 Keneth Iceland Kasozi
Keneth Iceland Kasozi Ewan Thomas MacLeod
Ewan Thomas MacLeod Ibrahim Ntulume
Ibrahim Ntulume Susan Christina Welburn
Susan Christina Welburn
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Vet. Sci. , 07 March 2022
Sec. Veterinary Pharmacology and Toxicology
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.828111
This article is part of the Research Topic Women in Veterinary Pharmacology and Toxicology: 2021 View all 7 articles
African trypanosomiasis is associated with Trypanosoma evansi, T. vivax, T. congolense, and T. brucei pathogens in African animal trypanosomiasis (AAT) while T. b gambiense and T. b rhodesiense are responsible for chronic and acute human African trypanosomiasis (HAT), respectively. Suramin sodium suppresses ATP generation during the glycolytic pathway and is ineffective against T. vivax and T. congolense infections. Resistance to suramin is associated with pathogen altered transport proteins. Melarsoprol binds irreversibly with pyruvate kinase protein sulfhydryl groups and neutralizes enzymes which interrupts the trypanosome ATP generation. Melarsoprol resistance is associated with the adenine-adenosine transporter, P2, due to point mutations within this transporter. Eflornithine is used in combination with nifurtimox. Resistance to eflornithine is caused by the deletion or mutation of TbAAT6 gene which encodes the transmembrane amino acid transporter that delivers eflornithine into the cell, thus loss of transporter protein results in eflornithine resistance. Nifurtimox alone is regarded as a poor trypanocide, however, it is effective in melarsoprol-resistant gHAT patients. Resistance is associated with loss of a single copy of the genes encoding for nitroreductase enzymes. Fexinidazole is recommended for first-stage and non-severe second-stage illnesses in gHAT and resistance is associated with trypanosome bacterial nitroreductases which reduce fexinidazole. In AAT, quinapyramine sulfate interferes with DNA synthesis and suppression of cytoplasmic ribosomal activity in the mitochondria. Quinapyramine sulfate resistance is due to variations in the potential of the parasite's mitochondrial membrane. Pentamidines create cross-links between two adenines at 4–5 pairs apart in adenine-thymine-rich portions of Trypanosoma DNA. It also suppresses type II topoisomerase in the mitochondria of Trypanosoma parasites. Pentamidine resistance is due to loss of mitochondria transport proteins P2 and HAPT1. Diamidines are most effective against Trypanosome brucei group and act via the P2/TbAT1 transporters. Diminazene aceturate resistance is due to mutations that alter the activity of P2, TeDR40 (T. b. evansi). Isometamidium chloride is primarily employed in the early stages of trypanosomiasis and resistance is associated with diminazene resistance. Phenanthridine (homidium bromide, also known as ethidium bromide) acts by a breakdown of the kinetoplast network and homidium resistance is comparable to isometamidium. In humans, the development of resistance and adverse side effects against monotherapies has led to the adoption of nifurtimox-eflornithine combination therapy. Current efforts to develop new prodrug combinations of nifurtimox and eflornithine and nitroimidazole fexinidazole as well as benzoxaborole SCYX-7158 (AN5568) for HAT are in progress while little comparable progress has been done for the development of novel therapies to address trypanocide resistance in AAT.
Trypanosomiasis is caused by the parasite of the genus Trypanosoma (T.) and the disease affects both humans and animals (1). In humans, the disease is generally categorized based on the protozoan species, i.e., Chagas disease (American trypanosomiasis) and sleeping sickness (human African trypanosomiasis). Both forms of the disease are prevalent in low-income countries of America, Africa, and Asia and it is of major concern since it affects wildlife (2) and livestock such as equines (horses and camels), and large and small ruminants (i.e., bovines, ovines, and caprines) (3). This affects livestock productivity especially among horses, donkeys, camels, sheep, goats, and buffalo due to its associated economic burden (4). Trypanosoma evansi, T. vivax, T. congolense, and T. brucei are the major protozoan species that cause animal trypanosomiasis notably African animal trypanosomiasis (AAT) (5).
In developing countries of Latin America, Africa, and South East Asia, over 31 species of Trypanosoma infected tsetse flies have been identified (6). This continues to put the lives of over 60 million humans and 160 million animals in peril following a wave of infection in the affected communities (7). In Africa (40/54 countries), 55 million animals in the tsetse belt are at risk of AAT with annual mortality rates in cattle of 55 million, 30 million in sheep, and 40 million in goats (3). Livestock losses reduce livestock productivity since they are used for farm traction (8), and also as sources of community livelihood (9). Furthermore, human African trypanosomiasis (HAT) is present in (36/54) African countries. Angola, Cameroon, Central African Republic, Chad, Congo, Guinea, Malawi, South Sudan, and Zambia reported approximately 10 to 100 new cases in 2019, while Côte d'Ivoire, Equatorial Guinea, Gabon, Uganda, United Republic of Tanzania, and Zimbabwe reported 1 to 10 new cases (10). Furthermore, in the last 10 years, isolated instances have been documented in Burkina Faso, Ghana, Kenya, and Nigeria. In countries such as Benin, Botswana, Burundi, Ethiopia, Gambia, Guinea Bissau, Liberia, Mali, Mozambique, Namibia, Niger, Rwanda, Senegal, Sierra Leone, Swaziland, and Togo, however, no new cases have been registered in over a decade (10, 11). In Ethiopia, 8.6% of cases are caused by T. congolense (82 cases) and a few cases (3) are attributed to T. vivax (7). However, Uganda is the only country globally where both T. brucei rhodesiense (rHAT) and T. brucei gambiense (gHAT) have been reported (12).
Strategic control measures may have been implemented in such countries to halt transmission; but, due to poor monitoring and diagnostic activities, as well as inaccessibility and unstable socioeconomic conditions in such areas, it is impossible to determine the true incidence of disease at a particular point in time. This is important due to the close interactions between animals and humans across the tsetse belt region for the promotion of socioeconomic transformation in the affected communities (6).
The tsetse insect is a well-known biological vector for trypanosomiasis transmission. Tsetse flies belong to the genus Glossina, which is divided into three subgroups: Glossina (G. morsitans group), Austenina (G. fusca group), and Nemorhina (G. palpalis group). The ability to become infected while feeding on a vertebrate host, as well as the ability to sustain the development of the infection and transmit trypanosomes to a new vertebrate host, indicates the vectorial potential of Glossina species. According to these criteria, the G. palpalis and G. morsitans groups are the only ones that include T. b. gambiense vector species and subspecies. G. palpalis palpalis is common in forested areas of west and central Africa while G. palpalis gambiensis is prevalent in the savannah belt. Both species of G. palpalis group are effective T. b. gambiense vectors. G.m. morsitans and G.m. centralis in East Africa (excluding Uganda and Kenya), G. pallidipes in eastern and southern Africa, and G. swynnertoni in Kenya and Tanzania are the most common vectors for T. b. rhodesiense and are all prevalent in the savannah region.
Flies frequently visit forested regions and thickets throughout the savannah in eastern Africa, and woods and vegetation near streams in western Africa (3). In this environment, they can become infected during a blood meal on wildlife (2), and subsequently introduce infections in domestic livestock. In addition, both tsetse fly sexes have the capacity to carry and transmit infection (13). The ability of trypanosomes to cross the placenta has led to maternal-fetal infections of HAT (3).
In animals, tsetse flies may mechanically spread trypanosomes by starting a blood meal on one infected host and ending it on another, as long as there is a short interval between the two meals for parasites to thrive in the insect mouthparts (3, 14).
Trypanosomes are a group of protozoan eukaryotes and their genomes and modes of gene expression differ in several important aspects from those of other eukaryotic model organisms (15). The African trypanosome, Trypanosoma brucei is 35 megabases with an extremely high diversity in different isolates. The chromosomal telomeres possess TTAGGG repeats and many of the telomeres of the megabases and intermediate chromosomes are linked to expression sites for genes encoding variant surface glycoproteins (VSG) showing that T. brucei has recent origins, and ancestral gene lineages have been repeatedly co-opted to novel functions (16). Furthermore, the C-terminal domain of T. brucei VSG plays a crucial role in facilitating exchange and metabolism, and acts as a barrier against chemotherapeutical agents (17, 18).
The minichromosomes serve as repositories for VSG genes since some of their telomeres are linked to non-transcribed copies of VSG genes and these expand the VSG gene pool, allowing the parasite to avoid elimination by the host immune system, a situation complicated further by the great VSG diversity in Trypanosoma brucei (19, 20). The multiplicity in the copies of VSG genes provides new insights since a single VSG protein is expressed from approximately 15 expression sites (ESs), proximal to the telomeres of megabase or intermediate chromosomes (21). T. brucei selective switching on and off of VSG involves converting VSG cassettes into active ES by diluting cell division and turnover being relatively much slower (22), demonstrating parasitic adaptations during host response to infection in an effort to evade the hosts immune defenses. The sequence of ES proteins showed mosaics and broad recombination, a strategy that has influenced its current evolutionary function and structure (23). Comparisons in T. b. gambiense and T. b. brucei sequences identified 99.2% similarity in coding regions and the gene order was collinear while a comparison in the VSG from both parasites showed that the structural repertoire of VSG domains was well-conserved across the two subspecies (24).
The “housekeeping” portion of the T. brucei genome is encompassed by 11 pairs of megabase-size chromosomes (MBCs) per genome (15, 21). Tolemere-associated chromosome fragmentation has shown that the GC-rich transcriptional “strand-swtich” is composed mainly of retrotransposons and these confer mitotic stability (25). In the MBCs of T. brucei, topoisomerase-II activity is also focused at single loci that encompass regions between directional gene clusters that contain transposable elements (21). T. brucei Topo-II nuclear isoform sequences have been found to be highly conserved over most of their length with differences in the carboxyl terminal regions (CTRs, i.e., amino acids 1165-1455) which are usually essential for enzyme activity (26). Furthermore, a lot remains to be known about minichromosomes which are composed of 177 base pair repeats and circular DNA of 400 kb, called NR (NlaIII repeat) elements which are common in many strains of T. brucei (21, 27, 28).
VSG recombination has been shown to rely on at least two distinct DNA-repair pathways, i.e., RMI1-TOPO3α to suppress recombination and the other which is dependent on RAD51 and RMI1, thus demonstrating their role in antigenic switching (29). Furthermore, the universal mini-circle sequence binding proteins (UMSBPs) conserved at the replication origins of the mitochondrial (kinetoplast) DNA of trypanosomatids have been found to be important in function and integrity of telomere (30). This is of importance because cell division in T. brucei is complex due to novel evolutionary and trypanosome-specific molecules acquired over the years, although proteins regulating cytokinesis initiation and completion have not changed (31, 32). Progress has been made on understanding the timing of these events in the cell cycle (33); however, identification of the important proteins involved remains to be established.
This is caused by trypanosome species, the salivaria group, i.e., Trypanosoma vivax—subgenus Duttonella, Trypanosoma congolense—subgenus Nannomonas and Trypanosoma brucei specie—subgenus Trypanozoon whose transmission to the animal host trails is through infected saliva of blood-sucking insects (3).
T. vivax can be found in the wildlife of South America, acting as a reservoir of infection (Table 1). As a result, T. vivax infection is considered an emerging illness in South America, although infections have been reported in Mauritius (beyond Africa) due to the proliferative livestock trade (34). It affects mainly bovines, ovines, caprines, Camelus dromedarius, and equines and it is genetically different from other animal trypanosomes. For example, T. vivax does not proliferate in the vector midgut, rather it prefers the proboscis where it completes its lifecycle. This implies that mechanical minges including Tabanus spp. and Stomoxys spp. can transmit T. vivax (3). Generally, T. vivax is maintained in the host vascular system; however, some few exceptions occur where it has been found in the lymphatics, cerebrospinal fluid, and eyes of infected patients (due to adaptions to low oxygen consumption), making chemotherapeutical treatment challenging and development of drug resistance (35).
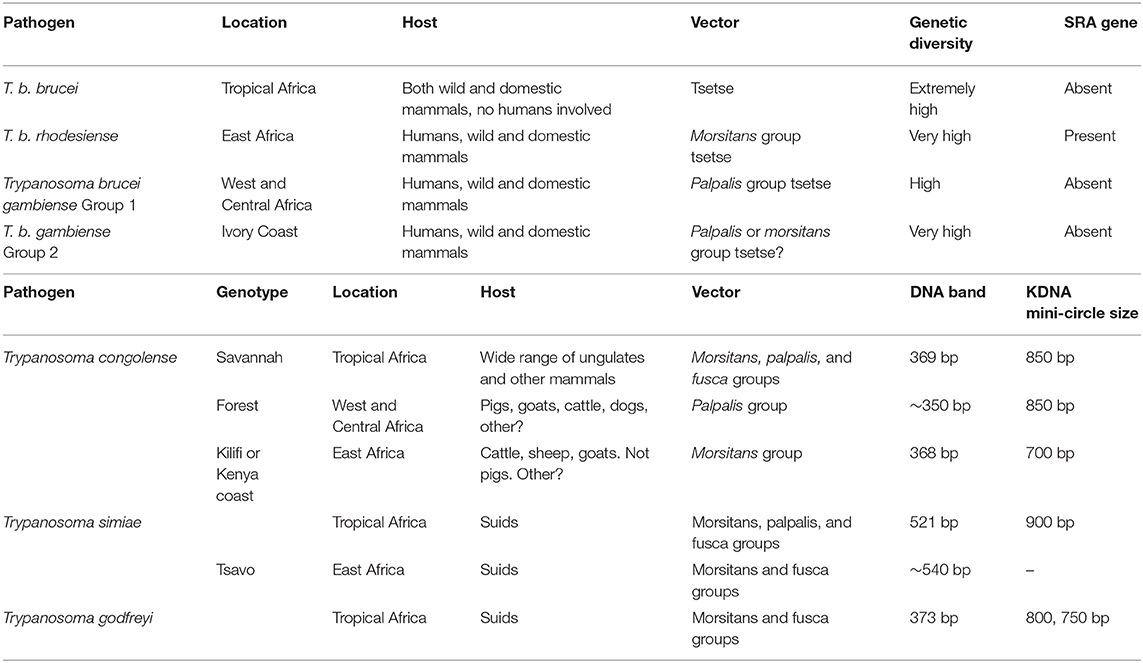
Table 1. Characterization of subspecies and subgroups within Trypanosoma brucei.
These are the smallest trypanosomes and are divided into 3 subgroups, i.e., Savananah, Forest, and Kilifi (36, 37). In cattle, the Savannah subgroup is the most virulent and clinically significant (3) than T. vivax. In tropical Africa, Trypanosoma (Nannomonas) congolense are the most common pathogenic trypanosomes in tsetse flies (38) and it has also been found in horses, sheep, goats, pigs, and dogs. T. congolense parasites are limited to the vascular system in their vertebrate hosts, where they use their flagellum to adhere to circulating erythrocytes and endothelial cells, inflicting damage at the adhesion point (3). T. congolense is a tiny trypanosome in the mammalian circulation, having a shorter length than T. brucei and no visible undulating membrane. Mechanical transmission of T. congolense has been demonstrated in laboratory conditions and hence cannot be ruled out as a factor in its spread in Africa (39, 40).
Resistance in T. congolense is associated with differences in carbohydrate metabolic pathways as observed in T. brucei. T. congolense has high comparable oxygen consumption rates as T. evansi and T. brucei groups although the rate of glucose consumption is lowest in T. congolense leading to the production of lactic acid, pyruvate, and carbon dioxide (3). Metabolic inhibitors, i.e., cyanide, malonate, antimycin A, and a combination of cyanide and malonate plus a combination of antimycin A and malonate can inhibit the rate of oxygen consumption by procyclic forms of T. congolense using proline as a substrate (41). Proline dehydrogenase, α-ketoglutarate dehydrogenase, succinate dehydrogenase, fumarase, NADP-linked malic enzyme, alanine aminotransferase, and malate dehydrogenase were among the enzymes engaged in proline catabolism with high activity. Alanine and glutamate are the end products of proline metabolism. Aspartate was absent for T. congolense unlike in T.b. brucei (42). Therefore, glycolysis is important in both T. congolense and T. brucei for energy metabolism (43). Furthermore, the electron transport chain (ETC) is not required in T. congolense followed by substantial resistance to fatty acid synthesis inhibitors (44, 45). These findings raise major challenges for chemotherapy development against drug resistance and host-pathogen interactions. Finally, T. congolense lacks an alternative for the T. brucei TbAT1 gene, which codes for the P2 nucleoside transporter which is essential for diminazene aceturate absorption (46).
T. simiae is part of the Nannomonas subgenus as is T. congolense and it is the only trypanosome species that is particularly harmful to pigs, causing hyperacute, lethal infection with death occurring within 2 days after the onset of symptoms (47). The parasite can also be found in other domestic animals (48), therefore all livestock on the farm should be treated prophylactically. The pathogenicity of trypanosome infections varies greatly based on a variety of factors, including parasite characteristics (species and virulence), host characteristics (species, breed, age, immunological status, nutritional status, presence of co-infection, and physical condition), vector characteristics (species, density, infection rate, and host preference), epidemiological situation (endemic or epidemic), and the environment (e.g., the availability of food and water and the season) (3).
There are three morphologically identical subspecies due to host range, diversity, and geographical dispersal other than biological variations, i.e., T.b. brucei, T. b. gambiense, and T. b. rhodesiense. The fact that T. b. brucei and T. b. rhodesiense might differ by as little as the expression of a single gene is particularly intriguing. In fact, there is more genetic variety among T. b. brucei isolates than there is between T. b. brucei and T. b. rhodesiense (49). This implies that a majority of T. b. gambiense constitute a homogenous group due to the narrow genetic range. In contrast to the normally fast-growing T. b. brucei/T. b. rhodesiense phenotype, T. b. gambiense group 1 adheres to the conventional understanding of T. b. gambiense as a slow-growing parasite in experimental mice (50).
Trypanosoma brucei species includes both animal (T. b. brucei, T. b. evansi, T. b. equiperdum) and human (T. b. rhodesiense, T. b. gambiense) infective subspecies unlike T. vivax (at least most strains) or T. congolense, which are prevalent in both the vascular system and other organs, and may parasitize the brain in experimental infections (2, 39). The bloodstream form of T. brucei, which causes sleeping sickness in people, and the procyclic form, which is found in tsetse flies, have two separate proliferative phases. The species' subspecies T. b. brucei predominantly affects cattle and occasionally other animals, and it does not infect humans under normal circumstances. T. brucei produces nagana, a wasting illness in cattle, but not in humans. Trypanosomes of the T. brucei group (T. b. brucei, T. b. evansi, T. b. equiperdum) are morphologically similar (except for the non-proliferative stumpy form in T. b. brucei). During chemotherapy, the treatment goal is the trypomastigote infective stage (3).
T. b. brucei may be found in several domestic ungulates, though more severe in horses, dogs, and camels. In areas where several trypanosome spp. exist, mixed infections in livestock are widespread, and modern genetic methods make speciation simpler. Many African wild animal species carry at least one species of trypanosome, thus serving as potential reservoirs for highly infectious trypanosomes in humans and livestock (Table 1).
T. b. evansi exhibits features of slender Trypanozoon parasites in fresh blood samples. It is small sized compared to T. theileri though it is larger than T. congolense. T. evansi has previously been understood as a monomorphic thin trypomastigote parasite once viewed on a Giemsa stained thin smear (51). T. b. brucei is assumed to be the origin of T. evansi, however, it cannot complete its cycle in Glossina due to the loss of the maxi-circles of kinetoplastic mitochondrial DNA (51). T. b. evansi has acquired a mechanical transmission method which has enabled its expansion and spread rapidly outside of Africa through the export of diseased animals. As a result, it is now the deadly animal trypanosome with the widest geographical range, spanning North-East Africa to most of East Asia and Latin America (51), and it is rapidly spreading. Recently, this infection has been imported in Europe with documented epidemics in Germany, Canary Islands, France, and mainland Spain. T. b. evansi may infect both wild and domestic animals (51), however, related studies show that T. b. evansi and other trypanosomes, such as T. lewisi, a rat pathogen also found in atypical human illnesses, both have a reservoir host in common, i.e., rodents (52). These discoveries have resurrected the importance of rodents as reservoirs of T. brucei. Rare incidences of T. b. evansi infections in humans have been described and also have the ability to infiltrate host tissues (53) indicating illness being linked to a null genetic mutation in the trypanosome lytic factor blood component Apolipoprotein L1 (APOL1). This generally shields humans against animal trypanosome infections (54), however, no alterations in APOL1 have been detected to elucidate atypical illness (53).
It is widespread throughout Africa and Asia, as well as the Middle East, South America, and Southeast Europe (Table 1). T. b. equiperdum has already been exterminated from Western Europe over the last century (55), however, there is still a danger of recurrence, as evidenced by a previous epidemic in Italy (56). The cycle of transmission for T. b equiperdum entirely excludes invertebrate vectors. Alternatively, it is passed on through generations of horses and other equids during mating. T. b equiperdum unlike other trypanosomes, has vertical or perinatal transmission via reproductive organs, resulting in a venereal illness known as dourine. T. b. equiperdum is a peculiar protozoan since it mostly invades host tissues and dwells in the blood capillaries of the urogenital tract and very occasionally in the peripheral circulation (57). As a result, parasite diagnosis, isolation, and therapy are all made more complex. Because of this peculiar transmission method and the lack of a reservoir in other species, the disease's management techniques differ from those for other arthropod forms of trypanosomiasis. Furthermore, pharmaceutical therapy is not recommended since it may only relieve clinical symptoms but not completely cure the parasite, thus turning the infected mammal into a possible parasite carrier. While there is no recognized therapy for dourine, studies have shown that melarsomine is effective in treating acute and chronic trypanosomiasis illnesses in horses caused by T. b. equiperdum (58).
These are considered as T. brucei mutants that have lost some portions of kinetoplast DNA (kDNA) (dyskinetoplastic) or all of it (akinetoplastic). The kDNA is a matrix of circular concatenated mini- and maxi-circles that makes up the mitochondrial genome. In the fly, T. b. equiperdum and T. b. evansi are unable to complete their life cycle and are trapped in the trypomastigote stage, a stage known to be reliant on glycolysis for ATP synthesis.
Because of a compensatory mutation in the nuclear genome-encoded-component of the ATP synthase, these species of trypanosomes can sustain their mitochondrial function in the absence of the F0–A6 subunit and hence live without the kinetoplast genome (59). As a result, these protozoa are no longer dependent on the tsetse fly for dissemination to their susceptible hosts.
Human African trypanosomiasis (HAT), often known as sleeping sickness, is a parasitic illness spread by vectors and is mainly endemic to sub-Saharan Africa. Humans get infected with the parasites through bites infected with tsetse flies infected with pathogenic parasites acquired either from infected human beings or from animals (3).
In humans, two subspecies of Trypanosoma brucei, i.e., T. b. gambiense and T. b. rhodesiense, are morphologically similar but induce unique illness patterns. T. b. gambiense and T. b. rhodesiense belong to the kingdom Protista, domain Eukarya, phylum Sarcomastigophora, subphylum Zoomastigophora, class Zoomastogophorea. All other animal-like (or non-photosynthetic) flagellates which locomote utilizing whip-like flagella and eat by pinocytosis or phagocytosis are members of this subphylum and class (3).
T. b. gambiense is a parasite that causes chronic African trypanosomiasis (better known as “West African sleeping sickness”) which is widespread in over 24 countries in central and west Africa (60). T. b. rhodesiense is widespread in 13 countries in eastern and southern Africa, accounting for <5% of all cases of acute African trypanosomiasis (also known as “East African sleeping sickness”) (60). This infection is known as “African sleeping sickness” in humans because it causes lethargy in the affected persons (3). It is only in Uganda where both kinds of the parasite illness may be found in separate parts of the country (61).
According to a review by Giordani et al. (3), T. b. gambiense parasites are mainly reserved in the human host; however, the infection has also been found in both domestic and wild animals. Cattle are the principal reservoir for T. b. rhodesiense infection. Other domestic animals (dogs, pigs, and sheep) as well as a variety of game species (warthogs, bushbuck, hartebeest, lions, zebras, impala, waterbuck, and hyenas) are also infected (2). The bloodstream form of trypanosomes is taken along with the blood meal when a tsetse fly bites an infected human (or animal). Trypanosomes in the fly travel to the midgut lumen, convert into the procyclic stage which then migrates to the salivary gland after 2 or 3 weeks. It is at this point that they undergo numerous developmental modifications before maturing into adult infectious metacyclic stage that are injected into the skin of a mammalian host while having a blood meal. The metacyclic trypanosomes ultimately become trypomastigotes spreading throughout the body circulatory and lymphatic systems.
Trypanosomes use a unique antigenic variation method to evade the immune system. Although the DNA contains up to 1,000 distinct variable surface glycoprotein (VSG) genes, every trypanosome typically encodes single VSG at a moment. The VSG forms a protective layer for the parasite's other invariant outer membrane elements. As antibodies to the VSG are produced, the parasite changes its VSG expression to a different one, still when more antibodies are created against the newly formed VSG, it shifts to another VSG form, and so forth (3). As a result, B cells are massively polyclonally activated, and an elevated IgM level is a characteristic sign of the disease. Immune activities of B-cells and T-cells are similarly suppressed, albeit without clinical implications. Hyperplasia of the lymph nodes and spleen is caused by high IgM levels and the resulting antigen-antibodies complex, along with lymphocytic proliferation. Trypanosomes invade the blood-brain barrier (BBB) and infect the central nervous system (CNS) at a certain stage throughout this protracted process, resulting in chronic lymphocytic meningo-encephalitis (3).
In 2006, 11,382 instances of Gambian trypanosomiasis were documented across the continent, as opposed to 486 cases of Rhodesian trypanosomiasis (62). The DRC continues to have the greatest prevalence of Gambian trypanosomiasis, with 8,023 cases reported in 2006 (up from 26,318 in 1998) (62, 63). In 2006, Angola recorded 1,105 instances (compared to 8,275 cases in 1997), while Sudan reported 809 victims. In 2006, Uganda recorded the largest prevalence of Rhodesian trypanosomiasis at a rate of 245 cases. Conversely, scarce occurrences of Gambian trypanosomiasis have been reported within developed countries mostly among migrants from Central Africa whereas Rhodesian trypanosomiasis has been reported in visitors traveling from East African game parks (64).
In the 26-megabase genome of this species, there are 9,068 projected genes, including roughly 900 pseudogenes and ~1,700 T. brucei-specific genes. According to huge subtelomeric arrays, the parasite employs 806 VSG genes to escape the mammalian host immune responses. Several VSG genes are pseudogenes that may be utilized to construct functional mosaic genes by ectopic recombination (17).
Hall et al. (65) revealed that the long-term T. brucei infection, transmission, and parasite success is all influenced by the interaction between host acquired immunity and antigenic polymorphism of the trypanosome's VSG coat. Around 0.1% of the parasite's cycle results in a transition to a new VSG due to fluctuating activation of hundreds of silent VSG genes and pseudogenes, and that distinct antigenic determinants “mosaic” VSG form by segmental gene transformation between donor variable surface glycoprotein genes or pseudogenes. Therefore, mosaic VSG are responsible for antigenic heterogeneity and long-term infection (65).
The VSG and procyclins are glycosylphosphatidylinositol (GPI)-anchored proteins that cover the surface of trypanosome cells. Despite this, the cellular membrane in the host bloodstream is heavily covered with roughly 10 million VSG molecules (parasites' genome comprises around 1,000 VSG genes). GPI anchoring is vital for parasite's endurance and establishment of infection through imitating the humoral immune response of the mammal.
Trypanocidal medications most often used in sub-Saharan Africa include: Isometamidium chloride, ethidium bromide, and diminazene aceturate accounting for 40, 26, and 33%, respectively (1). Isometamidium has equally prophylactic and therapeutic properties, whereas diminazene aceturate exclusively has curative capabilities. Suramin is employed in the management of T. b. evansi infections in comparison to other antibiotics (3). Homidium is one of the key medications currently available to regulate AAT according to Sahin et al. (66). The alarming increase of resistant cases to these small numbers of available trypanocides, particularly diminazene aceturate and isometamidium, is concerning because it implies that their future utility may be compromised.
It has been reported that diminazene and isometamidium drugs are the most commonly used medications to cure animal trypanosomes, however, these cannot penetrate the BBB, thus this might be a significant problem in the management of T. brucei parasites not within the circulatory system of its mammalian host (3). In addition, Cymelarsan (melarsamine hydrochloride) administered at a dosage of 0.25 and 0.5 mg/kg body weight has been successful in horses with acute and chronic dourine, respectively, due to its high curative effects (58). This revitalizes demand for research promoting the development of a wider spectrum of trypanocides in AAT.
International interest for the development of novel chemotherapeutical options is currently weak as observed from the limited drug options for the management of HAT (Table 2). This has subsequently led to the classification of trypanosomiasis as a Neglected Zoonotic Disease (NZD) by the U.S. Department of Health & Human Services through the Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO) (96–98).
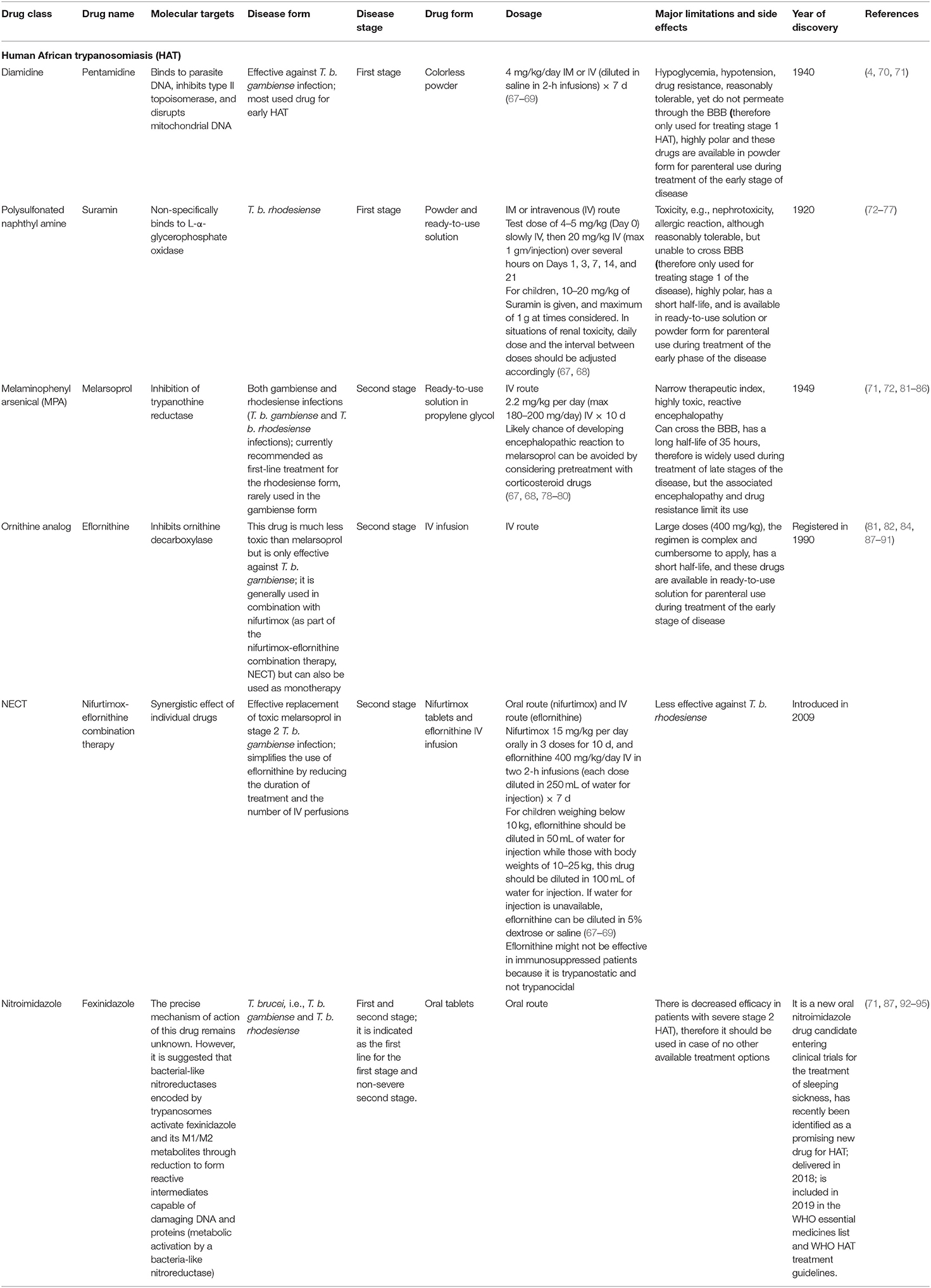
Table 2. Approved drugs for the treatment of human trypanosomiasis.
During treatment of trypanosomiasis, the major factors to be considered are: (1) the form of the disease and (2) the phase of the disease (Table 2). The prognosis is good if treatment is initiated in the initial phases of the disease (with early diagnosis), while the second stage requires drugs able to cross the BBB to reach the parasites (4, 67, 99). Currently, there are four approved drugs for treating HAT, i.e., suramin and pentamidine both used during the first (hemolymphatic) stage, and eflornithine and melarsoprol recommended for the second (meningo-encephalic) stage since this involves neuro-migration of the parasites to the CNS. Major challenges associated with these therapies include the high cost, poor oral bioavailability, toxicity, lack of efficacy, and prolonged treatment (87, 92, 100, 101). For example, Eflornithine involves multiple intravenous (IV) infusions for 2 weeks making it an expensive therapy especially for rural health centers, while melarsoprol is very toxic and usually causes reactive encephalopathy (encephalopathic syndrome) which has been associated with deaths in 3–10% of HAT patients (87). This has created a demand for novel therapeutical options which are more effective, safer, and easy to administer. To address challenges associated with monotherapies, approval of Nifurtimox-eflornithine combination therapy (NECT) is currently being promoted as the standard form of treatment. NECT has been associated with low toxicity, and a shortened therapeutical period unlike eflornithine or melarsoprol monotherapy. However, NECT requires 7-d IV infusions which can be a challenge to adhere to especially in resource limited settings (81). In addition, a 2-substituted 5-nitroimidazole (fexinidazole) is currently undergoing extensive preclinical and clinical trials for its use in HAT and preliminary studies have provided favorable results on safety, effectiveness, short-duration of treatment, oral use, and curing both acute and chronic HAT (93). The first all-oral treatment (fexinidazole) is ingested for 10 d for the treatment of both stages of the most common form of infection (T. b. gambiense). It is more advantageous than the previous standard therapy (NECT) since it eliminates frequent hospitalization and reduces the number of lumbar punctures (102, 103).
Suramin sodium is a symmetric polyanionic sulfonated naphthylamine drug. This trypanocide was launched in 1921 as a therapy of surra in camels and for the management of acute phase HAT. It is the oldest trypanocide currently in use (60). It is usually applied to treat HAT caused by T. b. rhodesiense, and as such it is still presently accessible. However, it has been superseded by pentamidine in the management of trypanosomiasis caused by T. b. gambiense. This drug is also the usual therapy for trypanosomiasis in horses mainly caused by T. brucei spp., outperforming diminazene since it is less toxic compared to quinapyramine.
Suramin forms a strong electrostatic bond with human blood proteins and several trypanosome enzymes. The medication was thought to penetrate trypanosomes via LDL receptor-mediated absorption and concentrate in the lysosome (104). This notion, on the other hand, appeared to be dubious and the exact mechanism of action has yet to be discovered. Suramin suppresses ATP generation during the glycolytic pathway in T. brucei through blockage of glycerol-3-phosphate oxidase and NAD+-dependent glycerol-3-phosphate dehydrogenase (105). In bloodstream T. brucei, 28 genes are involved in suramin action, including a surface glycoprotein family (ISG75), a known drug ligand; cathepsin L thought to deliver drug compounds from the ligand within the lysosomal system; a number of deubiquitinating enzymes, and numerous proteins participate in the endocytic pathway (106).
It is a member of the triazines family and is generated from arsenic. It has several negative toxic effects, including reactive encephalopathy (encephalopathic syndrome), which has a 3 to 10% mortality rate. It is now prescribed as a first-line therapy for rHAT, but it is only occasionally applied for the management of gHAT (60). This is the only currently available therapy for late-stage infections (parasitemia of the CNS) due to rHAT.
Melarsoprol is a prodrug that is converted to the active compounds melarsen oxide after delivery into the mammalian host. It operates by inhibiting the enzyme pyruvate kinase (PK) required during the parasite's aerobic metabolism of glucose. It works by binding irreversibly with PK protein sulfhydryl groups and neutralizing enzymes which interrupts the trypanosome ATP generation processes. Melarsenoxide (Mel Ox) interacts with trypanothione as well as spermidine-glutathione adduct that substitutes glutathione in parasite. Melarsen oxide-trypanothione adduct (Mel T) inhibits trypanothione reductase competitively, thereby extinguishing the parasite. Enough of the melarsoprol metabolite reaches the cerebrospinal fluid (CSF), where trypanosomes pick it up and concentrate it. This medication is very hazardous, with several adverse effects since it is incapable of discriminating the PK for host and that of the parasite (107).
It is prescribed for treatment of the second stage of sleeping sickness associated with T. b. gambiense. This trypanocide can be used in combination with nifurtimox. The lower toxicity exhibited by eflornithine to the host has led to its use as a substitute alternative to melarsoprol as the first-line treatment for HAT (108).
Eflornithine is substantially less toxic than melarsoprol and it is only effective against T. b. gambiense. Nonetheless, it is most commonly used in conjunction with nifurtimox (NECT), although it may also be administered alone, despite the fact that the treatment is complicated and time-consuming to administer (60). Due to the severe cytotoxicity associated with the use of this dual medication in the management of T. b. gambiense, melarsoprol has been restricted to the management of second-stage T. b. rhodesiense. It facilitates the administration of eflornithine by lowering therapeutic time and the number of IV perfusions required. Eflornithine works by inhibiting ornithine decarboxylase, an enzyme that catalyzes the formation of amine-based chemicals important in cell division and differentiation. Currently, combination therapies are already being explored to avert the occurrence of eflornithine-resistant trypanosomes, although little has yet been published in this regard.
Nifurtimox was first used in the treatment of the second stage of HAT as part of the nifurtimox/eflornithine combination therapy (NECT). However, when compared to other similar medications, nifurtimox alone is regarded as a poor trypanocide (109). Nifurtimox can treat both phases of T. b. gambiense including 60–90% of melarsoprol-resistant patients. In vitro and in vivo studies have revealed that nifurtimox-resistant (NfxR) T. brucei is cross-resistant to fexinidazole (110). The mechanism of action of nifurtimox has not been fully elucidated, however, it is believed that nifurtimox causes oxidative stress to the parasite (110). Inhibition of parasite dehydrogenase activity is another mode of action of nifurtimox that warrants further research. The Type 1 nitroreductases in the two-electron reduction of nitroheterocycles induces oxidative stress thus causing cellular death in the parasite (109, 111).
Fexinidazole is a medical breakthrough that is the first all-oral therapy for HAT caused by T. b. gambiense in patients aged 6 years and above who weigh at least 20 kg. It initially obtained a satisfactory scientific opinion from the European Medicines Agency in 2018 (60) and it is currently included in the WHO interim recommendations (102). This compound is recommended for first-stage and non-severe second-stage illnesses. It is administered within 30 min following a solid meal and under the supervision of a physician. A clinical investigation for its usage in rHAT is ongoing (60). The drug is also effective against T. cruzi (112), although no cases of American trypanosomiasis have been reported in Africa to this day probably due to limited movements and trade between Latin America and Africa.
In vitro studies carried out in T. b. gambiense have shown that fexinidazole and its two major metabolites, a sulfoxide (M1) and a sulfone (M2) are very crucial in its antitrypanocidal effects. Thus, it has been proposed that trypanosomes encode bacterial-like nitroreductases that reduce fexinidazole and its M1/M2 derivatives to generate reactive metabolites that can damage the trypanosome genome and its proteins (93). This implies that once the drug is absorbed by the parasite, its metabolites are the ones responsible for its therapeutical effects.
Pentamidine is exclusively utilized in the initial stages of T. b. gambiense illness, before transiting to the CNS. It is utilized as a backup alternative to suramin. Pentamidine works by disrupting the critical activities in DNA, RNA, phospholipid, and protein production. This drug creates cross-link between two adenines at 4–5 pairs apart in adenine-thymine-rich portions of Trypanosoma DNA. It also suppresses type II topoisomerase in the mitochondria of the parasites, culminating in a mitochondrial genome that is fragmented and unreadable (113).
African trypanosomiasis in animals has been associated with critical livestock production losses demonstrating the importance to shift policy on disease control to eliminate reservoir host species in endemic communities. The World Health Organization (WHO) has targeted the elimination of HAT “as a public health problem” by 2020 (114), however, this has remained challenging due to the evolving epidemiology pattern of HAT thus questioning the current status quo that the rhodesiense form is zoonotic while the gambiense form is the main reservoir in disease transmission (115).
The WHO 2020 target was associated with increased infrastructure and fewer cases being reported showing that its elimination was on track, however, the need to generate innovative tools for control of infections and elimination by 2030 cannot be taken for granted (116–118). This is because there has been slow progress in clinical isolates for gambiense HAT (114) and for rhodesiense HAT (117), however, this situation is geographically specific (for African countries) and not diffuse demonstrating challenges for the attainment of the 2030 target. Infections in domestic livestock [see Kasozi et al. (9)] in small ruminants which are never screened in several developing countries) will continue to act as sources of sporadic infections in humans creating a need to revise the current disease control strategy (Table 3). Furthermore, the narrow chemotherapeutical spectrum available for the control of AAT will continue to undermine WHO HAT targets unless policy is driven to promote innovations in the pharmaceutical industry to control infections.
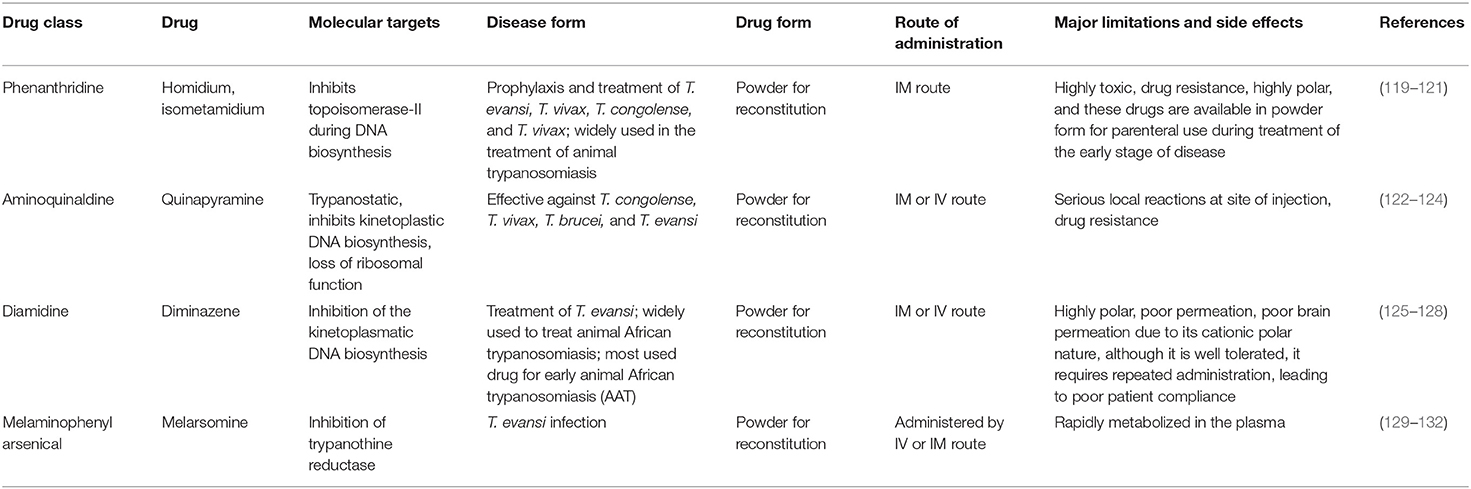
Table 3. Drugs for the treatment of animal trypanosomiasis.
Homidium bromide, also known as ethidium bromide, was launched as an advancement over earlier phenanthridine-based trypanocidal agents and it is also accessible as a chloride salt (133). Cross-resistances are attributed to the phenanthridine core. Homidium may perhaps be prescribed as a sanative pair with diminazene aceturate, but never with isometamidium (134). Treatment with this antitypanocide causes dyskinetoplasty in the same manner as several other phenanthridines and diamidines do (135) and alteration in gene activity has been believed to be a major attribute to its trypanocidal effects. Moreover, by twisting and altering the double helix structure, homidium inhibits both kinetoplast and nuclear DNA replication in T. brucei (136). At low dosages (0.02 g/ml), homidium was shown to eliminate dyskinetoplastic trypanosomes primarily through the breakdown of the kinetoplast network. However, at larger doses, homidium disrupts the parasite's gene, which explains its capability to kill dyskinetoplastic trypanosomes.
Isometamidium chloride hydrochloride contains therapeutic as well as preventative qualities used in the management of African trypanosomiasis. It is a blended phenanthridine with amphiphilic and cationic characteristics that are made by combining homidium with the diazotized p-aminobenzamide moiety of diminazene and then modifying it with the amidine group in the meta position. The phenanthridine isometamidium is primarily employed in the initial stages of AAT. The isometamidium chloride formulations mainly include a mixture of four phenanthridine compounds, namely; isometamidium chloride hydrochloride [8-(3-mamidinophenyl-2-triazeno)-3-amino-5-ethyl-6 phenylphenanthridinium chloride hydrochloride], the positional red isomer [3-(3-m-amidinophenyl-2- triazeno)-8-amino-5-ethyl-6-phenylphenanthridinium chloride hydrochloride], the blue isomer [7-(mamidinophenyldiazo)-3,8-diamino-5-ethyl-6 phenylphenanthridinium chloride hydrochloride], and the disubstituted compound [3,8-di(3-m amidinophenyltriazeno)-5-ethyl-6-phenylphenanthridinium chloride dihydrochloride]. Regrettably, this trypanocide is unsuccessful in the treatment of trypanosomiasis illnesses caused by T. b. evansi, hence it is less widely utilized beyond sub-Saharan Africa (60, 137). Isometamidium chloride works by forming an unconventional “sideways” geometry bond to kDNA of the trypanosome (138).
Surfen C is one of the early trypanocides from which quinapyramine sulfate was developed (139). The mechanism of action of quinapyramine is uncertain, however, two hypotheses have justified its interference with DNA synthesis and suppression of cytoplasmic ribosomes (hence, inhibit protein synthesis). However, like with phenanthridines and bis-benzamidines, its dicationic/aromatic properties imply mitochondrial drug accumulation as its mode of action (3).
Studies have shown significantly less efficiency of diminazene aceturate against the T. congolense group as compared to the T. b. group. This could be attributable to the absorption route used by latter parasites via the P2/TbAT1 transporter that permits for faster and concentrated absorption. Diminazene aceturate is promptly metabolized and eliminated, thus it is more ideal as a curative drug than prophylactic use (3). Diminazene aceturate binds to the minor groove of the DNA AT-rich locations of the parasite. kDNA is the recognized target of this drug where it binds, ultimately inducing inhibition of replication and kDNA loss. This may be worsened by the effects caused by suppression on mitochondrial type II topoisomerase (3), leading to a disruption of mitochondrial membrane transport protein activity. Furthermore, in vitro studies reported by Gould and Schnaufer (140) highlighted that trypanome dyskinetoplastic lines showed substantial resistance to diamidines, i.e., diminazene aceturate and phenanthridines.
Diminazene aceturate has been proposed to modify the reactions of the host's immunity by reducing pro-inflammatory cytokines and overwhelming immunological stimulation, which might impact the drug's in vivo properties (141). Diminazene aceturate can only pass through the cell membranes through specific transporters due to its charged composition, and this implies that: (a) The medicine is ineffective against infections that advance to the CNS because it cannot pass through the BBB; (b) trypanosomes express carrier proteins that particularly express genes to cause resistance; and (c) a loss of these specific transports by the parasite through VSG activity inevitably leads to induced resistance.
Melarsomine dihydrochloride is a melamino-phenylarsine that is produced by combining melarsen oxide with two cysteamine equivalents. Compared to melarsoprol, the chemical exhibits a higher water solubility (3). The drug (or, more precisely, its metabolite melarsen oxide) penetrates the cells of T. brucei via the same P2/ TbAT1 adenosine nucleoside transporter and TbAQP2 transporters that also deliver similar trypanocides; melaminophenyl arsenicals and diamidine (142, 143) highlighted that the majority of the cytotoxic activity of arsenicals is presumably due to selective absorption.
Close to 35 million doses of trypanocides are delivered in sub-Saharan Africa each year and are only adequately able to care for about one-third of the animals at risk (1). Remarkably, farmers in Africa have consistent access to a majority of the trypanocides which has culminated in widespread overuse and under-dosage of drugs without the guidance of veterinarians, resulting in the evolution of trypanocidal drug resistance. This situation has been precipitated by the lax legislation which promotes liberalization of the drug industry; however, this has led to increased drug abuse, wastage, and development of drug resistance.
At the moment, trypanocidal resistance has been registered in 21 African nations (1) including Ethiopia (6, 58). A study done between 1996 and 2003 in the Eastern Province of Zambia documented a five-fold significant increase of diminazene aceturate resistance to T. congolense over the 7-year study period (144). The usage of similar trypanocides over time has put selection pressure on drug-targeted trypanosome genes, resulting in genome mutations which promote trypanocide resistance (Table 4).
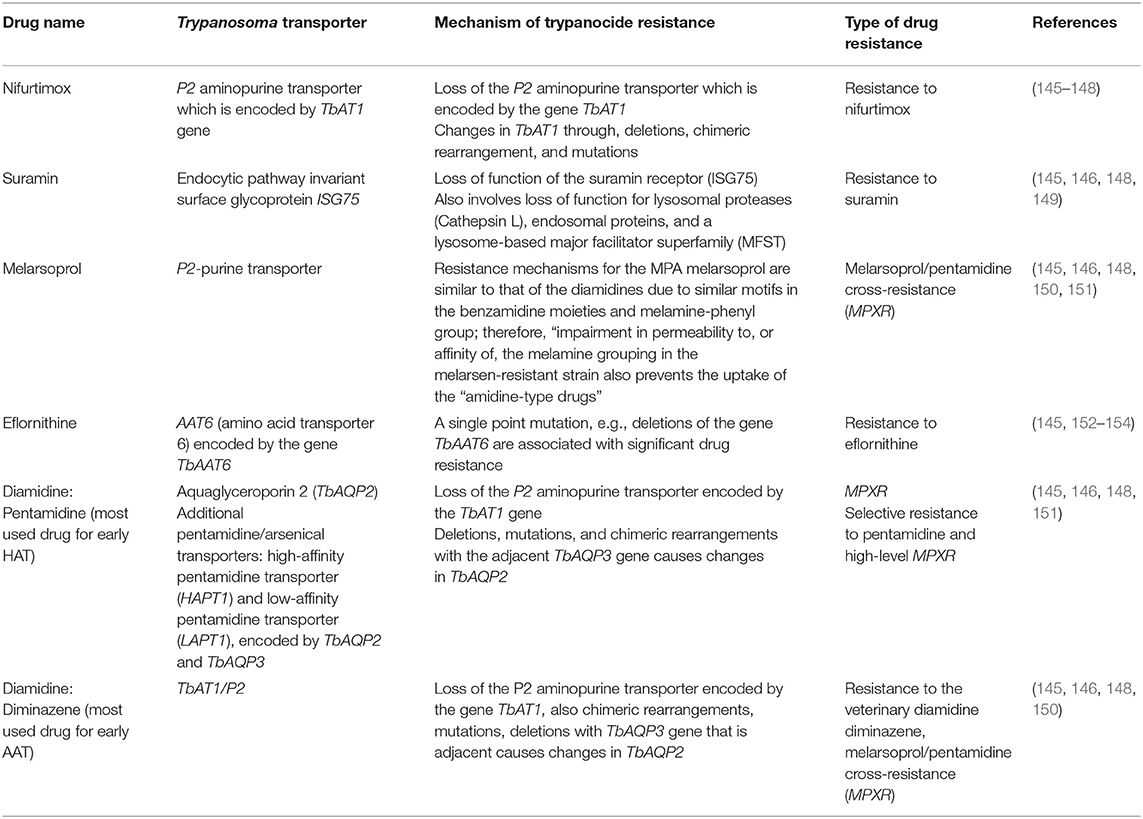
Table 4. Anti-trypanosomal drugs, transporters, and drug resistance.
Unfortunately, there has been a dearth of new trypanocides for decades, culminating in a condition where the limited number of drugs known to be accessible have diminishing efficacy as drug-resistant genes emerge (3). The drugs presently existing in communities have narrow therapeutic indices, yet a majority also cause local pain at the site of injection (especially with isometamidium). Remarkably, irrational usage of the trypanocides in the past has led to the development of drug resistant trypanosome species, and commencement of cross-resistance among almost all existing trypanocides that are chemically related, thus exacerbating the situation. Resistance has also been attributed to having therapeutic usage of most of the trypanocides rather than prophylactic application.
The choice between curative and prophylactic medications is influenced by a number of factors which include but is not limited to: the risk and exposure to the infection, accessibility of the drug, and logistics of delivery (155). Multiple-dose delivery regimens are generally not feasible in impoverished nations, and animal care facilities are often quite restricted. In management of trypanosomiasis in livestock, trypanocides prescribed as single dose required to cure and prevent the infection are usually favored. Unlike with HAT, where NECT is currently the standard first-line treatment for second-stage illness (81, 156), there is no other medication combination utilized in the treatment of AAT.
Henceforth, where possible, interchanging the use of available trypanocides with limited threat of cross-resistance, such as diminazene aceturate and isometamidium (known as a “sanative pair”), has been proposed, however, information on the therapeutics remains scarce to this date. This has subsequently led to medication errors reported in both West and East Africa, against T. congolense and T. vivax infections with isometamidium and diminazene. Moreover, cross-resistance profiles on diminazene aceturate and isometamidium had been thought to be an uncommon occurrence (3). These numerous resistant strains have been the consequence of distinct selection pressures created in the communities, justifying a need to revise policy to promote biomedical research to identify novel pathways which could widen the therapeutical options for the twenty-first century.
To preserve the efficacy of presently utilized drugs, it is critical to justify drug dose regimens based on whether the trypanosome species found in a specific region are phenotypically susceptible to trypanocide. Amid the continuing effort to develop new therapeutic and prophylactic trypanocidal drugs through Global Alliance for Livestock Veterinary Medicines (GALVmed), a public–private partnership with financial support from the Bill and Melinda Gates Foundation and the UK Department for International Development (http://www.galvmed.org/en/), no novel authorized drug has been developed for the last 50 years, necessitating the urgency to focus on innovative drug discoveries. Notwithstanding this need, a majority of the pharmaceutical industries have been discouraged to further invest drug discoveries due to the elevated expenses required in medicine development and the poor projected return on chemotherapy sales in developing countries (3).
Trypanosome survival within their mammalian and insect hosts has been associated with evolutionary changes involving restructuring of the VSG. Transitional changes during the trypanosome lifecycle implies that innate and acquired immune responses must be eluded through antigenic variation of the VSG which are present on the surface of the parasites. Currently, chemotherapy is the only available option against trypanosomes as efforts to develop a vaccine have been thwarted by the international community, however, the development of trypanocidal resistance has become a realistic threat especially for persons living in endemic regions (103, 145, 157, 158).
Trypanosoma transport proteins [on the surface of the parasite (see Table 4)] are responsible for pathogen survival; and trypanocidal resistance has been associated with changes in the pathogen transportome leading to dysfunction and a loss in therapeutical potential once drugs can no longer be absorbed by the parasite (152–154, 159). This justifies the need to promote further research for the discovery of novel chemotherapeutical options for the control of trypanosomiasis.
Drug resistance to diminazene aceturate is prevalent mostly in T. congolense infections, and it has been related to a mutation that alters the activity of a P2-type purine-transporter involved in drug absorption (1). Diminazene resistance was found in T. brucei subspecies brucei, evansi, and equiperdum when P2/TbAT1 expression was lost. Moreover, an additional gene called TeDR40 has been linked to resistance in T. b. evansi (160). TbAQP2 does not appear to be functional in T. congolense, however, TcoAT1, a potential P2/ TbAT1-type carrier protein with specific allele linked to diminazene aceturate resistance was discovered in T. congolense (161). Other chemically incomparable substances such as suramin and quinapyramine showed no cross-resistance. However, no new trypanocides have been developed despite the rising cases of trypanocide resistance.
Studies done on T. brucei show that homidium is assimilated into the kinetoplast and nucleus of trypanosomes. However, it is still unknown why trypanosomes establishes resistance to homidium, even though it is thought to be comparable with that of isometamidium, a compound related to it (136).
Isometamidium can be used in a sanative combination with diminazene, with the two medications being given in a certain order to reduce the chance of emerging resistance. Several African countries have demonstrated rising resistance to isometamidium especially in T. congolense, followed by T. brucei and T. vivax species and occasionally revealing cross-resistance with diminazene aceturate (162).
This is contrary to previous findings (1990s) when isometamidium was the most efficacious agent against AAT and no resistance was ever reported (163). After 15 years, extreme resistance to isometamidum chlodride in T. b. brucei without a functioning kinetoplast, as well as naturally occurring dyskinetoplastic T. b. evansi, has become rampant.
A study by Dean et al. (59) showed that the loss of the kinetoplast in trypanosome cells does not affect the mitochondrial membrane potential since a compensatory mutation in the F1F0-ATP synthase, a mutation in this ATP synthase subunit is adequate to generate a significant amount of resistance to isometamidium and homidium. The resistance mechanism in T. congolense has been associated with reductions in mitochondrial function. As a result, drug build-up in the mitochondrion is minimized, resulting in decreased absorption through the plasma membrane, due to the quick equilibration of drug quantities between the intracellular and extracellular composition of the cell as soon as the mitochondrial sink is removed. Furthermore, active expulsion of the drug via plasma membrane transporters is another mechanism of resistance (164), however, few studies have explored these mechanisms further.
Due to widespread resistance, the drug was discontinued from use in livestock until 1976 (165). It was then restored to use from around 1984 up to date in the management of T. b. evansi infections in horses and camels. The quinapyramine-resistant T. congolense have cross-resistance to isometamidium, homidium, and diminazene, and thus it is not indicated for usage in livestock (134). Even though the specific mechanism of quinapyramine resistance is uncertain, it is probable that most of these drugs have a mitochondrial target, and that any slight variation in the potential of the parasite's mitochondrial membrane, or when the inner mitochondrial membrane loses its organic cation carrier, multi-drug resistance might occur.
Although suramin is effective in the treatment of trypanosomiasis caused by T. simiae in pigs, it is unsuccessful in the management of T. vivax and T. congolense infections, owing to the metabolic physiological variations that differentiate T. brucei group species from other groups. By inhibiting suramin uptake, or its typical transit via the endocytic route after attaching to a particular receptor, it seems enough to make parasites resistant to the medication, although it is still uncertain exactly how suramin kills trypanosomes once aggregated intracellularly.
Reduced P2/TbAT1 activity is a recognized cause of cross-resistance in melarsomine dihydrochloride that penetrates the trypanosomes through this route. T. brucei strains resistant to melarsomine have a generally reduced susceptibility to diamidines and other arsenical medicines like melarsoprol, however, not to suramin (166, 167). T. congolense and T. vivax lack legitimate orthologs of TbAT1 and TbAQP2, which might suggest why the drug is less effective against such parasites (see Section Diminazene Aceturate Mode of Resistance).
The adenine-adenosine transporter, P2, is the source of resistance arising from point mutations within this transporter. The inactivation of this transporter is connected to trypanocide resistance (107). Furthermore, Koning (168) reported that over-expression of the gene encoding for TbMRPA, P-glycoprotein efflux pumps promote resistance to melamine-based arsenicals by causing the trypanothione adduct to develop within the cell to be excreted. Resistance is due to loss or point mutations associated with the P2 transporter (107). Furthermore, Barrett et al. (109) reported that for high levels of melaminophenyl arsenical resistance, both P2 and HAPT1 carrier proteins must be lost and of which the two resistance mechanisms were found to act independently but were rigorously complementary in studies.
Resistance to eflornithine is caused by the deletion or mutation of the TbAAT6 gene in many trypanosomes (108, 146). This gene is conserved throughout the Trypanosoma genome and encodes for the transmembrane amino acid transporter that delivers eflornithine into the cell, thus loss of the transporter protein results into eflornithine resistance. Resistance to eflornithine has potentiated the usage of melarsoprol which has later been associated with a 5% mortality rate among HAT recipients due to its toxic effects (109, 169).
The loss of a single copy of the genes encoding for nitroreductases enzymes (NTR) attributes to nifurtimox resistance (110, 111). Few studies have been conducted to explore resistance mechanism any further to date.
Pentamidine enters the mitochondria via the carrier proteins P2 and HAPT1 transporter proteins, thus loss of any of these carrier proteins results in resistance to pentamidine (109, 113).
The lack of a vaccine against HAT subspecies and antigenic variation of T. brucei makes treatment very challenging and expensive (170, 171). Limitations of monotherapies such as severe side effects and development of resistance imply that emphasis in chemotherapy is on combination therapy as observed with NECT which has continued to enjoy WHO approval (84, 170) (Table 5). Currently, new proposed candidates include nitroimidazole analog fexinidazole, i.e., 5-nitroimidazole, and its principal metabolites (fexinidazole sulfoxide and fexinidazole sulfone) have been characterized and shown to have potential for effective oral treatment against both stages of the T. b. gambiense and T. b. rhodesiense infections (93, 170, 172). In addition 1-aryl-4-nitro-1H-imidazoles (stage II HAT) have a genetic safety in mammalian cells (173–176), although information on clinical trials from endemic areas to revalidate this is scarce. Furthermore, another new drug on the horizon is Oxaborole SYX-7158, which is a benzoxaborole (stage II HAT) and has cleared the parasites in mice (170, 177).
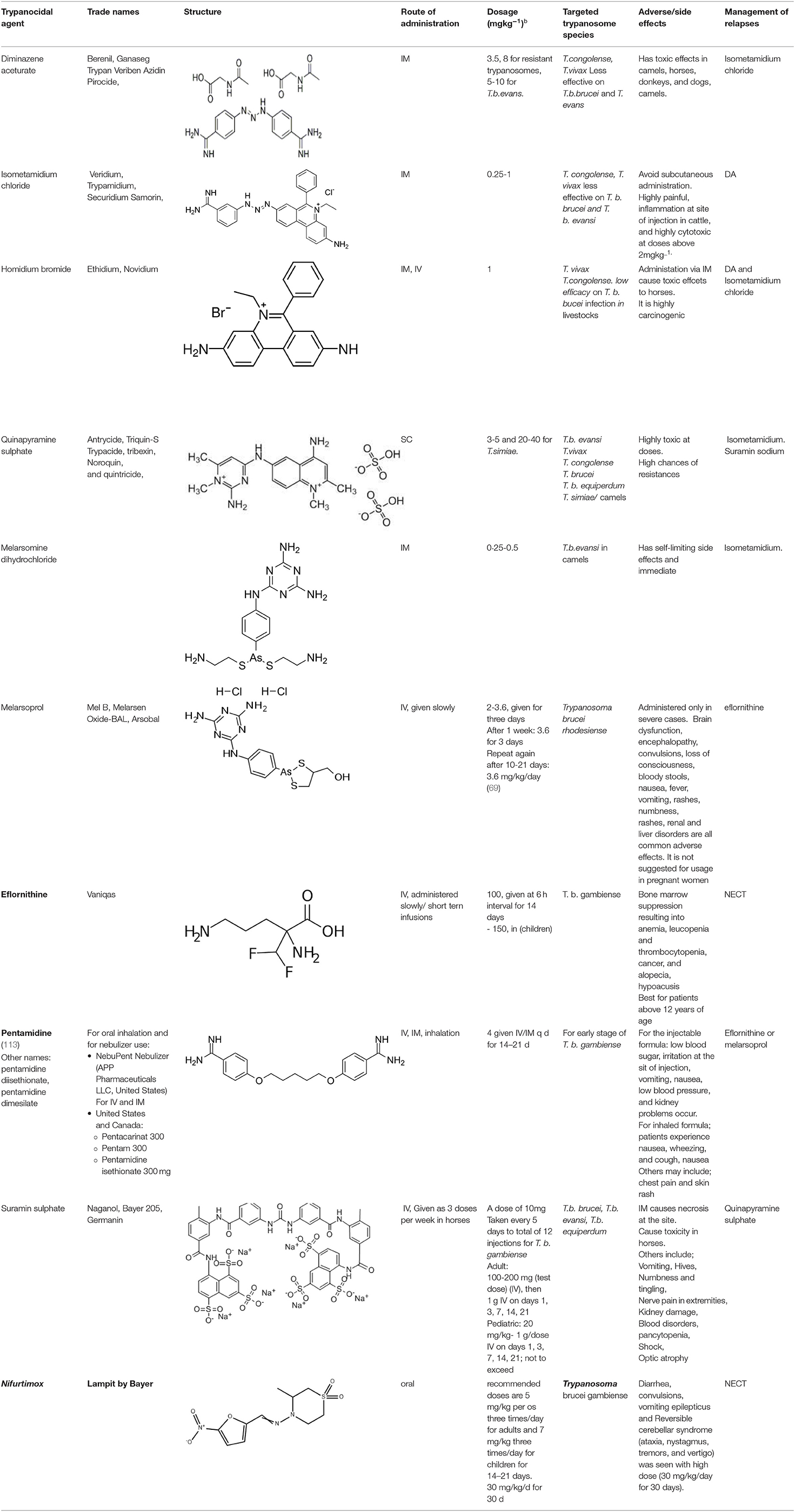
Table 5. Current alternative therapeutical options for management of trypanocidal resistance.
The Drugs for Neglected Diseases initiative (DNDi) has developed an efficacious mixture of two older drugs nifurtimox and eflornithine (81), as well as nitroimidazole fexinidazole (currently left with one clinical trial phase as an oral therapy for second phase HAT).
According to Steinmann et al. (178), a distinct benzoxaborole molecule similar to that used in humans is being developed and progressed for the treatment and prevention of AAT. Several similar agents are being added to the GALVmed portfolio as researchers look for therapies that meet the criteria outlined in a target product profile (TPP), which is applied to determine whatever attributes should trypanocidal agents have to provide a beneficial impact in AAT. The WHO's road map for NZD aimed to eliminate human African trypanosomiasis by 2020 and to cease transmission by 2030 (60).
African trypanosomiasis is a disease as old as colonial history in most African countries; however, political will both locally and internationally to control and eradicate the disease remains weak. In the previous two decades, great strides were made by global health partners, however, limited progressive research to identify and develop novel therapies has led to the development of trypanocide resistance against the limited chemotherapeutical options in the African market. The WHO has revived interest in the disease, and this has promoted the current ongoing clinical trials. However, reciprocal efforts from the Organization for Animal Health to engage global health partners continues to move at a snail's pace. The development of trypanocide resistance has been explored on the limited therapeutical options; however, a lot of opportunities arise for industry to develop drugs that could help control trypanosomiasis in livestock which are the reservoirs of HAT.
KK and SW conceptualized the study. KK, SW, and EM conducted the study design. KK collected the data. KK, EM, IN, and SW conducted data analysis. All authors reviewed, approved publication of the manuscript, and remain in agreement on all aspects of the work. All authors contributed to the article and approved the submitted version.
This research was supported by the National Institute for Health Research (NIHR) Global Health Research Programme (16/136/33) using UK aid from the UK government. This work was also supported by Zhejiang University Education Foundation Emergency Research Fund, Global Challenges Research Fund, and the University of Edinburgh (SW and KK). The work was also funded by the Commonwealth Scholarship Commission (grant ID number: UGCS-2021-447) in the UK (KK).
The views expressed in this publication are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chitanga S, Marcotty T, Namangala B, Van den Bossche P, Van Den Abbeele J, Delespaux V. High prevalence of drug resistance in animal trypanosomes without a history of drug exposure. PLoS Negl Trop Dis. (2011) 5:e1454. doi: 10.1371/journal.pntd.0001454
2. Kasozi KI, Zirintunda G, Ssempijja F, Buyinza B, Alzahrani KJ, Matama K, et al. Epidemiology of trypanosomiasis in wildlife—implications for humans at the wildlife interface in Africa. Front Vet Sci. (2021) 8:621699. doi: 10.3389/fvets.2021.621699
3. Giordani F, Morrison LJ, Rowan TIMG. The animal trypanosomiases and their chemotherapy : a review. Parasitology. (2016) 143:1862–89. doi: 10.1017/S0031182016001268
4. Prayag K, Surve DH, Paul AT, Kumar S, Jindal AB. Nanotechnological interventions for treatment of trypanosomiasis in humans and animals. Drug Deliv Transl Res. (2020) 10:945–61. doi: 10.1007/s13346-020-00764-x
5. Isaac C, Ohiolei JA, Ebhodaghe F, Igbinosa IB, Eze AA. Animal African trypanosomiasis in nigeria: a long way from elimination/eradication. Acta Tropica. (2017) 176:323–31. doi: 10.1016/j.actatropica.2017.08.032
6. Dagnachew S, Terefe G, Abebe G, Barry D, McCulloch R, Goddeeris B. In vivo experimental drug resistance study in Trypanosoma vivax isolates from tsetse infested and non-tsetse infested areas of Northwest Ethiopia. Acta Tropica. (2015) 146:95–100. doi: 10.1016/j.actatropica.2015.03.014
7. Tasew S, Duguma R. Cattle anaemia and trypanosomiasis in western Oromia State, Ethiopia. Revue Méd Vét. (2012) 163:581–8.
8. Okello WO, MacLeod ET, Muhanguzi D, Waiswa C, Shaw AP, Welburn SC. critical linkages between livestock production, livestock trade and potential spread of human african trypanosomiasis in uganda: bioeconomic herd modeling and livestock trade analysis. Front Vet Sci. (2021) 8:611141. doi: 10.3389/fvets.2021.611141
9. Kasozi KI, Namayanja M, Gaithuma AK, Mahero M, Matovu E, Yamagishi J, et al. Prevalence of hemoprotozoan parasites in small ruminants along a human-livestock-wildlife interface in western Uganda. Vet Parasitol Reg Stud Rep. (2019) 17:100309. doi: 10.1016/j.vprsr.2019.100309
10. World Health Organization. Trypanosomiasis, Human African (Sleeping Sickness). World Health Organization (2020). Available online at: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed October 10, 2021).
11. Franco JR, Cecchi G, Priotto G, Paone M, Diarra A, Grout L, et al. Monitoring the elimination of human African trypanosomiasis at continental and country level: update to 2018. PLoS Negl Trop Dis. (2020) 14:e0008261. doi: 10.1371/journal.pntd.0008261
12. Fyfe J, Picozzi K, Waiswa C, Bardosh KL, Welburn SC. Impact of mass chemotherapy in domestic livestock for control of zoonotic T. b. rhodesiense human African trypanosomiasis in Eastern Uganda. Acta Tropica. (2017) 165:216–29. doi: 10.1016/j.actatropica.2016.08.022
13. Geiger A, Ponton F, Simo G. Adult blood-feeding tsetse flies, trypanosomes, microbiota and the fluctuating environment in sub-Saharan Africa. ISME J. (2015) 9:1496–507. doi: 10.1038/ismej.2014.236
14. Baldacchino F, Muenworn V, Desquesnes M, Desoli F, Charoenviriyaphap T, Duvallet G. Transmission of pathogens by stomoxys flies (diptera, muscidae): a review. Parasite. (2013) 20:26. doi: 10.1051/parasite/2013026
15. Daniels JP, Gull K, Wickstead B. Cell biology of the trypanosome genome. Microbiol Mol Biol Rev. (2010) 74:552–69. doi: 10.1128/MMBR.00024-10
16. Jackson AP, Berry A, Aslett M, Allison HC, Burton P, Vavrova-Anderson J, et al. Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proc Natl Acad Sci USA. (2012) 109:3416–21. doi: 10.1073/pnas.1117313109
17. Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the african trypanosome Trypanosoma brucei. Science. (2005) 309:416–22. doi: 10.1126/science.1112642
18. Schwede A, Jones N, Engstler M, Carrington M. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol Biochem Parasitol. (2011) 175:201–4. doi: 10.1016/j.molbiopara.2010.11.004
19. Ersfeld K, Gull K. Partitioning of large and minichromosomes in Trypanosoma brucei. Science. (1997) 276:611–4. doi: 10.1126/science.276.5312.611
20. Mugnier MR, Cross GAM, Papavasiliou FN. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science. (2015) 347:1470–3. doi: 10.1126/science.aaa4502
21. Akiyoshi B, Gull K. Evolutionary cell biology of chromosome segregation: Insights from trypanosomes. Open Biol. (2013) 3:130023. doi: 10.1098/rsob.130023
22. Horn D. Molecular & biochemical parasitology antigenic variation in african trypanosomes. Mol Biochem Parasitol. (2014) 195:123–9. doi: 10.1016/j.molbiopara.2014.05.001
23. Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE. (2008) 3:e3527. doi: 10.1371/journal.pone.0003527
24. Jackson AP, Sanders M, Berry A, McQuillan J, Aslett MA, Quail MA, et al. The genome sequence of Trypanosoma brucei gambiense, causative agent of chronic human African trypanosomiasis. PLoS Negl Trop Dis. (2010) 4:e658. doi: 10.1371/journal.pntd.0000658
25. Obado SO, Bot C, Nilsson D, Andersson B, Kelly JM. Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi. Genome Biol. (2007) 8:R37. doi: 10.1186/gb-2007-8-3-r37
26. Obado SO, Bot C, Echeverry MC, Bayona JC, Alvarez VE, Taylor MC, et al. Centromere-associated topoisomerase activity in bloodstream form Trypanosoma brucei. Nucleic Acids Res. (2011) 39:1023–33. doi: 10.1093/nar/gkq839
27. Alsford NS, Navarro M, Jamnadass HR, Dunbar H, Ackroyd M, Murphy NB, et al. The identification of circular extrachromosomal DNA in the nuclear genome of Trypanosoma brucei. Mol Microbiol. (2003) 47:277–89. doi: 10.1046/j.1365-2958.2003.03266.x
28. Wickstead B. The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res. (2004) 14:1014–24. doi: 10.1101/gr.2227704
29. Kim HS, Cross GAM. Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS ONE. (2011) 6:e25313. doi: 10.1371/journal.pone.0025313
30. Klebanov-Akopyan O, Mishra A, Glousker G, Tzfati Y, Shlomai J. Trypanosoma bruceiUMSBP2 is a single-stranded telomeric DNA binding protein essential for chromosome end protection. Nucleic Acids Res. (2018) 46:7757–71. doi: 10.1093/nar/gky597
31. Hammarton TC. Cell cycle regulation in Trypanosoma brucei. Mol Biochem Parasitol. (2007) 153:1–8. doi: 10.1016/j.molbiopara.2007.01.017
32. Li Z. Regulation of the cell division cycle in Trypanosoma brucei. Eukaryotic Cell. (2012) 11:1180–90. doi: 10.1128/EC.00145-12
33. Benz C, Dondelinger F, McKean PG, Urbaniak MD. Cell cycle synchronisation of Trypanosoma brucei by centrifugal counter-flow elutriation reveals the timing of nuclear and kinetoplast DNA replication. Sci Rep. (2017) 7:17599. doi: 10.1038/s41598-017-17779-z
34. Osório ALAR, Rosa A, Madruga CR, Desquesnes M, Soares CO, Raquel L, et al. Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the new world - a review. Mem Inst Oswaldo Cruz. (2008) 103:1–13. doi: 10.1590/S.0074-02762008000100001
35. D'Archivio S, Cosson A, Medina M, Lang T, Minoprio P, Goyard S. Non-Invasive in vivo study of the Trypanosoma vivax infectious process consolidates the brain commitment in late infections. PLoS Negl Trop Dis. (2013) 7:e1976. doi: 10.1371/journal.pntd.0001976
36. Auty H, Torr SJ, Michoel T, Jayaraman S, Morrison LJ. Cattle trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev Sci Tech. (2015) 34:587–98. doi: 10.20506/rst.34.2.2382
37. Rodrigues AC, Ortiz PA, Costa-Martins AG, Neves L, Garcia HA, Alves JMP, et al. Congopain genes diverged to become specific to savannah, forest and kilifi subgroups of Trypanosoma congolense, and are valuable for diagnosis, genotyping and phylogenetic inferences. Infect Genet Evol. (2014) 23:20–31. doi: 10.1016/j.meegid.2014.01.012
38. Peacock L, Cook S, Ferris V, Bailey M, Gibson W. The life cycle of trypanosoma (nannomonas) congolense in the tsetse fly. Parasit Vectors. (2012) 5:109. doi: 10.1186/1756-3305-5-109
39. Radwanska M, Vereecke N, Deleeuw V, Pinto J, Magez S. Salivarian trypanosomosis: a review of parasites involved, their global distribution and their interaction with the innate and adaptive mammalian host immune system. Front Immunol. (2018) 9:2253. doi: 10.3389/fimmu.2018.02253
40. Sumba AL, Mihok S, Oyieke FA. Mechanical transmission of Trypanosoma evansi and T. congolense by Stomoxys niger and S. taeniatus in a laboratory mouse model. Med Vet Entomol. (1998) 12:417–22. doi: 10.1046/j.1365-2915.1998.00131.x
41. Hill GC. Electron transport systems in kinetoplastida. Biochim Biophys Acta. (1976) 456:149–93. doi: 10.1016/0304-4173(76)90011-2
42. Fargnoli L, Panozzo-zénere EA, Pagura L, Barisón MJ, Cricco JA, Silber AM, et al. Targeting L-proline uptake as new strategy for anti-chagas drug development. Front Chem. (2020) 8:696. doi: 10.3389/fchem.2020.00696
43. Alhadj M, Ibrahim M, Sophie J, Id W, Claudine S, Id HN, et al. Diversity of trypanosomes in humans and cattle in the HAT foci mandoul and maro, Southern Chad — a matter of concern for zoonotic potential? PLoS Negl Trop Dis. (2021) 15:e0009323. doi: 10.1371/journal.pntd.0009323
44. Baernstein HD. A review of electron transport mechanisms in parasitic protozoa. J Parasitol. (1963) 49:12. doi: 10.2307/3275663
45. Steketee PC, Dickie EA, Iremonger J, Crouch K, Paxton E, Jayaraman S, et al. Divergent metabolism between Trypanosoma congolense and Trypanosoma brucei results in differential sensitivity to metabolic inhibition. PLoS Pathog. (2021) 17:e1009734. doi: 10.1371/journal.ppat.1009734
46. Munday JC, Tagoe DNA, Eze AA, Krezdorn JAM, Rojas López KE, Alkhaldi AAM, et al. Functional analysis of drug resistance-associated mutations in the Trypanosoma brucei adenosine transporter 1 (TbAT1) and the proposal of a structural model for the protein. Mol Microbiol. (2015) 96:887–900. doi: 10.1111/mmi.12979
47. Hamill LC, Kaare MT, Welburn SC, Picozzi K. Domestic pigs as potential reservoirs of human and animal trypanosomiasis in Northern Tanzania. Parasit Vect. (2013) 6:322. doi: 10.1186/1756-3305-6-322
48. Maganga GD, Boundenga L, Ologui-Minkue-Edzo EJ, Bohou Kombila L, Ndong Mebaley TG, Kumulungui B, et al. Frequency and diversity of trypanosomes in sheep and goats from Mongo County in South Gabon, Central Africa. Vet World. (2020) 13:2502–7. doi: 10.14202/vetworld.2020.2502-2507
49. Echodu R, Sistrom M, Bateta R, Murilla G, Okedi L, Aksoy S, et al. Genetic diversity and population structure of Trypanosoma brucei in Uganda: implications for the epidemiology of sleeping sickness and nagana. PLOS Negl Trop Dis. (2015) 9:e0003353. doi: 10.1371/journal.pntd.0003353
50. Giroud C, Ottones F, Coustou V, Dacheux D, Biteau N, Miezan B, et al. Correction: murine models for Trypanosoma brucei gambiense disease progression—from silent to chronic infections and early brain tropism. PLOS Negl Trop Dis. (2016) 10:e0004645. doi: 10.1371/journal.pntd.0004645
51. Desquesnes M, Holzmuller P, Lai DH, Dargantes A, Lun ZR, Jittaplapong S. Trypanosoma evansi and surra: a review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, pathogenic effects. BioMed Res Int. (2013) 2013:194176. doi: 10.1155/2013/194176
52. Pumhom P, Morand S, Tran A, Jittapalapong S, Desquesnes M. Trypanosoma from rodents as potential source of infection in human-shaped landscapes of South-East Asia. Vet Parasitol. (2015) 208:174–80. doi: 10.1016/j.vetpar.2014.12.027
53. Van Vinh Chau N, Buu Chau L, Desquesnes M, Herder S, Phu Huong Lan N, Campbell JI, et al. A clinical and epidemiological investigation of the first reported human infection with the zoonotic parasite Trypanosoma evansi in Southeast Asia. Clin Infect Dis. (2016) 62:1002–8. doi: 10.1093/cid/ciw052
54. Truc P, Buscher P, Cuny G, Gonzatti MI, Jannin J, Joshi P, et al. Atypical human infections by animal trypanosomes. PLoS Negl Trop Dis. (2013) 7:e2256. doi: 10.1371/journal.pntd.0002256
55. Claes F, Büscher P, Touratier L, Goddeeris BM. Trypanosoma equiperdum: master of disguise or historical mistake? Trends Parasitol. (2005) 21:316–21. doi: 10.1016/j.pt.2005.05.010
56. Pascucci I, Di Provvido A, Cammà C, Di Francesco G, Calistri P, Tittarelli M, et al. Diagnosis of dourine in outbreaks in Italy. Vet Parasitol. (2013) 193:30–8. doi: 10.1016/j.vetpar.2012.12.006
57. Brun R, Kunz C. In vitro drug sensitivity test for Trypanosoma brucei subgroup bloodstream trypomastigotes. Acta Tropica. (1989) 46:361368.
58. Hagos A, Goddeeris BM, Yilkal K, Alemu T, Fikru R, Yacob HT, et al. Efficacy of cymelarsan and diminasan against Trypanosoma equiperdum infections in mice and horses. Vet Parasitol. (2010) 3–4:200–6. doi: 10.1016/j.vetpar.2010.03.041
59. Dean S, Gould MK, Dewar CE, Schnaufer AC. Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes. Proc Natl Acad Sci USA. (2013) 110:14741–6. doi: 10.1073/pnas.1305404110
60. CDC. Centers for Disease Control. African Trypanosomiasis. DPDx - Laboratory Identification of Parasites of Public Health Concern. (2020). Available online at: https://www.cdc.gov/dpdx/trypanosomiasisafrican/index.html (accessed September 10, 2021).
61. Silvester E, McWilliam KR, Matthews KR. The cytological events and molecular control of life cycle development of Trypanosoma brucei in the mammalian bloodstream. Pathogens. (2017) 6:29. doi: 10.3390/pathogens6030029
62. Simarro PP, Jannin JC. Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med. (2008) 5:e55. doi: 10.1371/journal.pmed.0050055
63. Pépin J, Méda HA. The epidemiology and control of human African trypanosomiasis. Adv Parasitol. (2001) 49:71–132. doi: 10.1016/S0065-308X(01)49038-5
64. Jelinek T, Bisoffi Z, Bonazzi L, van Thiel P, Bronner U, de Frey A, et al. Cluster of African trypanosomiasis in travelers to Tanzanian national parks. Emerg Infect Dis. (2002) 8:634–35. doi: 10.3201/eid0806.010432
65. Hall JP, Wang H, Barry JD. Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog. (2013) 9:e1003502. doi: 10.1371/journal.ppat.1003502
66. Sahin A, Asencio C, Izotte J, Pillay D, Coustou V, Karembe H, et al. The susceptibility of Trypanosoma congolense and Trypanosoma brucei to isometamidium chloride and its synthetic impurities. Vet Parasitol. (2014) 203:270–5. doi: 10.1016/j.vetpar.2014.04.002
67. Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet. (2017) 390:2397–409. doi: 10.1016/S0140-6736(17)31510-6
68. World Health Organization WHO Expert Committee on the Control Surveillance of Human African Trypanosomiasis. Control and Surveillance of Human African Trypanosomiasis: Report of a WHO Expert Committee. Geneva: World Health Organization (2013). Available online at: https://apps.who.int/iris/handle/10665/95732 (accessed October 10, 2021).
69. World Health Organization. WHO Interim Guidelines for the Treatment of Gambiense Human African Trypanosomiasis. Geneva: World Health Organization (2019). Available online at: https://apps.who.int/iris/handle/10665/326178 (accessed October 10, 2021).
70. Hafiz S, Kyriakopoulos C. Pentamidine. In: StatPearls. Treasure Island, F): StatPearls Publishing (2022). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK55 (accessed October 13, 2021)
71. WHO. Trypanosomiasis, Human African (Sleeping Sickness). World Health Organization (2021). Available online at: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness)
72. Docampo R, Moreno SNJ. Current chemotherapy of human African trypanosomiasis. Parasitol Res. (2003) 90:S10–3. doi: 10.1007/s00436-002-0752-y
73. Foulkes JR. Metronidazole and suramin combination in the treatment of arsenical refractory rhodesian sleeping sickness - a case study. Trans R Soc Trop Med Hyg. (1996) 90:422. doi: 10.1016/S0035-9203(96)90533-7
74. Hedley L, Fink D, Sparkes D, Chiodini PL. African sleeping sickness. Br J Hosp Med. (2016) 77:C157–60. doi: 10.12968/hmed.2016.77.10.C157
75. Kourbeli V, Chontzopoulou E, Moschovou K, Pavlos D, Mavromoustakos T, Papanastasiou IP. An overview on target-based drug design against kinetoplastid protozoan infections: human African trypanosomiasis, chagas disease and leishmaniases. Molecules. (2021) 26:4629. doi: 10.3390/molecules26154629
76. Nok AJ. Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human African trypanosomiasis. Parasitol Res. (2003) 90:71–9. doi: 10.1007/s00436-002-0799-9
77. Wiedemar N, Hauser DA, Mäser P. 100 years of suramin. Antimicrobiol Agents Chemother. (2020) 64:e01168–19. doi: 10.1128/AAC.01168-19
78. Schmid C, Nkunku S, Merolle A, Vounatsou P, Burri C. fficacy of 10-day melarsoprol schedule 2 years after treatment for late-stage gambiense sleeping sickness. Lancet. (2004) 364:789–90. doi: 10.1016/S0140-6736(04)16940-7
79. Schmid C, Richer M, Bilenge CM, Josenando T, Chappuis F, Manthelot CR, et al. Effectiveness of a 10-day melarsoprol schedule for the treatment of late-stage human African trypanosomiasis: confirmation from a multinational study (IMPAMEL II). J Infect Dis. (2005) 191:1922–31. doi: 10.1086/429929
80. Kuepfer I, Schmid C, Allan M, Edielu A, Haary EP, Kakembo A, et al. Safety and efficacy of the 10-day melarsoprol schedule for the treatment of second stage rhodesiense sleeping sickness. PLoS Negl Trop Dis. (2012) 6:e1695. doi: 10.1371/journal.pntd.0001695
81. Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma bruceigambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet. (2009) 374:56–64. doi: 10.1016/S0140-6736(09)61117-X
82. Balasegaram M. Melarsoprol versus eflornithine for treating late-stage Gambian trypanosomiasis in the Republic of Congo. Bull World Health Organ. (2006) 84:783–91. doi: 10.2471/BLT.06.031955
83. Blum A, Mudji J, Grize L, Burri C, Zellweger MJ, Blum J. Sleeping hearts: 12 years after a follow up study on cardiac findings due to sleeping sickness. One Health. (2020) 11:100182. doi: 10.1016/j.onehlt.2020.100182
84. Lutje V, Seixas J, Kennedy A. Chemotherapy for second-stage human african trypanosomiasis. Cochrane Database Syst Rev. (2021) 12:CD015374. doi: 10.1002/14651858.CD006201.pub3
85. Steverding D. Evaluation of trypanocidal activity of combinations of anti-sleeping sickness drugs with cysteine protease inhibitors. Exp Parasitol. (2015) 151–2:28–33. doi: 10.1016/j.exppara.2015.01.016
86. Steverding D, Rushworth SA. Front-line glioblastoma chemotherapeutic temozolomide is toxic to Trypanosoma brucei and potently enhances melarsoprol and eflornithine. Exp Parasitol. (2017) 178:45–50. doi: 10.1016/j.exppara.2017.05.006
87. Hidalgo J, Ortiz JF, Fabara SP, Eissa-Garcés A, Reddy D, Collins KD, et al. Efficacy and toxicity of fexinidazole and nifurtimox plus eflornithine in the treatment of African trypanosomiasis. Cureus. (2021) 13:e16881. doi: 10.7759/cureus.16881
88. Choudhury SD. Nano-Medicines a hope for chagas disease! Frontiers Mol Biosci. (2021) 8:655435. doi: 10.3389/fmolb.2021.655435
89. Kansiime F, Adibaku S, Wamboga C, Idi F, Kato CD, Yamuah L, et al. A multicentre, randomised, non-inferiority clinical trial comparing a nifurtimox-eflornithine combination to standard eflornithine monotherapy for late stage Trypanosoma brucei gambiense human African trypanosomiasis in Uganda. Parasit Vect. (2018) 11:105. doi: 10.1186/s13071-018-2634-x
90. Schmid C, Kuemmerle A, Blum J, Ghabri S, Kande V, Mutombo W, et al. In-Hospital safety in field conditions of nifurtimox eflornithine combination therapy (NECT) for T. b. gambiense sleeping sickness. PLoS Negl Trop Dis. (2012) 6:e1920. doi: 10.1371/journal.pntd.0001920
91. Yun O, Priotto G, Tong J, Flevaud L, Chappuis F. NECT is next: implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl Trop Dis. (2010) 4:e720. doi: 10.1371/journal.pntd.0000720
92. Kaiser M, Bray MA, Cal M, Bourdin Trunz B, Torreele E, Brun R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrobiol Agents Chemother. (2011) 55:5602–8. doi: 10.1128/AAC.00246-11
93. Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazué G, et al. Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis. (2010) 4:e923. doi: 10.1371/journal.pntd.0000923
94. Medscape. Fexinidazole Dosing, Indications, Interactions, Adverse Effects, and More. Medscape. (2021). Available online at: https://reference.medscape.com/drug/fexinidazole-4000234 (accessed October 9, 2021).
95. Mesu VKBK, Kalonji WM, Bardonneau C, Mordt OV, Blesson S, Simon F, et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet. (2018) 391:144–54. doi: 10.1016/S0140-6736(17)32758-7
96. CDC. Diseases: Neglected Tropical Diseases. Centers for Disease Control on Nov 20 (2020). Available online at: https://www.cdc.gov/globalhealth/ntd/diseases/index.html
97. WHO. Neglected Tropical Diseases: Sleeping Sickness (Human African Trypanosomiasis). Geneva: World Health Organization (2020). Available online at: https://www.who.int/news-room/questions-and-answers/item/neglected-topical-diseases-sleeping-sickness-(human-african-trypanosomiasis) (accessed October 9, 2021).
98. Zaidel EJ, Forsyth CJ, Novick G, Marcus R, Ribeiro ALP, Pinazo J, et al. COVID-19: implications for people with chagas disease. Global Heart. (2020) 15:69. doi: 10.5334/gh.891
99. Echeverria LE, Morillo CA. American trypanosomiasis (chagas disease). Infect Dis Clin N Am. (2019) 33:119–34. doi: 10.1016/j.idc.2018.10.015
100. Mills RM. Chagas disease: epidemiology and barriers to treatment. Am J Med. (2020) 133:1262–5. doi: 10.1016/j.amjmed.2020.05.022
101. Radisic MV, Repetto SA. American trypanosomiasis (Chagas disease) in solid organ transplantation. Transplant Infect Dis. (2020) 22:e13429. doi: 10.1111/tid.13429
102. Fairlamb AH. Fexinidazole for the treatment of human African trypanosomiasis. Drugs Today. (2019) 55:705. doi: 10.1358/dot.2019.55.11.3068795
103. Kande Betu Ku Mesu V, Mutombo Kalonji W, Bardonneau C, Valverde Mordt O, Ngolo Tete D, Blesson S, et al. Oral fexinidazole for stage 1 or early stage 2 African Trypanosoma brucei gambiense trypanosomiasis: a prospective, multicentre, open-label, cohort study. Lancet Glob Health. (2021) 9:e999–1008. doi: 10.1016/S2214-109X(21)00208-4
104. Vansterkenburg ELM, Coppens I, Wilting J, Bos OJM, Fischer MJE, Janssen LHM, et al. The uptake of the trypanocidal drug suramin in combination with low-density lipoproteins by Trypanosoma brucei and its possible mode of action. Acta Tropica. (1993) 54:237–50. doi: 10.1016/0001-706X(93)90096-T
105. Fairlamb AH, Bowman IB. Uptake of the trypanocidal drug suramin by bloodstream forms of Trypanosoma bruceia nd its effect on respiration and growth rate in vivo. Mol Biochem Parasitol. (1980) 1:315–33. doi: 10.1016/0166-6851(80)90050-X
106. Alsford S, Eckert S, Baker N, Glover L, Sanchez-Flores A, KaFai L, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. (2012) 482:232–6. doi: 10.1038/nature10771
107. Wetzel DM, Phillips MA. Chemotherapy of protozoal infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections. In: Brunton LL, editor. Goodman and Gilman's: The Pharmacological Basis of Therapeutics. 13ed. McGraw Hill (2017). Available online at: https://accessmedicine.mhmedical.com/content.aspx?bookid=2189§ionid=172484335
108. Vincent IM, Creek D, Watson DG, Kamleh MA, Woods DJ, Wong PE, et al. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. (2010) 6:E1001204. doi: 10.1371/journal.ppat.1001204
109. Barrett MP, Vincent IM, Burchmore RJS, Kazibwe AJN, Matovu E. Drug resistance in human African trypanosomiasis. Fut Microbiol. (2011) 6:1037–47. doi: 10.2217/fmb.11.88
110. Wyllie S, Foth BJ, Kelner A, Sokolova AY, Berriman M, Fairlamb AH. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J Antimicrobiol Chemother. (2016) 2015:625–34. doi: 10.1093/jac/dkv376
111. Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci USA. (2008) 105:5022–7. doi: 10.1073/pnas.0711014105
112. Watson JA, Strub-Wourgraft N, Tarral A, Ribeiro I, Tarning J, White NJ. Pharmacokinetic-Pharmacodynamic assessment of the hepatic and bone marrow toxicities of the new trypanoside fexinidazole. Antimicrobiol Agents Chemother. (2019) 63:e02515–e02518. doi: 10.1128/AAC.02515-18
113. Lemke TL, Williams D. Foye's Principles of Medicinal Chemistry. 7th edn. Philadelphia, PA: Lippincott Williams & Wilkins (2013).
114. Franco JR, Cecchi G, Priotto G, Paone M, Diarra A, Grout L, et al. Monitoring the elimination of human African trypanosomiasis: Update to 2016. PLoS Negl Trop Dis. (2018) 12:e0006890. doi: 10.1371/journal.pntd.0006890
115. Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of human African trypanosomiasis. Clin Epidemiol. (2014) 6:257–75. doi: 10.2147/CLEP.S39728
116. Bardosh KL, Scoones JC, Grace D, Kalema-Zikusoka G, Jones KE, De Balogh K, et al. Engaging research with policy and action: what are the challenges of responding to zoonotic disease in Africa? Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160172. doi: 10.1098/rstb.2016.0172
117. Mahamat MH, Peka M, Rayaisse JB, Rock KS, Toko MA, Darnas J., et al. Adding tsetse control to medical activities contributes to decreasing transmission of sleeping sickness in the mandoul focus (Chad). PLOS Negl Trop Dis. (2017) 11:e0005792. doi: 10.1371/journal.pntd.0005792
118. Welburn SC, Molyneux DH, Maudlin I. beyond tsetse - implications for research and control of human african trypanosomiasis epidemics. Trends Parasitol. (2016) 32:230–41. doi: 10.1016/j.pt.2015.11.008
119. Bengaly Z, Vitouley SH, Somda MB, Zongo A, Têko-Agbo A, Cecchi G, et al. Drug quality analysis of isometamidium chloride hydrochloride and diminazene diaceturate used for the treatment of African animal trypanosomosis in West Africa. BMC Vet Res. (2018) 14:361. doi: 10.1186/s12917-018-1633-7
120. Latif AA, Ntantiso L, Chantel DB. African animal trypanosomosis (nagana) in northern KwaZulu-Natal, South Africa: strategic treatment of cattle on a farm in endemic area. Onderstepoort J Vet Res. (2019) 86:e1–6. doi: 10.4102/ojvr.v86i1.1639
121. Thomas JA, Baker N, Hutchinson S, Dominicus C, Trenaman A, Glover L, et al. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLOS Negl Trop Dis. (2018) 12:e0006980. doi: 10.1371/journal.pntd.0006980
122. Davkharbayar B, Davaasuren B, Narantsatsral S, Battur B, Punsantsogvoo M, Battsetseg B, et al. Treatment efficiency of combination therapy with diminazene aceturate and quinapyramine sulfate in a horse with dourine. J Equine Vet Sci. (2020) 87:102905. doi: 10.1016/j.jevs.2019.102905
123. Echeverria JT, Soares RL, Crepaldi BA, Oliveira GG, de Silva PMP, et al. Clinical and therapeutic aspects of an outbreak of canine trypanosomiasis. Rev Bras Parasitol Vet. (2019) 28:320–4. doi: 10.1590/s1984-29612019018
124. Prayag KS, Paul AT, Ghorui SK, Jindal AB. Preparation and evaluation of quinapyramine sulphate-docusate sodium ionic complex loaded lipidic nanoparticles and its scale up using geometric similarity principle. J Pharm Sci. (2021) 110:2241–9. doi: 10.1016/j.xphs.2021.01.033
125. Cossic BGA, Adjahoutonon B, Gloaguen P, Dibanganga GL, Maganga G, Leroy P, et al. Trypanosomiasis challenge estimation using the diminazene aceturate (Berenil) index in Zebu in Gabon. Trop Anim Health Prod. (2017) 49:619–24. doi: 10.1007/s11250-017-1239-2
126. Fetene E, Leta S, Regassa F, Buscher P. Global distribution, host range and prevalence of Trypanosoma vivax: a systematic review and meta-analysis. Parasit Vect. (2021) 14:80. doi: 10.1186/s13071-021-04584-x
127. Mbwambo HA, Mella PNP, Lekaki KA. Berenil (diminazene aceturate)-resistant Trypanosoma congolense in cattle under natural tsetse challenge at Kibaha, Tanzania. Acta Tropica. (1988) 45:239–44.
128. Peregrine AS, Mamman M. Pharmacology of diminazene: a review. Acta Tropica. (1993) 54:185–203. doi: 10.1016/0001-706X(93)90092-P
129. ArYung N, TaeWon K, Qiang L, Rebhun RB, HwaYoung Y, KyoungWon S. Melarsomine suppresses canine osteosarcoma cell survival via inhibition of Hedgehog-GLI signaling. J Vet Med Sci. (2019) 81:1722–9. doi: 10.1292/jvms.19-0043
130. Hébert L, Guitton E, Madeline A, Géraud T, Zientara S, Laugier C, et al. Melarsomine hydrochloride (Cymelarsan®) fails to cure horses with Trypanosoma equiperdum OVI parasites in their cerebrospinal fluid. Vet Parasitol. (2018) 264:47–51. doi: 10.1016/j.vetpar.2018.11.005
131. Raftery AG, Jallow S, Rodgers J, Sutton DGM. Safety and efficacy of three trypanocides in confirmed field cases of trypanosomiasis in working equines in the Gambia: a prospective, randomised, non-inferiority trial. PLOS Negl Trop Dis. (2019) 13:e0007175. doi: 10.1371/journal.pntd.0007175
132. Savadelis MD, Day KM, Bradner JL, Wolstenholme AJ, Dzimianski MT, Moorhead AR. Efficacy and side effects of doxycycline versus minocycline in the three-dose melarsomine canine adulticidal heartworm treatment protocol. Parasit Vect. (2018) 11:671. doi: 10.1186/s13071-018-3264-z
133. Wainwright M. Dyes, trypanosomiasis and DNA: a historical and critical review. Biotech Histochem. (2010) 85:341–54. doi: 10.3109/10520290903297528
134. Peregrine AS, Gray MA, Moloo SK. Cross-resistance associated with development of resistance to isometamidium in a clone of Trypanosoma congolense. Antimicrobiol Agents Chemother. (1997) 41:1604–6. doi: 10.1128/AAC.41.7.1604
135. Shapiro TA, Englund PT. Selective cleavage of kinetoplast DNA minicircles promoted by antitrypanosomal drugs. Proc Natl Acad Sci USA. (1990) 87:950954.
136. Chowdhury RA, Bakshi R, Wang J, Yildirir G, Liu B, Pappas-Brown V, et al. The killing of African trypanosomes by ethidium bromide. PLoS Pathog. (2010) 6:e1001226. doi: 10.1371/journal.ppat.1001226
137. Stevenson P, Sones KR, Gicheru MM, Mwangi EK. Comparison of isometamidium chloride and homidium bromide as prophylactic drugs for trypanosomiasis in cattle at Nguruman, Kenya. Acta Tropica. (1995) 59:77–84. doi: 10.1016/0001-706X(94)00080-K
138. Dougherty G, Waring MJ. The interaction between prothidium dibromide and DNA at the molecular level. Biophys Chem. (1982) 15:27–40. doi: 10.1016/0301-4622(82)87014-2
139. Curd FH, Davey DG. Antrycide, a new trypanocidal drug. Br J Pharmacol Chemother. (1950) 5:25–3. doi: 10.1111/j.1476-5381.1950.tb00573.x
140. Gould MK, Schnaufer A. Independence from Kinetoplast DNA maintenance and expression is associated with multidrug resistance in Trypanosoma brucei in vitro. Antimicrobiol Agents Chemother. (2014) 58:2925–8. doi: 10.1128/AAC.00122-14
141. Kuriakose S, Muleme HM, Onyilagha C, Singh R, Ping J, Uzonna JE. Diminazene aceturate (Berenil) modulates the host cellular and inflammatory responses to Trypanosoma congolense infection. PLoS ONE. (2012) 7:e48696. doi: 10.1371/journal.pone.0048696
142. Koning HP, de Anderson LF, Stewart M, Burchmore RJS, Wallace LJM, Barrett MP. The trypanocide diminazene aceturate is accumulated predominantly through the TbAT1 purine transporter: additional insights on diamidine resistance in African trypanosomes. Antimicrobiaol Agents Chemother. (2004) 48:1515–9. doi: 10.1128/AAC.48.5.1515-1519.2004
143. Munday JC, Eze AA, Baker N, Glover L, Clucas C, Aguinaga Andrés D, et al. Trypanosoma bruceiaquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J Antimicrobiol Chemother. (2014) 69:651–63. doi: 10.1093/jac/dkt442
144. Delespaux V, Dinka H, Masumu J, Bossche P, van den, Geerts S. Five-fold increase in Trypanosoma congolense isolates resistant to diminazene aceturate over a seven-year period in Eastern Zambia. Drug Resist Updates. (2008) 11:205–9. doi: 10.1016/j.drup.2008.10.002
145. De Koning PH. The drugs of sleeping sickness: their mechanisms of action and resistance, and a brief history. Trop Med Infect Dis. (2020) 5:14. doi: 10.3390/tropicalmed5010014
146. Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol. (2007) 152:1155–71. doi: 10.1038/sj.bjp.0707354
147. Hulpia F, Campagnaro GD, Alzahrani KJ, Alfayez IA, Ungogo MA, Mabille D, et al. Structure–Activity relationship exploration of 3′-deoxy-7-deazapurine nucleoside analogues as anti-Trypanosoma brucei agents. ACS Infect Dis. (2020) 6:2045–56. doi: 10.1021/acsinfecdis.0c00105
148. Munday JC, Settimo L, de Koning HP. Transport proteins determine drug sensitivity and resistance in a protozoan parasite, Trypanosoma brucei. Front Pharmacol. (2015) 6:32. doi: 10.3389/fphar.2015.00032
149. Zoltner M, Leung KF, Alsford S, Horn D, Field MC. Modulation of the surface proteome through multiple ubiquitylation pathways in african trypanosomes. PLOS Pathog. (2015) 11:e1005236. doi: 10.1371/journal.ppat.1005236
150. Munday JC, Rojas López KE, Eze AA, Delespaux V, Van Den Abbeele J, Rowan T, et al. Functional expression of TcoAT1 reveals it to be a P1-type nucleoside transporter with no capacity for diminazene uptake. Int J Parasitol Drugs Drug Resist. (2013) 3:69–76. doi: 10.1016/j.ijpddr.2013.01.004
151. Quintana JF, Bueren-Calabuig J, Zuccotto F, de Koning HP, Horn D, Field MC. Instability of aquaglyceroporin (AQP) 2 contributes to drug resistance in Trypanosoma brucei. PLOS Negl Trop Dis. (2020) 14:e0008458. doi: 10.1371/journal.pntd.0008458
152. Haindrich AC, Ernst V, Naguleswaran A, Oliveres QF, Roditi I, Rentsch D. Nutrient availability regulates proline/alanine transporters in Trypanosoma brucei. J Biol Chem. (2021) 296:100566. doi: 10.1016/j.jbc.2021.100566
153. Landfear SM. Protean permeases: diverse roles for membrane transport proteins in kinetoplastid protozoa. Mol Biochem Parasitol. (2019) 227:39–46. doi: 10.1016/j.molbiopara.2018.12.006
154. Mathieu C, Salgado AG, Wirdnam C, Meier S, Grotemeyer MS, Inbar E, et al. Trypanosoma bruceieflornithine transporter AAT6 is a low-affinity low-selective transporter for neutral amino acids. Biochem J. (2014) 463:9–18. doi: 10.1042/BJ20140719
155. Gu Y, Gettinby G, McKendrick I, Murray M, Peregrine AS, Revie C. Development of a decision support system for trypanocidal drug control of bovine trypanosomosis in Africa. Vet Parasitol. (1999) 87:9–23. doi: 10.1016/S0304-4017(99)00156-9
156. Alirol E, Schrumpf D, Amici Heradi J, Riedel A, de Patoul C, Quere M, et al. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: médecins sans frontières experience in the democratic republic of the Congo. Clin Infect Dis. (2013) 56:195–203. doi: 10.1093/cid/cis886
157. Kannigadu C, N'Da DD. Recent advances in the synthesis and development of nitroaromatics as anti-infective drugs. Curr Pharmaceut Des. (2020) 26:4658–74. doi: 10.2174/1381612826666200331091853
158. Welburn SC, Maudlin I. Priorities for the elimination of sleeping sickness. Adv Parasitol. (2012) 79:299–337. doi: 10.1016/B978-0-12-398457-9.00004-4
159. Scott M. Drugs and transporters in kinetoplastid protozoa. Adv Exp Med Biol. (2008) 625:22–32. doi: 10.1007/978-0-387-77570-8_3
160. Witola WH, Tsuda A, Inoue N, Ohashi K, Onuma M. Acquired resistance to berenil in a cloned isolate of Trypanosoma evansi is associated with upregulation of a novel gene, TeDR40. Parasitology. (2005) 131:635–46. doi: 10.1017/S003118200500836X
161. Delespaux V, Chitanga S, Geysen D, Goethals A, Bossche P, van den, et al. SSCP analysis of the P2 purine transporter TcoAT1 gene of Trypanosoma congolense leads to a simple PCR-RFLP test allowing the rapid identification of diminazene resistant stocks. Acta Tropica. (2006) 100:96–102. doi: 10.1016/j.actatropica.2006.10.001
162. Mamoudou A, Delespaux V, Chepnda V, Hachimou Z, Andrikaye JP, Zoli A, et al. Assessment of the occurrence of trypanocidal drug resistance in trypanosomes of naturally infected cattle in the Adamaoua region of Cameroon using the standard mouse test and molecular tools. Acta Tropica. (2008) 106:11518. doi: 10.1016/j.actatropica.2008.02.003
163. Joshua RA, Obwolo MJ, Bwangamoi O, Mandebvu E. Resistance to diminazine aceturate by Trypanosoma congolense from cattle in the Zambezi Valley of Zimbabwe. Vet Parasitol. (1995) 60:1–6. doi: 10.1016/0304-4017(94)00780-G
164. Wilkes JM, Mulugeta W, Wells C, Peregrine AS. Modulation of mitochondrial electrical potential: a candidate mechanism for drug resistance in African trypanosomes. Biochem J. (1997) 326:755–61. doi: 10.1042/bj3260755
165. Connor RJ. The diagnosis, treatment and prevention of animal trypanosomiasis under field conditions. In: Programme for the Control of African Animal Trypanosomiasis and Related Development: Ecological and Technical Aspects. FAO Animal Production and Health Paper No. 100. Rome: Food and Agriculture Organisation of the United Nations (1992).
166. Zhang ZQ, Giroud C, Baltz T. In vivo and in vitro sensitivity of Trypanosoma evansi and T. equiperdum to diminazene, suramin, MelCy, quinapyramine and isometamidium. Acta Tropica. (1991) 50:101110.
167. Pospichal H, Brun R, Kaminsky R, Jenni L. Induction of resistance to melarsenoxide cysteamine (Mel Cy) in Trypanosoma brucei brucei. Acta Tropica. (1994) 58:187–97. doi: 10.1016/0001-706X(94)90013-2
168. Koning De HP. Ever-increasing complexities of diamidine and arsenical crossresistance in African trypanosomes. Trends Parasitol. (2018) 24:345–9. doi: 10.1016/j.pt.2008.04.006
169. Sayé M, Miranda MR, di Girolamo F, de los Milagros Cámara M, Pereira CA. Proline modulates the Trypanosoma cruzi resistance to reactive oxygen species and drugs through a novel D. L-proline transporter. PLoS ONE. (2014) 9:e92028. doi: 10.1371/journal.pone.0092028
170. Babokhov P, Sanyaolu AO, Oyibo WA, Fagbenro-Beyioku AF, Iriemenam NC. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog Glob Health. (2013) 107:242–52. doi: 10.1179/2047773213Y.0000000105
171. Baker CH, Welburn SC. The long wait for a new drug for human african trypanosomiasis. Trends Parasitol. (2018) 34:818–27. doi: 10.1016/j.pt.2018.08.006
172. Tweats D, Trunz BB, Torreele E. Genotoxicity profile of fexinidazole - a drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis. (2012) 27:523–32. doi: 10.1093/mutage/ges015
173. Hu L, Wu X, Han J, Chen L, Vass SO, Browne P, et al. Synthesis and structure-activity relationships of nitrobenzyl phosphoramide mustards as nitroreductase-activated prodrugs. Bioorgan Med Chem Lett. (2011) 21:3986–91. doi: 10.1016/j.bmcl.2011.05.009
174. Nesslany F, Brugier S, Mouriès MA, Le Curieux F, Marzin D. In vitro and in vivo chromosomal aberrations induced by megazol. Mutat Res. (2004) 560:147–58. doi: 10.1016/j.mrgentox.2004.02.013
175. Peacock L, Bailey M, Carrington M, Gibson W. Meiosis and haploid gametes in the pathogen Trypanosoma brucei. Curr Biol. (2014) 24:181–6. doi: 10.1016/j.cub.2013.11.044
176. Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homblé F, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. (2013) 501:460–4. doi: 10.1038/nature12516
177. Mäser P, Wittlin S, Rottmann M, Wenzler T, Kaiser M, Brun R. Antiparasitic agents: new drugs on the horizon. Curr Opin Pharmacol. (2012) 12:562–6. doi: 10.1016/j.coph.2012.05.001
Keywords: trypanocides, trypanosoma, trypanosomiasis, drug resistance, HAT, AAT
Citation: Kasozi KI, MacLeod ET, Ntulume I and Welburn SC (2022) An Update on African Trypanocide Pharmaceutics and Resistance. Front. Vet. Sci. 9:828111. doi: 10.3389/fvets.2022.828111
Received: 03 December 2021; Accepted: 12 January 2022;
Published: 07 March 2022.
Edited by:
María-Aránzazu Martínez, Complutense University of Madrid, SpainReviewed by:
Daniel A. Abugri, Alabama State University, United StatesCopyright © 2022 Kasozi, MacLeod, Ntulume and Welburn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keneth Iceland Kasozi, a2VuZXRoLmthc296aUBlZC5hYy51aw==; a2ljZWxhbmR5QGthYi5hYy51Zw==; Susan Christina Welburn, c3VlLndlbGJ1cm5AZWQuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.