
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 22 June 2022
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.923878
 Kyle S. Hoffman1
Kyle S. Hoffman1 Nicki L. Humphrey2John A. Korslund3
Nicki L. Humphrey2John A. Korslund3 Tavis K. Anderson1Kay. S. Faaberg1Kelly M. Lager1
Tavis K. Anderson1Kay. S. Faaberg1Kelly M. Lager1 Alexandra C. Buckley1*
Alexandra C. Buckley1*Vesicular disease caused by Senecavirus A (SVA) is clinically indistinguishable from foot-and-mouth disease (FMD) and other vesicular diseases of swine. When a vesicle is observed in FMD-free countries, a costly and time-consuming foreign animal disease investigation (FADI) is performed to rule out FMD. Recently, there has been an increase in the number of FADIs and SVA positive samples at slaughter plants in the U.S. The objectives of this investigation were to: (1) describe the environmental burden of SVA in sow slaughter plants; (2) determine whether there was a correlation between PCR diagnostics, virus isolation (VI), and swine bioassay results; and (3) phylogenetically characterize the genetic diversity of contemporary SVA isolates. Environmental swabs were collected from three sow slaughter plants (Plants 1-3) and one market-weight slaughter plant (Plant 4) between June to December 2020. Of the 426 samples taken from Plants 1-3, 304 samples were PCR positive and 107 were VI positive. There was no detection of SVA by PCR or VI at Plant 4. SVA positive samples were most frequently found in the summer (78.3% June-September, vs. 59.4% October-December), with a peak at 85% in August. Eighteen PCR positive environmental samples with a range of Ct values were selected for a swine bioassay: a single sample infected piglets (n = 2). A random subset of the PCR positive samples was sequenced; and phylogenetic analysis demonstrated co-circulation and divergence of two genetically distinct groups of SVA. These data demonstrate that SVA was frequently found in the environment of sow slaughter plants, but environmental persistence and diagnostic detection was not indicative of whether a sampled was infectious to swine. Consequently, a more detailed understanding of the epidemiology of SVA and its environmental persistence in the marketing chain is necessary to reduce the number of FADIs and aide in the development of control measures to reduce the spread of SVA.
Senecavirus A (SVA) is a non-enveloped, single-stranded, positive-sense RNA virus in the family Picornaviridae. It is the only species in the genus Senecavirus and is most closely related to viruses in the genus Cardiovirus (1). The SVA genome is approximately 7.2 kb long and encodes a single large polyprotein which is cleaved by the viral cysteine protease 3C (2). The polyprotein is composed of four structural proteins VP1, VP2, VP3 and VP4 and eight non-structural proteins L, 2A, 2B, 2C, 3A, 3B, 3C and 3D (1). Senecavirus A was discovered as a cell culture contaminate in 2002 and named Seneca Valley virus (SVV-001) (3). Between 1988 and 2005 picorna-like viruses were isolated from pigs displaying various clinical symptoms (4). Samples were partially sequenced and were similar to SVV-001, revealing that SVA has been present in swine populations in the United States for at least thirty years (4). Subsequently, SVA has been identified and described in swine from Brazil, Canada, China, Thailand, Vietnam and Colombia (5–10).
In 2015, experimental studies determined SVA was a causative agent for vesicular disease in pigs, which is characterized by blister-like lesions found on the snout and coronary band (11–13). Vesicular lesions resolve in 1–2 weeks, while viral nucleic acid can continue to be detected in oral/nasal and rectal swabs for several weeks after infection (13, 14). However, virus isolation from swab samples has typically only been reported during the first week after experimental infection. Recently, SVA was isolated from the tonsil of animals after a stressor event (i.e., transportation or parturition) 60 days after experimental infection, providing experimental evidence that SVA may establish a persistent infection in the tonsil of infected swine (15).
Both experimental and field studies have demonstrated transmission of SVA from pig-to-pig through direct contact (15, 16). In addition, viable SVA has been recovered from environmental samples and mouse feces, and viral nucleic acids have been recovered from house flies, potentially providing other routes for transmission of SVA to susceptible hosts (17). SVA has been detected in both swine arriving to slaughter plants as well as assembly yards (18, 19). The presence of SVA in areas where swine from multiple origins congregate may have broad impacts, as contaminated transport vehicles could be a source of viral transmission back to swine farms (20–22). Additional evidence suggests that transportation stress may increase clinical disease severity and this may be relevant in sows shipped to slaughter plants (15).
SVA and foot-and-mouth disease (FMD) cause clinically identical vesicular disease, and the presence of vesicular lesions on swine requires a foreign animal disease investigation (FADI) in FMD free countries (23). With an increase in the number of SVA cases in the U.S. since 2015, particularly centered at slaughter plants, there has been a corresponding increase in FADIs. These investigations impart a substantial burden on local and federal resources and can disrupt market supply chains, thus further adding to the cost (24). Therefore, a better understanding of SVA ecology at slaughter plants could inform measures to reduce the number of FADIs due to SVA. The objectives for this study were to understand the environmental burden of SVA in sow slaughter plant lairages; to determine whether there was a correlation between PCR detection, viral isolation (VI), and swine bioassay results; and to genetically characterize 2020 SVA sequences from environmental samples to prior SVA isolates circulating in the United States.
Environmental samples were collected between June and December 2020 from three U.S. sow slaughter plants in the Midwest that process between 500 and 2,800 head/day. Plant 4 was a market-weight slaughter plant also in the Midwest that processes ~12,000 head/day. Plants 1-3 had previously experienced a high incidence of FADIs and Plant 4 had no reports of FADIs. Cotton-tipped swabs used for environmental collection were pre-moistened with tris-buffered tryptose broth (TBTB, 1.21 g/L Tris-base, 26 g/L Tryptose broth). Individual swabs were gently rubbed on surfaces exposed to animals that included flooring, waterers, gating, side panels of trailers and the coronary bands of pigs without vesicular lesions and then placed into 2 mL of TBTB media. Samples were shipped to the National Animal Disease Center (NADC) in Ames, IA. Tubes containing environmental swabs were clarified to pellet residual debris and supernatant was filtered through a 0.45 μm filter, aliquoted, and frozen at−80°C for future testing.
Environmental swab samples and samples from the swine bioassay were tested for SVA nucleic acids by reverse transcription-quantitative PCR (RT-qPCR) as previously described (14). Briefly, RNA was extracted from samples using the MagMAX Pathogen RNA/DNA kit (Applied Biosystems, Waltham, MA) following the manufacturers' recommendations. Subsequently, 5 μL was added to 20 μL of the Path-ID Multiplex One-Step RT-PCR reaction master mix for rectal swabs and environmental swabs or 20 μL AgPath-ID One-Step RT-PCR kit (Applied Biosystems) for oral swabs and sera. The primers and probe were designed to target a conserved region containing nucleotides 602-710 of the SVA genome. The forward primer sequence was 5′-TGCCTTGGATACTGCCTGATAG-3′, the reverse primer sequence was 5′-GGTGCCAGAGGCTGTATCG-3′, and the probe sequence was 5′-CGACGGCCTAGTCG GTCGGTT-3′. RNA copies were determined using a dilution series with a plasmid containing the target region and cycle threshold (Ct) values >35 were considered negative.
Swine testicular (ST) cells (National Veterinary Services Laboratory, Ames, IA) were grown in minimum essential media (MEM, Gibco, Waltham, MA) supplemented with 10% fetal bovine serum (FBS, Atlanta Bio, Flowerly Way, GA), 1% L-glutamine (Life Technologies, Carlsbad, CA) and 50 mg/L gentamicin at 37°C and 5% CO2. Monolayers of ST cells were grown until confluent in 24 well plates, MEM was removed and replaced with 1 mL serum-free MEM. Cells were inoculated with 100 μL of filtered environmental samples and allowed to incubate at 37°C and 5% CO2 for 1 h, after which the inoculum was removed and replaced with 1 mL MEM. Negative control wells were present on each plate. Plates were microscopically observed daily for 4 days for cytopathic effect (CPE). Two additional blind passages were performed on each environmental sample.
All animal research was performed in accordance with an Animal Care and Use Protocol (ACUP ARS-2018-750) approved by the NADC Animal Care and Use Committee. At the end of the study, all animals were humanely euthanized with an intravenous administration of a barbiturate (Fatal Plus, Vortech Pharmaceuticals, Dearborn, MI) following the label dose (1 mL/4.45 kg).
Eighteen SVA PCR positive environmental samples were selected for swine bioassay (Table 1). Samples were representative of the various slaughter plants, sample types, and dates of collection. Two samples, one VI positive and one VI negative, were selected for each Ct value between 23 and 31. The exception was the Ct value of 23 which did not have any VI negative samples. Environmental samples were diluted 1:5 with serum free MEM to generate material for the swine bioassay, PCR, and VI.
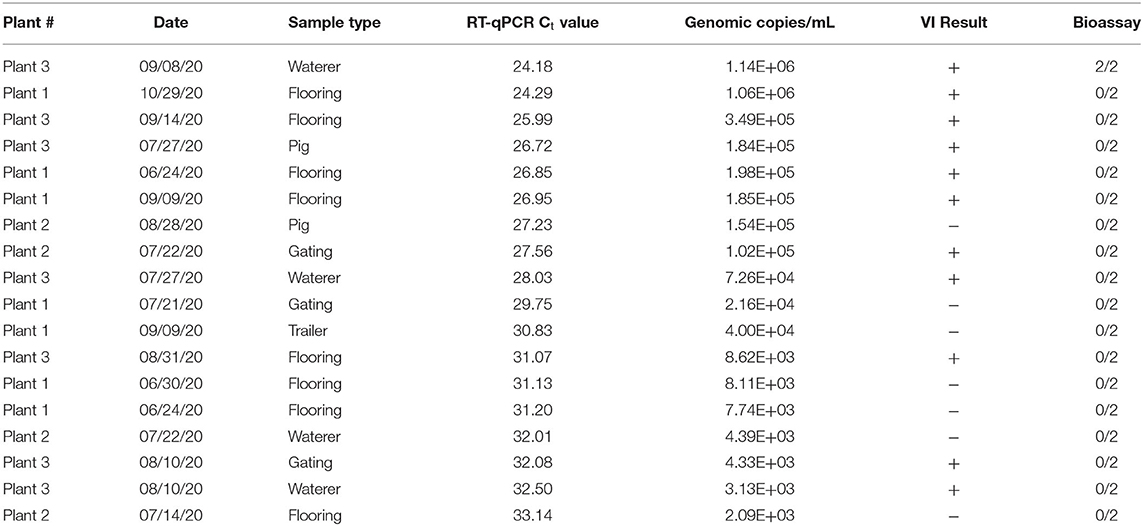
Table 1. RT-qPCR, virus isolation (VI), and swine bioassay results of selected SVA samples from Plants 1-3.
Piglets were weaned between 3 and 5 days-of-age and placed into individual isolator cages with two cages per ABSL-2 animal space. After 24 h of acclimation, piglets were bled, oral and rectal swabbed, and inoculated (n = 2/sample) with 2 mL orally on 0 days post inoculation (dpi). All animals were observed daily for clinical signs including lameness, lethargy, inappetence and diarrhea. Blood, oral swabs, and rectal swabs were collected on 6 dpi when piglets were removed from isolation cages and placed on raised decks in the same ABSL-2 animal space, as well as on 10 and 14 dpi (12, 13). Blood was collected in serum separator tubes (BD Vacutainer, Franklin Lakes, NJ) and centrifuged to separate the serum for storage. Oral and rectal swabs were collected using a sterile polyester tipped applicator (Puritan Medical Products, Guilford, ME) immersed in 3 mL of MEM. All samples were frozen at −80°C prior to testing.
Serum samples from 0, 4, 6 and 10 dpi were heat inactivated at 56°C for 30 min. Samples were run in quadruplicate, diluted four-fold from 1:4 to 1:4096 in MEM. Cross-neutralizing antibodies have been demonstrated with different geographic and temporal isolates of SVA (25), as such a reference isolate of SVA (SVA/KS/2018) was selected for the VN assay. An equal volume of serum and SVA isolate (~200 TCID50) were mixed and incubated for 1 h at 37°C. The virus-serum mixture was transferred to 96-well plates of confluent ST cells with growth media replaced by MEM supplemented with 2% FBS. Plates were microscopically evaluated daily for 4 days. The titer was recorded as the highest dilution of serum where viral infectivity was completely neutralized in 50% of the inoculated wells. A back titration of the SVA isolate was performed for each run. Virus neutralization titers of ≤ 1:16 were considered negative.
Six environmental samples were selected for whole genome sequencing. Two samples with the lowest Ct values were selected from Plants 1, 2 and 3 and 100 μL of each sample was used to inoculate ST cell monolayers in 25 cm2 flasks for 1.5 h. Flasks were decanted, replaced with serum-free MEM, and incubated for 24–48 h. Flasks were frozen at−80°C and three freeze/thaw cycles were performed prior to clarification at 3,000 XG for 10 min. Clarified cell culture supernatant was submitted to the Iowa State University Veterinary Diagnostic Laboratory for next generation sequencing using the Illumina platform. Sample library preparation and sequencing were performed following previously described methods (26). The accession numbers for the SVA genome sequences are as follows: NADC1 (MZ733980), NADC2 (MZ733979), NADC3 (MZ733978), NADC4 (MZ733977), NADC5 (MZ73396) and NADC6 (MZ733975).
Illumina sequence reads from environmental samples were imported into Geneious Prime® 2021.2.2 and were paired using default conditions. Paired-end reads were mapped to a reference SVA sequence (MT360257.1). Publicly available full-length SVA genome sequences (>7,100 bp) with location and date of collection were downloaded from NCBI GenBank on August 14 2021 (27). The complete genome sequences of SVA isolated in this study along with the full-length SVA genomes available on GenBank were aligned using MAFFT with default settings (28). A maximum-likelihood phylogenetic tree for the SVA genomes was inferred using IQ-TREE following automatic model selection on the IQ-TREE web server (29): statistical support was assessed with 1,000 ultrafast bootstrap iterations.
To estimate the evolutionary dynamics of the SVA genome sequences collected in the United States after 2002, we implemented a time-scaled Bayesian approach. All SVA genome sequences collected in the U.S. were downloaded from NCBI GenBank on October 15, 2021 and aligned using default settings in MAFFT. These data were screened for evidence of recombination in RDP5 with the application of the following algorithms RDP, MaxChi, 3seq, GENECOV, Bootscan, SiScan, and Chimera (30). Genomes were considered to have a recombination signal when more than four of the above methods had statistically significant support for recombination, when the identified breakpoints and breakpoint regions were longer than 100 nucleotides and when the parental and recombinant strains exhibited spatial and temporal concordance [e.g., (31)]. Recombinant sequences were removed from subsequent Bayesian analyses. A maximum likelihood phylogenetic tree was inferred with FastTree (32) from the resultant dataset (n = 110), and screened in TempEst (33) to assess whether there were any sequences with incongruent dates and divergence resulting in a final dataset of 89 sequences. A time-scaled Bayesian phylogenetic tree was inferred in BEAST v1.10.4 (34) employing a strict molecular clock, a coalescent-based constant size demographic model (35) and a HKY85 + Γ substitution model. The Markov chain Monte Carlo (MCMC) was run for 100 million steps with sampling every 10,000 generations. Two independent analyses were performed, and convergence was assessed in Tracer v1.7.2. Evolutionary history was summarized and visualized using an annotated maximum clade credibility tree using TreeAnnotator v1.8.4 and FigTree v1.4.4.
In this study, environmental swab samples were collected from three sow slaughter plants, with high incidence of FADIs (Plants 1-3) and a fourth market-weight slaughter plant with no reported FADIs (Plant 4). All samples (n = 69) tested from Plant 4 were negative for SVA by RT-qPCR. From Plants 1-3, 426 swab samples were taken from the environment including flooring (n = 92), waterers (n = 92), gating (n = 91), trailers (n = 22) and from the coronary bands of pigs (n = 129) that did not have vesicular lesions. Data for Plants 1–3 were combined for subsequent analyses. For each sample type, <50% of the samples collected across the sampling period were RT-qPCR positive for SVA. Samples collected from the flooring of the lairage had the highest number of percent positives (93.5% positive), while samples collected from gating (61.5% positive) and trailers (54.6% positive) were the two lowest percent positive sample types (Figure 1A).
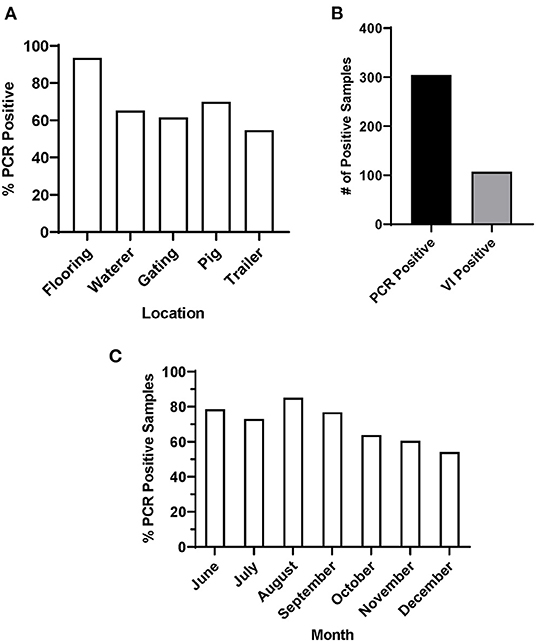
Figure 1. PCR and VI results of environmental samples from Plants 1-3. (A) Percentage of SVA positive samples by sample location, (B) total number of SVA PCR positive and VI positive samples and (C) percentage of SVA positive samples by month.
Of the 71.4% of environmental samples that were SVA RT-qPCR positive, 35.2% were also VI positive (Figure 1B). Samples with a lower Ct value were more likely to be VI positive. 67.6% of all samples with a Ct value below 30 were VI positive, while only 18.8% of samples with a Ct value between 30 and 35 were VI positive. These data were subsequently separated by season to determine whether there was any pattern in SVA percent positive across the sampling period. When all sample data were combined by month, the percentage of positive samples for Plants 1-3 SVA was higher throughout the summer, with a percent positive peak in August (85%), that then declined through the fall and early winter (Figure 1C).
A swine bioassay was performed to determine whether there was a correlation between PCR Ct values, virus isolation and SVA infectivity in swine. The inoculum was tested by PCR and VI the same time as animals were inoculated. SVA RT-qPCR Ct values for the inoculums ranged from 24 to 33 and 11/18 were VI positive (Table 1). Lower Ct values were more likely to be VI positive, but some inoculums with higher Ct values (~32) were also VI positive.
Pigs used in the swine bioassay tested negative for SVA nucleic acids and neutralizing antibodies prior to inoculation. Only one set of piglets were infected with SVA and had positive swabs and serum at 6 dpi when removed from isolation cages. These two pigs also developed neutralizing antibody titers of 256 by 14 dpi. The inoculum material had the lowest Ct value (24.18), tested positive for virus isolation, and came from an environmental sample collected from a waterer in the lairage (Table 1). Remaining pigs were RT-qPCR negative for SVA in all samples collected and there was no evidence of a neutralizing antibody response in 14 dpi sera.
Six environmental samples (n = 2/plant) with the lowest Ct values were selected for whole genome sequencing. A complete SVA polyprotein and the accompanying 5′ and 3′ UTR nucleotides were determined for each of the six samples with no insertions or deletions. The pairwise identity between all six sequences was 98.2%. Percent identity between the nucleotide and amino acid sequences of the six SVA isolates ranged from 96.5 to 99.9% and 98.9 to 99.9%, respectively. NADC1 was most dissimilar from the other five isolates sequenced with approximately 250 nucleotide differences across the genome (Table 2). When excluding NADC1, the remaining isolates had 99.0 and 99.7% identity at the nucleotide and amino acid level, respectively.
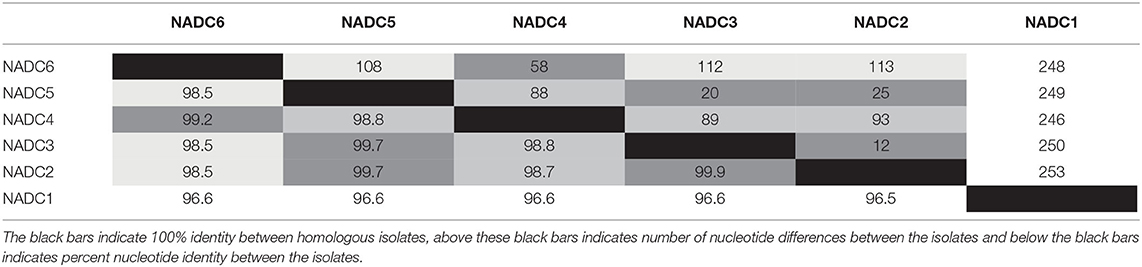
Table 2. Complete genome sequence distance matrix and heat map for the six sequenced SVA isolates.
There was a significant increase in the detection of SVA cases in the U.S. during 2015. To identify if any conserved non-synonymous amino acid changes had occurred since 2015, the 2020 SVA isolate genomes were compared to 2015 U.S. isolates. Six conserved amino acid changes from the 2015 isolates were found in the mature viral proteins (VP3, 2B, 2C, 3A and 3D) in all 2020 isolates, excluding NADC1 (Table 3). The 12 protein coding regions in the six SVA isolates had completely identical protein sequences for VP4, VP2, 2A and 3B.
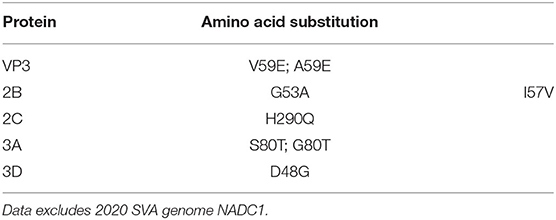
Table 3. Non-synonymous amino acid changes detected in 2020 SVA isolates as compared to 2015 U.S. isolates.
The complete genome sequences of the six isolates in this study and 233 SVA whole genomes available from the U.S. Swine Pathogen Database (36) were analyzed. The analysis revealed that the historical SVA isolates from 1988 to 2006 formed a single monophyletic clade, and the contemporary SVA isolates were in a separate monophyletic clade with tree topology reflecting geographic origin and year of collection (Figure 2). Of the six SVA sequences collected in this study, five were located in a single clade with other 2020 strains: the sixth strain (NADC1) shared an evolutionary history with U.S. SVA strains isolated in 2017 (Figure 3). The most recent common ancestor (MRCA) for the six strains collected in this study was 2011.9 (2011.82–2012.3 95% HPD), and the MRCA for the five similar 2020 isolates was 2016.7 (2013.47–2021.06 95% HPD).
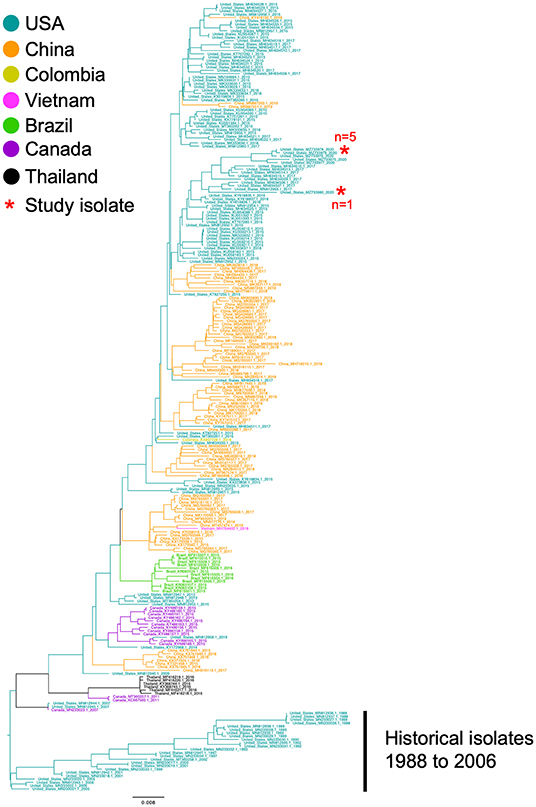
Figure 2. Phylogenetic analysis of 233 SVA whole genomes and the six 2020 SVA isolates from Plants 1-3. The tips of the tree are color coded to the country of sequence origin. The six SVA genome sequences analyzed in this study are noted with a red asterisk.
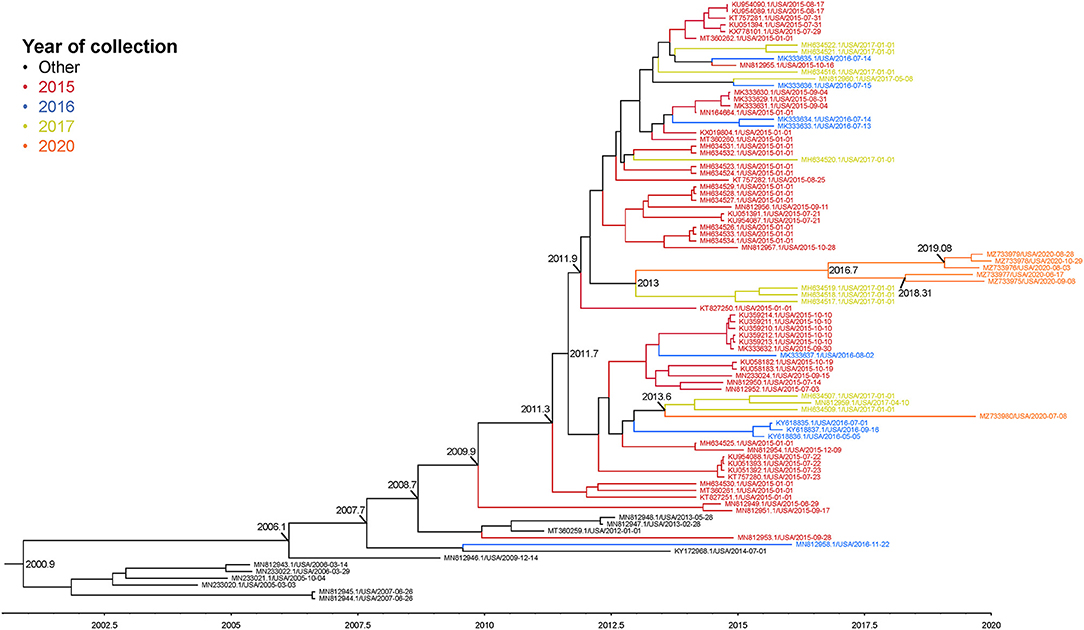
Figure 3. Maximum clade credibility (MCC) phylogenetic tree representing the evolutionary history of 83 SVA genome sequences and the six 2020 isolates. Colors indicate year of sample collection date. The x-axis indicates a timescale in years. Nodes are labeled with years of an estimated common ancestor.
Vesicular lesions caused by SVA cannot be differentiated from those of FMD. As SVA continues to be endemic in the U.S, the number of FADIs related to swine vesicular lesions has been increasing (37). We investigated the presence of SVA in the environment of select U.S. sow slaughter plants, and quantified genetic diversity in the SVA genome in 2020 isolates relative to isolates collected in 2015.
SVA was found on multiple surfaces within slaughter plants with the flooring containing the highest percentage of SVA positive samples. Experimentally SVA has been demonstrated to be shed in feces multiple weeks after infection; therefore, it is likely that infected sows are shedding SVA in fecal material after arriving in holding pens at slaughter plants (14, 15). This could also explain why samples collected higher at the pig level like the gating and trailer swabs were less likely to be RT-qPCR positive for SVA. About a third of the SVA RT-qPCR positive environmental samples contained infectious SVA as determined by virus isolation. The lower number of VI positive samples compared to PCR positive samples may be the result of low levels of virus or inactivated virus due to time virus has been in the environment exposed to the elements, or the application of disinfectants in lairage. Numerically, a higher percentage of SVA RT-qPCR positive samples were found during warmer months. In contrast, other swine pathogens especially those involved with respiratory disease such as porcine respiratory and reproductive syndrome virus are diagnosed in a higher percentage during colder months in the fall and winter (38). Previous work has reported that house flies (Musca domestica) tested positive for SVA nucleic acids (17, 39); therefore, they could serve as a mechanical vector to spread SVA, which has been shown for other viruses and bacteria (40–42).
Previous studies have isolated SVA from environmental samples; however, there is limited information assessing the correlation between RT-qPCR results and virus isolation from environmental samples with infectivity of a pig (17, 18). Although samples with Ct values >30 were more likely to be VI negative there were several samples with high Ct values that were also VI positive. Since these samples were collected from lairage, they often contained fecal material; therefore, it is possible PCR inhibitors were present that may have impacted PCR Ct values (43). In this study, only one sample tested (Ct = 24.18) was positive by swine bioassay and capable of infecting piglets. A recent publication demonstrated that neonates are readily susceptible to SVA and had a minimum infectious dose of approximately 630 TCID50 for a 2011 SVA isolate (44). Due to the number of VI positive samples that did not infect piglets, the data in this study suggests that cell culture is more sensitive than swine bioassay. One evident explanation for VI being more sensitive than bioassay is the absence of innate immune barriers including mucus membranes, enzymes, stomach acid, and phagocytic cells. A better understanding of the infectivity of SVA found in the environment can help inform measures to reduce transmission and spread of SVA. In addition, previous work has shown the efficacy of some disinfectants including bleach (sodium hypochlorite) and accelerated hydrogen peroxide for inactivating SVA on various surfaces (45, 46).
Phylogenic analysis from this work showed one isolate (NADC1) did not group with the other five sequenced environmental samples. Our time-scaled phylogenetic analysis estimated the NADC1 sequence shared a most recent common ancestor with the other 2020 isolates in late 2011. The abundance of SVA positive samples from swine farms found throughout the United States combined with a range of reported sequences from 2015 until now suggest that a diversity of SVA may be cocirculating in U.S. swine herds (19, 47). Comparisons of the five most similar 2020 SVA isolates with the U.S. outbreak 2015 SVA isolates found six non-synonymous amino acid changes. Only the single amino acid change in the VP3 surface loop, “the knob,” is unique to the 2020 isolates (48). The other five amino acid changes were also identified in several 2017 U.S. SVA isolates suggesting these changes emerged prior to 2017. Experimentally infected pigs were shown to produce SVA specific IgG and IgM responses directed primarily against VP2 and VP3 (49). Although these amino acid changes could alter structural proteins, serum from experimentally inoculated pigs has been shown to have cross-neutralizing activity against geographically and temporally distinct SVA isolates (25).
While the majority of market-weight hogs are transported directly to slaughter plants, the sow market may include shipment to one or more collection points prior to reaching the slaughter plant (50). These points of congregation potentially housing sows from multiple sites likely act as areas of pathogen transmission and could explain the greater amounts of SVA found in the sow slaughter plants compared to the market-weight slaughter plant in this study (50). Since 2016, a majority of the vesicular disease FADIs were related to swine, with a large proportion of these due to the continued circulation of SVA in the U.S. (37). The presence of an endemic vesicular disease of swine is of great concern for growing complacency in FMD free countries (17–19). As such, it remains essential that producers and the market chain remain vigilant to avoid dismissing a case of FMD.
These data demonstrate that SVA can be detected in the environment of sow slaughter plants and these environmental isolates have the potential to infect naïve pigs. Our data on contemporary 2020 isolates demonstrated limited genetic diversity change relative to isolates from 2015; however, our surveillance of three sow slaughter plants detected two co-circulating genetic clades that diverged over ten years ago, and the observed genetic diversity between these strains suggests that phenotypic assessment and efficacy of vaccine control measures may need to be assessed relative to strains collected within the region. Future studies looking at SVA presence throughout the cull sow market chain and to sow farms of origin could provide valuable information about the continued circulation and transmission of SVA and guide measures to reduce the spread of SVA in the swine industry.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MZ733975-MZ733980.
The animal study was reviewed and approved by NADC Animal Care and Use Committee in accordance with the Animal Care and Use Protocol (ACUP ARS-2018-750).
NH, JK, KL, and AB were responsible for the conception and design of the study. NH and AB were responsible for acquisition of the data. AB was responsible for animal and laboratory experiments. KH, TA, KF, and AB were responsible for data analysis and interpretation. KH was responsible for drafting the manuscript. AB and TA supervised manuscript editing. All authors read, revised, and approved the final manuscript.
This work was supported in part by: the U.S. Department of Agriculture (USDA) Agricultural Research Service [ARS project number 5030-32000-230-000-D]; the U.S. Department of Agriculture (USDA) Animal and Plant Health Inspection Service, Veterinary Services [ARS project number 5030-32000-118-072-I]; and the USDA Agricultural Research Service Research Participation Program of the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and USDA Agricultural Research Service [contract number DE-AC05- 06OR23100].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors wish to thank all the Animal and Plant Health Inspection Service Veterinary Service personnel involved in the environmental sample collection at the slaughter plants as well as the enrolled slaughter plants for their willingness to participate in the study. Dr. Li and his staff at the Iowa State University Veterinary Diagnostic Laboratory for the next-generation sequencing and support. Additionally, we would like to thank Deb Adolphson and Sarah Anderson for their technical support and Jason Huegel, Justin Miller, Alyssa Dannon, Randy Leon, Adam Hartfiel, and Nate Horman for animal care and handling.
1. Hales LM, Knowles NJ, Reddy PS, Xu L, Hay C, Hallenbeck PL. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol. (2008) 89:1265–75. doi: 10.1099/vir.0.83570-0
2. Fernandes MHV, Maggioli MF, Otta J, Joshi LR, Lawson S, Diel DG. Senecavirus A 3C protease mediates host cell apoptosis late in infection. Front Immunol. (2019) 10:363. doi: 10.3389/fimmu.2019.00363
3. Segalés J, Barcellos D, Alfieri A, Burrough E, Marthaler D. Senecavirus A. An emerging pathogen causing vesicular disease and mortality in pigs? Vet Pathol. (2017) 54:11–21. doi: 10.1177/0300985816653990
4. Knowles NJ, Hales LM, Jones BH, Landgraf JG, House JA, Skele KL, et al. Epidemiology of seneca valley virus: identification and characterization of isolates from pigs in the united states. In: XIV Meeting of the European Study Group on Molecular Biology of Picornaviruses. Saariselkä (2006).
5. Vannucci FA, Linhares DCL, Barcellos DESN, Lam HC, Collins J, Marthaler D. Identification and complete genome of Seneca Valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg Dis. (2015) 62:589–93. doi: 10.1111/tbed.12410
6. Pasma T, Davidson S, Shaw SL. Idiopathic vesicular disease in swine in Manitoba. Can Vet J. (2008) 49:84–5.
7. Wu Q, Zhao X, Bai Y, Sun B, Xie Q, Ma J. The first identification and complete genome of Senecavirus A affecting pig with idiopathic vesicular disease in China. Transbound Emerg Dis. (2017) 64:1633–40. doi: 10.1111/tbed.12557
8. Saeng-Chuto K, Rodtian P, Temeeyasen G, Wegner M, Nilubol D. The first detection of Senecavirus A in pigs in Thailand, 2016. Transbound Emerg Dis. (2018) 65:285–8. doi: 10.1111/tbed.12654
9. Arzt J, Bertram MR, Vu LT, Pauszek SJ, Hartwig EJ, Smoliga GR, et al. First detection and genome sequence of Senecavirus A in Vietnam. Microbiol Resour Announc. (2019) 8:e01247–18. doi: 10.1128/MRA.01247-18
10. Sun D, Vannucci F, Knutson TP, Corzo C, Marthaler DG. Emergence and whole-genome sequence of Senecavirus A in Colombia. Transbound Emerg Dis. (2017) 64:1346–9. doi: 10.1111/tbed.12669
11. Guo B, Pineyro PE, Rademacher CJ, Zheng Y, Li G, Yuan J, et al. Novel Senecavirus A in swine with vesicular disease, United States, July 2015. Emerg Infect Dis. (2016) 22:1325–7. doi: 10.3201/eid2207.151758
12. Montiel N, Buckley A, Guo B, Kulshreshtha V, VanGeelen A, Hoang H, et al. Vesicular disease in 9-week-old pigs experimentally infected with Senecavirus A. Emerg Infect Dis. (2016) 22:1246–8. doi: 10.3201/eid2207.151863
13. Joshi LR, Fernandes MHV, Clement T, Lawson S, Pillatzki A, Resende TP, et al. Pathogenesis of Senecavirus A infection in finishing pigs. J Gen Virol. (2016) 97:3267–79. doi: 10.1099/jgv.0.000631
14. Buckley A, Montiel N, Guo B, Kulshreshtha V, Van Geelen A, Hoang H, et al. Dexamethasone treatment did not exacerbate Seneca valley virus infection in nursery-age pigs. BMC Vet Res. (2018) 14:1–10. doi: 10.1186/s12917-018-1693-8
15. Maggioli MF, Fernandes MHV, Joshi LR, Sharma B, Tweet MM, Noll JCG, et al. Persistent infection and transmission of Senecavirus A from carrier sows to contact piglets. J Virol. (2019) 93:e00819–19. doi: 10.1128/JVI.00819-19
16. Tousignant SJP, Bruner L, Schwartz J, Vannucci F, Rossow S, Marthaler DG. Longitudinal study of Senecavirus a shedding in sows and piglets on a single United States farm during an outbreak of vesicular disease. BMC Vet Res. (2017) 13:277. doi: 10.1186/s12917-017-1172-7
17. Joshi LR, Mohr KA, Clement T, Hain KS, Myers B, Yaros J, et al. Detection of the emerging picornavirus Senecavirus A in pigs, mice, and houseflies. J Clin Microbiol. (2016) 54:1536. doi: 10.1128/JCM.03390-15
18. Xu W, Hole K, Goolia M, Pickering B, Salo T, Lung O, et al. Genome wide analysis of the evolution of Senecavirus A from swine clinical material and assembly yard environmental samples. PLoS ONE. (2017) 12:e0176964. doi: 10.1371/journal.pone.0176964
19. Silva GS, Graham K, Novak V, Holtkamp DJ, Linhares DCL. A retrospective investigation of risk factors associated with loads of pigs positive for Senecavirus A at a midwestern US packing plant during the summer of 2017. J Swine Health Prod. (2020) 28:87–93.
20. Lowe J, Gauger P, Harmon K, Zhang J, Connor J, Yeske P, et al. Role of transportation in spread of porcine epidemic diarrhea virus infection, United States. Emerg Infect Dis. (2014) 20:872–4. doi: 10.3201/eid2005.131628
21. Lowe J, McCann R, Greiner L. Factors that influence mechanical transmission of porcine reproductive and respiratory syndrome virus at the time of unloading animals into slaughter plant lairage. J Swine Health Prod. (2017) 25:19–23.
22. Elbers ARW, Stegeman JA, de Jong MCM. Factors associated with the introduction of classical swine fever virus into pig herds in the central area of the 1997/98 epidemic in the Netherlands. Vet Rec. (2001) 149:377–82. doi: 10.1136/vr.149.13.377
23. Hause BM, Myers O, Duff J, Hesse RA. Senecavirus A in pigs, United States, 2015. Emerg Infect Dis. (2016) 22:1323–5. doi: 10.3201/eid2207.151591
24. Buckley A, Kulshreshtha V, van Geelen A, Montiel N, Guo B, Yoon KJ, et al. Experimental seneca valley virus infection in market-weight gilts. Vet Microbiol. (2019) 231:7–10. doi: 10.1016/j.vetmic.2019.02.019
25. Buckley AC, Michael DD, Faaberg KS, Guo B, Yoon KJ, Lager KM. Comparison of historical and contemporary isolates of Senecavirus A. Vet Microbiol. (2021) 253:108946. doi: 10.1016/j.vetmic.2020.108946
26. Zhang J, Zheng Y, Xia XQ, Chen Q, Bade SA, Yoon KJ, et al. High-throughput whole genome sequencing of Porcine reproductive and respiratory syndrome virus from cell culture materials and clinical specimens using next-generation sequencing technology. J Vet Diagn Invest. (2017) 29:41–50. doi: 10.1177/1040638716673404
27. Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res. (2020) 48:D84–6. doi: 10.1093/nar/gkaa1023
28. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. (2002) 30:3059–66. doi: 10.1093/nar/gkf436
29. Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. (2016) 44:W232–5. doi: 10.1093/nar/gkw256
30. Martin DP, Varsani A, Roumagnac P, Botha G, Maslamoney S, Schwab T, et al. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. (2021) 7:veaa087. doi: 10.1093/ve/veaa087
31. van Geelen AGM, Anderson TK, Lager KM, Das PB, Otis NJ, Montiel NA, et al. Porcine reproductive and respiratory disease virus: evolution and recombination yields distinct ORF5 RFLP 1-7-4 viruses with individual pathogenicity. Virology. (2018) 513:168–79. doi: 10.1016/j.virol.2017.10.002
32. Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. (2009) 26:1641–50. doi: 10.1093/molbev/msp077
33. Rambaut A, Lam TT, Carvalho LM, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. (2016) 2:vew007. doi: 10.1093/ve/vew007
34. Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. (2018) 4:vey016. doi: 10.1093/ve/vey016
35. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. (2005) 22:1185–92. doi: 10.1093/molbev/msi103
36. Anderson TK, Inderski B, Diel DG, Hause BM, Porter EG, Clement T, et al. The United States swine pathogen database: integrating veterinary diagnostic laboratory sequence data to monitor emerging pathogens of swine. Database. (2021) 2021:baab078. doi: 10.1093/database/baab078
37. Summary of Recent FAD Investigations. In: USDA, editor. Riverdale, MD: Animal and Plant Health Inspection Service (2020). p. 1–11.
38. Trevisan G, Linhares LCM, Crim B, Dubey P, Schwartz KJ, Burrough ER, et al. Macroepidemiological aspects of porcine reproductive and respiratory syndrome virus detection by major United States veterinary diagnostic laboratories over time, age group, and specimen. PLoS ONE. (2019) 14:e0223544. doi: 10.1371/journal.pone.0223544
39. Turner JH, Paim WP, Maggioli MF, Peter CM, Miknis R, Talley J, et al. Prolonged viability of Senecavirus A in exposed house flies (Musca domestica). Viruses. (2022) 14:127. doi: 10.3390/v14010127
40. Pitkin A, Deen J, Otake S, Moon R, Dee S. Further assessment of houseflies (Musca domestica) as vectors for the mechanical transport and transmission of porcine reproductive and respiratory syndrome virus under field conditions. Can J Vet Res. (2009) 73:91–6.
41. Wang YC, Chang YC, Chuang HL, Chiu CC, Yeh KS, Chang CC, et al. Transmission of Salmonella between Swine Farms by the Housefly (Musca domestica). J Food Protect. (2011) 74:1012–6. doi: 10.4315/0362-028X.JFP-10-394
42. Medveczky I, Kovacs L, Kovacs F, Papp L. The role of the housefly, Musca domestica, in the spread of Aujeszky's disease (pseudorabies). Med Vet Entomol. (1988) 2:81–6. doi: 10.1111/j.1365-2915.1988.tb00052.x
43. Song Y, Hampson DJ. Development of a multiplex qPCR for detection and quantitation of pathogenic intestinal spirochaetes in the faeces of pigs and chickens. Vet Microbiol. (2009) 137:129–36. doi: 10.1016/j.vetmic.2008.12.020
44. Buckley A, Lager K. Infectious dose of Senecavirus A in market weight and neonatal pigs. PLoS ONE. (2022) 17:e0267145. doi: 10.1371/journal.pone.0267145
45. Hole K, Ahmadpour F, Krishnan J, Stansfield C, Copps J, Nfon C. Efficacy of accelerated hydrogen peroxide[(R)] disinfectant on foot-and-mouth disease virus, swine vesicular disease virus and Senecavirus A. J Appl Microbiol. (2017) 122:634–9. doi: 10.1111/jam.13361
46. Singh A, Mor SK, Aboubakr H, Vannucci F, Patnayak DP, Goyal SM. Efficacy of three disinfectants against Senecavirus A on five surfaces and at two temperatures. J Swine Health Prod. (2017) 25:64–8.
47. Houston E, Gimenez-Lirola LG, Magtoto R, Mora-Diaz JC, Baum D, Pineyro PE. Seroprevalence of Senecavirus A in sows and grower-finisher pigs in major swine producing-states in the United States. Prev Vet Med. (2019) 165:1–7. doi: 10.1016/j.prevetmed.2019.01.012
48. Jayawardena N, Burga LN, Easingwood RA, Takizawa Y, Wolf M, Bostina M. Structural basis for anthrax toxin receptor 1 recognition by Seneca Valley Virus. Proc Natl Acad Sci U S A. (2018) 115:E10934–E40. doi: 10.1073/pnas.1810664115
49. Maggioli MF, Lawson S, de Lima M, Joshi LR, Faccin TC, Bauermann FV, et al. Adaptive immune responses following Senecavirus A infection in pigs. J Virol. (2018) 92:e01717–17. doi: 10.1128/JVI.01717-17
Keywords: Senecavirus A, genetic diversity, slaughter plants, bioassay, SVA, swine
Citation: Hoffman KS, Humphrey NL, Korslund JA, Anderson TK, Faaberg KS, Lager KM and Buckley AC (2022) Characterization of Senecavirus A Isolates Collected From the Environment of U.S. Sow Slaughter Plants. Front. Vet. Sci. 9:923878. doi: 10.3389/fvets.2022.923878
Received: 19 April 2022; Accepted: 16 May 2022;
Published: 22 June 2022.
Edited by:
Zhenhai Chen, Yangzhou University, ChinaReviewed by:
Charles K. Nfon, National Centre for Foreign Animal Disease (NCFAD), CanadaCopyright © 2022 Hoffman, Humphrey, Korslund, Anderson, Faaberg, Lager and Buckley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra C. Buckley, YWxleGFuZHJhLmJ1Y2tsZXlAdXNkYS5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.