
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Vet. Sci., 15 December 2022
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.1079359
This article is part of the Research TopicGenomics And Proteomics Approaches for The Improved Biologics Against Infectious AgentsView all 6 articles
 Muhammad Zubair1Jia Wang1
Muhammad Zubair1Jia Wang1 Yanfei Yu1,2,3Muhammad Faisal4
Yanfei Yu1,2,3Muhammad Faisal4 Mingpu Qi5
Mingpu Qi5 Abid Ullah Shah6
Abid Ullah Shah6 Zhixin Feng1
Zhixin Feng1 Guoqing Shao1,2Yu Wang7*
Guoqing Shao1,2Yu Wang7* Qiyan Xiong1,3,8*
Qiyan Xiong1,3,8*Proteomics is playing an increasingly important role in identifying pathogens, emerging and re-emerging infectious agents, understanding pathogenesis, and diagnosis of diseases. Recently, more advanced and sophisticated proteomics technologies have transformed disease diagnostics and vaccines development. The detection of pathogens is made possible by more accurate and time-constrained technologies, resulting in an early diagnosis. More detailed and comprehensive information regarding the proteome of any noxious agent is made possible by combining mass spectrometry with various gel-based or short-gun proteomics approaches recently. MALDI-ToF has been proved quite useful in identifying and distinguishing bacterial pathogens. Other quantitative approaches are doing their best to investigate bacterial virulent factors, diagnostic markers and vaccine candidates. Proteomics is also helping in the identification of secreted proteins and their virulence-related functions. This review aims to highlight the role of cutting-edge proteomics approaches in better understanding the functional genomics of pathogens. This also underlines the limitations of proteomics in bacterial secretome research.
The total protein content of an organism is referred to as the proteome. The proteome, particularly of prokaryotic cells, has a wide range of roles and pathogenic properties, and proteomics is the study of these functions and characteristics (1). Proteomics has contributed not only to the discovery of pathogen virulence components, but also to the research of pathogen structural makeup, pathogenesis, disease diagnosis, and vaccine development or design (2–4). Proteins from bacteria and viruses act as virulent agents in the transmission of diseases in humans and animals. Membrane proteins (5), cell surface proteins, and secreted proteins are among the most important, as they play a crucial role in pathogenicity and have been extensively researched utilizing proteomics techniques (6–8). These proteins function as enzymes, transport molecules, toxins, adhesins, invasive, evasive, and receptors, and hence play a crucial role in the initiation and course of disease. Proteomics methods have vastly improved in the recent decade, making it possible to search for these critical proteins and examine their structures, molecular functions, and role in disease. Proteomics has been useful in identifying the microorganisms that cause various diseases and their architecture. Because genomics can only provide information on the pathophysiology of a disease, it is unable to expound on the cell state and pathogenic actions of the molecules that cause illness onset. Proteins are well-recognized for depicting the state of a disease by informing the pathogenic components that are the foundations for illness initiation (9). As a result, understanding the functions of such proteins is critical for understanding the pathophysiology, diagnosis, control, and therapy of infectious illnesses. Many proteomics technologies have been created over time and have shown to be invaluable in the study of pathogens and course of illnesses. Traditional proteomics techniques such as chromatography and western blotting have been utilized for a long time. Gel-based techniques such as 1-DE (1-Dimensional Gel Electrophoresis), 2-DE (2-Dimensional Gel Electrophoresis), and 2-DDGE (2-dimensional Differential Gel Electrophoresis) assisted in protein separation and identification (10). Low abundant proteins in the sample can be fractioned by using isoelectric fractionators followed by 2-D gels. Depending on the isoelectric focusing (IEF), low abundant proteins are concentrated making identification and quantification more reliable. Some commonly used fractionators are Rotofor (BioRad) and Zoom IEF fractionator (Invitrogen) (11). The combination of Gel Electrophoresis with Mass Spectrometry (2-DE-MS) improved the accuracy of protein identification. Isotope-Coded Affinity Tag (ICAT), Stable Isotopic Labeling with Amino Acids (SILAC), and Isobaric tag for relative and absolute quantification (iTRAQ) are some of the new quantitative approaches that have emerged as a result of advances in proteomics (1, 12). These modern quantitative approaches include surface plasmon resonance (SPR) for protein-protein interaction and Multidimensional protein identification technology (MudPIT), both of which are label-free tools commonly utilized for protein identification (13, 14). Absolute quantification of proteins can be accomplished using a variety of strategies, including absolute quantification using protein epitope signature tags (PrEST), protein standard absolute quantification (PSAQ), and intensity-based absolute quantification (iBAQ) (15–17). Membrane coated nanosponges paired with quantitative proteomics methods have recently been discovered to be a powerful source for identifying virulence factors (18). Types of proteomics techniques and their sub-divisions are depicted in Figure 1. Our understanding of infectious diseases, causative agents, and their diagnosis has increased over time due to advances in proteomics. As a result, the goal of this study was to shed light on the function of various proteomics methods in elucidating the pathophysiology, diagnosis, and causative agents of infectious diseases that affect humans and animals. In addition, we discussed the limitations of proteomics in terms of identifying pathogenic secreted proteins, as well as its future prospects.
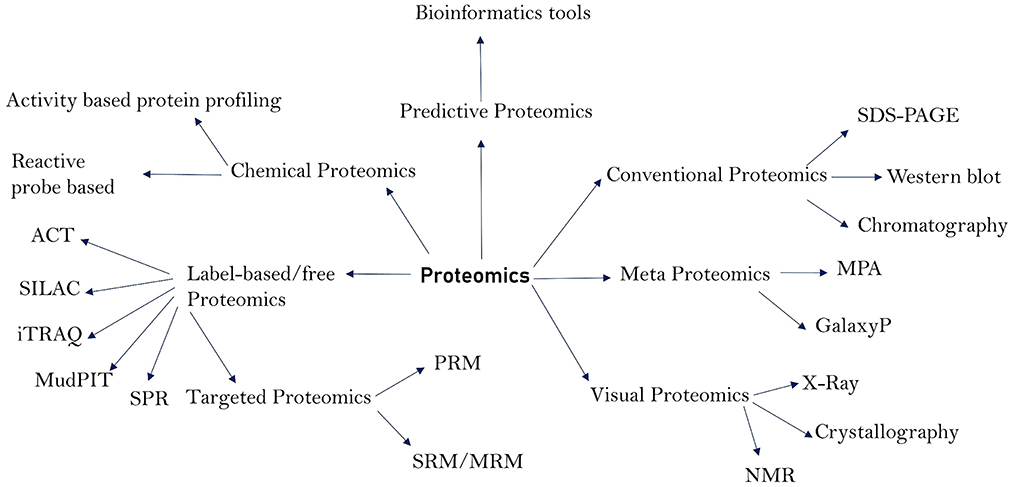
Figure 1. Major proteomics techniques and their subdivisions.
The first stage in diagnosing a disease is to identify the causal agent, since their precise and detailed identification and confirmation aids in the prevention of illness transmission and knowledge of its epidemiology (2). Biochemical features, Gram staining, and carbohydrate metabolism are some of the traditional methods for identifying bacteria that have been used for a long time. Proteomics technologies, such as Mass Spectrometry (MS), have recently become popular for precisely identifying and confirming bacterial infections (19, 20). Proteomics methods are commonly used to identify pathogen structure and other components that contribute to virulence. Proteomics methods are being used to describe the structures of both bacterial and viral pathogens, with the goal of not only identifying structural and non-structural proteins involved in virulence, but also investigating metabolic and physiological factors. Classification of un-sequenced microorganisms has been made easier by using capLC-MS/MS on an Orbitrap (21).
Proteomics has been used to identify bacterial infections that cause various disorders. The use of proteomics methods to identify bacterial communities in surface and soil samples has also been done. Samples were gathered from children's books in Texas and California libraries, and the Orbitrap FusionTM TribridTM mass spectrometer identified a variety of non-pathogenic and harmful bacteria species. S. haemolyticus, S. pneumoniae, and A. baumannii were the most commonly discovered pathogenic species, causing skin infections and Multidrug-Resistant Tuberculosis (MDR) correspondingly (22). Streptomyces violaceoruber, Streptomyces albus, and Streptomyces badius were identified using MALDI-ToF-MS from soil samples collected in Algeria's Sahara (23). Mass spectrometry's most important and revolutionary role is in clinical microbiology, where it has shown to be a useful tool for rapidly identifying infectious pathogens at species level. Traditional methods for identifying a pathogen take longer, resulting in a serious illness condition before it can be treated (24). Antibiotic resistance develops as a result of the use of broad-spectrum antibiotics prior to the identification of the causative agent, as well as a detrimental influence on the patient's health (25). Forensic proteomics is another growing tool to identify bacterial species in a given sample. This method is based on identification of unique peptides and facing few challenges i.e., signature erosion (loss of signature sequences due to the addition of new sequences of identified species in database), absence of statistical precision and limited database (26).
Body fluids such as urine, milk, and blood are the most acceptable samples for microbe identification, and proteomics has done an excellent job of identifying microbes from these samples (27–29), as well as others such as cerebrospinal fluid, joint cavity fluid, vitreous fluid, and pleural fluid (27, 30).
By creating a specific reference urine database called Urinf, 90% of 500 samples were accurately diagnosed using MALDI-ToF (31). MALDI-ToF-MS was used to successfully identify Corynebacterium rigelii, a pathogen that causes urinary tract infections, from a case of urosepsis in a 67-year-old female patient (32). The urine-short incubation MALDI-TOF (U-si-MALDI-ToF) method was created mainly for the detection of E. coli, a bacteria that causes urinary tract infections. Using this technology, 86% of Gram-negative bacteria responsible for urinary tract infections, such as E. coli, Klebsiella pneumoniae, and Enterobacteriaceae, were discovered (33). Mass spectrometry has recently been combined with other technologies to improve identification accuracy. The Alfred 60 method was used in conjunction with MALDI-ToF-MS to detect bacteria that cause urinary tract infections. For most positive samples, combined technique proved more reliable and accurate in identifying uropathogens (25). Combining mass spectrometry with other screening technologies like flow cytometry saves time while improving identification quality (34). Urine samples were initially screened using a Sysmex (UF-1000i) flow 36 cytometer before being sent to the MALDI-ToF-MS. This method correctly detected 86.1% Gram-negative bacteria without any microorganism misidentification (35). Combining flow cytometry, such as the UF-5000i, with mass spectrometry reduces the time it takes to identify etiological agents responsible for urinary tract infections from 24 to 1 h (36). When MALDI-ToF-MS was paired with Urine Analysis (93.4 and 96.3%), sensitivity and specificity for the detection of urinary pathogens from urine samples were enhanced and more reliable than when MALDI-ToF-MS was used alone (86.6 and 91.5%) (37). Leptospires that cause leptospirosis were discovered using mass spectrometry and whole cell protein spectra. MALDI-ToF-MS also identified whole cells of leptospires with spikes in urine samples (38). Because of its greater sensitivity and specificity, researchers prefer LC-MS-MS to MALDI-ToF-MS. In light of this, a method for identifying urinary tract pathogens utilizing specific LC-MS-MS peptide signatures was devised. This targeted proteomics technique identified urinary tract infections in 97% of patients without the need for a culture and in < 4 h, proving to be the most rapid and reliable method for pathogen identification in urinary tract infections (39). Although it is clear that the proteomics tool of mass spectrometry has evolved as a viable approach for identifying urinary tract pathogens, the culture-independent MALDI-ToF approach can only identify pathogens in single microbial urine samples (40).
In the same way that mass spectrometry has made it easier and faster to identify pathogens in blood, its combination with other technologies has made it considerably more effective in urine analysis. The bacteria in blood culture samples were identified using a comparative analysis. In comparison to the SepsiTyper kit approach, which identified 99% (184/186) isolates, MALDI-TOF-MS was able to identify 90% (168/186) of them. As a result, it was determined that MALDI-ToF-MS analysis is recommended for bacterial identification in blood cultures due to its speed and ease of use (41). MALDI-ToF-MS identified 93.43% (185/198) Gram-negative germs and 78.43% (275/350) Gram-positive bacteria from blood cultures, with specificity and sensitivity of 84.7 and 77.5%, respectively, in another investigation (42). Positive blood cultures were quickly cultured on solid media before being identified using MALD-ToF-MS, which proved to be a reliable method for bacterial identification. At 3, 5, and 24 h, this approach correctly identified bacteria at the species level with 64.1, 85.0, and 94.1%, respectively. This is thought to be a viable method for identifying bacteria directly (43). Bacteria were enhanced in a blood sample using magnetite (Fe3O4) magnetic beads modified with human IgG (IgG@Fe3O4) and MALDI-ToF-MS, which showed to be a more sensitive method with less time than other culture-based methods. Bacteria with a concentration of 105 CFU/100 μl whole blood sample were identified quickly (44). MALDI-ToF-MS was used to analyze spiked blood culture samples, and the efficiency was found to be comparable to SepsiTyper (94.4%). This approach identified 82% Gram-positive bacteria in blood samples and was more sensitive (92.8%) for Gram-negative bacteria (45). The combination of MALDI-ToF-MS with immune-affinity has yielded highly consistent findings for bacterial identification at low concentrations (500 cells/ml for blood serum and 8,000 cells/ml for whole blood samples). Within roughly 4 h, this combination technique was able to identify S. aureus and E. coli in clinical samples (29).
Another essential body fluid for detecting germs that cause diseases in humans and animals is milk. The only reliable source for identifying bacterial infections that cause mastitis is milk. From a human milk sample, MALDI-ToF-MS successfully identified 56 (53.3%) streptococcal isolates at the species level (46). MALDI-ToF-MS was used to identify microbial diversity in 647 milk samples from women who had clinical symptoms of mastitis. In milk samples, the most common pathogens were Staphylococcus epidermidis (87.6%) and Staphylococcus aureus (22.1%), with Streptococcus (68.6%) being the second most common species (47). Colony culture of milk samples from cows with subclinical mastitis followed by MALDI-ToF-MS identified 106/120 (88.3%) at the genus and species level (score 2.0) and it was found to be more reliable than direct MALDI-ToF-MS after pre-incubation (48). Mass spectrometry alone is insufficientfor accurate and rapid pathogen detection,; a combined approach has proven to be more trustworthy while saving time. Consequently, three methods for identifying bacteria in milk samples from calved cows or with clinical mastitis were evaluated as agreement approaches: biochemical method, MALDI-ToF-MS, and 16S rRNA partial genomic sequence analysis. At the species level, E. coli and S. aureus were recognized, while others were identified at the genus level. Positive agreement was determined to be 94% among three approaches, and 95–98% between each pair of methods (49). With time, mass spectrometry has become more capable, and some laboratories are replacing biochemical approaches with MALDI-ToF-MS for the detection of microorganisms in milk samples. MALDI-ToF-MS was utilized by the researchers to match the bacterial isolates from the udder with other species in the database. Five hundred isolates were processed as bacterial colony material for this study, and 93.5% of them were recognized at the species level, while 6.5% were identified at the genus level. Those that were unable to be recognized at the species level were submitted to 16S rDNA sequencing. Streptococci, Staphylococci, Enterobacteriaceae, and Coryneforme are the most common bacteria (50). Wald et al. recently detected and distinguished S. aureus and coagulase negative Staphylococci in 200 milk samples from animals with clinical and subclinical mastitis, as well as cows with a somatic cell count of < 100,000 cells/ml (51). From subclinical mastitis milk samples, MLADI-ToF-MS found S. argenteus in seven isolates and S. aureus in eight (52). When MALDI-ToF-MS was compared to PCR-RFLP for detecting streptococci from milk samples, it was discovered that PCR-RFLP was more efficient and repeatable (53). Alnakip et al., on the other hand, recently compared MALDI-ToF-MS with 16S rRNA gene sequencing study to distinguish streptococci responsible for bovine mastitis. MALDI-ToF-MS was found to have a wide range of variability for detecting streptococcus at the species and sub-species level. It is clear that MALDI-ToF-MS is as powerful as 16S rRNA gene sequencing analysis, but it takes less time and is easier to do (54). Microbes can also be identified by mass spectrometry in other body fluids such as saliva, cerebrospinal fluids, and synovial fluids from humans and animals (55–57). It is past time to develop a combined MALDI-ToF-MS with instruments that will make it a standard and universal approach for the accurate detection of bacterial infections in all types of body fluids in clinical laboratories.
Proteomics tools are also contributing and improving with time in order to better understand the etiology of practically all bacterial illnesses. In fact, this technique has transformed this field by providing a straightforward and diverse way to learn about pathogenesis. Proteomics methods are commonly used to investigate virulence-related variables, oxidative stress, and the role of proteins in the host-pathogen interaction. Proteomics advancements have made it possible to investigate the hidden mechanisms of infections and identify the proteins involved. Pérez-Llarena and Bou (58), Katsafadou et al. (59), Yang et al. (60) have written some review studies in this area. We will highlight recent advancements in understanding bacterial pathogenesis in this portion of the review.
Bacterial proteins are widely known for their roles in virulence and other processes that aid bacteria in their pathogenicity (5). Proteomics' role in identifying virulent factors of key human pathogens such Mycobacterium TB, Streptococcus pneumoniae, and Staphylococcus aureus has been summarized (13, 61, 62). Gel-based proteomics, such as SDS-PAGE, 2-Dimensional gel electrophoresis (2-DE), and 2-Dimensional Differential Gel Electrophoresis (2-DDGE), are still popular methods for separating proteins before mass spectrometry analysis. They appear to be irreplaceable but have been improved with the addition of modern techniques. SDS-PAGE was used to segregate the whole cell proteome of B. abortus and B. mellitensis, which was then reacted with field sera from buffalo, cow, sheep, and goat. MALDI-ToF-MS was used to identify various important proteins such as heat shock proteins, binding proteins, hypothetical proteins, and enzymes. It was hypothesized that the antigens listed play a vital role in the pathogen's survival in the host cell environment (63). SDS-PAGE was used to isolate the phage protein PA-PP, which was then characterized using mass spectrometry (64). Li et al. created agarose native gel electrophoresis, which has been effectively applied to the characterization of antibodies in serum (65) as well as western blotting (66). SMA-PAGE, a technology combining styrene maleic acid lipid particles with this technique, was developed specifically for the separation of membrane proteins (67). Khan et al. employed 2-Dimensional gel electrophoresis to separate whole cell and membrane proteins extracted from M. bovis (68). The proteomes of high pathogenic (Staph 38) and less virulent (8325-4) strains of Staph aureus, which causes keratitis, were compared. Four binding proteins were discovered in less virulent strains using 2-DE and mass spectrometry, but many adhesions were found in staph 38, indicating its high virulence on the host cell surface (69). Streptococcus suis is a zoonotic bacterium that causes infections in pigs and humans, with symptoms such as meningitis, arthritis, and pneumonia. Two-Dimensional Differential Gel Electrophoresis (2-DDGE) was used to segregate the proteomes of two mutant strains, and differential proteins were discovered using label-free analysis. SBP2, or putative pilus protein, was discovered to be a novel pathogenic component of S. suis (serotype 2) that functions as a fibronectin and laminin adhesin (70). Nascimento Filho et al. summarized the role of proteomics methods in determining the virulence of the Leptospira pathogenic sp. that causes human leptospirosis (71). Many studies show that SDS-PAGE or 2-DE can be used to separate bacterial proteins and determine virulence factors. Proteome of Compylobacter jejuni, exo-proteome of Clostridium difficle, biofilm and adherence mechanism of Vibrio parahaemolyticus, identification of C. jejuni adhesion protein attached to the skin of slaughtered chicken, and fibronectin binding proteins in enteropathogenic E. coli O55:H7 are among the more recent studies (72–75). Pathogenesis is investigated by discovering the adhesion function of pathogenic proteins, as adhesion is the first step for bacteria to commence infection. Following predicted and applied proteomics, M. bovis nuclease demonstrated the ability to attach to macrophages and invade cells, as well as being cytotoxic to the host cells (76). Chlamydia trachomatis is a sexually transmitted disease that affects both men and women. Quantitative proteomics was used to detect its proteome during its replicative and infective stages in order to better understand its pathophysiology. Several proteins with metabolic activities were discovered using reverse phase two-dimensional UPLC followed by mass spectrometry (77). The persistence of B. suis in a host cell environment with reduced oxygen supply was investigated using the proteome and transcriptome. RegA was discovered to regress genes and proteins involved in metabolism and energy synthesis, particularly the Isocitrate Lyase gene (ICL). RegA's regression action inhibits pathogen metabolism, ensuring the infection's long-term survival in the host cell. ICL was discovered to be important in B. suis virulence and pathogenicity (78).
Quantitative proteomics is gaining popularity as a way to identify a group of proteins linked to a disease and achieve good results if the proteins aren't already separated on a gel. The proteomes of individuals with atopic dermatitis and healthy people were studied using the LC-MS-MS method. Some bacterial species, such as Aeromonas hydrophila, Staphylococcus aureus, and Shewanella sp., have been found to play a role in disease. Glyceraldehyde-3-phosphate, enolase, and chaperons like DnaK and HtpG were among the proteins found to be important in pathogenesis (79). Four acetyltransferases were discovered and described by mass spectrometry in E. coli (RimI, YiaC, YjaB, and PhnO). YiaC was a new protein discovered to play a role in flagellar motility and bacterial pathogenicity (80). By constructing the phosphoproteome followed by LC-MS-MS, the mechanism of protein phosphorylation related with S. aureus pathogenicity was elucidated. In comparison to previously reported mechanisms, Ser/Thr kinase signaling was found to be more efficient in virulence (81).
Another proteomic investigation analyzed the proteomes of ESBL and non-ESBL Klabsiella pneumoniae strains using nano LC-MS-MS. Stress proteins G and A, Lon proteases, and ElaB proteins were found to be shared between the two strains' proteomes. Furthermore, virulence-associated proteins such as lyase, oxidoreductase, catalase, and isochoristamase were discovered in ESBL K. pneumoniae, indicating that it is a more virulent strain (82). The quantity of pathogenic factors such as adenylate cyclase and O antigen was found to vary dramatically in the Bordetella parapertussis proteome using nano LC-MS-MS in limited iron circumstances. The research was expanded to look for proteins that were missing or thought to be pseudogenes in Bordetella pertussis in order to distinguish between the two species that cause whooping cough based on their virulence associated proteins (83).
The proteomes of Salmonella typhimurium wild type and fnr null mutant were characterized using label-free mass spectrometry. There were 153 significantly diverse proteins among the 1,798 discovered proteins, each responsible for a different metabolic activity. The fumarate nitrate reduction pathway in Salmonella regulates fis (DNA binding protein), a virulence related protein in Salmonella typhimurium, according to the findings (84). The extracellular and cell associated proteome profile of mutant and wild type strains of Mycobacterium avium hominissuis responsible for human infections was identified via label free analysis utilizing an LTQ Orbitrap Velos mass spectrometer. The lysX gene in mutant strains was discovered to be responsible for pathogen metabolic and virulence functions, as well as intracellular cell survival (85). Sputum and saliva from tuberculosis patients were exposed to quantitative proteomics utilizing the LTQ-Orbitrap technology in order to learn more about the processes that occur throughout the course of the disease. Proteins implicated in immunological regulation, complement activation, and inflammation were found in both samples. Uninfected people's samples contained a collection of proteins involved in pathogen protection and the innate immune response (86).
Two proteins (PRRC2C and RAB14) were identified using iTRAQ to have higher levels among 606 proteins, and three bacterial taxa (Streptococcus, Veillonella, and Haemophilus) were reported to have a tight relationship with chronic rhinosinusitis. Proteins linked with these bacteria were in short supply and served a variety of roles related to virulence and pathogenicity (61). In another investigation, iTRAQ was utilized to discover the Lactobacillus acidophilus differently expressed proteins at pH 7.4. A total of 207 proteins were found to be involved in carbohydrate and amino acid metabolism, as well as peptidoglycan production. At pH 7.5, adhesion-related proteins fmtB and PrtP were found to be increased, while anti-adhesion protein pyruate kinase was downregulated (87). In humans, Acinetobacter baumannii is known to cause nosocomial infections such as bacteremia, pneumonia, and meningitis, all of which have significant mortality and morbidity rates. Differential proteins were discovered using iTRAQ after infecting pigs' intestines with enterotoxigenic E. coli F4 (producing diarrhea in piglets) and pre-treating them with Lactobacillus plantrum. Cell division, differentiation, and cell cycle regulation were revealed to be connected with differentially expressed proteins between two bacterial species. The findings revealed ETEC intestinal epithelial cell processes and the protective effect of L. plantrum (88). Clearly, iTRAQ is the preferable technology for quantitative proteome analysis, as it provides a more trustworthy and comprehensive result. Another experiment measured the quantity of Salmonella enteritidis proteins in LB media supplemented with egg white and entire egg white. Protein abundance was observed to decrease as the amount of egg white was reduced using iTRAQ. Some virulence-related proteins were downregulated, while ABC transporters and co-factors were predominantly increased (89).
The abundance of ABC transporters and adhesion-related proteins in high pathogenic strains was discovered using LC-MS-MS and iTRAQ. It was also established that the sbp protein was implicated in the pathophysiology of the disease (90). iTRAQ coupled with 2D LC-MS-MS was used to investigate the proteome of A. baimannii standard strain and tigecycline-resistant strain. A total of 3,639 proteins were found, with 961 of them being differentially expressed. Differential proteins were linked to cellular component organization, stress responses, protein synthesis, protein degradation, and related functions, according to functional analysis. There were also some pathways linked to tigecycline resistance discovered (91).
However, additional quantitative proteome techniques have lately been applied. The abundance of outer membrane vesicles in coccoid was discovered utilizing a comparative proteome study of coccoid and spiral shaped Helicobacter pylori (gastric cancer) using the SILAC (stable isotopic labeling by amino acids in cell culture) proteome technique. Some proteins were discovered to be down regulated, including CagA, arginase RocF, and TNF-inducers (92). Another method for identifying isotope-labeled proteins is isotope dilution mass spectrometry. An isotope-labeled 15N-Cys C protein in E. coli was effectively discovered using this method (93). TMT (94, 95) is a method for quantifying proteins/peptides using tissue, serum, plasma, or other body fluid samples from the affected/diseased area. This has proven to be a reliable method for identifying proteins that are expressed at different phases of disease, allowing researchers to track disease pathophysiology and development (96). The aforementioned technique has recently been substituted by membrane coated nanosponges paired with quantitative proteomics technologies. This enhanced method proved to be quite beneficial in identifying bacterial toxins and/or pathogenic components (18). There are several mass spectrometry-based and isotopic labeled/label-free proteomics methods that have aided in the better understanding of the etiology of important bacterial diseases in humans and animals. Protein microarray is one of the advanced proteomics techniques of recent era. This includes antibody microarray in which proteins are labeled with captured antibodies, functional microarray uses purified proteins for various interactions and reverse-phase protein microarray finds its application in probing the target protein from cell lysates using antibodies (1).
This method entails the use of bioinformatics tools to identify and test proteins based on their unique nature, structure, and functions. It is commonly used to anticipate the proteins produced by a certain bacterial pathogen and to identify the most important proteins linked to virulence. Gene ontology and enrichment analyses for function and pathway studies, as well as visualization tools to portray data in the form of graphs and charts, are the most crucial tools (97). This method was used to predict Chlamydia pneumonia nuclear targeting proteins that may play a role in lung cancer genesis (98). Computational biology and chemoinformatics were used to predict new therapeutic targets for A. baumannii (99). Baarda et al. compiled a list of tools that were useful in identifying vaccine candidates for N. gonorrhoeae (100). Using several bioinformatics tools available as web servers, secretory proteins of M. bovis were recently examined. Two proteins (MbovP274 and MbovP570) were chosen from the secretome data and experimentally confirmed to be immunogenic proteins (101). Many bacterial pathogens (C. botulinum, C. defficile, Y. pseudotuberclosis, S. saprophyticus, and Legionella sp.) have been studied in silico in order to find therapeutic targets and vaccine candidates (102–105). I-TASSER (https://seq2fun.dcmb.med.umich.edu//I-TASSER/) is an extensively used web server for the prediction of structure and function of the given protein (106) and Phyre2 (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index) for protein modeling analysis (107). ConSurf web server (https://consurf.tau.ac.il/consurf_index.php) is usually used to identify the functional regions in protein (108). STRING (https://string-db.org/) is another online tool used for protein-protein interaction and functional predictions (109).
Proteins are a significant source of biomarkers and are used for illness diagnosis, prognosis, staging, and monitoring. Hormones, carbohydrate epitopes, enzymes, genetic alterations, and receptors are examples of biomarkers (110). Pathogen proteins have been shown to be responsible for virulence and infections, and hence can be used to find useful biomarkers for illness detection (111). The fact that they are important diagnostic indicators has piqued the interest of scientists all around the world in using proteomics technologies to uncover specific disease markers. Pasteurellosis and pneumonia in sheep have been proven to have biomarkers in the form of proteins and cytokines (112). Proteomics, both traditional and modern, is playing an increasingly important role in diagnostics, providing trustworthy and meaningful results. Since the last decade, mass spectrometry-based techniques have advanced significantly and are becoming increasingly useful in the search for promising diagnostic markers. Recent advances in quantitative proteomics, as well as increased accuracy, have paved the road for the discovery of effective diagnostic markers for a variety of disorders (113). The LC-MS-MS method is commonly used to diagnose diseases such as TB and periodontitis (114, 115). Table 1 depicts the many proteomics methodologies used to identify diagnostic markers for a certain disease.
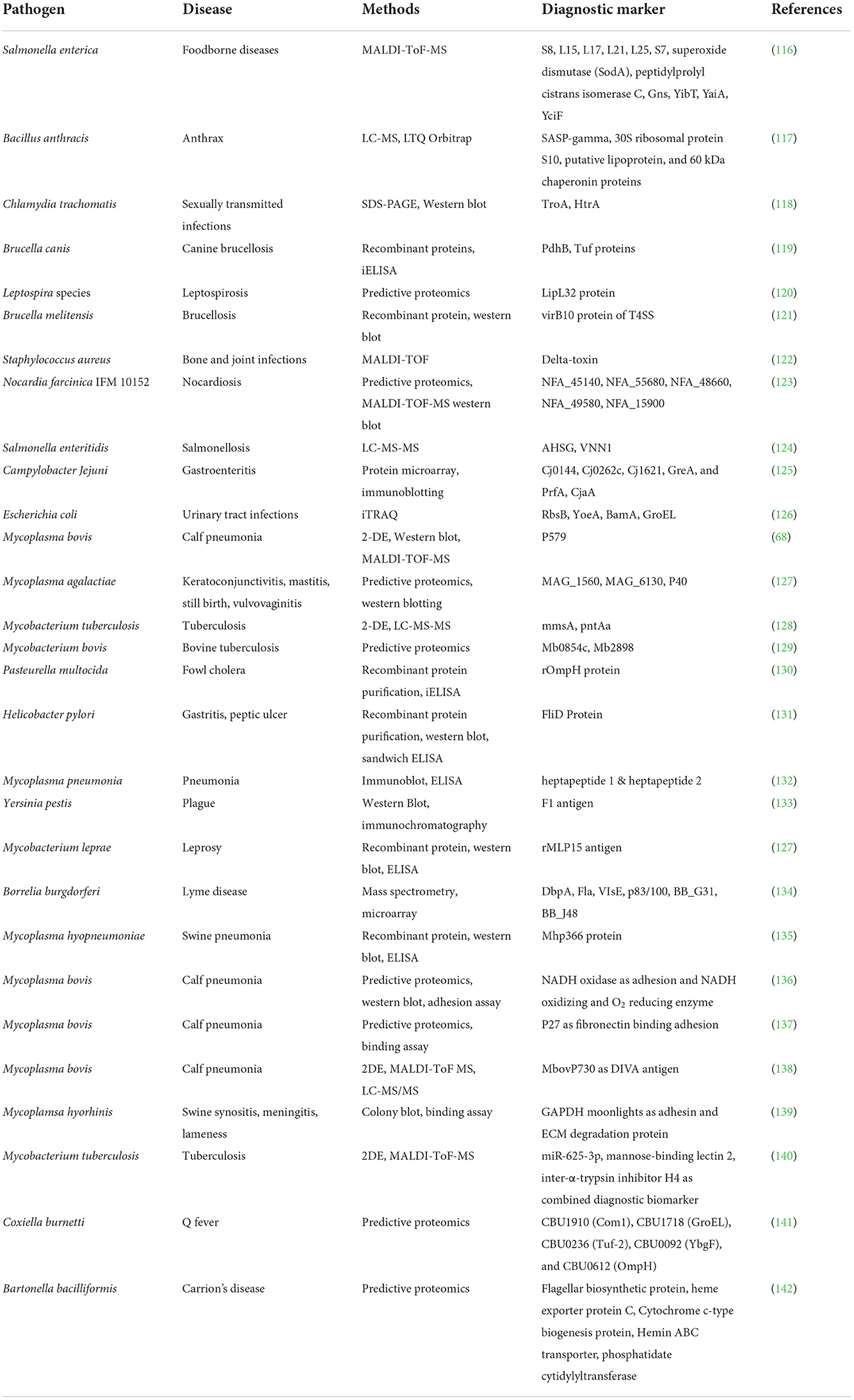
Table 1. Diagnostic markers of various important bacterial diseases using proteomics tools.
The improved mass spectrometry approach for the absolute detection of biomarkers from Salmonella serotypes was introduced by Fukuyama and colleagues (116). In a cohort research, quantitative proteomics was used to uncover distinct biomarkers in the plasma of individuals with active tuberculosis. Five proteins, CFHR5, LRG1, CRP, LBP, and SAA1, have been discovered to clearly distinguish tuberculosis patients from those with other respiratory illnesses (143). The protein microarray technique was utilized to identify diagnostic indicators for Salmonella typhi, and it was found to be highly repeatable (144). In another cohort investigation, the whole proteome microarray approach was employed to identify protein biomarkers from Chlamydia trachomatis. A total of 121 antigens were discovered, 18 of which might be used as diagnostic markers. Furthermore, the antigens CT 858, CT 813, and CT 142 were thought to represent possible disease markers in the future (145). Considering the findings of recent studies, it is clear that proteomics technologies are playing an important role in illness diagnoses.
The secretome is a collection of proteins that are either released in soluble form or overlapped by vesicles from bacterial cells. These secreted proteins play a critical role in bacterial virulence, and their characterization has become increasingly relevant in the quest better understanding bacterial virulence. Proteomics technologies have proven to be extremely useful in this procedure, from secretome extraction to characterization (146). Bioinformatics and other proteomics methods, including as mass spectrometry and immunoproteomics, have contributed in the discovery of antigenic secreted proteins and vaccine candidates (147). Table 2 illustrates the secretomes of pathogenic bacteria and the proteins that have been identified as virulent factors, protective antigens, or vaccine candidates.
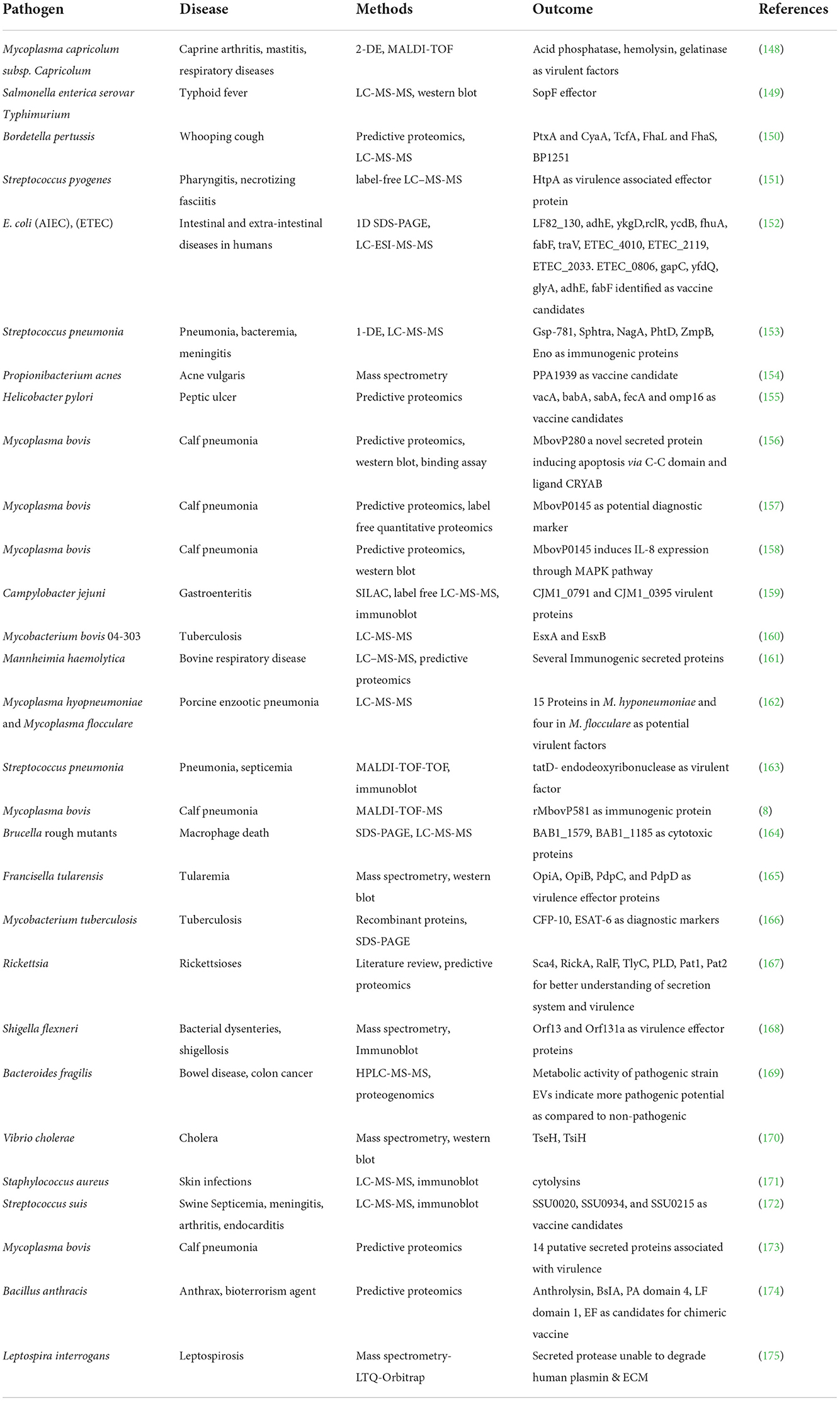
Table 2. Secreted proteins from important bacterial pathogens using proteomics tools.
It is clear that proteomics methods are extremely useful for studying and characterizing the bacterial secretome in depth. The limits for identifying really secreted proteins are a crucial point to make here. Bioinformatics alone is insufficient to achieve this goal (176). A highly advanced proteomics technique is required to identify proteins that were originally produced by specific bacteria. Many in silico studies have been carried out, however there is still some confusion concerning protein secretion and the mechanisms involved, such as classical and non-classical secretion (167, 173, 177–179). Another area of debate that gets mixed up with the bacterial secretome is cell lysis and secretion of non-classical proteins (180, 181). Visual proteomics is a viable approach for identifying bacterial extracellular vesicles in a sample, but due to their small size, released soluble proteins are not visible by SEM or TEM. Because proteins are only projected to be secreted via multiple secretion pathways, there is a pressing need to develop a better proteomics tool. This allows one to identify truely secreted proteins as well as their secretion pathways, removing the ambiguities associated with prediction tools. A review gives light on the challenges of extracting and characterizing the bacterial secretome, particularly in the case of Mycoplasma sp. (176). Because serum in growth media interferes with secreted proteins, many people utilize media with lower serum concentrations (8, 162, 182). It is now recommended that serum-free media be used for Mycoplasma sp. culture in order to discover proteins of interest without disrupting serum proteins. If this can be accomplished without affecting growth or cell lysis, it will be a significant contribution to the field of proteomics for Mycoplasma sp. In terms of proteomics, Mycoplasma bovis has been a widely investigated bacterium in recent years. Using entire cell proteins, membrane proteins, and secreted proteins, effective research has recently been published in order to uncover diagnostic markers and vaccine candidates (8, 68, 101, 173). Profiling core secretome proteins among different strains of pathogenic bacteria might be significant to future studies as supported by the recent core genome studies (183). Figure 2 depicts the advancement of the proteome of Mycoplasma bovis in a schematic manner, which could be extremely useful in filling gaps in proteomics study of other significant Mycoplasma sp. such as M. hyorhinis, M. hyopneumoniae, M. agalactiae, and M. mycoides sub sp mycoides.
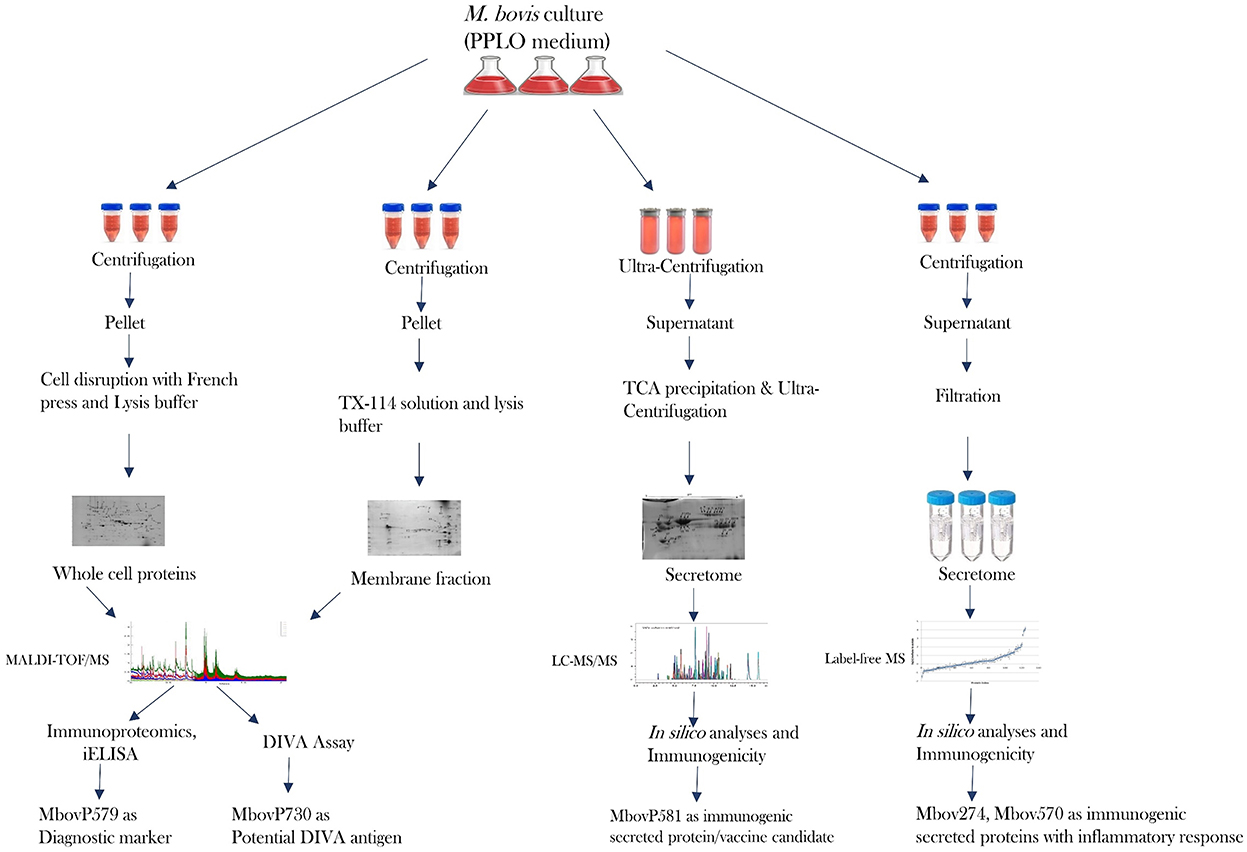
Figure 2. Recent progress in the proteome and secretome of M. bovis and its outcome.
Proteomics has played a vital role in identifying and distinguishing bacterial infections, as well as understanding and diagnosing their pathophysiology. Using a combination of methods, researchers were able to more effectively detect infections as well as identify and characterize the proteins involved in pathogenicity. Proteomics enabled to detect the secretome of bacterial pathogens, in addition to entire cell and membrane proteins, and gave a new platform for the field of preventive medicine. In order to confirm and describe the secretory nature of proteins implicated in bacterial pathogenicity, more progress must be made.
MZ wrote the manuscript. YY, JW, and AS collected the literature. MF set the tables. MQ designed the figures. ZF and GS revised the manuscript. YW and QX organized the contents and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Key R&D program of China (2022YFD1800903), Jiangsu Agricultural Science and Technology Fund [CX(22)3195], and National Natural Science Foundation of China (32102675 and 32172860).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Aslam B, Basit M, Nisar MA, Khurshid M, Rasool MH. Proteomics: technologies and their applications. J Chromatogr Sci. (2017) 55:182–96. doi: 10.1093/chromsci/bmw167
2. Chen B, Zhang D, Wang X, Ma W, Deng S, Zhang P, et al. Proteomics progresses in microbial physiology and clinical antimicrobial therapy. Eur J Clin Microbiol Infect Dis. (2017) 36:403–13. doi: 10.1007/s10096-016-2816-4
3. Venkatesh A, Gil C, Fuentes M, LaBaer J, Srivastava S, A. Perspective on proteomics of infectious diseases. Proteom Clin Appl. (2018) 12:1–7. doi: 10.1002/prca.201700139
4. Ye Y, Mar EC, Tong S, Sammons S, Fang S, Anderson LJ, et al. Application of proteomics methods for pathogen discovery. J Virol Methods. (2010) 163:87–95. doi: 10.1016/j.jviromet.2009.09.002
5. Khan FA, Rasheed MA, Faisal M, Menghwar H, Zubair M, Sadique U, et al. Proteomics analysis and its role in elucidation of functionally significant proteins in Mycoplasma bovis. Microb Pathog. (2017) 111:50–9. doi: 10.1016/j.micpath.2017.08.024
6. Adamu JY, Wawegama NK, Browning GF, Markham PF. Membrane proteins of mycoplasma bovis and their role in pathogenesis. Res Vet Sci. (2013) 95:321–5. doi: 10.1016/j.rvsc.2013.05.016
7. Foster TJ. Surface proteins of Staphylococcus epidermidis. Front Microbiol. (2020) 11:1–22. doi: 10.3389/fmicb.2020.01829
8. Zubair M, Muhamed SA, Khan FA, Zhao G, Menghwar H, Faisal M, et al. Identification of 60 secreted proteins for Mycoplasma bovis with secretome assay. Microb Pathog. (2020) 143:104135. doi: 10.1016/j.micpath.2020.104135
9. Mora-Montes HM. A perspective on the role of proteins and peptides in the virulence and pathogenesis. Curr Protein Pept Sci. (2019) 20:960–1. doi: 10.2174/1389203720999190722164728
10. Khan FA, Chen X, Shoaib M, Shah M, Ahmad F, Khan H, et al. Two dimensional gel electrophoresis (2-DE) for high-throughput proteome analyses of Mycoplasma bovis. Acta Biochim Pol. (2019) 66:321–7. doi: 10.18388/abp.2019_2794
11. Chandramouli K, Qian P-Y. Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum Genom. Proteom. (2009) 1. doi: 10.4061/2009/239204
12. Cordwell SJ. Technologies for bacterial surface proteomics. Curr Opin Microbiol. (2006) 9:320–9. doi: 10.1016/j.mib.2006.04.008
13. Bonar E, Wójcik I, Wladyka B. Proteomics in studies of Staphylococcus aureus virulence. Acta Biochim Pol. (2015) 62:367–81. doi: 10.18388/abp.2015_1083
14. Douzi B. Chapter 21: Protein-Protein interactions: surface plasmon resonance. Bact Protein Secret Syst Methods Protoc. (2017) 1615:257–75. doi: 10.1007/978-1-4939-7033-9_21
15. Hou TY, Chiang-Ni C, Teng SH. Current status of MALDI-TOF mass spectrometry in clinical microbiology. J Food Drug Anal. (2019) 27:404–14. doi: 10.1016/j.jfda.2019.01.001
16. Soufi B, Macek B. Global analysis of bacterial membrane proteins and their modifications. Int J Med Microbiol. (2015) 305:203–8. doi: 10.1016/j.ijmm.2014.12.017
17. Zeiler M, Straube WL, Lundberg E, Uhlen M, Mann M. A protein epitope signature tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol Cell Proteom. (2012) 11:1–13. doi: 10.1074/mcp.O111.009613
18. Distler U, Tenzer S. Tools for pathogen proteomics: fishing with biomimetic nanosponges. ACS Nano. (2017) 11:11768–72. doi: 10.1021/acsnano.7b07363
19. Mutters NT, Hodiamont CJ, De Jong MD, Overmeijer HPJ, Van Den Boogaard M, Visser CE. Performance of kiestra total laboratory automation combined with MS in clinical microbiology practice. Ann Lab Med. (2014) 34:111–7. doi: 10.3343/alm.2014.34.2.111
20. Sabbagh B, Mindt S, Neumaier M, Findeisen P. Clinical applications of MS-based protein quantification. Proteom Clin Appl. (2016) 10:323–45. doi: 10.1002/prca.201500116
21. Wynne C, Edwards NJ, Fenselau C. Phyloproteomic classification of unsequenced organisms by top-down identification of bacterial proteins using capLC-MS/MS on an Orbitrap. Proteomics. (2010) 10:3631–43. doi: 10.1002/pmic.201000172
22. Jung RH, Kim M, Bhatt B, Choi JM, Roh JH. Identification of pathogenic bacteria from public libraries via proteomics analysis. Int J Environ Res Public Health. (2019) 16. doi: 10.3390/ijerph16060912
23. Mohamed H, Miloud B, Zohra F, García-Arenzana JM, Veloso A, Rodríguez-Couto S. Isolation and characterization of actinobacteria from Algerian Sahara soils with antimicrobial activities. Int J Mol Cell Med. (2017) 6:109–20. doi: 10.22088/acadpub.BUMS.6.2.5
24. Angeletti S, Ciccozzi M. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry in clinical microbiology: an updating review. Infect Genet Evol. (2019) 76:104063. doi: 10.1016/j.meegid.2019.104063
25. Athamna A, Zbriger A, Avadov S, Shapira M, Tal Y, Freimann S. Rapid identification of uropathogens by combining Alfred 60 system with matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry technology. Eur J Clin Microbiol Infect Dis. (2020) 39:1855–63. doi: 10.1007/s10096-020-03919-3
26. Merkley ED, Wunschel DS, Wahl KL, Jarman KH. Applications and challenges of forensic proteomics. Forensic Sci Int. (2019) 297:350–63. doi: 10.1016/j.forsciint.2019.01.022
27. Barreiro JR, Gonçalves JL, Braga PAC, Dibbern AG, Eberlin MN, Veiga dos Santos M. Non-culture-based identification of mastitis-causing bacteria by MALDI-TOF mass spectrometry. J Dairy Sci. (2017) 100:2928–34. doi: 10.3168/jds.2016-11741
28. Tang M, Yang J, Li Y, Zhang L, Peng Y, Chen W, et al. Diagnostic accuracy of MALDI-TOF mass spectrometry for the direct identification of clinical pathogens from urine. Open Med. (2020) 15:266–73. doi: 10.1515/med-2020-0038
29. Zhu Y, Qiao L, Prudent M, Bondarenko A, Gasilova N, Möller SB, et al. Sensitive and fast identification of bacteria in blood samples by immunoaffinity mass spectrometry for quick BSI diagnosis. Chem Sci. (2016) 7:2987–95. doi: 10.1039/C5SC04919A
30. Tian Y, Zheng B, Wang B, Lin Y, Li M. Rapid identification and multiple susceptibility testing of pathogens from positive-culture sterile body fluids by a combined MALDI-TOF mass spectrometry and Vitek susceptibility system. Front Microbiol. (2016) 7:523. doi: 10.3389/fmicb.2016.00523
31. Pinault E, Didier Raoult, Florence Fenollarb L. Direct identification of pathogens in urine by use of a specific. J Clin Microbiol. (2019) 57:1–9. doi: 10.1128/JCM.01678-18
32. Pichon M, Micaelo M, Longuet P, Plantefève G, Abderrahmane M, Wifaq B, et al. A rare case of Corynebacterium riegelii urosepsis: role of the MALDI-TOF mass spectrometry in the identification of emerging pathogens. Med Mal Infect. (2019) 49:474–7. doi: 10.1016/j.medmal.2019.06.005
33. Haiko J, Savolainen LE, Hilla R, Pätäri-Sampo A. Identification of urinary tract pathogens after 3-hours urine culture by MALDI-TOF mass spectrometry. J Microbiol Methods. (2016) 129:81–4. doi: 10.1016/j.mimet.2016.08.006
34. Zboromyrska Y, Rubio E, Alejo I, Vergara A, Mons A, Campo I, et al. Development of a new protocol for rapid bacterial identification and susceptibility testing directly from urine samples. Clin Microbiol Infect. (2016) 22:561.e1–e6. doi: 10.1016/j.cmi.2016.01.025
35. Íñigo M, Coello A, Fernández-Rivas G, Rivaya B, Hidalgo J, Quesada MD, et al. Direct identification of urinary tract pathogens from urine samples, combining urine screening methods and matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. (2016) 54:988–93. doi: 10.1128/JCM.02832-15
36. Sun C, Zhang X, Wang J, Cheng C, Kang H, Gu B, et al. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry combined with UF-5000i urine flow cytometry to directly identify pathogens in clinical urine specimens within 1 hour. Ann Transl Med. (2020) 8:602. doi: 10.21037/atm.2019.10.73
37. Huang B, Zhang L, Zhang W, Liao K, Zhang S, Zhang Z, et al. Direct detection and identification of bacterial pathogens from urine with optimized specimen processing and enhanced testing algorithm. J Clin Microbiol. (2017) 55:1488–95. doi: 10.1128/JCM.02549-16
38. Sonthayanon P, Jaresitthikunchai J, Mangmee S, Thiangtrongjit T, Wuthiekanun V, Amornchai P, et al. Whole cell matrix assisted laser desorption/ ionization time-of-flight mass spectrometry (MALDI-TOF MS) for identification of Leptospira spp. in Thailand and Lao PDR. PLoS Negl Trop Dis. (2019) 13:1–16. doi: 10.1371/journal.pntd.0007232
39. Roux-Dalvai F, Gotti C, Leclercq M, Hélie MC, Boissinot M, Arrey TN. Fast and accurate bacterial species identification in urine samples using LC-MS/MS mass spectrometry and machine learning. Mol Cell Proteom. (2019) 18:2492–505. doi: 10.1074/mcp.TIR119.001559
40. Oros D, Ceprnja M, Zucko J, Cindric M, Hozic A, Skrlin J, et al. Identification of pathogens from native urine samples by MALDI-TOF/TOF tandem mass spectrometry. Clin Proteomics. (2020) 17:1–9. doi: 10.1186/s12014-020-09289-4
41. Azrad M, Keness Y, Nitzan O, Pastukh N, Tkhawkho L, Freidus V, et al. Cheap and rapid in-house method for direct identification of positive blood cultures by MALDI-TOF MS technology. BMC Infect Dis. (2019) 19:1–7. doi: 10.1186/s12879-019-3709-9
42. Barberino MG, Silva M de O, Arraes ACP, Correia LC, Mendes AV. Direct identification from positive blood broth culture by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS). Brazilian J Infect Dis. (2017) 21:339–42. doi: 10.1016/j.bjid.2017.03.007
43. Curtoni A, Cipriani R, Marra ES, Barbui AM, Cavallo R, Costa C. Rapid identification of microorganisms from positive blood culture by MALDI-TOF MS after short-term incubation on solid medium. Curr Microbiol. (2017) 74:97–102. doi: 10.1007/s00284-016-1161-2
44. Zhu YL, Lian YM, Wang JK, Chen ZP Yu RQ. Ultrasensitive detection of protein biomarkers by MALDI-TOF mass spectrometry based on ZnFe2O4 nanoparticles and mass tagging signal amplification. Talanta. (2021) 224:121848. doi: 10.1016/j.talanta.2020.121848
45. Zhou X, Xing X, Hou J, Liu J. Quantitative proteomics analysis of proteins involved in alkane uptake comparing the profiling of Pseudomonas aeruginosa SJTD-1 in response to n-octadecane and n-hexadecane. PLoS ONE. (2017) 12:1–13. doi: 10.1371/journal.pone.0179842
46. Martín V, Mediano P, Del Campo R, Rodríguez JM, Marín M. Streptococcal diversity of human milk and comparison of different methods for the taxonomic identification of streptococci. J Hum Lact. (2016) 32:NP84–94. doi: 10.1177/0890334415597901
47. Marín M, Arroyo R, Espinosa-Martos I, Fernández L, Rodríguez JM. Identification of emerging human mastitis pathogens by MALDI-TOF and assessment of their antibiotic resistance patterns. Front Microbiol. (2017) 8:1–13. doi: 10.3389/fmicb.2017.01258
48. Barreiro JR, Gonçalves JL, Grenfell R, Leite RF, Juliano L, Santos M V. Direct identification of bovine mastitis pathogens by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry in pre-incubated milk. Brazilian J Microbiol. (2018) 49:801–7. doi: 10.1016/j.bjm.2018.04.012
49. Wilson DJ, Middleton JR, Adkins PRF, Goodell GM. Test agreement among biochemical methods, matrix-assisted laser desorption ionization–time of flight mass spectrometry, and 16S rRNA sequencing for identification of microorganisms isolated from bovine milk. J Clin Microbiol. (2019) 57:1–22. doi: 10.1128/JCM.01381-18
50. Nonnemann B, Lyhs U, Svennesen L, Kristensen KA, Klaas IC, Pedersen K. Bovine mastitis bacteria resolved by MALDI-TOF mass spectrometry. J Dairy Sci. (2019) 102:2515–24. doi: 10.3168/jds.2018-15424
51. Wald R, Hess C, Urbantke V, Wittek T, Baumgartner M. Characterization of staphylococcus species isolated from bovine quarter milk samples. Animals. (2019) 9:1–16. doi: 10.3390/ani9050200
52. Pumipuntu N. Staphylococcus argenteus: an emerging subclinical bovine mastitis pathogen in Thailand. Vet World. (2019) 12:1940–4. doi: 10.14202/vetworld.2019.1940-1944
53. Rosa NM, Agnoletti F, Lollai S, Tola S. Comparison of PCR-RFLP, API® 20 Strep and MALDI-TOF MS for identification of Streptococcus spp. collected from sheep and goat milk samples. Small Rumin Res. (2019) 180:35–40. doi: 10.1016/j.smallrumres.2019.09.023
54. Alnakip MEA, Rhouma NR, Abd-Elfatah EN, Quintela-Baluja M, Böhme K, Fernández-No I, et al. Discrimination of major and minor streptococci incriminated in bovine mastitis by MALDI-TOF MS fingerprinting and 16S rRNA gene sequencing. Res Vet Sci. (2020) 132:426–38. doi: 10.1016/j.rvsc.2020.07.027
55. Pappa E, Vougas K, Zoidakis J, Vastardis H. Proteomic advances in salivary diagnostics. Biochim Biophys Acta Proteins Proteom. (2020) 1868:140494. doi: 10.1016/j.bbapap.2020.140494
56. Liu WT, Lv YJ, Yang RC, Fu JY, Liu L, Wang H, et al. New insights into meningitic Escherichia coli infection of brain microvascular endothelial cells from quantitative proteomics analysis. J Neuroinflamm. (2018) 15:1–19. doi: 10.1186/s12974-018-1325-z
57. Lallemand E, Coiffier G, Arvieux C, Brillet E, Guggenbuhl P, Jolivet-Gougeon A, et al. Performance compared to direct examination, culture, and 16S rDNA PCR for the rapid diagnosis of bone and joint infections. Eur J Clin Microbiol Infect Dis. (2016) 35:857–66. doi: 10.1007/s10096-016-2608-x
58. Pérez-Llarena FJ, Bou G. Proteomics as a tool for studying bacterial virulence and antimicrobial resistance. Front Microbiol. (2016) 7:1–21. doi: 10.3389/fmicb.2016.00410
59. Katsafadou AI, Tsangaris GT, Billinis C, Fthenakis GC. Use of proteomics in the study of microbial diseases of small ruminants. Vet Microbiol. (2015) 181:27–33. doi: 10.1016/j.vetmic.2015.07.017
60. Yang Q, Farooq U, Chen W, Ullah MW, Wang S. Fluorimetric detection of single pathogenic bacterium in milk and sewage water using ph-sensitive fluorescent carbon dots and MALDI-TOF MS. Microorganisms. (2020) 8. doi: 10.3390/microorganisms8010053
61. Biswas K, Mackenzie BW, Waldvogel-Thurlow S, Middleditch M, Jullig M, Zoing M, et al. Differentially regulated host proteins associated with chronic rhinosinusitis are correlated with the sinonasal microbiome. Front Cell Infect Microbiol. (2017) 7:1–11. doi: 10.3389/fcimb.2017.00504
62. Bittaye M, Cash P. Streptococcus pneumoniae proteomics: determinants of pathogenesis and vaccine development. Expert Rev Proteomics. (2015) 12:607–21. doi: 10.1586/14789450.2015.1108844
63. Wareth G, Melzer F, Weise C, Neubauer H, Roesler U, Murugaiyan J. Proteomics-based identification of immunodominant proteins of Brucellae using sera from infected hosts points towards enhanced pathogen survival during the infection. Biochem Biophys Res Commun. (2015) 456:202–6. doi: 10.1016/j.bbrc.2014.11.059
64. Al-Wrafy F, Brzozowska E, Górska S, Drab M, Strus M, Gamian A. Identification and characterization of phage protein and its activity against two strains of multidrug-resistant Pseudomonas aeruginosa. Sci Rep. (2019) 9:1–14. doi: 10.1038/s41598-019-50030-5
65. Li C, Akuta T, Nakagawa M, Sato T, Shibata T, Maruyama T, et al. Agarose native gel electrophoresis for characterization of antibodies. Int J Biol Macromol. (2020) 151:885–90. doi: 10.1016/j.ijbiomac.2020.02.185
66. Sakuma C, Sato T, Shibata T, Nakagawa M, Kurosawa Y, Okumura CJ, et al. Western blotting analysis of proteins separated by agarose native gel electrophoresis. Int J Biol Macromol. (2021) 166:1106–10. doi: 10.1016/j.ijbiomac.2020.10.265
67. Pollock NL, Rai M, Simon KS, Hesketh SJ, Teo ACK, Parmar M, et al. SMA-PAGE: A new method to examine complexes of membrane proteins using SMALP nano-encapsulation and native gel electrophoresis. Biochim Biophys Acta Biomembr. (2019) 1861:1437–45. doi: 10.1016/j.bbamem.2019.05.011
68. Khan FA, Faisal M, Chao J, Liu K, Chen X, Zhao G, et al. Immunoproteomic identification of MbovP579, a promising diagnostic biomarker for serological detection of Mycoplasma bovis infection. Oncotarget. (2016) 7:39376–95. doi: 10.18632/oncotarget.9799
69. Khan S, Cole N, Hume EBH, Garthwaite LL, Nguyen-Khuong T, Walsh BJ, et al. Identification of pathogenic factors potentially involved in Staphylococcus aureus keratitis using proteomics. Exp Eye Res. (2016) 151:171–8. doi: 10.1016/j.exer.2016.08.016
70. Yu Y, Qian Y, Du D, Xu C, Dai C, Li Q, et al. SBP2 plays an important role in the virulence changes of different artificial mutants of Streptococcus suis. Mol BioSyst. (2016) 12:1948–62. doi: 10.1039/C6MB00059B
71. Nascimento Filho EG, Vieira ML, Teixeira AF, Santos JC, Fernandes LGV, Passalia FJ, et al. Proteomics as a tool to understand Leptospira physiology and virulence: recent advances, challenges and clinical implications. J Proteomics. (2018) 180:80–7. doi: 10.1016/j.jprot.2018.02.025
72. Turonova H, Haddad N, Hernould M, Chevret D, Pazlarova J, Tresse O. Profiling of campylobacter jejuni proteome in exponential and stationary phase of growth. Front Microbiol. (2017) 8:1–12. doi: 10.3389/fmicb.2017.00913
73. Guo L, Wang J, Gou Y, Tan L, Liu H, Pan Y, et al. Comparative proteomics reveals stress responses of Vibrio parahaemolyticus biofilm on different surfaces: Internal adaptation and external adjustment. Sci Total Environ. (2020) 731:138386. doi: 10.1016/j.scitotenv.2020.138386
74. Taniguchi T, Ohki M, Urata A, Ohshiro S, Tarigan E, Kiatsomphob S, et al. Detection and identification of adhesins involved in adhesion of Campylobacter jejuni to chicken skin. Int J Food Microbiol. (2021) 337:108929. doi: 10.1016/j.ijfoodmicro.2020.108929
75. Quesada-Gómez C, Murillo T, Arce G, Badilla-Lobo A, Castro-Peña C, Molina J, et al. Proteogenomic analysis of the Clostridium difficile exoproteome reveals a correlation between phylogenetic distribution and virulence potential. Anaerobe. (2020) 62:1–8. doi: 10.1016/j.anaerobe.2020.102151
76. Zhang H, Zhao G, Guo Y, Menghwar H, Chen Y, Chen H, et al. Mycoplasma bovis MBOV_RS02825 encodes a secretory nuclease associated with cytotoxicity. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17050628
77. Skipp PJS, Hughes C, McKenna T, Edwards R, Langridge J, Thomson NR, et al. Quantitative proteomics of the infectious and replicative forms of Chlamydia trachomatis. PLoS ONE. (2016) 11:1–17. doi: 10.1371/journal.pone.0149011
78. Abdou E, de Bagüés MPJ, Martínez-Abadía I, Ouahrani-Bettache S, Pantesco V, Occhialini A, et al. RegA plays a key role in oxygen-dependent establishment of persistence and in isocitrate lyase activity, a critical determinant of in vivo Brucella suis pathogenicity. Front Cell Infect Microbiol. (2017) 7:1–19. doi: 10.3389/fcimb.2017.00186
79. Kandil M, Khalil G, El-Attar E, Shehata G, Hassan S. Accuracy of heparin binding protein: as a new marker in prediction of acute bacterial meningitis. Brazilian J Microbiol. (2018) 49:213–9. doi: 10.1016/j.bjm.2018.05.007
80. Christensen DG, Meyer JG, Baumgartner JT, D'Souza AK, Nelson WC, Payne SH, et al. Identification of novel protein lysine acetyltransferases in Escherichia coli. bioRxiv. (2018) 9:1–23. doi: 10.1128/mBio.01905-18
81. Prust N, van der Laarse SAM, van den Toorn H, van Sorge NM, Lemeer S. In depth characterization of the Staphylococcus aureus phosphoproteome reveals new targets of Stk1. Mol Cell Proteom. (2020) 20:1–27. doi: 10.1074/mcp.RA120.002232
82. Enany S, Zakeer S, Sayed AA, Magdeldin S. Shotgun proteomic analysis of ESBL-producing and non-ESBL-producing Klebsiella Pneumoniae clinical isolates. Microbiol Res. (2020) 234:126423. doi: 10.1016/j.micres.2020.126423
83. Oviedo JM, Surmann K, Gorgojo JP, Valdez H, Dhople VM, Lamberti Y, et al. Shotgun proteomic analysis of Bordetella parapertussis provides insights into the physiological response to iron starvation and potential new virulence determinants absent in Bordetella pertussis. J Proteomics. (2019) 206:103448. doi: 10.1016/j.jprot.2019.103448
84. Behera P, Nikhil KC, Kumar A, Gali JM, De A, Mohanty AK, et al. Comparative proteomic analysis of Salmonella Typhimurium wild type and its isogenic fnr null mutant during anaerobiosis reveals new insight into bacterial metabolism and virulence. Microb Pathog. (2020) 140. doi: 10.1016/j.micpath.2019.103936
85. Kirubakar G, Schäfer H, Rickerts V, Schwarz C, Lewin A. Mutation on lysX from Mycobacterium avium hominissuis impacts the host–pathogen interaction and virulence phenotype. Virulence. (2020) 11:132–44. doi: 10.1080/21505594.2020.1713690
86. Mateos J, Estévez O, González-Fernández Á, Anibarro L, Pallarés Á, Reljic R, et al. High-resolution quantitative proteomics applied to the study of the specific protein signature in the sputum and saliva of active tuberculosis patients and their infected and uninfected contacts. J Proteomics. (2019) 195:41–52. doi: 10.1016/j.jprot.2019.01.010
87. Wu Z, Wang G, Wang W, Pan D, Peng L, Lian L. Proteomics analysis of the adhesion activity of Lactobacillus acidophilus ATCC 4356 upon growth in an intestine-like pH environment. Proteomics. (2018) 18:1–29. doi: 10.1002/pmic.201700308
88. Zhu C, Lv Y, Yang J, Bai Y, Ye J, Wang Z, et al. Proteomic alteration of porcine intestinal epithelial cells after pretreatment with Lactobacillus plantarum followed by infection with enterotoxigenic Escherichia coli F4. Vet Immunol Immunopathol. (2020) 222:109943. doi: 10.1016/j.vetimm.2019.109943
89. Qin X, He S, Zhou X, Cheng X, Huang X, Wang Y, et al. Quantitative proteomics reveals the crucial role of YbgC for Salmonella enterica serovar Enteritidis survival in egg white. Int J Food Microbiol. (2019) 289:115–26. doi: 10.1016/j.ijfoodmicro.2018.08.010
90. Wang Y, Li J, Zhang A, Zhu W, Zhang Q, Xu Z, et al. iTRAQ-based quantitative proteomic analysis reveals potential virulence factors of Erysipelothrix rhusiopathiae. J Proteomics. (2017) 160:28–37. doi: 10.1016/j.jprot.2017.03.004
91. Yang N, Liu Y, He P, Ke R, Zhao Y, Feng Y, et al. ITRAQ-based differential proteomic analysis reveals the pathways associated with tigecycline resistance in Acinetobacter baumannii. Cell Physiol Biochem. (2018) 51:1327–39. doi: 10.1159/000495551
92. Müller SA, Pernitzsch SR, Haange SB, Uetz P, von Bergen M, Sharma CM, et al. Stable isotope labeling by amino acids in cell culture based proteomics reveals differences in protein abundances between spiral and coccoid forms of the gastric pathogen Helicobacter pylori. J Proteomics. (2015) 126:34–45. doi: 10.1016/j.jprot.2015.05.011
93. Zhang Q, Cai Z, Lin H, Han L, Yan J, Wang J, et al. Expression, purification and identification of isotope-labeled recombinant cystatin C protein in Escherichia coli intended for absolute quantification using isotope dilution mass spectrometry. Protein Expr Purif. (2021) 178:105785. doi: 10.1016/j.pep.2020.105785
94. Kuleš J, Bilić P, Horvatić A, Kovačević A, Guillemin N, Ljubić BB, et al. Serum proteome profiling in canine chronic valve disease using a TMT-based quantitative proteomics approach. J Proteomics. (2020) 223:103825. doi: 10.1016/j.jprot.2020.103825
95. Chai YN, Qin J, Tong YL, Liu GH, Wang XR, Liu CY, et al. TMT proteomics analysis of intestinal tissue from patients of irritable bowel syndrome with diarrhea: implications for multiple nutrient ingestion abnormality. J Proteomics. (2021) 231:103995. doi: 10.1016/j.jprot.2020.103995
96. García-Hernández V, Sánchez-Bernal C, Schvartz D, Calvo JJ, Sanchez JC, Sánchez-Yagüe J, et al. Tandem mass tag (TMT) proteomic analysis during the early phase of experimental pancreatitis reveals new insights in the disease pathogenesis. J Proteomics. (2018) 181:190–200. doi: 10.1016/j.jprot.2018.04.018
97. Patel K, Singh M, Gowda H. Bioinformatics methods to deduce biological interpretation from proteomics data. Methods Mol Biol. (2017) 1549:147–61. doi: 10.1007/978-1-4939-6740-7_12
98. Khan S, Imran A, Khan AA, Kalam MA, Alshamsan A. Systems biology approaches for the prediction of possible role of Chlamydia pneumoniae proteins in the etiology of lung cancer. PLoS ONE. (2016) 11:1–13. doi: 10.1371/journal.pone.0148530
99. Uddin R, Masood F, Azam SS, Wadood A. Identification of putative non-host essential genes and novel drug targets against Acinetobacter baumannii by in silico comparative genome analysis. Microb Pathog. (2019) 128:28–35. doi: 10.1016/j.micpath.2018.12.015
100. Baarda BI, Martinez FG, Sikora AE. Proteomics, bioinformatics and structure-function antigen mining for gonorrhea vaccines. Front Immunol. (2018) 9:2793. doi: 10.3389/fimmu.2018.02793
101. Shirani I, Zhang H, Zhao G, Lu S, Marawan MA, Dawood A, et al. In silico identification of novel immunogenic secreted proteins of mycoplasma bovis from secretome data and experimental verification. Pathogens. (2020) 9:1–20. doi: 10.3390/pathogens9090770
102. Aswal M, Garg A, Singhal N, Kumar M. Comparative in-silico proteomic analysis discerns potential granuloma proteins of Yersinia pseudotuberculosis. Sci Rep. (2020) 10:1–13. doi: 10.1038/s41598-020-59924-1
103. Bhardwaj T, Haque S, Somvanshi P. Comparative assessment of the therapeutic drug targets of C. botulinum ATCC 3502 and C. difficile str. 630 using in silico subtractive proteomics approach. J Cell Biochem. (2019) 120:16160–84. doi: 10.1002/jcb.28897
104. Shahid F, Ashfaq UA, Saeed S, Munir S, Almatroudi A, Khurshid M. In silico subtractive proteomics approach for identification of potential drug targets in Staphylococcus saprophyticus. Int J Environ Res Public Health. (2020) 17:1–10. doi: 10.3390/ijerph17103644
105. Sohrabi SM, Mohammadi M, Tabatabaiepour SN, Tabatabaiepour SZ, Hosseini-Nave H, Soltani MF, et al. A systematic in silico analysis of the Legionellaceae family for identification of novel drug target candidates. Microb Drug Resist. (2019) 25:157–66. doi: 10.1089/mdr.2017.0328
106. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. (2014) 12:7–8. doi: 10.1038/nmeth.3213
107. Kelley L a, Mezulis S, Yates CM, Wass MN, Sternberg MJE. Europe PMC Funders Group The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc. (2015) 10:845–58. doi: 10.1038/nprot.2015.053
108. Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, et al. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. (2003) 19:163–4. doi: 10.1093/bioinformatics/19.1.163
109. von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, et al. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. (2005) 33:433–7. doi: 10.1093/nar/gki005
110. Aronson JK, Ferner RE. Biomarkers—a general review. Curr Protoc Pharmacol. (2017) 76:9.23.1–9.23.17. doi: 10.1002/cpph.19
111. Alharbi RA. Proteomics approach and techniques in identification of reliable biomarkers for diseases. Saudi J Biol Sci. (2020) 27:968–74. doi: 10.1016/j.sjbs.2020.01.020
112. El-Deeb WM, Elmoslemany AM. The diagnostic accuracy of acute phase proteins and proinflammatory cytokines in sheep with pneumonic pasteurellosis. PeerJ. (2016) 2016:1–12. doi: 10.7717/peerj.2161
113. Cifani P, Kentsis A. Towards comprehensive and quantitative proteomics for diagnosis and therapy of human disease. Proteomics. (2017) 17:1–24. doi: 10.1002/pmic.201600079
114. Li C, He X, Li H, Zhou Y, Zang N, Hu S, et al. Discovery and verification of serum differential expression proteins for pulmonary tuberculosis. Tuberculosis. (2015) 95:547–54. doi: 10.1016/j.tube.2015.06.001
115. Rizal MI, Soeroso Y, Sulijaya B, Assiddiq BF, Bachtiar EW, Bachtiar BM. Proteomics approach for biomarkers and diagnosis of periodontitis: systematic review. Heliyon. (2020) 6:e04022. doi: 10.1016/j.heliyon.2020.e04022
116. Fukuyama Y, Ojima-Kato T, Nagai S, Shima K, Funatsu S, Yamada Y, et al. Improved MALDI-MS method for the highly sensitive and reproducible detection of biomarker peaks for the proteotyping of Salmonella serotypes. J Mass Spectrom. (2019) 54:966–75. doi: 10.1002/jms.4469
117. Chenau J, Fenaille F, Caro V, Haustant M, Diancourt L, Klee SR, et al. Identification and validation of specific markers of bacillus anthracis spores by proteomics and genomics approaches. Mol Cell Proteom. (2014) 13:716–32. doi: 10.1074/mcp.M113.032946
118. Hokynar K, Korhonen S, Norja P, Paavonen J, Puolakkainen M. Antibody to Chlamydia trachomatis proteins, TroA and HtrA, as a biomarker for Chlamydia trachomatis infection. Eur J Clin Microbiol Infect Dis. (2017) 36:49–56. doi: 10.1007/s10096-016-2769-7
119. Sánchez-Jiménez MM, de la Cuesta Zuluaga JJ, Garcia-Montoya GM, Dabral N, Alzate JF, Vemulapalli R, et al. Diagnosis of human and canine Brucella canis infection: development and evaluation of indirect enzyme-linked immunosorbent assays using recombinant Brucella proteins. Heliyon. (2020) 6. doi: 10.1016/j.heliyon.2020.e04393
120. Kumaran SK, Bakar MFA, Mohd-Padil H, Mat-Sharani S, Sakinah S, Poorani K, et al. 3D modelling of the pathogenic Leptospira protein LipL32: a bioinformatics approach. Acta Trop. (2017) 176:433–9. doi: 10.1016/j.actatropica.2017.09.011
121. Pathak P, Kumar A, Sarangi PP, Bhagyawant S, Thavaselvam D. Cloning, expression and purification of virB10 protein of Brucella melitensis and evaluation of its role as a serological marker for Brucella infection in experimental and natural host. Protein Expr Purif. (2018) 145:53–8. doi: 10.1016/j.pep.2017.12.014
122. Valour F, Rasigade JP, Trouillet-Assant S, Gagnaire J, Bouaziz A, Karsenty J, et al. Delta-toxin production deficiency in Staphylococcus aureus: a diagnostic marker of bone and joint infection chronicity linked with osteoblast invasion and biofilm formation. Clin Microbiol Infect. (2015) 21:568.e1–e11. doi: 10.1016/j.cmi.2015.01.026
123. Xu S, Hou X, Sun L, Zhang J, Ji X, Wang X, et al. An immunoproteomic approach to identify antigenic proteins in Nocardia farcinica IFM 10152. Microb Pathog. (2019) 137:103705. doi: 10.1016/j.micpath.2019.103705
124. Polansky O, Seidlerova Z, Faldynova M, Sisak F, Rychlik I. Protein expression in the liver and blood serum in chickens in response to Salmonella Enteritidis infection. Vet Immunol Immunopathol. (2018) 205:10–6. doi: 10.1016/j.vetimm.2018.10.006
125. Liu J, Parrish JR, Hines J, Mansfield L, Finley RL, A. proteome-wide screen of Campylobacter jejuni using protein microarrays identifies novel and conformational antigens. PLoS ONE. (2019) 14:1–16. doi: 10.1371/journal.pone.0210351
126. Hong J, Dauros-Singorenko P, Whitcombe A, Payne L, Blenkiron C, Phillips A, et al. Analysis of the Escherichia coli extracellular vesicle proteome identifies markers of purity and culture conditions. J Extracell Vesicles. (2019) 8. doi: 10.1080/20013078.2019.1632099
127. Barbosa MS, dos Santos Alves RP, de Souza Rezende I, Pereira SS, Campos GB, Freitas LM, et al. Novel antigenic proteins of Mycoplasma agalactiae as potential vaccine and serodiagnostic candidates. Vet Microbiol. (2020) 251. doi: 10.1016/j.vetmic.2020.108866
128. Kai-Cheen A, Lay-Harn G. Comparison of aqueous soluble proteins profile of Mycobacterium tuberculosis H37Rv and H37Ra and a Malaysian clinical isolate. Biotechnol Appl Biochem. (2018) 12:1–18. doi: 10.1002/bab.1687
129. Cho YS, Jang YB, Lee SE, Cho JY, Ahn JM, Hwang I, et al. Short communication: proteomic characterization of tuberculin purified protein derivative from Mycobacterium bovis. Res Vet Sci. (2015) 101:117–9. doi: 10.1016/j.rvsc.2015.06.003
130. Liu R, Chen C, Cheng L, Lu R, Fu G, Shi S, et al. Ducks as a potential reservoir for Pasteurella multocida infection detected using a new rOmpH-based ELISA. J Vet Med Sci. (2017) 79:1264–71. doi: 10.1292/jvms.17-0124
131. Khalifeh Gholi M, Kalali B, Formichella L, Göttner G, Shamsipour F, et al. Helicobacter pylori FliD protein is a highly sensitive and specific marker for serologic diagnosis of H. pylori infection. Int J Med Microbiol. (2013) 303:618–23. doi: 10.1016/j.ijmm.2013.08.005
132. Shi W, Zhao L, Li S, Xu G, Zeng Y. Serological diagnosis of Mycoplasma pneumoniae infection by using the mimic epitopes. World J Microbiol Biotechnol. (2018) 34:1–9. doi: 10.1007/s11274-018-2467-y
133. Tsui PY, Tsai HP, Chiao DJ, Liu CC, Shyu RH. Rapid detection of Yersinia pestis recombinant fraction 1 capsular antigen. Appl Microbiol Biotechnol. (2015) 99:7781–9. doi: 10.1007/s00253-015-6663-5
134. Pflughoeft KJ, Mash M, Hasenkampf NR, Jacobs MB, Tardo AC, Mitchell Magee D, et al. Multi-platform approach for microbial biomarker identification using Borrelia burgdorferi as a model. Front Cell Infect Microbiol. (2019) 9:1–10. doi: 10.3389/fcimb.2019.00179
135. Ding H, Zhou Y, Wang H. Development of an indirect ELISA for detecting humoral immunodominant proteins of Mycoplasma hyopneumoniae which can discriminate between inactivated bacterin-induced hyperimmune sera and convalescent sera. BMC Vet Res. (2019) 15:1–8. doi: 10.1186/s12917-019-2077-4
136. Zhao G, Zhang H, Chen X, Zhu X, Guo Y, He C, et al. Mycoplasma bovis NADH oxidase functions as both a NADH oxidizing and O 2 reducing enzyme and an adhesin. Sci Rep. (2017) 7:1–13. doi: 10.1038/s41598-017-00121-y
137. Chen X, Huang J, Zhu H, Guo Y, Khan FA, Menghwar H, et al. P27 (MBOV_RS03440) is a novel fibronectin binding adhesin of Mycoplasma bovis. Int J Med Microbiol. (2018) 308:848–57. doi: 10.1016/j.ijmm.2018.07.006
138. Khan FA, Zhao G, Guo Y, Faisal M, Chao J, Chen X, et al. Proteomics identification and characterization of MbovP730 as a potential DIVA antigen of Mycoplasma bovis. Oncotarget. (2018) 9:28322–36. doi: 10.18632/oncotarget.22265
139. Wang J, Li Y, Pan L, Li J, Yu Y, Liu B, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) moonlights as an adhesin in Mycoplasma hyorhinis adhesion to epithelial cells as well as a plasminogen receptor mediating extracellular matrix degradation. Vet Res. (2021) 52:1–14. doi: 10.1186/s13567-021-00952-8
140. Wang J, Zhu X, Xiong X, Ge P, Liu H, Ren N, et al. Identification of potential urine proteins and microRNA biomarkers for the diagnosis of pulmonary tuberculosis patients. Emerg Microbes Infect. (2018) 7. doi: 10.1038/s41426-018-0066-5
141. Gerlach C, Škultéty L, Henning K, Neubauer H, Mertens K. Coxiella burnetii immunogenic proteins as a basis for new Q fever diagnostic and vaccine development. Acta Virol. (2017) 61:377–90. doi: 10.4149/av_2017_320
142. Gul H, Ali SS, Saleem S, Khan S, Khan J, Wadood A, et al. Subtractive proteomics and immunoinformatics approaches to explore Bartonella bacilliformis proteome (virulence factors) to design B and T cell multi-epitope subunit vaccine. Infect Genet Evol. (2020) 85:104551. doi: 10.1016/j.meegid.2020.104551
143. Garay-Baquero DJ, White CH, Walker NF, Tebruegge M, Schiff HF, Ugarte-Gil C, et al. Comprehensive plasma proteomic profiling reveals biomarkers for active tuberculosis. JCI Insight. (2020) 5:1–20. doi: 10.1172/jci.insight.137427
144. Liang L, Juarez S, Nga TVT, Dunstan S, Nakajima-Sasaki R, Huw Davies D, et al. Immune profiling with a Salmonella Typhi antigen microarray identifies new diagnostic biomarkers of human typhoid. Sci Rep. (2013) 3:1–10. doi: 10.1038/srep01043
145. Hufnagel K, Hoenderboom B, Harmel C, Rohland JK, van Benthem BHB, Morré SA, et al. Chlamydia trachomatis whole-proteome microarray analysis of the netherlands chlamydia cohort study. Microorganisms. (2019) 7:1–18. doi: 10.3390/microorganisms7120703
146. Mukherjee P, Mani S. Methodologies to decipher the cell secretome. Biochim Biophys Acta Proteins Proteom. (2013) 1834:2226–32. doi: 10.1016/j.bbapap.2013.01.022
147. Dwivedi P, Alam SI, Tomar RS. Secretome, surfome and immunome: emerging approaches for the discovery of new vaccine candidates against bacterial infections. World J Microbiol Biotechnol. (2016) 32:1–9. doi: 10.1007/s11274-016-2107-3
148. Voros A, Delongchamp J, Saleh M. The Secretome of Mycoplasma capricolum subsp. capricolum in neutral and acidic media. J Proteomics Bioinforma. (2015) 8:155–63.
149. Cheng S, Wang L, Liu Q, Qi L, Yu K, Wang Z, et al. Identification of a novel Salmonella type III effector by quantitative secretome profiling. Mol Cell Proteom. (2017) 16:2219–28. doi: 10.1074/mcp.RA117.000230
150. Luu LDW, Octavia S, Zhong L, Raftery M, Sintchenko V, Lan R. Characterisation of the Bordetella pertussis secretome under different media. J Proteomics. (2017) 158:43–51. doi: 10.1016/j.jprot.2017.02.010
151. Wen YT, Wang JS, Tsai SH, Chuan CN, Wu JJ, Liao PC. Label-free proteomic analysis of environmental acidification-influenced Streptococcus pyogenes secretome reveals a novel acid-induced protein histidine triad protein A (HtpA) involved in necrotizing fasciitis. J Proteomics. (2014) 109:90–103. doi: 10.1016/j.jprot.2014.06.026
152. Boysen A, Borch J, Krogh TJ, Hjernø K, Møller-Jensen J. SILAC-based comparative analysis of pathogenic Escherichia coli secretomes. J Microbiol Methods. (2015) 116:66–79. doi: 10.1016/j.mimet.2015.06.015
153. Choi CW, Lee YG, Kwon SO, Kim HY, Lee JC, Chung YH, et al. Analysis of Streptococcus pneumoniae secreted antigens by immuno-proteomic approach. Diagn Microbiol Infect Dis. (2012) 72:318–27. doi: 10.1016/j.diagmicrobio.2011.12.013
154. Yu Y, Champer J, Kim J. Analysis of the surface, secreted, and intracellular proteome of Propionibacterium acnes. EuPA Open Proteom. (2015) 9:1–7. doi: 10.1016/j.euprot.2015.06.003
155. Naz A, Awan FM, Obaid A, Muhammad SA, Paracha RZ, Ahmad J, et al. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect Genet Evol. (2015) 32:280–91. doi: 10.1016/j.meegid.2015.03.027
156. Zhao G, Zhu X, Zhang H, Chen Y, Schieck E, Hu C, et al. Novel secreted protein of Mycoplasma bovis mbovp280 induces macrophage apoptosis through CRYAB. Front Immunol. (2021) 12:1–15. doi: 10.3389/fimmu.2021.619362
157. Zhang H, Hu G, Lu D, Zhao G, Zhang Y, Zubair M, et al. Comparative secretome analyses of mycoplasma bovis virulent and attenuated strains revealed MbovP0145 as a promising diagnostic biomarker. Front Vet Sci. (2021) 8:666769. doi: 10.3389/fvets.2021.666769
158. Lu D, Zhang H, Zhang Y, Zhao G, Khan FA, Chen Y, et al. Secreted mbovp0145 promotes il-8 expression through its interactive β-actin and mapk activation and contributes to neutrophil migration. Pathogens. (2021) 10. doi: 10.3390/pathogens10121628
159. Scanlan E, Yu L, Maskell D, Choudhary J, Grant A. A quantitative proteomic screen of the Campylobacter jejuni flagellar-dependent secretome. J Proteomics. (2017) 152:181–7. doi: 10.1016/j.jprot.2016.11.009
160. Vargas-Romero F, Mendoza-Hernández G, Suárez-Güemes F, Hernández-Pando R, Castañón-Arreola M. Secretome profiling of highly virulent Mycobacterium bovis 04-303 strain reveals higher abundance of virulence-associated proteins. Microb Pathog. (2016) 100:305–11. doi: 10.1016/j.micpath.2016.10.014
161. Ayalew S, Confer AW, Hartson SD, Canaan PJ, Payton M, Couger B. Proteomic and bioinformatic analyses of putative Mannheimia haemolytica secretome by liquid chromatography and tandem mass spectrometry. Vet Microbiol. (2017) 203:73–80. doi: 10.1016/j.vetmic.2017.02.011
162. Paes JA, Lorenzatto KR, de Moraes SN, Moura H, Barr JR, Ferreira HB. Secretomes of Mycoplasma hyopneumoniae and Mycoplasma flocculare reveal differences associated to pathogenesis. J Proteomics. (2017) 154:69–77. doi: 10.1016/j.jprot.2016.12.002
163. Jhelum H, Sori H, Sehgal D. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci Rep. (2018) 8:1–17. doi: 10.1038/s41598-018-25865-z
164. Li P, Tian M, Hu H, Yin Y, Guan X, Ding C, et al. Lable-free based comparative proteomic analysis of secretory proteins of rough Brucella mutants. J Proteom. (2019) 195:66–75. doi: 10.1016/j.jprot.2019.01.008
165. Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, et al. Secreted effectors encoded within and outside of the francisella pathogenicity island promote intramacrophage growth. Anal Chem. (2017) 25:368–79.
166. Tripathi DK, Srivastava K, Nagpal KL, Shukla PK, Srivastava KK. Exploration of some new secretory proteins to be employed for companion diagnosis of Mycobacterium tuberculosis. Immunol Lett. (2019) 209:67–74. doi: 10.1016/j.imlet.2019.03.010
167. Gillespie JJ, Kaur SJ, Sayeedur Rahman M, Rennoll-Bankert K, Sears KT, Beier-Sexton M, et al. Secretome of obligate intracellular Rickettsia. FEMS Microbiol Rev. (2015) 39:47–80. doi: 10.1111/1574-6976.12084
168. Pinaud L, Ferrari ML, Friedman R, Jehmlich N, Von Bergen M, Phalipon A, et al. Identification of novel substrates of Shigella T3SA through analysis of its virulence plasmid-encoded secretome. PLoS ONE. (2017) 12:1–23. doi: 10.1371/journal.pone.0186920
169. Zakharzhevskaya NB, Vanyushkina AA, Altukhov IA, Shavarda AL, Butenko IO, Rakitina DV, et al. Outer membrane vesicles secreted by pathogenic and nonpathogenic Bacteroides fragilis represent different metabolic activities. Sci Rep. (2017) 7:1–16. doi: 10.1038/s41598-017-05264-6
170. Altindis E, Fu Y, Mekalanos JJ. Proteomic analysis of Vibrio cholerae outer membrane vesicles. Proc Natl Acad Sci USA. (2014) 111:E1548–56. doi: 10.1073/pnas.1403683111
171. Wang X, Thompson CD, Weidenmaier C, Lee JC. Release of Staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat Commun. (2018) 9. doi: 10.1038/s41467-018-03847-z
172. Gómez-Gascón L, Luque I, Tarradas C, Olaya-Abril A, Astorga RJ, Huerta B, et al. Comparative immunosecretome analysis of prevalent Streptococcus suis serotypes. Comp Immunol Microbiol Infect Dis. (2018) 57:55–61. doi: 10.1016/j.cimid.2018.06.006
173. Rasheed MA Qi J, Zhu X, Chenfei H, Menghwar H, Khan FA, et al. Comparative genomics of mycoplasma bovis strains reveals that decreased virulence with increasing passages might correlate with potential virulence-related factors. Front Cell Infect Microbiol. (2017) 7:1–14. doi: 10.3389/fcimb.2017.00177
174. Gupta N, Khatoon N, Mishra A, Verma VK, Prajapati VK. Structural vaccinology approach to investigate the virulent and secretory proteins of Bacillus anthracis for devising anthrax next-generation vaccine. J Biomol Struct Dyn. (2020) 38:4895–905. doi: 10.1080/07391102.2019.1688197
175. da Silva LB, Menezes MC, Kitano ES, Oliveira AK, Abreu AG, Souza GO, et al. Leptospira interrogans secreted proteases degrade extracellular matrix and plasma proteins from the host. Front Cell Infect Microbiol. (2018) 8:92. doi: 10.3389/fcimb.2018.00092
176. Zubair M, Khan FA, Menghwar H, Faisal M, Ashraf M, Rasheed MA, et al. Progresses on bacterial secretomes enlighten research on Mycoplasma secretome. Microb Pathog. (2020) 144:104160. doi: 10.1016/j.micpath.2020.104160
177. Juibari AD, Ramezani S, Rezadoust MH. Bioinformatics analysis of various signal peptides for periplasmic expression of parathyroid hormone in E. coli. J Med Life. (2019) 12:184–91. doi: 10.25122/jml-2018-0049
178. Nguyen TT, Chon TS, Kim J, Seo YS, Heo M. Comparative and bioinformatics analyses of pathogenic bacterial secretomes identified by mass spectrometry in Burkholderia species. J Microbiol. (2017) 55:568–82. doi: 10.1007/s12275-017-7085-0
179. Santos AR, Carneiro A, Gala-García A, Pinto A, Barh D, Barbosa E, et al. The corynebacterium pseudotuberculosis in silico predicted pan-exoproteome. BMC Genomics. (2012) 13:S6. doi: 10.1186/1471-2164-13-S5-S6
180. Zhao L, Chen J, Sun J, Zhang D. Multimer recognition and secretion by the non-classical secretion pathway in Bacillus subtilis. Sci Rep. (2017) 7:1–18. doi: 10.1038/srep44023
181. Kim JH, Lee J, Park J, Gho YS. Gram-negative and gram-positive bacterial extracellular vesicles. Semin Cell Dev Biol. (2015) 40:97–104. doi: 10.1016/j.semcdb.2015.02.006
182. Chernov VM, Chernova OA, Mouzykantov AA, Efimova IR, Shaymardanova GF, Medvedeva ES, et al. Extracellular vesicles derived from Acholeplasma laidlawii PG8. Sci World J. (2011) 11:1120–30. doi: 10.1100/tsw.2011.109
Keywords: proteomics, secretome, diseases, pathogenesis, diagnostics
Citation: Zubair M, Wang J, Yu Y, Faisal M, Qi M, Shah AU, Feng Z, Shao G, Wang Y and Xiong Q (2022) Proteomics approaches: A review regarding an importance of proteome analyses in understanding the pathogens and diseases. Front. Vet. Sci. 9:1079359. doi: 10.3389/fvets.2022.1079359
Received: 26 October 2022; Accepted: 28 November 2022;
Published: 15 December 2022.
Edited by:
Farhan Anwar Khan, University of Agriculture, PakistanReviewed by:
Rajwali Khan, University of Agriculture, PakistanCopyright © 2022 Zubair, Wang, Yu, Faisal, Qi, Shah, Feng, Shao, Wang and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Wang, bGF0YXNhMDIyN0B5ZWFoLm5ldA==; Qiyan Xiong, cWl5YW54aW9uZ25qQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.