
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 12 May 2022
Sec. Animal Nutrition and Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.824785
This article is part of the Research Topic Regulation of Mitochondrial Function on Animal Diseases View all 13 articles
 Xing Gao1†Dongjing Wang2,3†Zhao Zhang1Chuxian Quan1Shimeng Zhou1Kewei Li1Yan Li1Suonan Zhao4Xiangying Kong4
Xing Gao1†Dongjing Wang2,3†Zhao Zhang1Chuxian Quan1Shimeng Zhou1Kewei Li1Yan Li1Suonan Zhao4Xiangying Kong4 Muhammad Fakhar-e-Alam Kulyar1Jiangyong Zeng2,3*
Muhammad Fakhar-e-Alam Kulyar1Jiangyong Zeng2,3* Jiakui Li1,5*
Jiakui Li1,5*The present study determined the complete mitochondrial DNA (mt DNA) sequence of Fasciola intermediate (isolated from yaks) based on gene content and genome organization. According to our findings, the genome of Fasciola intermediate was 13,960 bp in length, containing 2 ribosomal RNA (rRNA) genes, 12 protein-coding genes (PCGs), and 22 transfer RNA (tRNA) genes. The A+T content of genomes was 63.19%, with A (15.17%), C (9.31%), G (27.51%), and T as the nucleotide composition (48.02%). Meanwhile, the results showed negative AT-skew (-0.52) and positive GC-skew (0.494). The AT bias significantly affected both the codon usage pattern and amino acid composition of proteins. There were 2715 codons in all 12 protein-coding genes, excluding termination codons. Leu (16.72%) was the most often used amino acid, followed by Val (12.74%), Phe (10.90%), Ser (10.09%), and Gly (8.39%). A phylogenetic tree was built using Maximum-Likelihood (ML) through MEGA 11.0 software. The entire mt DNA sequence of Fasciola intermediate gave more genetic markers for investigating Trematoda population genetics, systematics, and phylogeography. Hence, for the first time, our study confirmed that yaks on the Qinghai-Tibet plateau have the infestation of Fasciola intermediate parasite.
Yak is a unique bovine specie on the Qinghai-Tibet plateau, China. The Qinghai-Tibet plateau is home to around 14 million yaks (a small amount of distribution in India, Bhutan, Sikkim, Afghanistan and Pakistan) (1). The yaks are necessary for herders in this area because of their milk, wool, and meat (2). Fasciolosis is one of the most important parasitic zoonotic issue, mainly caused by Fasciola hepatica (F. hepatica), Fasciola gigantica (F. gigantica) (3, 4). The infection occurs mostly through the oral route (5), causing emaciation, fever, hepatomegaly, cholangitis, and jaundice, and even death (5). Approximately 2.4 million humans and more than 600 million animals are infected per year, causing serious public health threats and considerable economic loss to the livestock industry (6, 7).
The sequence of mitochondrial DNA (mt DNA) is an extrachromosomal genome, commonly used as an informative genetic marker for various evolutionary studies among species due to the maternal inheritance. It includes molecular evolution, comparative population genetics, phylogenetics, and evolutionary genomics (8, 9). In most parasites, mitogenomes are ~13 kilobases (Kb) in size as closed circular molecules (10). The mt DNA contains 36 genes, including 12 protein coding genes (PCGs) (subunits 6 of the ATPase (atp6), cytochrome B (cob), cytochrome c oxidase subunits 1–3 (cox1–cox3), NADH dehydrogenase subunits 1–6 and 4 L (nad1–6 and nad4L), two ribosomal RNA genes encoding the small and large subunit rRNAs (rrnL and rrnS), 22 transfer RNA (tRNA) genes and a control region (CR) of variable length, known as the A+T-rich region (11, 12).
Previous research has identified the species of Fasciola in buffaloes and sheep, but not in yaks of the Qinghai-Tibet plateau. Therefore, in this study, the complete mitogenome of F. intermediate from yaks was initially sequenced and compared with other Trematoda mitogenomes (4). The available complete mitogenomes were used to provide insight into the phylogenetic relationship of F. intermediate. These findings would be valuable in order to have a better understanding of the F. intermediate mitogenome and the evolutionary relationships within Trematoda. The characteristics of F. intermediate mitogenome might be used to build appropriate genetic markers to detect Fasciola in yaks. This study might provide a basis for the accurate prevention, diagnosis, and treatment of Fasciolosis in yaks for studying the population genetic structure of F. intermediate in China.
The F. intermediate samples used in this study were collected from the chopped liver of yaks (Xiahua slaughter house in Tibetan Autonomous Prefecture of Haibe, Qinghai Province, China). The samples were fixed in 75% alcohol and stored at −20°C until used for DNA extraction. Total genomic DNA was isolated by using TIANcombi DNA Lyse&Det PCR Kit [TIANGEN biotech (Beijing) Co., Ltd.].
DNA samples were randomly interrupted, the required length of DNA fragments were collected, the base “A” to 3′-end was added, DNA fragments and 3′-end were connected with “T” base special joint, and finally used for cluster preparation and sequencing.
The mitogenome of F. intermediate was sequenced by next-generation sequencing (NGS). Two lanes for F. intermediate were sequenced as 400 bp reads using Illumina MiSeq (1 GB raw). The raw data was saved by Paired-End FASTQ and generated high-quality sequences by using AdapterRemoval (version 2) and SOAPec (version 2.01) based on K-mer distribution (13).
A5-miseqv20150522 and SPAdesv3.9.0 were used to assemble high-quality second-generation sequencing data to construct contig and scaffold sequences (14, 15). Mitochondrial sequences of each splicing result were selected by Blastn (BLAST v2.2.31+), compared between the sequences with high sequencing depth and the NT library on NCBI. By mummerv3.1 software. The results of mitochondrial splicing were used for collinearity analysis. The location relationship was determined, and then gap between contig was filled (16).
The complete mitochondrial genome sequences of splicing were uploaded to MITOS web server (http://mitos.bioinf.uni-leipzig.de/) for functional annotation (17). The Genetic Code was set to 05-invertebrate; the rest was set as the default parameter by MITOS. The secondary structure of each predicted tRNA can be obtained in the MITOS web server. The complete genome circle map of mitochondria was drawn using CGview visualization software (18).
The taxonomic status of F. intermediate with available Trematoda was estimated by reconstructing phylogenetic trees. The complete nucleotide sequences of 36 Trematoda and one outgroup are available at GenBank (https://www.ncbi.nlm.nih.gov/genbank/).
Nucleotide sequences of each gene and their deduced amino acid sequences were aligned separately by using https://www.novoprolabs.com/ and MEGA 11.0 (19). The phylogenetic tree was built using Maximum-Likelihood (ML) through MEGA 11.0 software. The genome information used in this study is shown in Table 1.
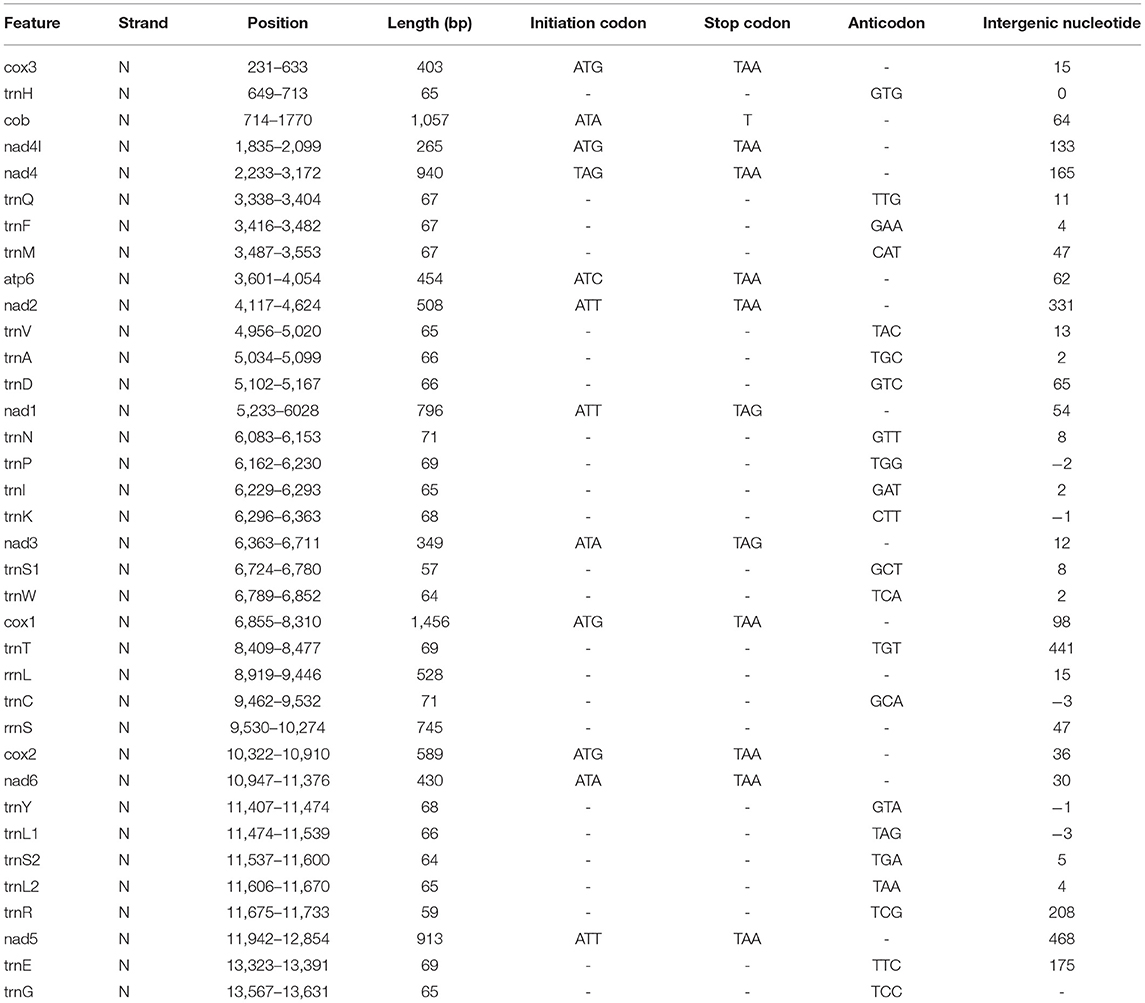
Table 1. Position and nucleotide sequence lengths of mitochondrial genomes of F. intermediate, and start and stop codons for protein-coding genes as well as their tRNA gene anticodons.
In our study, the mitogenome of F. intermediate was a closed circular molecule of 13,960 bp in length (Figure 1). The mitogenome contained 36 typical mitochondrial genes [12 PCGs (cox1-3, nad1-6, nad4L, cob and atp6), 22 tRNAs, 2 rRNAs (rrnS and rrnL)] (Table 2). The mitogenome of F. intermediate had been submitted to the NCBI GenBank under the accession number MH621335.1. The nucleotide compositions of the mitogenome of F. intermediate were as follows: A = 15.17%; T = 48.02%; G = 27.51%; and C = 9.31%. The whole mitogenome of F. intermediate was biased toward AT nucleotides (63.19%) (Table 3). All genes were encoded on the minority (N) strand (Table 2). There was a 10 bp overlap between genes in five locations: trnP/trnI, trnK/nad3, trnC/rrnS, trnY/trnL1, and trnL1/trnS2.
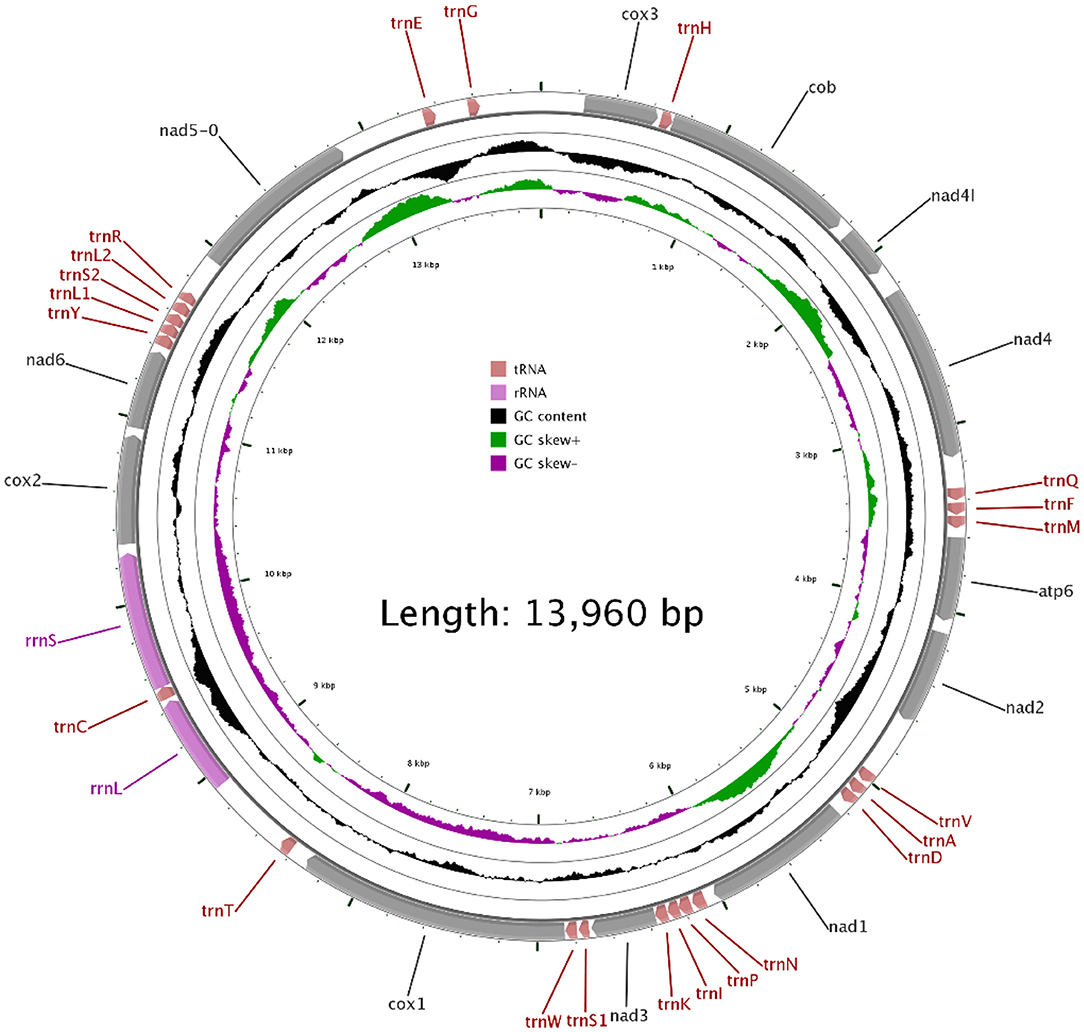
Figure 1. The mitochondrial genome of F. intermediate from yak.
Table 2. Mitochondrial genome sequences of F. intermediate.
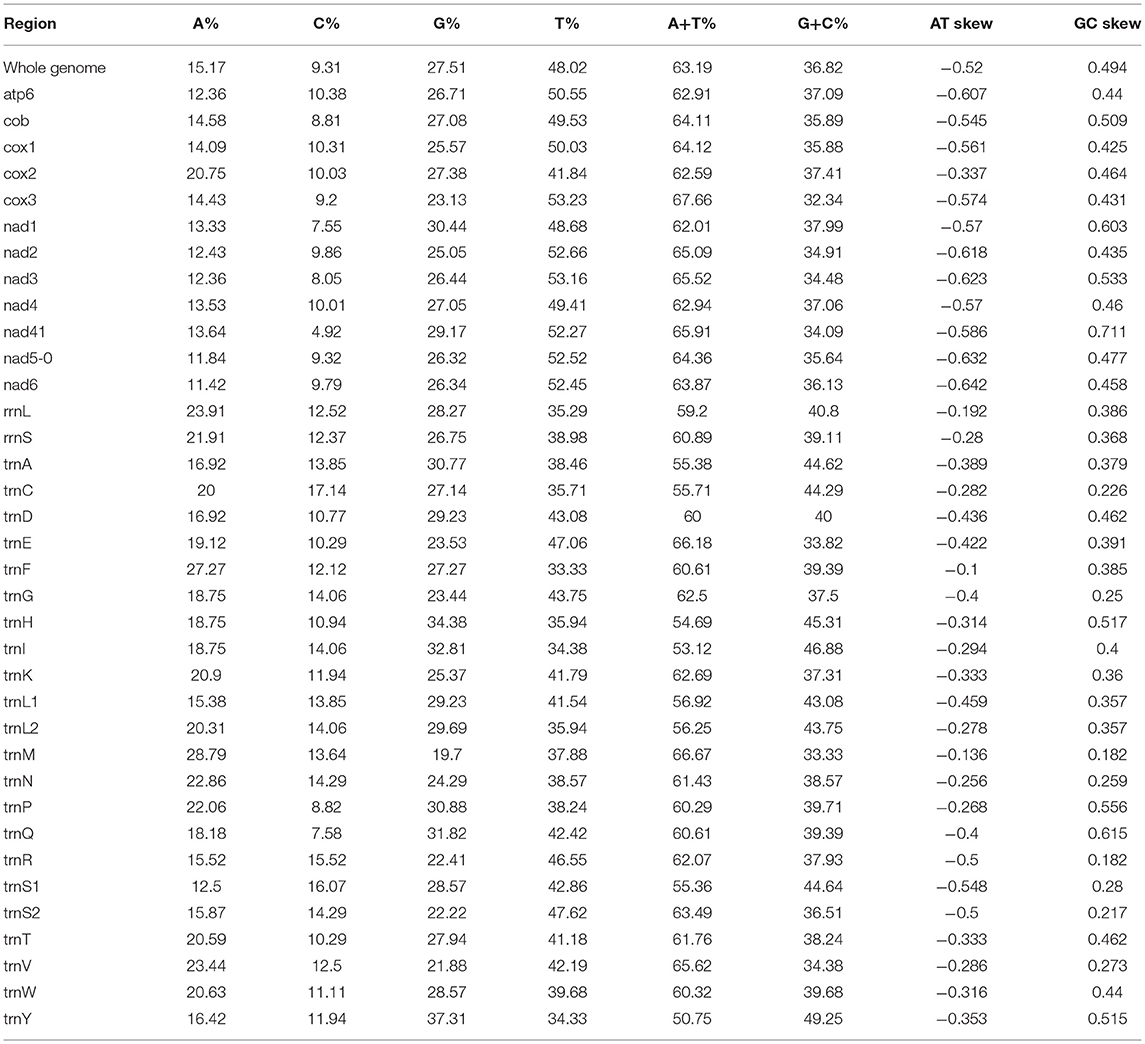
Table 3. Composition and skewness of F. intermediate mitogenome.
The mitogenome of F. intermediate had 12 typical PCGs, containing seven NADP genes (nad1-6 and nad4L), one ATP gene (atp6) and four cytochrome genes (cox1-3 and cob) (Figure 1). The region of PCGs was 8,160 bp in size. The PCGs started with ATG (cox1-3, nad4L), ATA (cob, nad3, nad6), TAG (nad4), ATC (atp6), ATT (nad1-2, nad5), and nine PCGs, terminated by TAA (cox1-3, atp6, nad2, nad4, nad5, nad6 and nad4L). The nad1 and nad3 used TAG as a stop codon whereas the cob terminated by a single T (Table 2).
The pattern of codon usage and relative synonymous codon usage (RSCU) in the F. intermediate mt DNA was studied. A total of 2,716 amino acids were encoded by the F. intermediate mitogenome, including the termination codes. The most frequently used amino acids were Leu (16.72%), followed by Val (12.74%), Phe (10.90%), Ser (10.09%) and Gly (8.39%) (Table 4 and Figure 2).
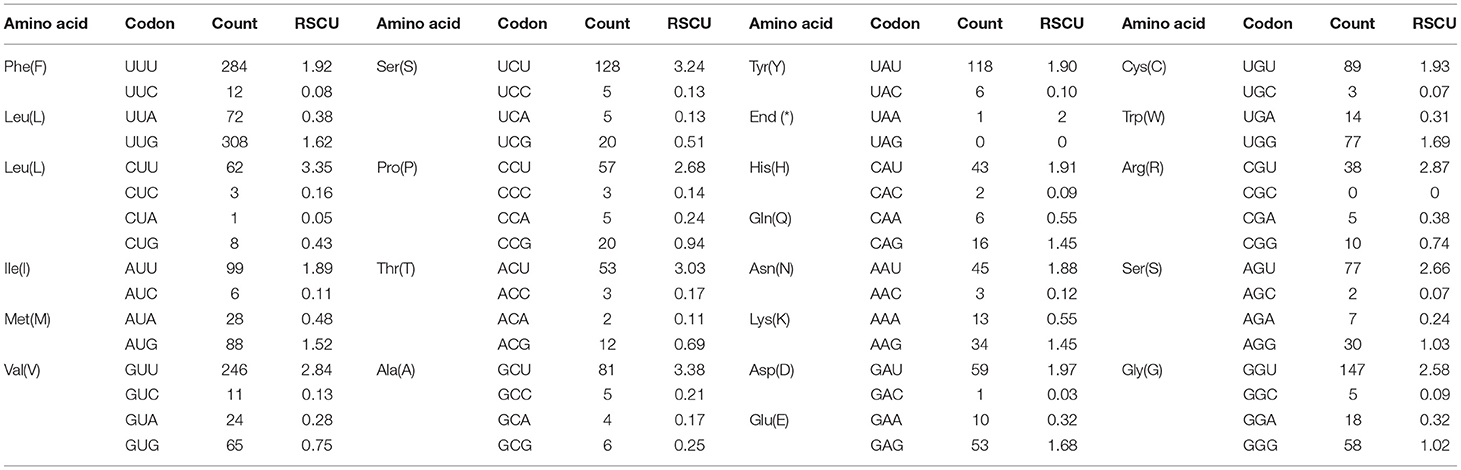
Table 4. The codon number and relative synonymous codon usage in F. intermediate mitochondrial protein coding genes.
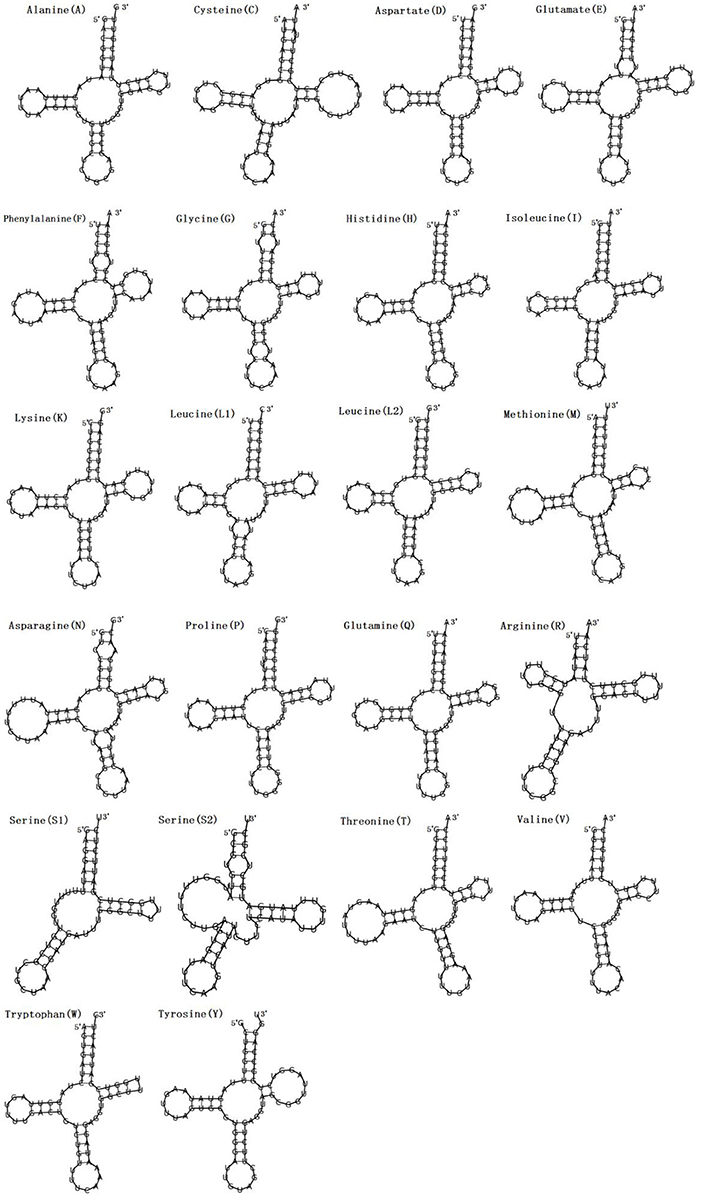
Figure 2. Secondary structures of the 22 transfer RNA genes of F. intermediate tRNAs (labeled with the abbreviations of their corresponding amino acids).
In the mitogenome of F. intermediate, the skew of AT was negative and the skew of GC was positive, indicating an obvious bias toward the use of T and A (Table 3). Like most mt DNA, the F. intermediate mitogenome contained a set of 22 tRNAs genes. The tRNAs ranged in size from 57 to 71 bp. All the tRNA genes were present on the N strand and all the tRNA genes had the typical cloverleaf structure (Figure 3).
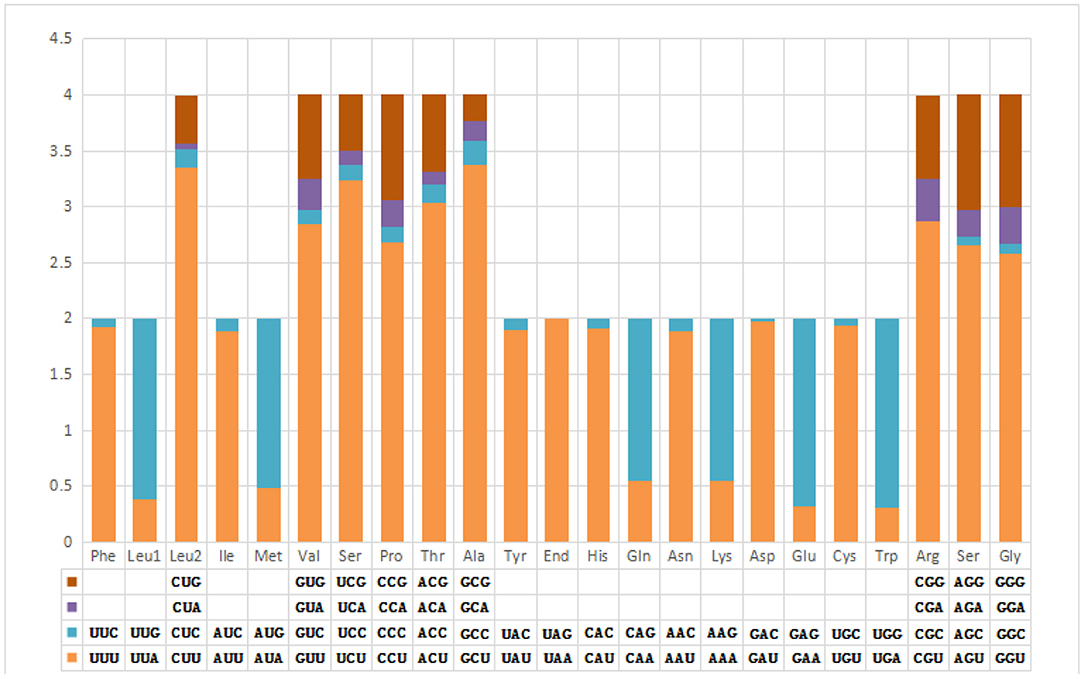
Figure 3. Relative synonymous codon usage in F. intermediate mitogenome.
The phylogenetic relationship was analyzed based on the concatenated nucleotide sequences of 12 PCGs from 36 Trematoda and one outgroup. The result of analyses, generated a consistent tree topologies (Figure 4).
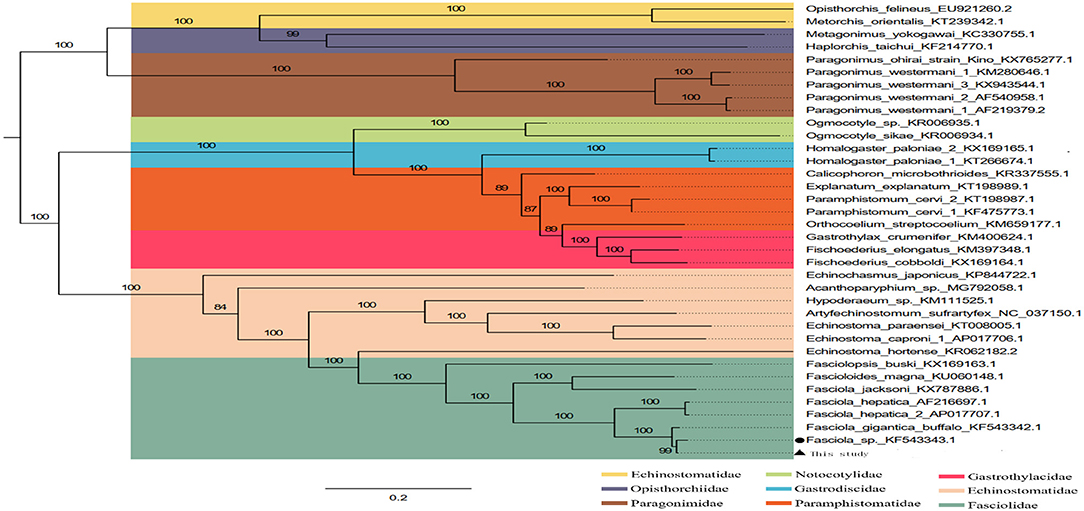
Figure 4. Inferred phylogenetic relationship among the F. intermediate. The phylogenetic tree was inferred from the nucleotide sequences of mitogenome by using ML methods (Numbers on branches indicate ML).
In this study, the ML analyses showed that each superfamily in the tree formed a monophyletic clade. Obviously, F. hepatica, F. intermediate, and F. gigantica clustered in one branch in the phylogenetic tree with high nodal support values (Figure 4), indicating that F. hepatica, F. intermediate, and F. gigantica have a sister group relationship. Additionally, the phylogenetic analyses revealed that F. hepatica, F. intermediate, and F. gigantica were grouped into one clade in fascioliases within Trematoda, which was consistent with a previous study (4).
The complete mitogenome sequences in this study (F. intermediate, Fi, MH621335.1) were 493, 414, and 518 bp shorter than F. intermediate from bovine (fin, KF543343.1, 14453 bp), F. hepatica (Fh, AP017707.1, 14374 bp), and F. gigantica (Fg, KF543342.1, 14478 bp), respectively. A comparison of the nucleotide sequences of each mt gene and the amino acid sequences, conceptually translated from all mt protein-encoding genes of the four flukes, as shown in Tables 5, 6. The sequence difference across the entire mitogenome was 182 nucleotide substitutions between Fi and Fin, 1, 432 nucleotide substitutions between Fi and Fh, and 197 nucleotide substitutions between Fi and Fg. The difference across amino acid sequences of the 12 protein-coding was 42 amino acid substitutions between the Fi and Fin; 270 amino acid substitutions between Fi and Fh; and 44 amino acid substitutions between Fi and Fg, respectively.
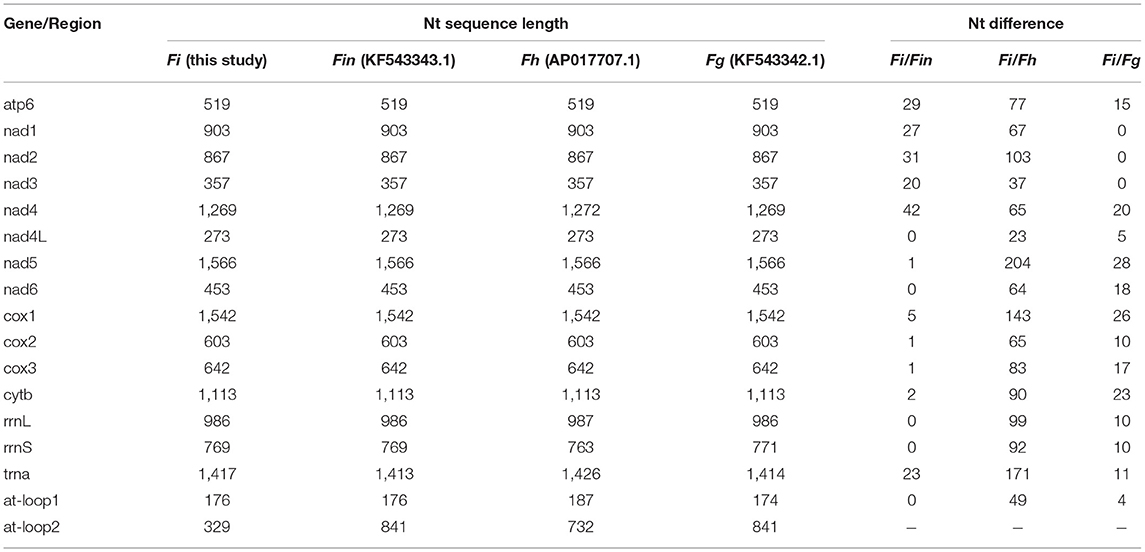
Table 5. The sequence differences of nucleotide (nt) in each mt gene among F. intermediate, F. gigantica and F. hepatica.
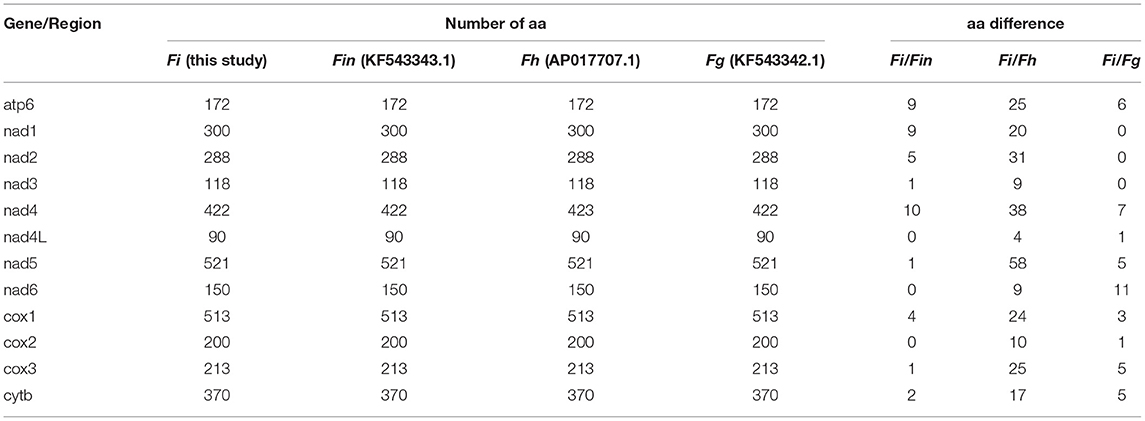
Table 6. Amino acid (aa) sequence differences in each mt gene among F. intermediate, F. gigantica, and F. hepatica.
Fasciolosis is a zoonotic disease belonging to water-borne trematodes. The previous studies showed the prevalence of fasciolosis in Africa with the highest range in cattle (1.2–91.0%) and the lowest in sheep (0.19–73.7%). In America the highest range was in goats (24.5–100%) and the lowest in cattle (3.0–66.7%) in America. In Asia the highest range in cattle (0.71–69.2%) and lowest in goat (0.0–47.0%). In Australia, the highest range was in cattle (26.5–81.0%), and the lowest range was in sheep (5.5–52.2%). While in Europe, the highest range was in cattle (0.12–86.0%) and the lowest in goats (0.0–0.8%) (20). Annually 2000 million dollars are lost because of helminthic infection (21).
F. hepatica and F. gigantica are considered two effective species in the genus Fasciola. However, the researchers found that there was also an “intermediate type” of Fasciola (F. intermediate). This “intermediate type” of Fasciola was first discovered in Japan and was subsequently reported in China and South Korea (22). Many studies on Fascioliasis have limited information about the Fasciola types in yaks. Genome-based molecular identification has been more commonly employed to identify biological types in recent years due to advances in biotechnology. mt DNA is an ideal genetic marker due to the simple and stable structure, reflecting the maternal genetic background (23).
Our study showed that F. intermediate from yak and F. intermediate from bovine belonged to the same branch and it was to be similar to F. gigantica compared with the F. hepatica through phylogenetic analysis. We compared the whole mitochondrial genome between Fi, Fin, Fg, and Fh. The results showed that Fi was no base site mutations in rrnL and rrnS compared with Fin, 10 base site mutations in rrnL and rrnS compared with Fg. Compared with Fh, Fi there were 99 base site mutations in rrnL and 92 base site mutations in rrnS, respectively. Hence, it would be the ideal genetic marker to distinguish the three species of Fasciola. Meanwhile, Fi had a deletion of nearly 500 bp in the AT-loop area, compared with Fin, Fg, and Fh, which may be due to the adaptability to plateau for Fi.
In this study, we sequenced the complete 13,960 bp mitogenome of F. intermediate from yaks, in which 36 genes (12 PCGs, 22tRNA genes and 2 rRNA genes) were located as a typical of Trematoda mitogenome. All PCGs were initiated by ATN codon, the cob genes had incomplete stop codons consisting of just T, and the other 11 PCGs stop with the canonical TAA or TAG. The AT-skew were negative, and the GC-skew were positive in the mitogenomes of F. intermediate, consistent with most sequenced Trematoda. The phylogenetic analyses support that F. intermediate from yaks was the same as the F. intermediate from bovine. This study would provide a basis for the accurate prevention, diagnosis, and treatment of Fasciolosis in yaks.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The samples were collected under the permission of the relevant institutions. All procedures were approved and performed by Laboratory Animals Research Centre of Hubei province and Qinghai province in P.R. China, and the Ethics Committee of Huazhong Agricultural University, China (Permit number: 4200695757). All animal experiments and procedures were conducted under the relevant procedures of Proclamation of the Standing Committee of Hubei People's congress (PSCH No. 5), China.
XG, JZ, and JL conceived and designed the study. XG, SZ, ZZ, KL, and XK executed the experiment and analyzed the sera and tissue samples. XG, SZ, CQ, and YL analyzed the data. XG and DW finished the first draft. MK finished the revision. All authors interpreted the data, critically revised the manuscript for important intellectual contents, and approved the final version.
This study was supported by the Chinese Agricultural Research Systems (CARS-37) and the major science and technology projects of Tibet Autonomous Region (XZ202101ZD0002N).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li JK, Li K, Shahzad M, Han ZQ, Nabi F, Gao JF, et al. Seroprevalence of Bluetongue virus in domestic yaks (Bos grunniens) in Tibetan regions of China based on circulating antibodies. Trop. Anim. Health Prod. (2015) 47:1221–3. doi: 10.1007/s11250-015-0853-0
2. Li K, Gao J, Shahzad M, Han Z, Nabi F, Liu M. Seroprevalence of Toxoplasma gondii infection in yaks (Bos grunniens) on the Qinghai-Tibetan Plateau of China. Vet Parasitol. (2014) 205:354–6. doi: 10.1016/j.vetpar.2014.07.014
3. Nyindo M, Lukambagire AH. Fascioliasis: an ongoing zoonotic trematode infection. Biomed Res Int. (2015) 2015:1–8. doi: 10.1155/2015/786195
4. Liu GH Gasser RB Young ND Song HQ Ai L and Zhu XQ. Complete mitochondrial genomes of the ‘intermediate form' of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasites & Vectors. (2014) 7:150. doi: 10.1186/1756-3305-7-150
5. Biu AA, Ahmed MI, Mshelia SS. Economic assessment of losses due to parasitic diseases common at the Maiduguri abattoir, Nigeria. African Scientist. (2006).
6. Piedrafita D, Spithill TW, Smith RE, Raadsma HW. Improving animal and human health through understanding liver fluke immunology. Parasite Immunol. (2010) 32:572–581 doi: 10.1111/j.1365-3024.2010.01223.x
7. Chen JX, Chen MX, Ai L, Xu XN, Jiao JM, Zhu TJ. An outbreak of human fascioliasis gigantica in Southwest China. PLoS ONE. (2013) 8: e71520 doi: 10.1371/journal.pone.0071520
8. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. (1999) 27:1767–80. doi: 10.1093/nar/27.8.1767
9. Liu QN Zhu BJ Dai LS Wei GQ and Liu CL. The complete mitochondrial genome of the wild silkworm moth, Actias selene. Gene. (2012) 505:291–9. doi: 10.1016/j.gene.2012.06.003
10. Dai LS Zhu BJ Liu QN Wei GQ and Liu CL. Characterization of the complete mitochondrial genome of Bombyx mori strain H9 (Lepidoptera: Bombycidae). Gene. (2013) 519:326–34. doi: 10.1016/j.gene.2013.02.002
11. Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. (1992) 141:173–216. doi: 10.1016/S0074-7696(08)62066-5
12. Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. (2008) 101:301–20. doi: 10.1038/hdy.2008.62
13. Mikkel S. Adapter Removal v2: rapid adapter trimming, identification, read merging. BMC Res Notes. (2016) 9:88. doi: 10.1186/s13104-016-1900-2
14. McLean JS, Lombardo MJ, Ziegler MG, Novotny M, Yee-Greenbaum J, Badger JH. Genome of the pathogen Porphyromonas gingivalis recovered from a biofilm in a hospital sink using a high-throughput single-cell genomics platform. Genome Res. (2013) 23:867–77. doi: 10.1101/gr.150433.112
15. Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. (2015) 31:587–9. doi: 10.1093/bioinformatics/btu661
16. Toro M, Philippou A, Arboleda S, Puerta M. Mean-field semantics for a process calculus for spatially-explicit ecological models. Electron Proc Theor Comput. (2016) 79–94. doi: 10.4204/EPTCS.204.7
17. Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, et al. (2013). MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–9. doi: 10.1016/j.ympev.2012.08.023
18. Beatson SA, das Graças de Luna M, Bachmann NL, Alikhan NF, Hanks KR, Sullivan MJ. Genome sequence of the emerging pathogen Aeromonas caviae. J Bacteriol. (2011) 193:1286–7. doi: 10.1128/JB.01337-10
19. Tamura K, Stecher G, Kumar S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. (2021). doi: 10.1093/molbev/msab120
20. Mehmood K, Zhang H, Sabir AJ, Abbas RZ, Ijaz M, Durrani AZ. A review on epidemiology, global prevalence and economical losses of fasciolosis in ruminants[J]. Microb Pathog. (2017) 109:253–62. doi: 10.1016/j.micpath.2017.06.006
21. Mungube EO, Bauni SM, Tenhagen BA, Wamae LW, Nginyi JM, Mugambi JM. The prevalence and economic significance of Fasciola gigantica and Stilesia hepatica in slaughtered animals in the semi-arid coastal Kenya. Trop Anim Health Prod. (2006) 38:475–83. doi: 10.1007/s11250-006-4394-4
22. Zeng MH, Lan Z, Guo XR. Analysis of polymorphic sites in the large and small ribosomal subunit sequences of the “intermediate” Fasciola [J]. Heilongjiang Animal Husbandry and Veterinary Medicine. (2020)09:85−8.
Keywords: yaks, Fasciola intermediate, mitogenome, gene order, phylogenetic
Citation: Gao X, Wang D, Zhang Z, Quan C, Zhou S, Li K, Li Y, Zhao S, Kong X, Kulyar MF, Zeng J and Li J (2022) Genetic Characterization and Phylogenetic Analysis of Fasciola Species Isolated From Yaks on Qinghai-Tibet Plateau, China. Front. Vet. Sci. 9:824785. doi: 10.3389/fvets.2022.824785
Received: 29 November 2021; Accepted: 22 February 2022;
Published: 12 May 2022.
Edited by:
Yung-Fu Chang, Cornell University, United StatesReviewed by:
Quan Liu, Foshan University, ChinaCopyright © 2022 Gao, Wang, Zhang, Quan, Zhou, Li, Li, Zhao, Kong, Kulyar, Zeng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangyong Zeng, emVuZ2ppYW5neW9uZ0Bob3RtYWlsLmNvbQ==; Jiakui Li, bGlqazIxMEBzaW5hLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.