter, the quality and quantity of RNA were assessed using a Nano Photometer® spectrophotometer (IMPLEN, CA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). High-quality RNA samples were subsequently submitted to Sangon Biotech (Shanghai) Co., Ltd. for library preparation and sequencing.
For miRNA sequencing, libraries were generated using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) according to the manufacturer's recommendations. Using total RNA as the starting sample, small RNA ends were directly connected to the adapter and synthesized by reverse transcription into cDNA. DNA fragments of 140–150 bp were separated by PAGE gel electrophoresis and the cDNA library was recovered. Finally, library quality was assessed on the Agilent Bioanalyzer 2100 system, and the libraries were sequenced on an Illumina NextSeq 500 platform.
For RNA-seq analysis, sequencing libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) according to the manufacturer's recommendations. Sequencing was performed using the Illumina Novaseq 6000 platform, generating 150 bp paired-end reads. The Illumina sequencing data used in this study could be found in Sequence Read Archive data base with project accession number: PRJNA832521.
Paired-end clean reads were aligned to our newly generated reference genome of the E. tenella H strain (deposited into CNGB Sequence Archive of China National GeneBank DataBase with accession number CNP0003153) using hisat2 software version 2.2.1 (23). Htseq-count version 0.13.5 (24) was used to count the reads. Differential expression analysis was performed for both conditions (Comparison of endogenous and exogenous developmental stages, and comparison between USO and SO) using the DESeq2 R package (1.20.0) (25). P-values were adjusted using the Benjamini & Hochberg method. Corrected P < 0.01 and log2 (Fold change) > 1 were considered significantly different. The FPKM values of each gene were calculated as described previously (26), and the FPKM values of the selected genes were used for clustered heatmap drawing via Pretty heatmaps in the R package.
Clean reads were obtained by removing reads that contained ploy-N, with 5′ adapter contaminants, without 3′ adapter or the insert tag, reads that contained ploy A or T or G or C, and low-quality reads from raw data. Then, a certain range of lengths was chosen from the clean reads to perform all downstream analyses. Using Bowtie software (27), clean reads were searched against several databases such as Silva, GtRNAdb, Rfam, and Repbase to filter for ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA) and other ncRNAs and repeats. The remaining reads were used to detect known miRNAs and novel miRNAs by comparison with our reference genome and known miRNAs from miRBase. Randfold software (28) was used for secondary structure prediction. Target gene prediction was performed by psRobot_tar in miRanda (29). Gene function was annotated based on the following databases: Nr, Pfam, KOG/COG, Swiss-Prot, KEGG, and GO.
DESeq2 (25) was used to compare expression levels between sample pairs. We estimated library size correction factors with median of ratios method, based on the number of mapped reads, in DESeq2 and used those factors in further models analyzing differential abundance. We used fold change > 2 and P < 0.05 as thresholds to define a significant differentially expressed miRNA. To provide an overview of the different states of gene expression, we used volcano plots to present genes with differential expression between paired samples. Gene Ontology (GO) enrichment analysis was used on target gene candidates of differentially expressed miRNAs. Goseq (30) was implemented for GO enrichment analysis. Transcripts per million (TPM) were calculated for each miRNA for data normalization (TPM = Readcounts * 1,000,000/Mapped Reads), and used for clustered heatmap drawing.
Key domains are involved in miRNA biogenesis, including Piwi-Argonaute-Zwille (PAZ, Pfam accession number: PF02170), Piwi (Pfam accession number: PF02171), Ribonuclease 3 (RNase III, Pfam accession number: PF00636), double-stranded RNA binding motif (Pfam accession number: PF00035) and double stranded RNA binding domain (DSRM, Pfam accession number: PF03368), whose models were obtained from the Pfam database (http://pfam.sanger.ac.uk/). Local HMMER3 (31) was used to find protein sequences containing domains targeting E. tenella and several other species by default parameters. Multiple alignments and maximum likelihood phylogenetic trees were built by muscle v3.8 (32) and IQ-TREE2 (33) based on Dicer and Argonaut sequences, respectively. A thousand bootstrap replicates were computed for each phylogenetic tree.
Previous analyses have shown that plant and animal Dicer proteins have divergent origins (34). Sequence analysis revealed that the E. tenella genome encodes only a Dicer-like protein, Et-ECL, which exhibits significant variability in protein sequence and domain organization compared to higher eukaryotes (Figure 1A and Supplementary Dataset 1). Et-ECL has two RNase III catalytic domains (RNase IIIa and RNase IIIb), but lacks a recognizable DSRM, PAZ, or RNA helicase domain (Figure 1B). These features clearly distinguish Et-DCL from Dicer of another coccidian, T. gondii, which has 4,447 amino acids and contains an additional helicase domain and a DEXDc domain (19). This structure is similar to the Dicer-like protein of the Sporozoa parasite Cyclospora cayetanensis (Figures 1B,C) and belongs to a specific branch supported by a strong bootstrap score (Figure 1A). Dicers from fungi, plants and vertebrates show similarity in domain organization and are located in their own clade in the phylogenetic tree. However, we were unable to identify any Et-ECL orthologs in other chicken coccidia even under a high cutoff (E-value = 1.0), and we speculate that this may be due to the lack of genome completeness or the absence of RNA silencing machinery in these species (Supplementary Dataset 2).
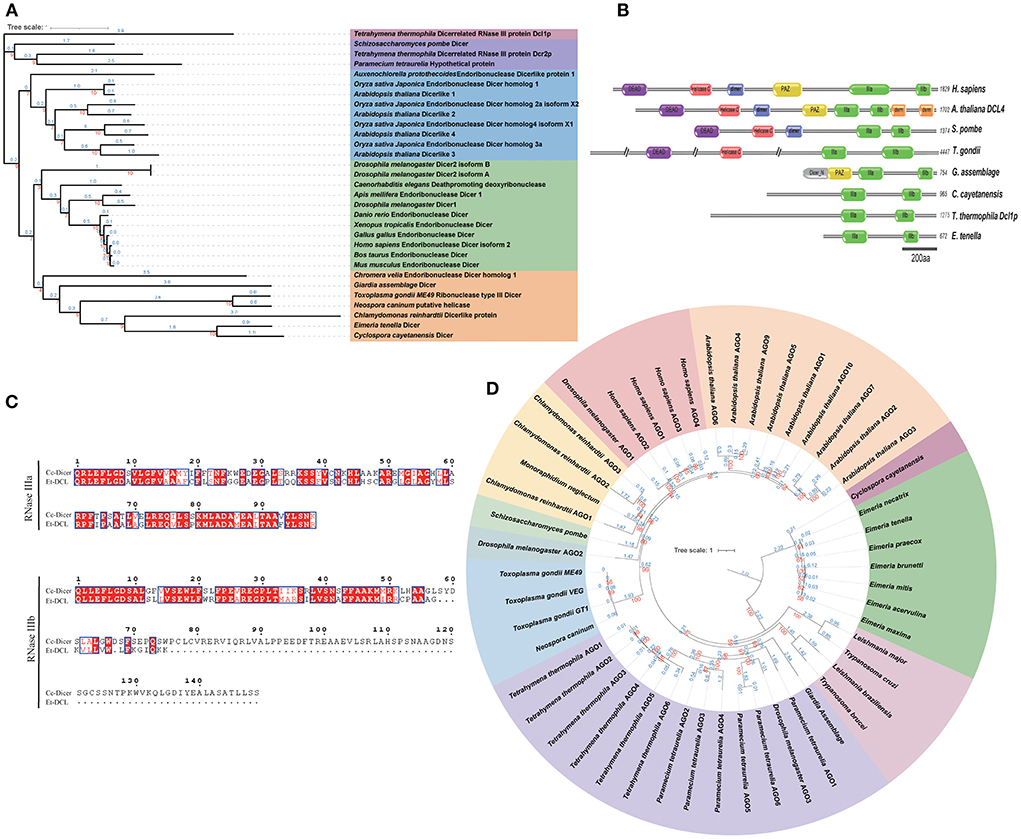
Figure 1. Domain organization and phylogenetic analysis of Dicer and Ago proteins. (A) Dicer-based maximum likelihood tree showing the evolutionary relationships between Eimeria and other species. Protein sequences were aligned by Muscle software, and the unrooted maximum likelihood tree was constructed using IQ-TREE2 with 1,000 bootstrap replicates. (B) Domain organization of Dicer proteins. IIIa, RNase IIIa; IIIb, RNase IIIb; PAZ, Piwi-Argonaute-Zwille domain; dsrm, recognizable dsRNA binding domain; Helicase C, RNA helicase domain; DEAD, DEAD box helicase; dimer, Dicer dimerisation domain. (C) Sequence alignment of conserved RNase domains of Dicer proteins from E. tenella and C. cayetanensis. (D) Argonaute-based maximum likelihood tree (rooted at midpoint) showing the evolutionary relationships between Eimeria and other species. Red and blue numbers on both phylogenetic trees represent the bootstrap values (%) and branch lengths, respectively. Protein accession ID shown in Supplementary Datasets 10, 11.
Interestingly, despite the lack of Dicer-like proteins in other chicken coccidia, a single Argonaute protein (Ago) was found in each Eimeria parasite by HMMER searches using the PAZ domain (PF02170) and the Piwi domain (PF02171) (Supplementary Dataset 1). By phylogenetic analysis, the Ago protein of E. tenella is located in the same branch with several other species of Eimeria and Cyclospora with long branch length. T. gondii and N. caninum Agos fall in a clade with algae (Chlamydomonas and Monoraphidium), plants and animals (Figure 1D). These results suggest that Eimreia's Ago proteins are divergent from Tg-Ago and others.
As Dicer-like protein was only found in E. tenella among chicken coccidia, we are interested to know its characteristics and roles in gene regulation during different life stages. To this end, miRNAs from USO, SO, Mer108, and Mer120 were identified by Illumina sequencing. After removal of low-quality reads, a total of 58 million clean reads were obtained (Supplementary Dataset 3). Unannotated reads containing miRNAs were obtained after filtering for ncRNAs such as ribosomal RNA (rRNA), transfer RNA (tRNA), intranuclear small RNA (snRNA), nucleolar small RNA (snoRNA), and repetitive sequences (the proportions of small ncRNAs in the four samples are given in Supplementary Dataset 4). In the merozoite samples (Mer108 and Mer120), only ~23% of the unannotated reads could be aligned with the E. tenella genome, and ~50% in the USO and SO samples (Supplementary Dataset 5). Comparable reads in each sample (ranged from 0.7 to 1.3 M, and with an average of 0.76 M for Mer108, 1.1 M for Mer120, 1.23 M for SO, and 0.83 M for USO) were used for differential expression analysis.
In total, we obtained 392 miRNAs with length distributions ranging from 18 to 25 nt across all samples, of which 18 nt represents the major size class (Figures 2A,B and Supplementary Dataset 6), which is significantly different from the length distribution and major size of T. gondii miRNAs (ranging from 17 to 33 nt with a major size of 25 nt). The development of miRNAs from precursors to mature bodies was accomplished by Dicer enzyme cutting. The specificity of enzyme cutting points gives a strong bias to the first base of its mature body sequence. By base-preference of the first base at the 5' end and at each site of the miRNA, we found that 18–20 nt of the miRNA first bases were biased toward G, except for miRNAs of 24 nt length that were biased toward U and others that were biased toward C (Figure 2C). This finding is less consistent with the previously reported first base bias toward the use of U (35–37), which is speculated to be either species-specific or related to the quality of sequencing, for reasons that require further investigation.
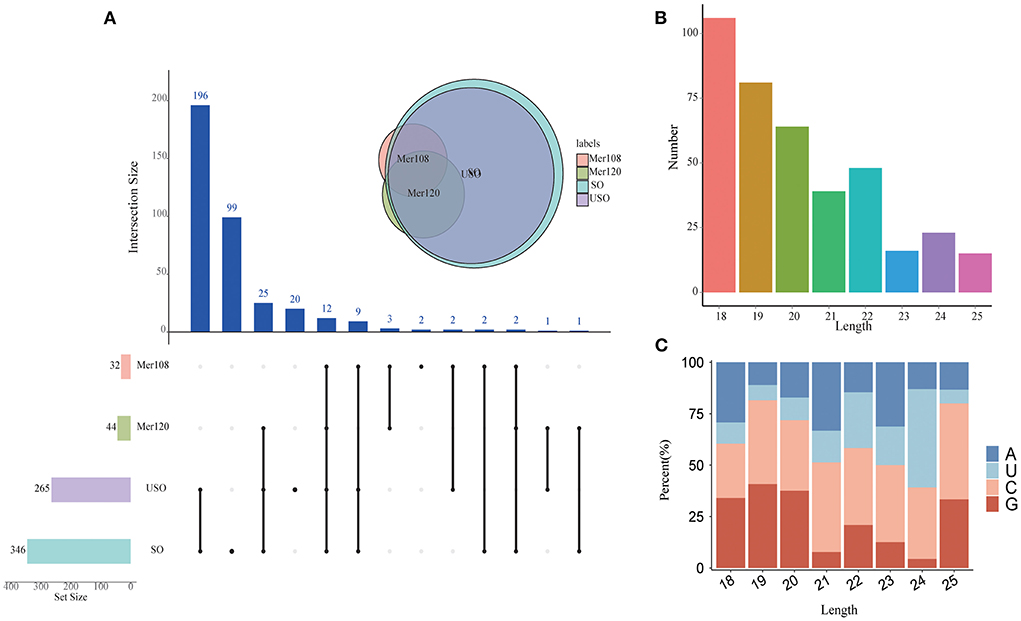
Figure 2. Characterization of the novel miRNAs in E. tenella. (A) An UpSet plot showing the set of all miRNAs from four different developmental stages. Vertical bars show the number of intersecting miRNAs between stages, denoted by the connected black circles below the histogram. The horizontal bars show the size of the transcript sets between stages. The Venn diagram shows the numbers and overlapping situation of miRNAs with different transcripts in different groups. (B) Length distribution of the novel E. tenella miRNAs. (C) Codon usage preference of the first base of E. tenella miRNAs of different lengths.
Eimeria parasites undergo endogenous and exogenous development, interacting with totally different environments. To understand whether miRNAs have a regulatory effect on the parasite when dealing with different environments, we compared miRNA expression between endogenous (Mer108 and Mer120) and exogenous (USO and SO) developmental stages. A total of 24 differential miRNAs were identified, six of which were up-regulated and 18 were down-regulated (Figure 3A). In addition, two miRNAs were unique at the endogenous stage and 124 were unique at the exogenous developmental stage. We found that the target genes of differential miRNAs were mainly enriched for protein kinase activity (GO: 0004672, p = 0.0041), including CDPKs, CMGCs and ribosomal protein S6 kinase, which were up-regulated at the endogenous schizogony stages (Figures 3B–D and Supplementary Dataset 7). Apart from the protein kinases, protein translation related proteins (mainly ribosomal proteins) were also significantly upregulated at schizogony stages (Supplementary Dataset 8). CDPKs participate in many processes, such as parasite invasion and egress from host cells (38). Therefore, we speculate that these miRNAs may be involved in the parasitism of E. tenella during schizogony stage.
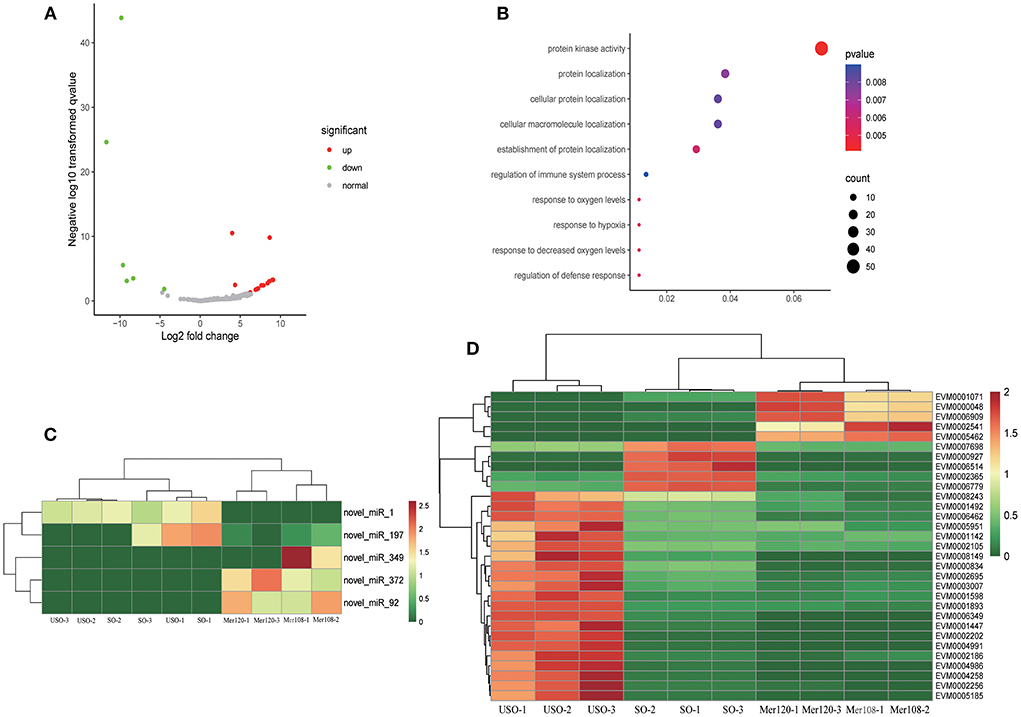
Figure 3. Target gene analysis of differentially expressed miRNAs at exogenous and endogenous stages. (A) Volcano plot showing differentially expressed miRNAs between endogenous developmental stages (Mer108 and Mer120) and exogenous stages (USO and SO). Gray dots represent non-differentially expressed miRNAs, red dots represent up-regulated miRNAs, and green dots represent down-regulated miRNAs. |log2 (FC) | ≥ 1.00 and FDR ≤ 0.05 were regarded as statistically significant. (B) Go enrichment plot of differentially expressed miRNA-targeted genes in exogenous and endogenous stages. Only the 10 GO functions with the smallest p-value were taken for statistical purposes. (C) Heatmap showing differentially expressed miRNA-targeted genes enriched in the protein kinase activity pathway. (D) Heatmap showing differentially expressed miRNAs enriched in the protein kinase activity pathway.
Eimeria parasites undergo drastic chromatin rearrangements during the sporulation process though meiosis and mitosis, leading to dramatic changes in gene expression through complex epigenetic reprogramming events, as well as variations in cellular morphogenesis (39). Since miRNAs play a key role during development, it is reasonable to assume that they could play a relevant role in sporulation.
In the comparison between USO and SO, we obtained 56 differentially expressed miRNAs, of which 38 were up-regulated and 18 were down-regulated (Figure 4A). The 56 differentially expressed miRNAs targeted 3,442 differentially expressed mRNAs. By GO enrichment analysis, we found that most of the target genes (86 genes) were enriched in the regulation of gene expression (GO: 0010468, p = 8.77E−10) and chromatin binding (GO: 0003682, p = 1.74E−5) pathways (Figure 4B). After clustering all differentially expressed target genes enriched in this pathway (Figure 4C and Supplementary Dataset 9), it was observed that genes highly expressed in USO were mainly associated with gene expression regulation (Figure 4D). For example, the chromo domain-containing protein (EVM0000869) is associated with epigenetic chromatin remodeling and operation (40), while ApiAp2 transcription factors (e.g., EVM0006489) are reported as master regulators in many cellular processes. This result reveals a comprehensive chromatin remodeling and gene expression regulation during sporulation in Eimeria, and the involvement of these miRNAs in this process.
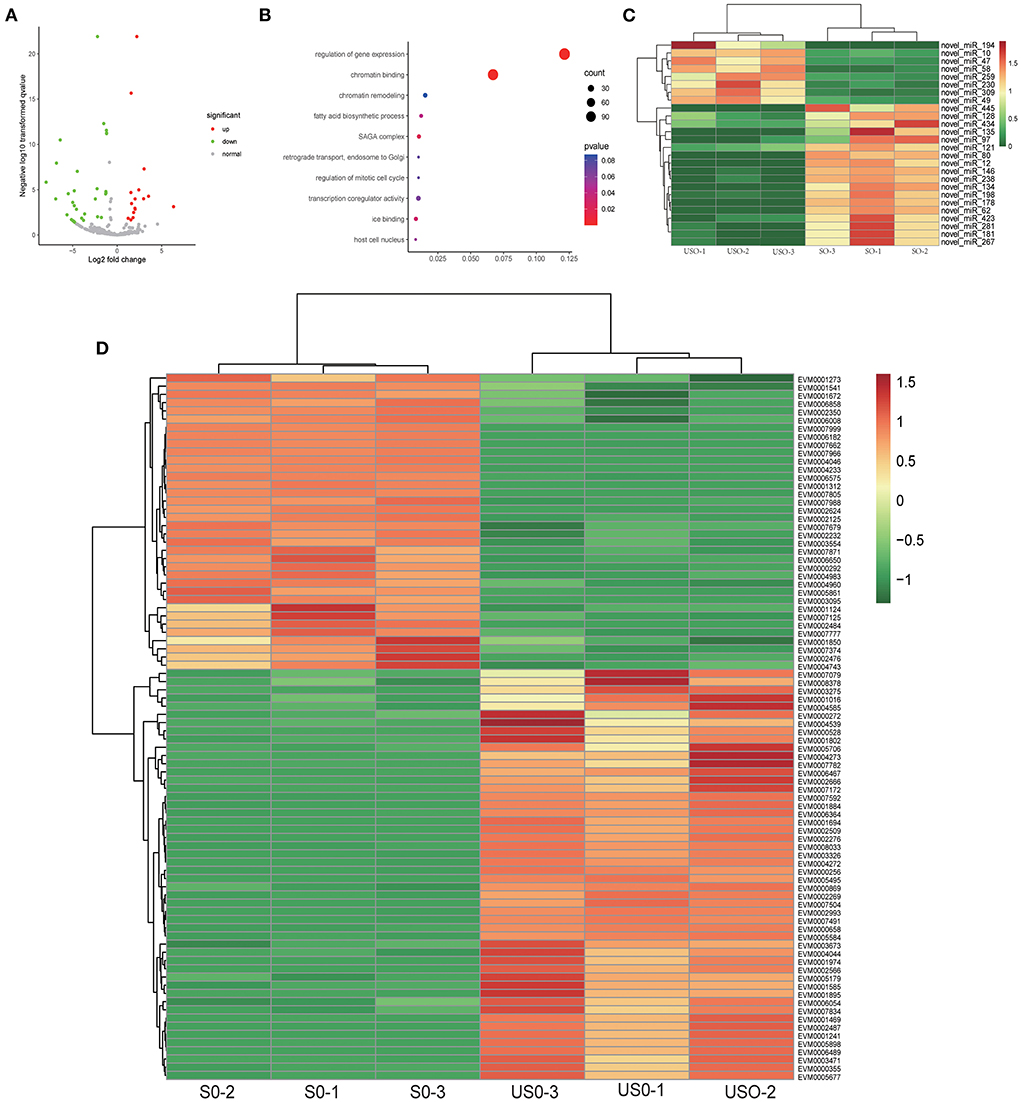
Figure 4. Target gene analysis of differentially expressed miRNAs in USO vs. SO. (A) Volcano plot showing differentially expressed miRNAs between USO (unsporulated oocysts) and SO (sporulated oocysts). (B) Go enrichment plot of target genes for differentially expressed miRNAs in USO vs. SO. Only the 10 GO functions with the smallest p-value were taken for statistical purposes. (C) Heatmap showing DEGs (differentially expressed genes of targets) of differentially expressed miRNAs enriched in gene expression regulation and chromatin binding pathway. (D) Heatmap showing differentially expressed miRNAs enriched in gene expression regulation and chromatin binding pathway.
Numerous reports have found that miRNAs lead to translational repression or mRNA degradation through complementary binding to the 3' UTR of target genes. In animals, most miRNAs are partially complementary to target mRNA sequences and usually repress translation of target genes, implying that miRNA expression is negatively correlated with mRNA expression (41, 42). Based on the regulation of negative regulation by miRNAs in animals, it is possible to predict the target genes that are potentially negatively regulated by miRNAs by combining miRNA target gene data and transcriptome data, greatly narrowing down the number of crude genes underlying differential miRNAs. By miRNA-mRNA association analysis, twenty-six differential miRNAs were obtained to be negatively correlated with 101 genes (Figure 5). However, not all differential miRNAs had negatively correlated mRNAs. It is speculated that it may be related to the positive regulation of mRNAs, but the mechanism of this positive regulation remains unclear (43). This suggests that the manipulation of miRNAs on target genes needs to be verified by a large number of experiments.
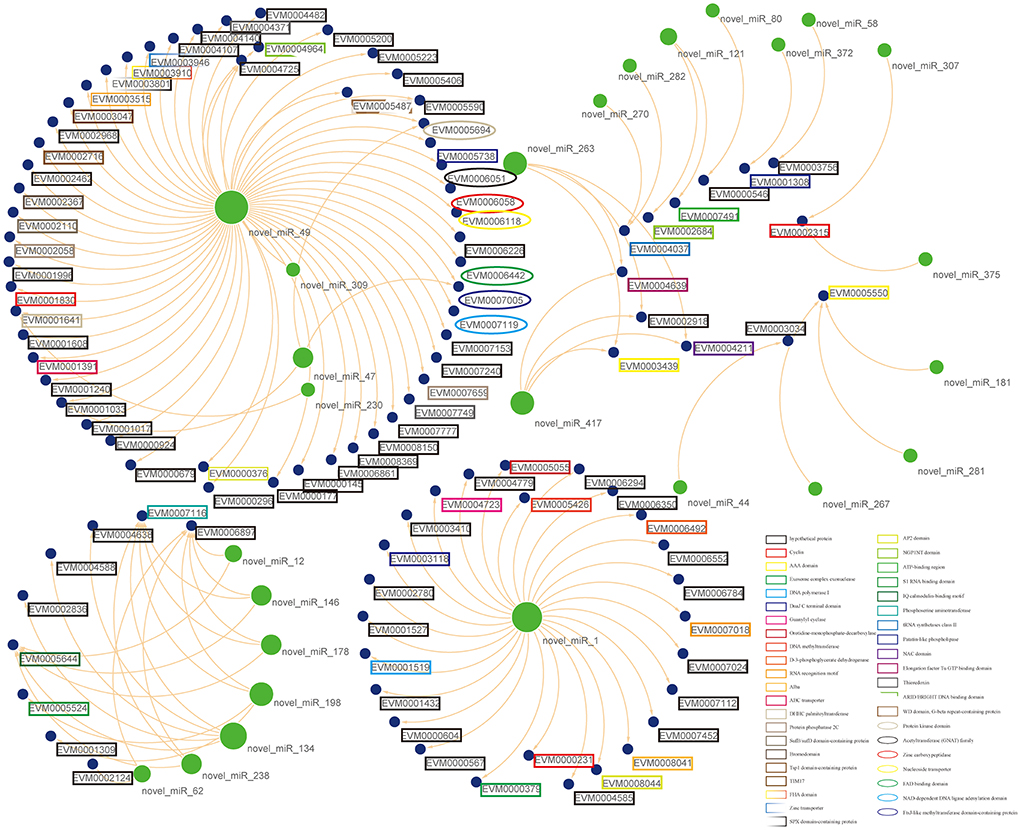
Figure 5. Putative functional interactions. All the co-expressed interactions found in this study are shown as a network by using Cytoscape. The size of the node represents its outdegree, and the functional annotations for mRNAs were also marked in the lable.
miRNAs are considered key regulators of gene expression at the post-transcriptional level. Previous studies have found that T. gondii has a complete RNA silencing pathway, suggesting that small non-coding RNAs may play a key role in parasite development and its parasitism in the host (19, 44). In this study, we characterized the RNA silencing mechanism of E. tenella, and also investigated the distribution of miRNA population at different periods of the life cycle.
We identified Dicer-like protein and Argonaute protein in the genome of E. tenella, suggesting the potential of a complete RNA silencing machinery. However, we did not find the Dicer homolog in other chicken coccidia, but instead found the Argonaute protein. It is speculated that this may be due to the lack of genome completeness or the loss of RISC-related genes in a similar evolutionary process as in Leishmania species, in which L. braziliensis do exist RISC proteins and others (e.g., L. major and L. donovani) do not (16, 34). By phylogenetic analysis, we found that the Et-ECL and Et-Ago are not, as in T. gondii, patched together by proteins of plant and fungal origin. Interestingly, Et-ECL has only two RNase domains, which suggests that it may function like Tetrahymena thermophila, requiring coupling of other proteins for pre-miRNA clipping (8).
By high-throughput sequencing, we identified a total of 392 miRNAs, and there were far more miRNAs in sporulated and unsporulated oocysts than in merozoites. We checked the purity after harvesting the merozoites and concluded that the final sample had over 90% merozoites except for some cell fragments (data not shown). Since chickens are abundant in miRNAs and Eimeria are relatively poor in miRNAs, therefore, a small amount of host cell contamination may result in a smaller number of Eimeria miRNA reads. However, comparable mapped reads in each sample were obtained and used for miRNA prediction and further analysis (Supplementary Dataset 5). Thus, we believe the different miRNA numbers in the different life stage samples should be its natural characteristics. Through the first base bias analysis, we found that the first base of the remaining miRNAs was not U, except for miRNAs with a length of 24 nt. Whereas, in other eukaryotes, such as Drosophila, Penicillium marneffei, Swarnaprabha rice and Rat, there is a high proportion of miRNAs whose first base is U (35–37, 45–47). Considering the cutting point preference of Dicer on the first base of miRNA, this different characteristics may result in significant differences in miRNAs of E. tenella compared to other organisms (48).
Previous studies reported that 336 miRNAs and 201 miRNAs were found in the tachyzoites of T. gondii RH strain and ME49 strain (44), respectively, while 300 miRNAs were found in the tachyzoites of N. caninum (49). More than 200 miRNAs were found in the green alga C. reinhardtii (50–52) and 148 miRNAs were found in Drosophila melanogaster (53). These results show that the number of miRNAs seems to be independent of the evolution of the species. By analyzing the differential genes and miRNAs, we found that miRNAs may be involved in the regulation of coccidia growth and development, which is consistent with the findings in animals and plants (54, 55).
By comparing endogenous and exogenous development stages, we found that the target genes of differential miRNAs were mainly enriched for protein kinase activity. EVM0005185 and EVM0001071 annotated as P. cynomolgi circumsporozoite protein and CMGC kinase, respectively, are both highly expressed at endogenous developmental stages. The former gene associated with cell movement (56) and the latter related to cell division (38). These are both crucial processes for merozoites development in vivo. Meanwhile, EVM0002541, annotated as CDPK1, was highly expressed in the endogenous stage. Previous report on T. gondii showed that CDPK1 has an important role in regulating parasite motility and host cell invasion (38). Thus, we speculate that these differentially expressed miRNAs may be involved in parasite invasion or egress by regulating its target genes. In addition, we found only one differentially expressed miRNA (novel_miR_400) in the comparison between two merozoite stages, suggesting relatively consistency of miRNA expression in merozoites. However, the target mRNA of the miRNA was not differentially expressed (data not shown).
The target gene functions of the differentially expressed miRNAs in USO and SO are significantly enriched in two pathways, namely gene expression regulation and chromatin binding, which are consistent with the biological process of unsporulated oocysts undergoing meiosis and mitosis to give rise to eight haploid sporozoites. EVM0000869, annotated as a Chromo domain-containing protein, is evolutionarily conserved and plays an important role in regulating gene activation, genome recombination and repair, and chromatin remodeling in different organisms. These proteins regulate epigenetic processes through various signaling pathways (40). The gene is also targeted by three differentially expressed miRNAs (two down-regulated and one up-regulated), making it difficult to find the specific functions of these miRNAs. Interestingly, the target gene EVM0007966 had only one differentially expressed miRNA, namely novel_miR_423. There was a positive correlation between them. During the whole life cycle, EVM0007966 had the lowest expression in the unsporulated oocysts, while all miRNAs of the target gene had the highest expression in sporulated and unsporulated oocysts, and no miRNA of the target gene was present in the merozoite. This also suggests that the regulatory mechanism of miRNAs is complex and that the same miRNA may play different roles at different times. Further work is needed to better understand the exact roles of differentially expressed genes, miRNAs, and miRNA interactions at different developmental stages.
In this study, we characterized the RNA silencing machinery in E. tenella and investigated the miRNA distribution of E. tenella at different time periods by high-throughput sequencing. We identified 58 miRNAs that were differentially expressed between US and SO and potentially regulate Ap2 transcription factors and epigenetic modulators. Twenty-four miRNAs were differentially expressed between endogenous and exogenous development stages, and may be involved in protein kinase activities. Our results might lead to functional understanding of gene regulation in E. tenella, and also has the potential to provide basic information to inform research on treatment against the parasite.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The animal study was reviewed and approved by Administration Committee of Laboratory Animals in Guangxi University (Approval No: Gxu-2021-013).
LZ: investigation, writing—original draft, and visualization. LC: investigation. HZ: validation and visualization. HS: supervision. XL: writing—review and editing. XS: conceptualization and writing—review and editing. DH: software, funding acquisition, and supervision. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (Grant No. 32102694), the Natural Science Foundation of Guangxi Zhuang Autonomous region (Grant No. 2021GXNSFBA220057), and the Specific Research Project of Guangxi for Research Base and Talents (Grant No. AD21075028).
We are grateful to Zehua Lin and Yue Hou for their technical help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.954725/full#supplementary-material
Supplementary Dataset 1. HMMER search results for key RNAi pathway proteins in Eimeria species. Thirteen protozoan reference genomes were downloaded from vEuPathDB (release 57), and new assemblies of E. tenella, E. maxima and E. acervulina from our unpublished data were built as a database. Hmm model of PAZ, Piwi, Ribonuclease 3, double-stranded RNA binding motif and double stranded RNA binding domain were downloaded from Pfam database and used as query against the protozoan genome database by hmmsearch using default parameters.
Supplementary Dataset 2. Sequence alignment files of Dicer and Ago proteins. Amino acid sequences were trimmed manually and then were aligned by Muscle v3.8 with default parameters.
Supplementary Dataset 3. Statistical table of sequencing data.
Supplementary Dataset 4. SRNA annotation classification statistics for different samples.
Supplementary Dataset 5. Statistical table of comparative information with the reference genome.
Supplementary Dataset 6. Statistics of the amount of miRNA expression in each sample, and normalization of the amount of expression by TPM.
Supplementary Dataset 7. Genes targeted by differentially expressed miRNAs enriched in the protein kinase activity pathway.
Supplementary Dataset 8. Table of differential mRNA statistics for differential miRNA targets in endogenous and exogenous stages.
Supplementary Dataset 9. DEGs (differentially expressed genes for targets) of differentially expressed miRNAs enriched in gene expression regulation and chromatin binding pathway.
Supplementary Dataset 10. Protein accession ID of Dicer proteins in different species.
Supplementary Dataset 11. Protein accession ID of AGO proteins in different species.
1. Chapman HD. Milestones in avian coccidiosis research: a review. Poult Sci. (2014) 93:501–11. doi: 10.3382/ps.2013-03634
2. Blake DP, Knox J, Dehaeck B, Huntington B, Rathinam T, Ravipati V, et al. Re-calculating the cost of coccidiosis in chickens. Vet Res. (2020) 51:115. doi: 10.1186/s13567-020-00837-2
3. Mesa-Pineda C, Navarro-Ruiz JL, Lopez-Osorio S, Chaparro-Gutierrez JJ, Gomez-Osorio LM. Chicken coccidiosis: from the parasite lifecycle to control of the disease. Front Vet Sci. (2021) 8:787653. doi: 10.3389/fvets.2021.787653
4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
5. Davalos V, Esteller M. MicroRNAs and cancer epigenetics: a macrorevolution. Curr Opin Oncol. (2010) 22:35–45. doi: 10.1097/CCO.0b013e328333dcbb
6. Wang W, Wang Y, Liu W, van Wijnen AJ. Regulation and biological roles of the multifaceted miRNA-23b (MIR23B). Gene. (2018) 642:103–9. doi: 10.1016/j.gene.2017.10.085
7. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
8. Lee SR, Collins K. Physical and functional coupling of RNA-dependent RNA polymerase and Dicer in the biogenesis of endogenous siRNAs. Nat Struct Mol Biol. (2007) 14:604–10. doi: 10.1038/nsmb1262
9. Kutter C, Svoboda P. miRNA, siRNA, piRNA: knowns of the unknown. RNA Biol. (2008) 5:181–8. doi: 10.4161/rna.7227
10. Wang B, Li S, Qi HH, Chowdhury D, Shi Y, Novina CD. Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins. Nat Struct Mol Biol. (2009) 16:1259–66. doi: 10.1038/nsmb.1712
11. Nahar S, Nayak AK, Ghosh A, Subudhi U, Maiti S. Enhanced and synergistic downregulation of oncogenic miRNAs by self-assembled branched DNA. Nanoscale. (2017) 10:195–202. doi: 10.1039/C7NR06601E
12. Baum J, Papenfuss AT, Mair GR, Janse CJ, Vlachou D, Waters AP, et al. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. (2009) 37:3788–98. doi: 10.1093/nar/gkp239
13. Mueller AK, Hammerschmidt-Kamper C, Kaiser A. RNAi in Plasmodium. Curr Pharm Des. (2014) 20:278–83. doi: 10.2174/13816128113199990027
14. Castellanos-Gonzalez A. A novel method to silence genes in cryptosporidium. Methods Mol Biol. (2020) 2052:193–203. doi: 10.1007/978-1-4939-9748-0_11
15. Xue X, Zhang Q, Huang Y, Feng L, Pan W. No miRNA were found in Plasmodium and the ones identified in erythrocytes could not be correlated with infection. Malar J. (2008) 7:47. doi: 10.1186/1475-2875-7-47
16. Ullu E, Tschudi C, Chakraborty T. RNA interference in protozoan parasites. Cell Microbiol. (2004) 6:509–19. doi: 10.1111/j.1462-5822.2004.00399.x
17. Kolev NG, Tschudi C, Ullu E. RNA interference in protozoan parasites: achievements and challenges. Eukaryot Cell. (2011) 10:1156–63. doi: 10.1128/EC.05114-11
18. Djikeng A, Shi H, Tschudi C, Ullu E. RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24-26-nucleotide RNAs. RNA. (2001) 7:1522–30. doi: 10.1017.S1355838201014522
19. Braun L, Cannella D, Ortet P, Barakat M, Sautel CF, Kieffer S, et al. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. (2010) 6:e1000920. doi: 10.1371/journal.ppat.1000920
20. Eckert J, Coudert P, Shirley MW, Braun R. Guidelines on Techniques in Coccidiosis Research. Brussels: Office for Official Publications of the European Communities (1995).
21. Novaes J, Rangel LT, Ferro M, Abe RY, Manha AP, de Mello JC, et al. A comparative transcriptome analysis reveals expression profiles conserved across three Eimeria spp. of domestic fowl and associated with multiple developmental stages. Int J Parasitol. (2012) 42:39–48. doi: 10.1016/j.ijpara.2011.10.008
22. Schwarz RS, Fetterer RH, Rosenberg GH, Miska KB. Coccidian merozoite transcriptome analysis from Eimeria maxima in comparison to Eimeria tenella and Eimeria acervulina. J Parasitol. (2010) 96:49–57. doi: 10.1645/GE-2253.1
23. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. (2019) 37:907–15. doi: 10.1038/s41587-019-0201-4
24. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
26. Hu S, Mei S, Liu N, Kong X. Molecular genetic characterization of cblC defects in 126 pedigrees and prenatal genetic diagnosis of pedigrees with combined methylmalonic aciduria and homocystinuria. BMC Med Genet. (2018) 19:154. doi: 10.1186/s12881-018-0666-x
27. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. (2009) 10:R25. doi: 10.1186/gb-2009-10-3-r25
28. Mathews DH. Revolutions in RNA secondary structure prediction. J Mol Biol. (2006) 359:526–32. doi: 10.1016/j.jmb.2006.01.067
29. Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. (2008) 36:D149–53. doi: 10.1093/nar/gkm995
30. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. (2010) 11:R14. doi: 10.1186/gb-2010-11-2-r14
31. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. (2011) 7:e1002195. doi: 10.1371/journal.pcbi.1002195
32. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. (2004) 32:1792–7. doi: 10.1093/nar/gkh340
33. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
34. Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. (2006) 50:81–99. doi: 10.1007/s00294-006-0078-x
35. Du J, Wu Y, Zhang Y, Wu L, Wang X, Tao S. Large-scale information entropy analysis of important sites in mature precursor miRNA sequences. Sci China C Life Sci. (2009) 52:771–9. doi: 10.1007/s11427-009-0099-z
36. Seitz H, Tushir J, Zamore P. A 5′-uridine amplifies miRNA/miRNA* asymmetry in Drosophilaby promoting RNA-induced silencing complex formation. Silence. (2011) 2:4. doi: 10.1186/1758-907X-2-4
37. Meijer HA, Smith EM, Bushell M. Regulation of miRNA strand selection: follow the leader? Biochem Soc Trans. (2014) 42:1135–40. doi: 10.1042/BST20140142
38. Wei F, Wang W, Liu Q. Protein kinases of Toxoplasma gondii: functions and drug targets. Parasitol Res. (2013) 112:2121–9. doi: 10.1007/s00436-013-3451-y
39. del Cacho E, Pages M, Gallego M, Barbero JL, Monteagudo L, Sanchez-Acedo C. Meiotic chromosome pairing and bouquet formation during Eimeria tenella sporulation. Int J Parasitol. (2010) 40:453–62. doi: 10.1016/j.ijpara.2009.09.008
40. Iyer LM, Anantharaman V, Wolf MY, Aravind L. Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol. (2008) 38:1–31. doi: 10.1016/j.ijpara.2007.07.018
41. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
42. Riedmann LT, Schwentner R. miRNA, siRNA, piRNA and argonautes: news in small matters. RNA Biol. (2010) 7:133–9. doi: 10.4161/rna.7.2.11288
43. Dragomir MP, Knutsen E, Calin GA. SnapShot: unconventional miRNA functions. Cell. (2018) 174:1038–1038.e1. doi: 10.1016/j.cell.2018.07.040
44. Wang J, Liu X, Jia B, Lu H, Peng S, Piao X, et al. A comparative study of small RNAs in Toxoplasma gondii of distinct genotypes. Parasit Vectors. (2012) 5:186. doi: 10.1186/1756-3305-5-186
45. Lau S, Chow W, Wong A, Yeung J, Bao J, Zhang N, et al. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl Trop Dis. (2013) 7:e2398. doi: 10.1371/journal.pntd.0002398
46. Dong J, Gu W, Yang X, Zeng L, Wang X, Mu J, et al. Crosstalk between polygonatum kingianum, the miRNA, and gut microbiota in the regulation of lipid metabolism. Front Pharmacol. (2021) 12:740528. doi: 10.3389/fphar.2021.740528
47. Panigrahy M, Panigrahi K, Poli Y, Ranga A, Majeed N. Integrated expression analysis of small RNA, degradome and microarray reveals complex regulatory action of miRNA during prolonged shade in swarnaprabha rice. Biology. (2022) 11:798. doi: 10.3390/biology11050798
48. Li Y, Baptista R, Kissinger J. Noncoding RNAs in apicomplexan parasites: an update. Trends Parasitol. (2020) 36:835–49. doi: 10.1016/j.pt.2020.07.006
49. Liu G, Jia L, Shao Q, Lu H, Zhao J, Yin J. MicroRNA profiling of Neospora caninum tachyzoites (NC-1) using a high-throughput approach. Parasitol Res. (2021) 120:2165–74. doi: 10.1007/s00436-021-07155-2
50. Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. (2007) 21:1190–203. doi: 10.1101/gad.1543507
51. Shu L, Hu Z. Characterization and differential expression of microRNAs elicited by sulfur deprivation in Chlamydomonas reinhardtii. BMC Genomics. (2012) 13:108. doi: 10.1186/1471-2164-13-108
52. Gao X, Zhang F, Hu J, Cai W, Shan G, Dai D, et al. MicroRNAs modulate adaption to multiple abiotic stresses in Chlamydomonas reinhardtii. Sci Res. (2016) 6:38228. doi: 10.1038/srep38228
53. Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. (2007) 17:1850–64. doi: 10.1101/gr.6597907
54. Li C, Zhang B. MicroRNAs in control of plant development. J Cell Physiol. (2016) 231:303–13. doi: 10.1002/jcp.25125
55. Alberti C, Cochella L. A framework for understanding the roles of miRNAs in animal development. Development. (2017) 144:2548–59. doi: 10.1242/dev.146613
Keywords: Eimeria tenella, miRNA, dicer, Argonaute, gene expression
Citation: Zhang L, Chen L, Zhang H, Si H, Liu X, Suo X and Hu D (2022) A comparative study of microRNAs in different stages of Eimeria tenella. Front. Vet. Sci. 9:954725. doi: 10.3389/fvets.2022.954725
Received: 27 May 2022; Accepted: 30 June 2022;
Published: 22 July 2022.
Edited by:
Damer Blake, Royal Veterinary College (RVC), United KingdomReviewed by:
Emanuel Heitlinger, Leibniz Institute for Zoo and Wildlife Research (LG), GermanyCopyright © 2022 Zhang, Chen, Zhang, Si, Liu, Suo and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dandan Hu, aHVkYW5kYW5AZ3h1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.