
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 24 November 2022
Sec. Livestock Genomics
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.1068882
 Shihao Wei†
Shihao Wei† Xiaolong Kang†
Xiaolong Kang† Chaoyun Yang†
Chaoyun Yang† Feng WangTianshu DaiXingru GuoZiming Ma
Feng WangTianshu DaiXingru GuoZiming Ma Chenglong Li
Chenglong Li Hongxi Zhao*
Hongxi Zhao* Xingang Dan*
Xingang Dan*Seasonal estrus is an important factor limiting the fertility of some animals such as sheep. Promoting estrus in the anestrus season is one of the major ways in improving the fecundity of seasonally breeding animals. The pineal-hypothalamus-pituitary-ovary (PHPO) axis plays a decisive role in regulating animal reproduction. However, the molecular mechanisms by which the PHPO axis regulates seasonal reproduction in animals are not well understood, especially in Tan sheep. To this end, we collected pineal, hypothalamus, pituitary and ovary tissues from Tan sheep during estrus and anestrus for RNA-Sequencing, and performed bioinformatics analysis on the entire regulatory axis of the pineal-hypothalamic-pituitary-ovary (PHPO). The results showed that 940, 1,638, 750, and 971 DEGs (differentially expressed genes, DEGs) were identified in pineal, hypothalamus, pituitary and ovary, respectively. GO analysis showed that DEGs from PHPO axis-related tissues were mainly enriched in “biological processes” such as transmembrane transport, peptide and amide biosynthesis and DNA synthesis. Meanwhile, KEGG enrichment analysis showed that the bile acid secretion pathway and the neuroactive ligand-receptor interaction pathway were significantly enriched. Additionally, four potential candidate genes related to seasonal reproduction (VEGFA, CDC20, ASPM, and PLCG2) were identified by gene expression profiling and protein-protein interaction (PPI) analysis. These findings will contribute to be better understanding of seasonal reproduction regulation in Tan sheep and will serve as a useful reference for molecular breeding of high fertility Tan sheep.
Some animals in nature undergo some behavioral changes with the change of seasons, such as fish migration, turtle hibernation, and bird migration. It has been found that animal reproduction activity varies with the seasons, which is not only related to the ambient temperature and light, but also closely related to the endogenous regulation of animals themselves. Studies have shown that genes involved in the regulation of animal reproduction change rhythmically with the seasons and play an important role in the regulation of seasonal reproduction (1). The reproductive activity of seasonal breeding animals is governed by a regulatory network of the Pineal—Hypothalamus—Pituitary—Ovary (PHPO) axis. The mammalian pineal is a neuroendocrine sensor whose primary function is to convert photoperiodic information into nocturnal hormonal signals for melatonin synthesis and secretion (2). The hormonal activity of the pineal is influenced by the light and dark cycles, as well as seasonal cycle, and it plays an important role in the seasonal reproductive shift of animals (3). The hypothalamus, an important part of the central nervous system, senses changes in the light and dark signals from the external environment via the retina-supraoptic nucleus bundle, synchronizing the biological rhythms with environmental light and dark changes (4). The pituitary, an important reproductive endocrine regulatory organ in animals, secretes adrenocorticotropic hormone, thyroid stimulating hormone, luteinizing hormone, follicle stimulating hormone, growth hormone and prolactin (5). Among them, LH and FSH play a key role in the seasonal breeding of animals. Furthermore, the pituitary gland is not only controlled by the hypothalamus, but also influences ovarian function through follicle stimulating hormone and luteinizing hormone, and it serves as a link in the hypothalamic-pituitary-ovarian (HPO) axis. The ovary is an important reproductive organ in female mammals and its function is regulated by the HPO axis (6). Moreover, the ovary can regulate the synthesis and secretion of reproduction-related hormones in the hypothalamus and pituitary via positive and negative feedback mechanisms (7).
In the past, studies have found a number of important genes and pathways regulating animal reproduction by high-throughput sequencing in the HPO axis of female animals (8, 9). In sheep, many studies have focused on transcriptome sequencing analysis of muscle tissue (10), adipose tissue (11) or skin tissue (12) and have identified a number of candidate markers. Meanwhile, RNA-Seq was also been used to investigating reproductive regulation mechanism of animals. Ullah et al. (13) have discovered six miRNAs associated with estrus including miR-143 and miR-199a after transcriptome sequencing analysis of the pituitary of sheep during estrus and anestrus. Also, Di et al. (14) used transcriptome sequencing to screen for differentially expressed miRNAs between the pineal of estrus and anestrus sheep and showed that miR-89 negatively regulates the expression of mRNA involved in the melatonin synthesis rate-limiting enzyme arylalkylamine N-acetyltransferase, which in turn regulates reproduction in sheep. None of these studies, however, examined the pineal along with the hypothalamus, pituitary and ovary, which might have missed some novel findings in the regulation of seasonal reproduction in animals. In this study, mRNA from the pineal, hypothalamus, pituitary, and ovarian tissues of female Tan sheep during estrus and anestrus were first sequenced separately. These sequencing results were then analyzed by bioinformatics method to uncover the key genes and signal pathways that regulate seasonal reproduction in the entire PHPO axis of Tan sheep.
All methods of this study were performed in accordance with the Regulations for the Administration of Laboratory Animal Affairs (revised, 2017) providing animal experiments and animal care. All experimental animal studies were reviewed and approved by the Animal Welfare Committee of Ningxia University (permit number NXUC20180306).
In this study, 12 Tan sheep (14 months old) in similar body condition during the anestrus season (early April) were selected and kept individually for 1 week at a Tan sheep breeding farm in Ningxia Autonomous Region, and three female Tan sheep were proved not to be in heat by test rams again. Subsequently, frozen tubes of pineal, hypothalamus, pituitary and ovarian tissues were collected from each of the three Tan sheep after humanitarian execution at the slaughterhouse and immediately cast into liquid nitrogen for backup until RNA extraction. At the same time, three female Tan sheep were screened from the remaining nine Tan sheep during the same year's anestrus season (late August, 18 months old) by means of a test ram, and the pineal, hypothalamus, pituitary gland and ovary tissues were collected in frozen tubes after slaughter at the slaughterhouse, and immediately placed in liquid nitrogen until RNA extraction.
Total RNA was extracted from all samples according to the instructions of TRIZOL RNA Extraction Kit (Invitrogen, Carlsbad, CA, USA). The integrity and quality of sample RNA were assessed by the Agilent 2,100 bio analyze system (Agilent Technologies, Santa Clara, CA, USA), and samples RNA that passed the test were used for subsequent library construction.
The NEBNext® Ultra™ RNA Library Prep Kit was used for library construction. mRNAs with polyA tails was enriched by Oligo (dT) magnetic beads and resulting RNA was subsequently randomly interrupted with divalent cations in NEB Fragmentation Buffer. The fragmented mRNA was used as a template, and random oligonucleotides were used as primers to synthesize the first strand of cDNA in M-MuLV reverse transcriptase system, followed by RNaseH degradation of RNA strands and dNTP synthesis in DNA polymerase I system. The purified double-stranded cDNAs were end-repaired, A-tailed and connected to sequencing adapters, and the cDNAs of about 200 bp were screened for PCR amplification using AMPure XP beads. After that, the PCR products were purified again using AMPure XP beads to finally obtain the library.
After constructing the library, it was quantified with a Qubit 2.0 Fluorometer, diluted to 1.5 ng/μl, and tested for the insert size with an Agilent 2,100 bioanalyzer. Subsequently, qRT-PCR was used to accurately quantify the effective concentration of the library (above 2 nM) to ensure the library's quality.
The sequenced fragments were converted into fastq format reads by CASAVA base identification of the images from the high-throughput sequencer. After obtaining the raw data to ensure quality and reliability of the data, Trim-galore software (version 0.6.6, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used to remove N-containing reads, filter reads with adapters and remove low-quality reads. To obtain the comparison efficiency of each sample reads and the position of reads on the genome, all trimmed data were mapped to the sheep reference genome Ovis aries (ftp:/ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/298/735/ GCA000298735.2Oarv4.0).
Gene expression levels were quantified using the feature Counts (version 1.5.0-p3, http://subread.sourceforge.net/) tool in subread software, and gene expression values for RNA-seq were calculated using FPKM. DEGs were detected using DESeq2 (version1.24.0, http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) with differential gene screening criteria of |log2(FoldChange)| ≥ 3 and padj ≤ 0.01. GO and KEGG analyzes were performed on the DAVID (version 2021, https://david.ncifcrf.gov/) website.
The STEM (Short Time-series Expression Miner, version 1.3.13, http://www.sb.cs.cmu.edu/stem) analysis program was used to identify significant gene expression profiles and to compare and visualize clusters of genes with similar expression trends in estrus and anestrus. Each gene was assigned to the closest profile using a Pearson correlation-based distance metric. To determine the significance level of the clustering profiles, alignment-based tests were used to quantify the expected number of genes that would be assigned to each profile. The gene expression levels used for analysis are expressed as FPKM values, with a P value < 0.05 in the clustering profile considered significantly enriched. Where the standardization method used for STEM analysis was chosen as “Log normalize data” and the number of trends for analysis using the “TEM Clustering Method” was chosen as “50,” other parameters were selected as default values.
To elucidate the potential relationships between DEGs and different tissues over time, we analyzed colored Clusters using STEM. DEGs with significant differences and similar trends contained in colored clusters (25, 24, 8, 35) were then subjected to protein-protein interaction (PPI) analysis in the STRING database (version 11.5, https://string-db.org/). Cluster 25, 24, 8, and 35 were examined with confidence criteria of 0.7, 0.92, 0.5, and 0.9, respectively. The Cytoscape tool (version 3.9.0, https://cytoscape.org/) was used to further visualize the interaction networks. The CytoHubba plugin's network topology analysis approach (Degree) was used to determine the core genes.
RNA was extracted from four tissue samples of six Tan sheep stored in liquid nitrogen, and after determination of satisfactory concentration and purity, diluted to 1 μg-μl−1 with double-distilled water. First strand of cDNA was prepared using the PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Perfect Real Time, Cat#RR047A) kit. For each tissue, six DEGs were randomly chosen to design unique primers, with the β-actin gene serving as an internal reference gene (Supplementary Table S1). RT-qPCR analysis of DEGs from four Tan sheep tissue was performed with the TB Green® Premix Ex Taq™ II (Takara, Tli RNaseH Plus, Cat#RR820A) kit. The reaction system was: 2 × TB Green Premix Ex Taq II 10 μl, Primer F 0.8 μl, Primer R 0.8 μl, RNase Free ddH2O 6.4 μl, cDNA template 2 μl; reaction conditions: pre-denaturation at 95 °C for 30 s, denaturation at 95 °C for 10 s, annealing at 60 °C 30 s, total 40 cycles. Three replicates of each sample were used to calculate the relative expression by the 2−ΔΔCt method. In order to confirm the accuracy of the RNA-Seq results, their results of RT-qPCR were compared with those of RNA-Seq.
Statistical processing of RT-qPCR results was performed using SPSS (version 19.0, International Business Machines Corporation). Student's t-test for independent samples was used to test for differences between the two groups. The differences were statistically significant (P < 0.05). Data are expressed as mean ± standard deviation.
The average number of reads for raw and clean reads was over 76 million and 73.5 million, respectively. After alignment with the Ovis aries genome, an average alignment rate of 89.49% was obtained for all samples, with a minimum value of 88.52% and a maximum value of 92.31% (Supplementary Table S2). Therefore, the quality of data can meet the requirements for the subsequent analysis of DEGs between the estrus and anestrus groups of Tan sheep.
To investigate DEGs in four tissues of Tan sheep during estrus and anestrus, gene expression levels were quantified by FPKM in the pineal, hypothalamus, pituitary and ovary. A total of 940 (up-regulated 779, down-regulated 161), 1,638 (up-regulated 1,214, down-regulated 424), 750 (up-regulated 482, down-regulated 268), and 971 (up-regulated 364, down-regulated 607) DEGs were found in the pineal, hypothalamus, pituitary and ovary tissues in estrus and anestrus periods, respectively (Figure 1, Supplementary Table S3). According to Venn diagram analysis, a total of 25 DEGs were co-expressed in the four tissues, with the number of tissue-specific DEGs being 381, 923, 363, and 649 in the pineal, hypothalamus, pituitary and ovary, respectively. This indicates significant changes in gene transcription on the PHPO axis during the transition between breeding seasons in Tan sheep (Figure 2). In order to examine the expression patterns of DEGs in different samples, hierarchical clustering analysis was employed (Figure 3). The results showed similar expression patterns in the pineal, hypothalamus and pituitary tissues, but distinct expression patterns in the ovarian tissue, which could be explained by the specificity of ovarian tissue and variations in ovarian development and ovulatory cycles in Tan sheep.
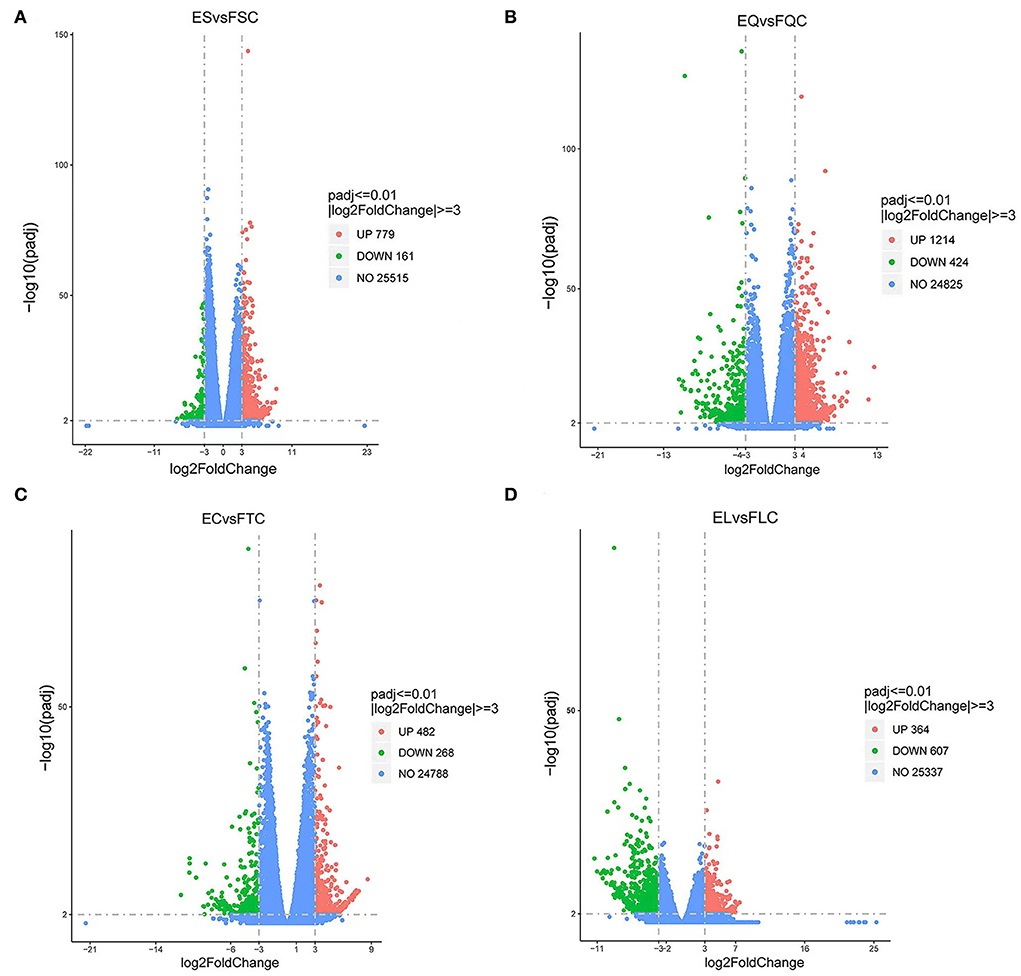
Figure 1. Volcano map of differentially expressed genes. (A) Pineal, (B) Hypothalamus, (C) Pituitary, and (D) Ovary map of differentially expressed genes in the ovary. Blue dots indicate genes that did not show significant changes between estrus group (EG) and anestrus group (AG) samples. Green dots represent down-regulated genes and red dots represent up-regulated genes.
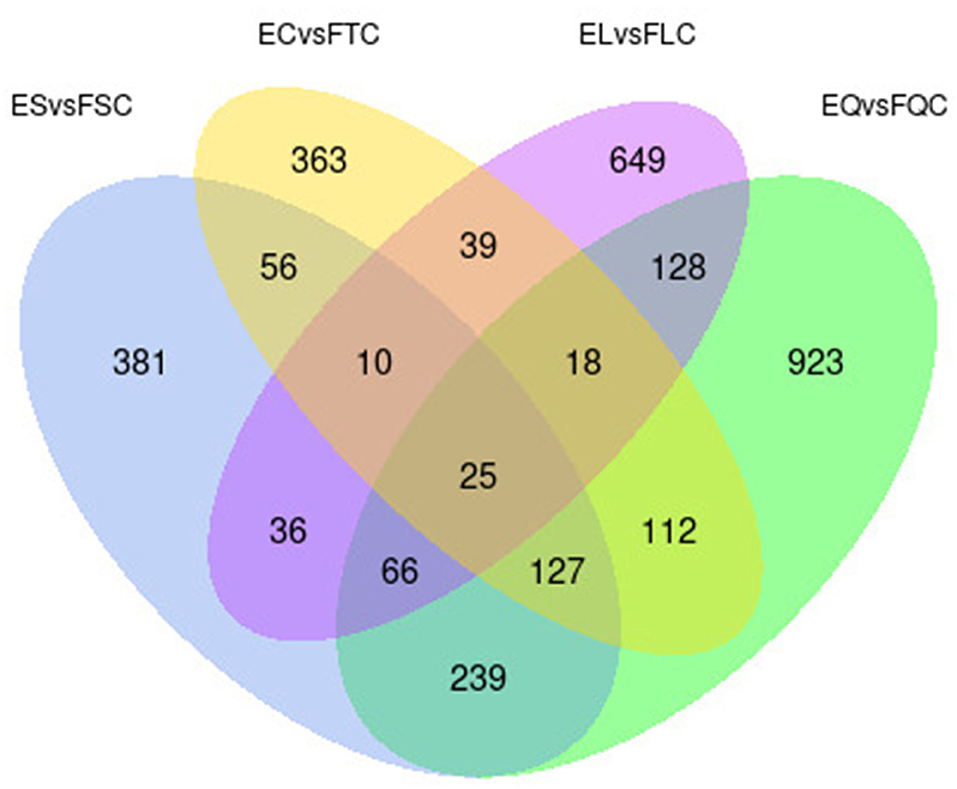
Figure 2. Venn diagram of differentially expressed genes in four tissues.
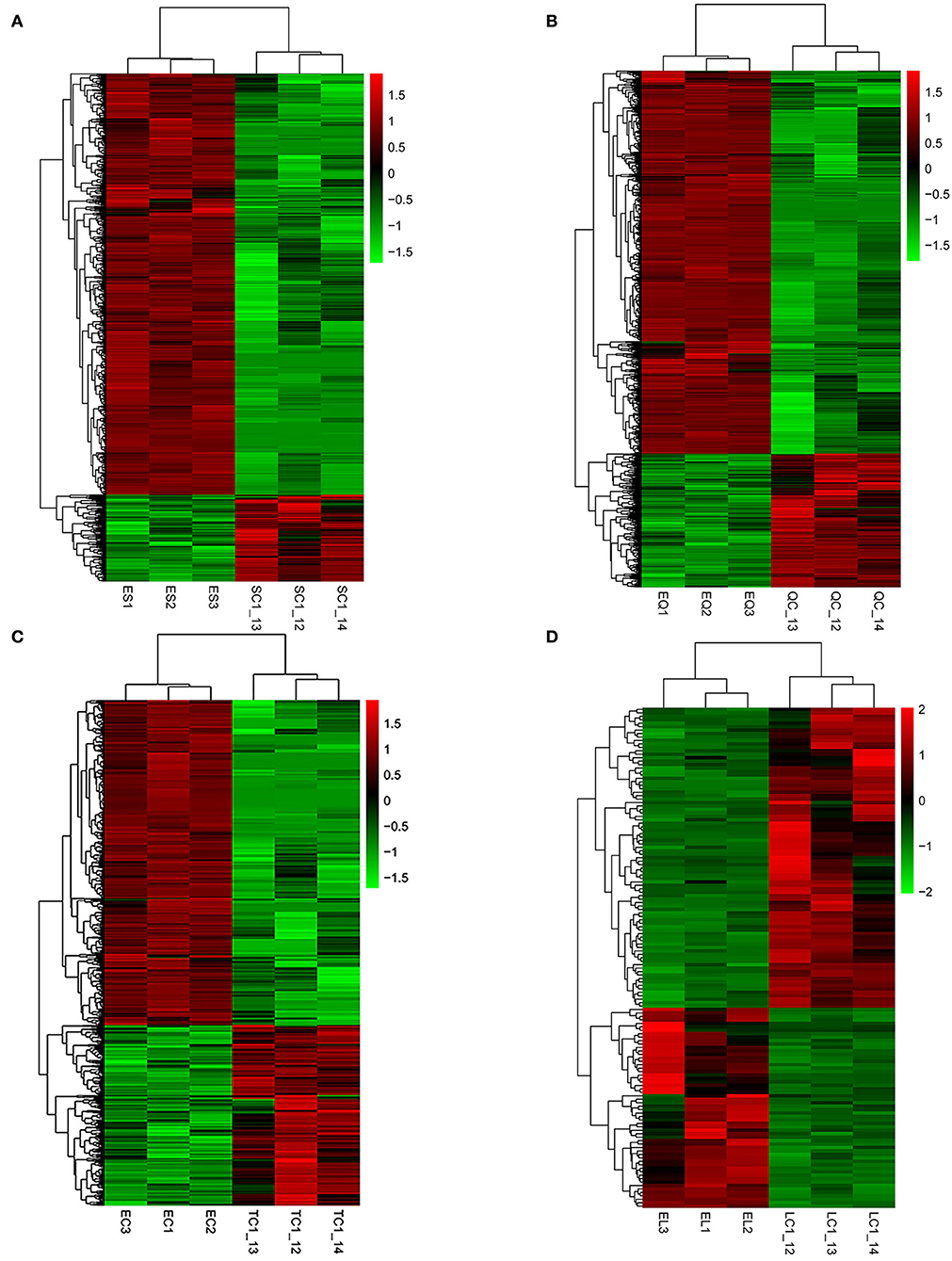
Figure 3. Hierarchical clustering analysis of differentially expressed genes. (A–D) Represent the pineal, hypothalamus, pituitary and ovarian tissue, respectively. The colored bars indicate the expression level. Green represents genes with low expression levels and red represents genes with high expression.
To understand the molecular functions of DEGs at the transcriptional level of the PHPO axis, DEGs were annotated using GO enrichment (Figure 4, Supplementary Table S4). The 940 DEGs on the pineal gland were found to be enriched to 502 GO entries, including 258 biological processes (BP), 63 cellular components (CC), and 181 molecular functions (MF). Processes such as DNA synthesis, translation and metabolism, peptide and amide synthesis and metabolism were predominant on the BP. It is primarily connected to the mitochondrial proton transport ATP synthase complex, riboprotein complex, virion, extracellular area, and other parts in the CC. In MF, it mostly relates to the activities of ribonucleases, endonucleases, endogenous enzyme activity, structural components of ribosomes, hormone action, etc.
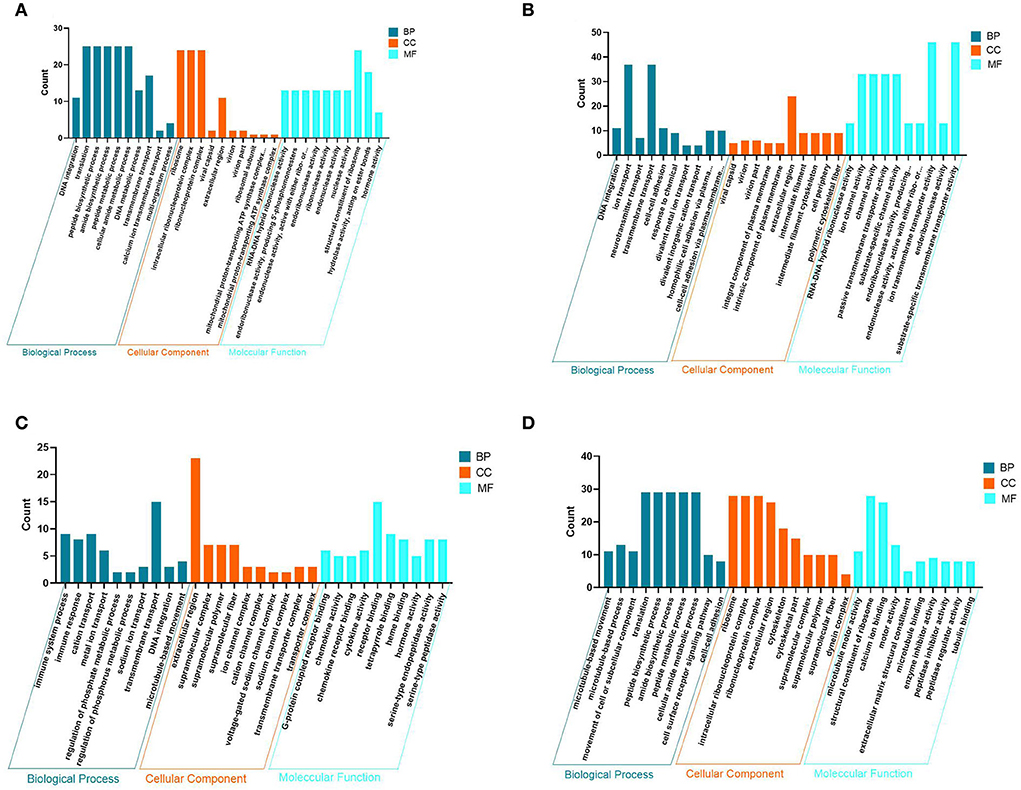
Figure 4. Map of GO entries for differentially expressed genes. (A–D) Represent the pineal, hypothalamus, pituitary and ovarian tissue, respectively. Dark blue indicates biological processes; yellow indicates cellular components; light blue indicates molecular functions.
There are 1,638 DEGs enriched to 646 GO entries in the hypothalamus, including 325 BP, 84 CC and 237 MF. The enrichment of DEGs on the hypothalamus is associated with processes such as transport of neurotransmitters, ions and other substances, cell adhesion and other processes. on the CC, there are mainly components of the virion, shell, intrinsic and intrinsic components of the plasma membrane, extracellular regions, intermediate filaments and intermediate filament cytoskeleton. The MF category is associated with RNA-DNA hybrid ribonuclease activity, endonuclease activity, ion channel activity, substrate specific channel activity, and transmembrane transporter activity.
In the pituitary, 750 DEGs were enriched to 514 GO entries, including 225 BP, 72 CC, and 187 MF. BP was mainly enriched to processes such as immune response, ion transport, DNA synthesis, regulation of phosphate metabolic processes and microtubule movement. It was mainly associated with components of ion and sodium channel transporter complexes, extracellular regions, supramolecular complexes, fibers and polymers On the CC. Additionally, there are some category mainly associated with G-protein coupled receptors, chemokine receptors, tetrapyrrole and heme binding, chemokines, cytokines, hormones and serine-type endopeptidase activity in the MF.
In addition, 971 DEGs were enriched to 561 GO entries, including 259 BP, 89 CC, 213 MF in the ovaries. In BP, it was mainly enriched in processes such as movement of microtubules and cellular or subcellular components, peptide and amide synthesis and metabolism, cell surface receptor signaling pathways, cell-cell adhesion. It was mainly associated with ribosomes, ribonucleoproteins, and Intracellular ribonucleoprotein complexes, extracellular regions, skeletal fractions, supramolecular fibers, complexes and polymers, and other components in the CC. While in MF, DEG category is mainly enriched to molecular functions such as serine-type peptidase, hydrolase endopeptidase and endopeptidase inhibitor and regulator activity, nucleoside-triphosphatase and transmembrane receptor protein kinase activity. As a result, it is clear that DNA synthesis, extracellular regions, enzyme and hormone activities are the most common GO entries in the four tissues of the PHPO axis.
To further determine the role of specific signaling pathways in the regulation of seasonal reproduction in Tan sheep, we completed an analysis of the KEGG pathway enrichment of DEGs in the PHPO axis of Tan sheep during estrus and anestrus, and the top 20 pathways in the four tissues are displayed in the graphs (Figure 5, Supplementary Table S5). The ribosome signaling pathway had the most DEGs (14) in the pineal gland, and the Ras signaling pathway, relaxin signaling, and circadian regulatory pathways are likely to be important in the regulation of seasonal anestrus in Tan sheep. The neuroactive ligand-receptor pathway has the most DEGs (15) in the hypothalamus, followed by the Notch signaling pathway, ovarian steroidogenesis, and the calcium signaling pathway, which were related to reproduction. In the pituitary, the cytokine-cytokine receptor interaction pathway had the most DEGs (12), whereas the P53 signaling pathway, cell adhesion molecules (CAMs), the NF-kappa B signaling system, and oocyte meiosis were involved in reproduction regulation. In addition, the largest number of DEGs in the ovary was in the human papillomavirus infection pathway (16), and the reproduction-related pathways included the “Hippo” signaling pathway, CAMs, arachidonic acid metabolism, and AMPK signaling pathway. Notably, the bile secretion pathway was the only pathway that was co-enriched in the four tissues during estrus and diestrus (Table 1). The neuroactive ligand-receptor pathway was also found to be co-enriched in pineal, hypothalamic and pituitary tissues (Table 2). Additionally, we also discovered that adenosine triphosphate-binding transporter protein and the ECM-receptor interaction pathway were co-enriched in hypothalamic, pineal and ovarian tissues.
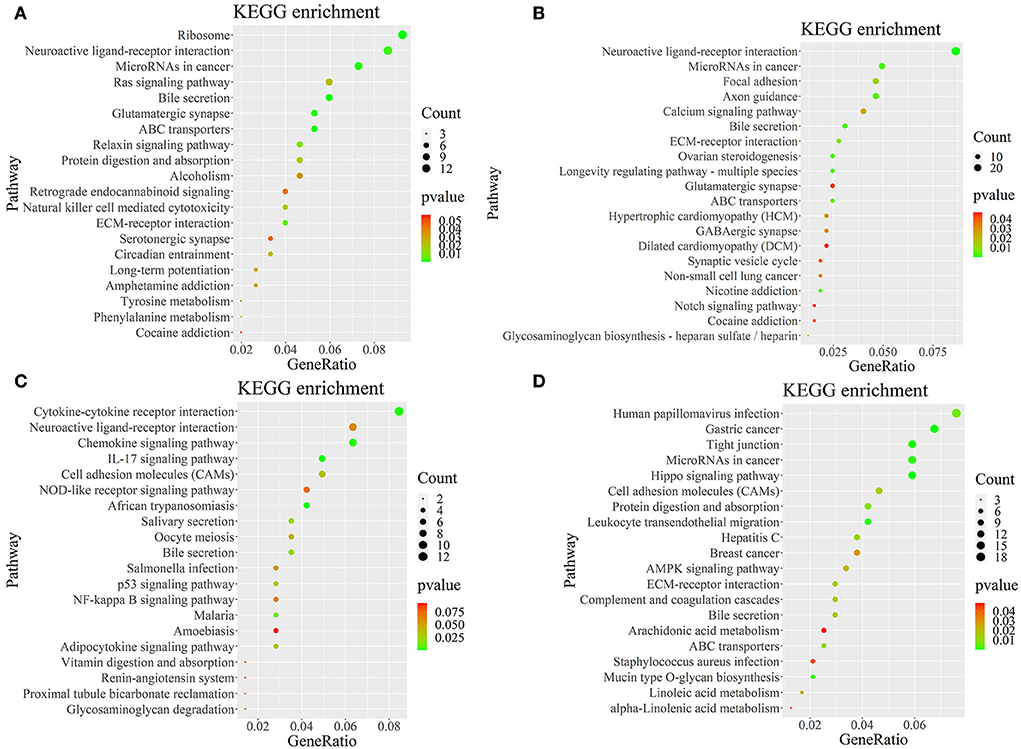
Figure 5. KEGG pathway map of differentially expressed genes. (A–D) Represent the pineal, hypothalamus, pituitary and ovarian tissue, respectively. Horizontal coordinates indicate enrichment factors and vertical coordinates indicate GO terms. The color of the dots corresponds to the range of P-value, and the size of the dots indicates how many differentially expressed genes are in the pathway.
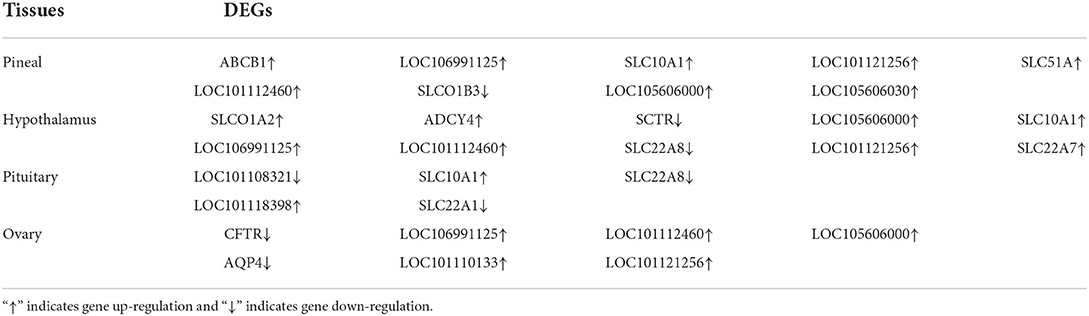
Table 1. DEGs significantly enriched in bile secretion pathway.
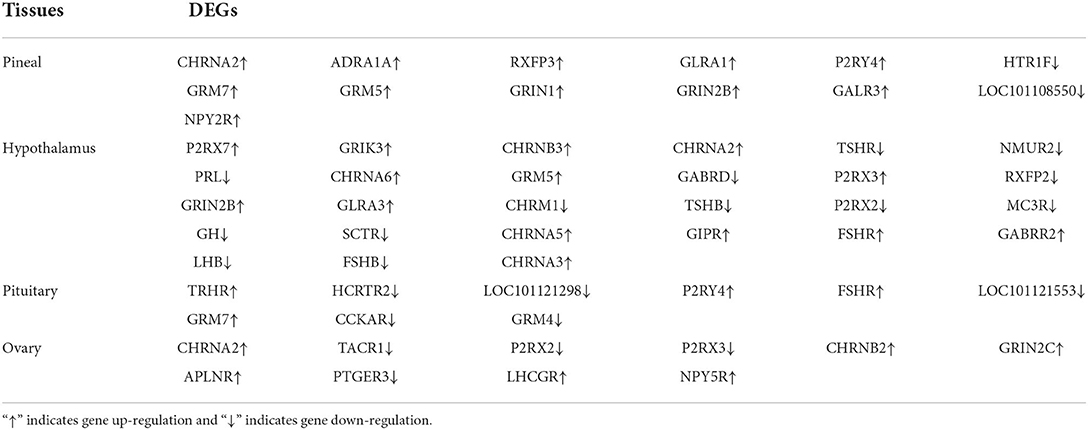
Table 2. DEGs significantly enriched in neuroactive ligand-receptor interaction pathway.
Analysis of gene expression profiles using the STEM program allowed identification of potential pathways and DEGs within the PHPO axis that regulate estrus in Tan sheep. A total of 50 modules were obtained from 0 to 49 after the analysis, and these modules could be rearranged according to the number of genes assigned and the significance P value (Figure 6, Supplementary Table S6). The top seven colored modules in the top were significantly different (P < 0.05), these seven significant modules were 25, 24, 8, 35, 39, 33, and 12, while the remaining white modules were not significantly different (P > 0.05). The gene expression trends in the colored modules 25, 24, 8, and 35 were similar, while those in the colored modules 8 and 39 were opposite. As a result, the DEGs discovered from the colored modules 25, 24, 8, and 35 with similar and considerably divergent gene expression trends can be subjected to bioinformatics analysis to further investigate the core genes regulating seasonal reproduction and core genes in Tan sheep (Supplementary Figure S1).
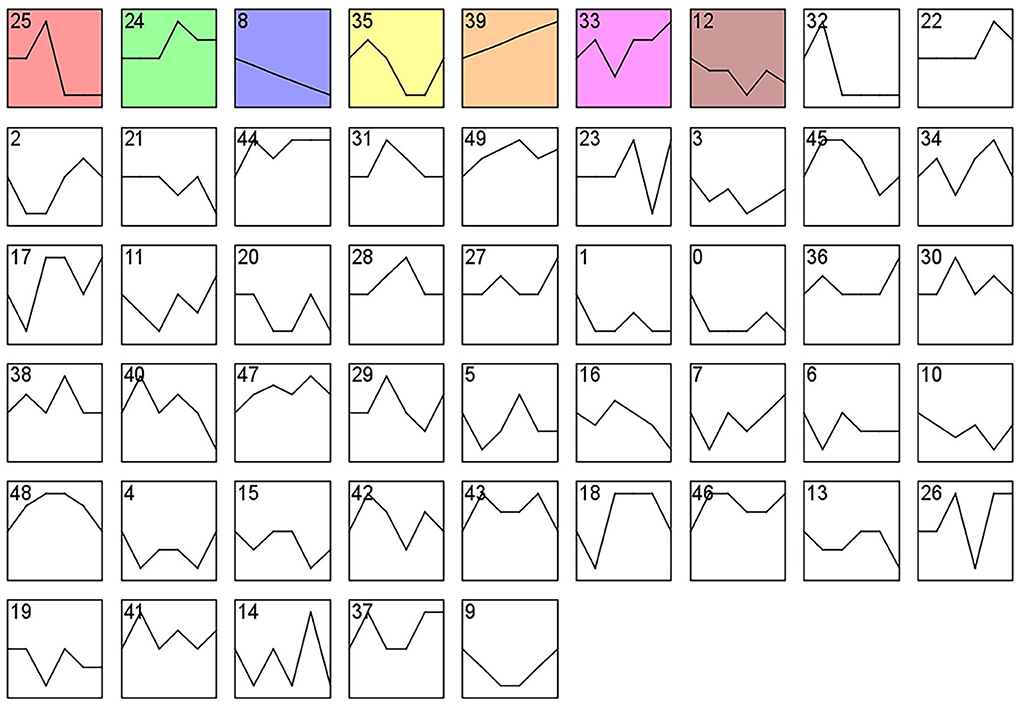
Figure 6. Gene expression profile clusters. Differential gene expression patterns on PHPO axis-related tissues. The vertical axis indicates gene expression levels. Blocks represent differential genes with the same expression pattern. White blocks indicate P-values >0.05 and colored blocks indicate P-values < 0.05.
We constructed four separate protein interaction networks (Figure 7) from the DEGs identified in the colored Cluster (25, 24, 8, 35). The DEGs encoding proteins in the PHPO axis tissue were visualized by PPI to identify regulatory networks within the PHPO axis that may regulate seasonal breeding of Tan sheep (Table 3). Cluster 25 generates a PPI network with 10 nodes and 10 edges, Cluster 24 generates a PPI network with 16 nodes and 41 edges, and Cluster 8 and 35 both generate a PPI network with 11 nodes and 12 edges. Subsequently, the core genes were screened from the top-ranked DEGs by using the Degree analysis method in the CytoHubba plugin. As a result, there core genes including VEGFA, CDC20, PLCG2, ASPM were identified as key candidate DEGs within the PHPO axis in Cluster 25, 24, 8, and 35. VEGFA was found in the pituitary and VEGFA is able to regulate the development of blood vessels near the corpus luteum and follicles. CDC20 was found to inhibit oocyte meiosis in the pituitary gland and ASPM in the ovary, while ASPM promoted oocyte meiosis. In addition, PLCG2 was found in the hypothalamus, and PLCG2 regulates the development of the uterine muscles and epididymis in animals.
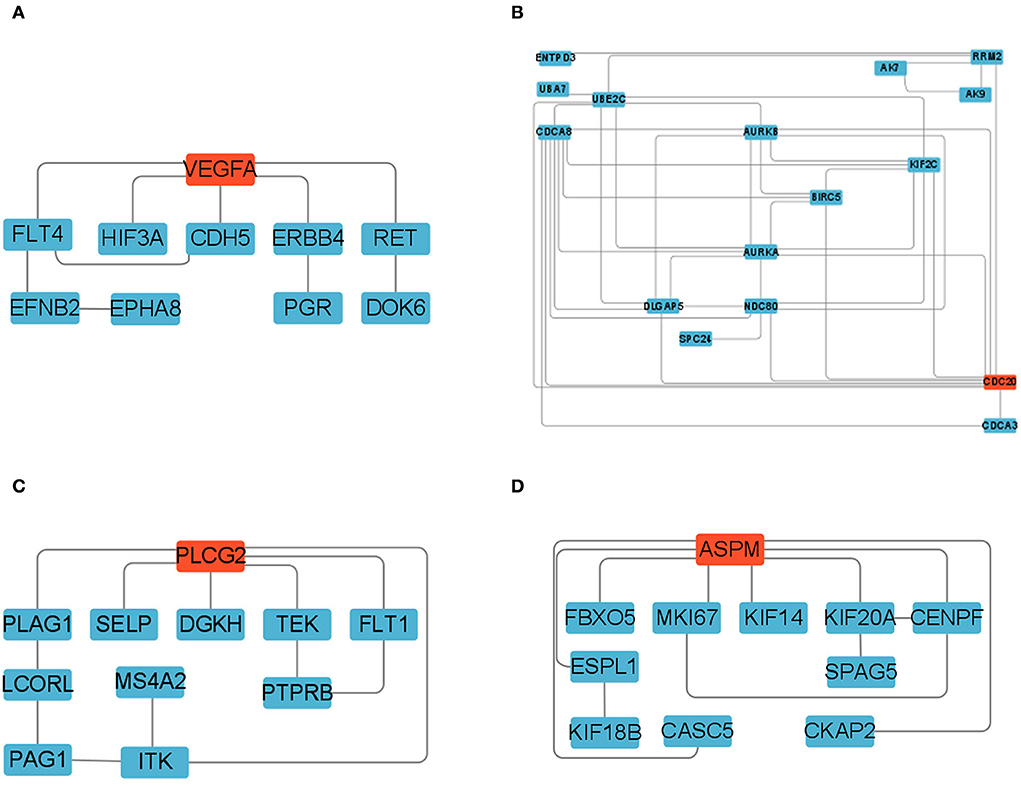
Figure 7. Protein-protein interaction network diagram. (A–D) Represent clusters 25, 24, 8, and 35, respectively. Key candidate genes are indicated in red, and differentially expressed genes in the network are indicated in blue.
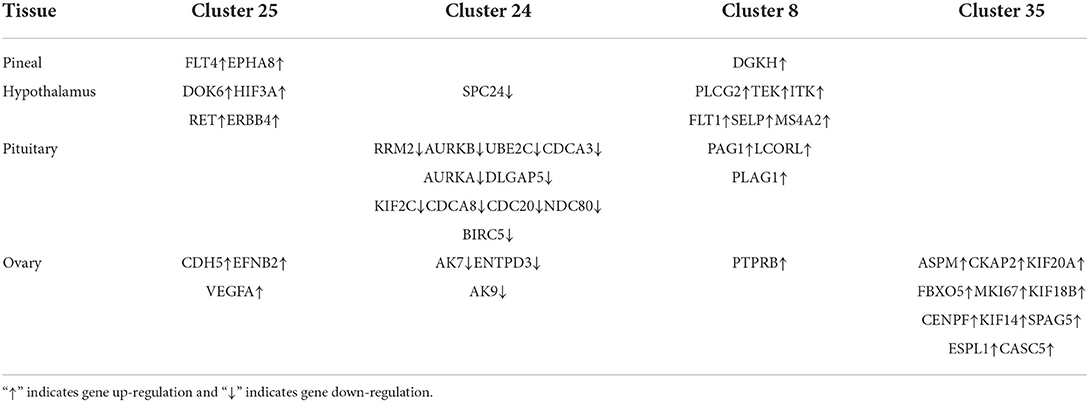
Table 3. The key DEGs enriched in PHPO axis-related tissues.
To verify the differentially expressed genes identified by transcriptome sequencing, six differentially expressed genes were randomly selected on each of the four tissues of the PHPO axis, and gene expression was confirmed by RT-qPCR using β-actin as an internal reference gene. The results showed consistent gene expression trends between both RT-qPCR and RNA-Seq (Figure 8). Therefore, it indicates that the RNA-Seq results are reliable and can be used for bioinformatics analysis of sequencing results.
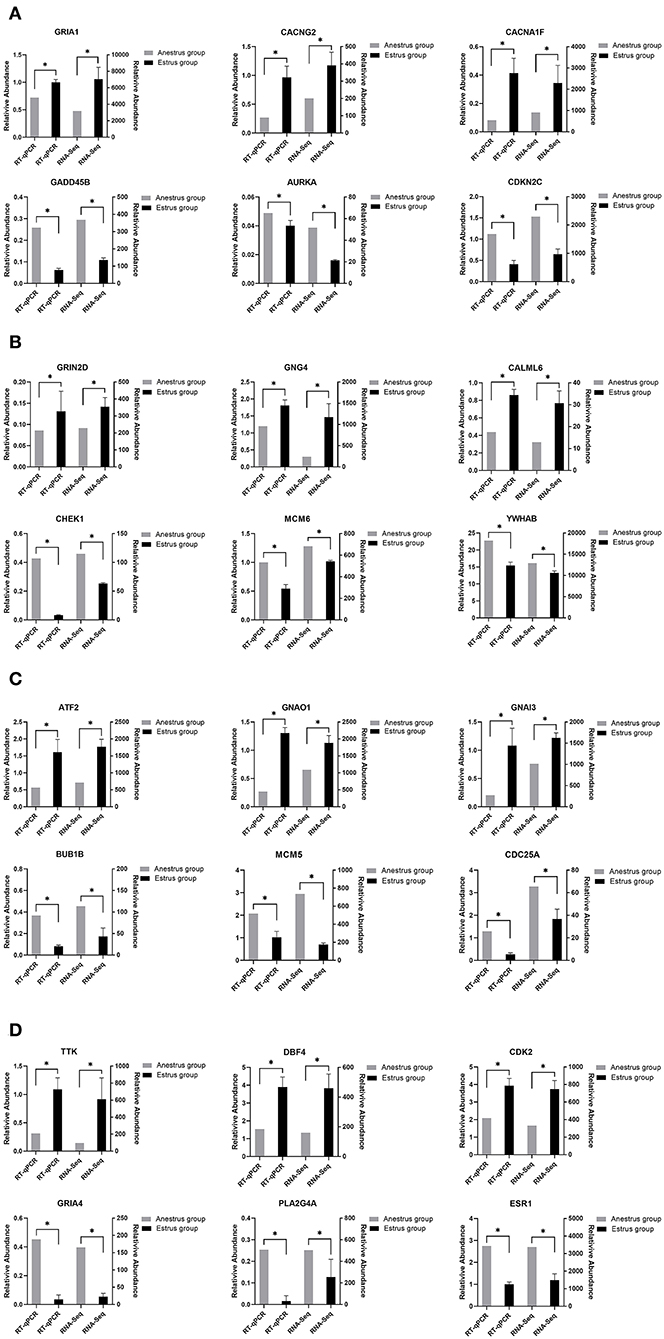
Figure 8. This figure shows the gene expression levels between RNA-Seq and qRT-PCR in four tissues: (A) Pineal, (B) Hypothalamus, (C) Pituitary, and (D) Ovary.
It is critical to investigate the seasonal estrus regulating mechanism in Tan sheep in order to improve their reproductive efficiency. The gene regulatory network is a continuous and complicated dynamic system, and gene regulation is a dynamic event that varies with the environment. Researchers can use RNA-Seq to uncover the gene regulatory network of the animal organism (17). The network pathways of gene regulation can be identified by sequencing the transcriptome of tissues or cells under various conditions, which allows biological analysis of the data to look for differential genes and important signaling pathways that are enriched. Transcriptome sequencing can provide insight into the variations in gene expression levels in the pineal, hypothalamus, pituitary, and ovarian tissues of Tan sheep during estrus and anestrus, which can serve as foundation for elucidating the molecular mechanisms of seasonal breeding regulation in Tan sheep.
The length of external light and the hormones generated by the reproductive system have an impact on the reproductive activity of Tan sheep. Previous research on sheep mainly focused on adipose tissue (18) or skin tissue (19) to identify the molecular pathways related with economic features. Another studies also determined some key genes and signaling pathways relating to the reproductive regulation of Tan sheep in pituitary (16) or ovarian (20) in sheep. In contrast, we first performed a combined transcriptome sequencing analysis across the pineal-hypothalamic-pituitary-ovarian axis and identified some hub genes and key signaling pathways regulating seasonal breeding in Tan sheep, this can provide some insight into the molecular regulation mechanism of seasonal reproduction in Tan sheep. We screened 940, 1,638, 750, and 971 DEGs in the pineal, hypothalamus, pituitary and ovary of Tan sheep during estrus and anestrus, respectively. This is the first study that, to our knowledge, examined the variations in gene expression and regulatory networks across the entire PHPO axis by RNA-seq during estrus and anestrus in Tan sheep.
In the pineal gland, hypothalamus, pituitary, and ovarian tissues of Tan sheep, the bile acid secretion pathway was identified as a common signaling pathway by the KEGG pathway analysis. Nuclear receptors (NRs) are a family of transcription factors that include endocrine receptors activated by steroids and thyroid hormones, which regulate target genes though binding its ligands and can play a role in biological processes such as energy metabolism, reproduction, and development (21). FXR, the bile acid nuclear receptor, has been found to be expressed in the mouse testis and to influence reproduction in male mice by regulating the expression of the pluripotency marker Lin28 in germ cells (22). It has also been demonstrated that FXR is also expressed in mouse ovarian granulosa cells and that FXR can regulate the function of granulosa cells by acting on target genes such as SHP and FABP6 (23). Another study discovered that the bile acids can be transported from the blood to the follicular fluid and have an impact on the reproductive system (24). Schuermann et al. (25) used metabolomics to show that metabolites like bile acids are present in the follicular fluid of cows and suggested that these metabolites may be involved in regulating reproductive performance at different physiological stages in cows. Also, studies in humans have found that total bile acid levels in human follicular fluid are twofold higher than in serum, and that bile acid synthesis may compete with steroid hormone synthesis in follicular and granulosa cells (26). Furthermore, total bile acid levels in dominant follicle follicular fluid were higher than in non-pregnant cows, and bile acid transport proteins and receptors were higher in dominant follicle granulosa cells (27). Yang et al. (28) also found differences in bile acid metabolites in follicular fluid between healthy human ovaries and polycystic ovary syndrome, suggesting that bile acid metabolism in follicles may affect follicular development. Similar to the above results, we found a common bile acid secretion pathway in the pineal-hypothalamic-pituitary-ovarian axis between the estrus group and anestrus group in Tan sheep, as well as enrichment of DEGs in the pathway, suggesting that the bile acid secretion pathway is also likely to be involved in relevant reproductive events and may regulate seasonal reproduction across the PHPO axis in Tan sheep.
In addition, we found that the neuroactive ligand-receptor interaction pathway was enriched in three tissues: pineal, hypothalamus and pituitary. The neuroactive ligand-receptor interaction pathway regulates reproductive activity according to transcriptomic studies on ducks (15, 29), chickens (30), and fish (31, 32). Another study found that nutritional restriction affects sow estrus via the neuroactive ligand-receptor interaction pathway (33). Similarly, mRNA transcriptome sequencing of the goat hypothalamus revealed that the neuroactive ligand-receptor interaction pathway influences sexual maturation (34). As a result, we propose that the neuroactive ligand-receptor interaction pathway may also be an important in the pineal-hypothalamic-pituitary (PHP) axis of Tan sheep, which may regulate seasonal breeding in Tan sheep.
STEM allows for the analysis of biological data gene expression clustering in a short period of time (35). Pizzi et al. (36) suggested that genes with similar expression have similar biological functions and work in concert. Fan et al. (37) used STEM to examine genes associated with mammary development at peak, mid and late lactation in dairy cows and found that DEGs are mainly involved in apoptosis and energy metabolism, suggesting the reliability of the STEM analysis method. In the present study, we obtained four colored Clusters with the same trend (25, 24, 8, and 35) by STEM analysis of DEGs cross PHPO axis in Tan sheep. PPI analysis was performed on the DEGs contained in the four Clusters, and four key core genes were displayed, namely VEGFA, CDC20, ASPM, and PLCG2.
We discovered VEGFA in the ovary, which, along with its receptor FLT4, is the central protein in Cluster 25 protein network. The network includes six DEGs encoding proteins of pineal and hypothalamic, and three DEGs encoding proteins of ovarian. The establishment of the peripheral vascular network is tightly linked to the growth and development of associated tissues in animal reproduction (38). VEGFA, as one of the VEGF family, is mainly involved in angiogenesis, and VEGFA can control the growth of blood vessels close to follicles and corpus luteum in the ovary (39–41). Numerous studies have shown that the development of vasculature in reproduction-related tissues helps to enhance the synthesis and transmission of endocrine factors, promoting follicular development and luteal formation (42, 43). Castle-Miller et al. (44) discovered that melatonin might modulate gonadotropin gene expression during the breeding and non-breeding seasons by regulating VEGF synthesis. Additionally, it was discovered that the localization of VEGFA and its receptor FLT4 within the relevant tissues can alter with different estrous cycles of animals (45, 46). This further suggests that VEGFA genes may play an important role in regulating seasonal breeding of animals. In our study, VEGFA and the FLT4 were found to have comparable expression profiles in Cluster 25, and the expression of DEGs was upregulated in the PPI network formed by Cluster 25, indicating that these DEGs may be crucial in the regulation of animal seasonal breeding. In addition, three signal transduction routes are also involved in the VEGF signaling system (Supplementary Figure S2), the first of which is the Ca2+ signaling pathway, which controls mesenchymal cell production of steroid hormones as well as cell autophagy, apoptosis, and proliferation (47, 48). Another is the MAPK/extracellular signal-regulated kinase pathway, which is essential for oocyte maturation (49, 50). There is also a phosphatidylinositol 3-kinase/protein kinase signaling pathway that promotes primordial follicle activation and reduces apoptosis in sheep (51, 52). As a result, we hypothesize that there may be a network of VEGFA signal pathway regulating seasonal breeding in the PHO axis of Tan sheep. Of course, more research is needed to uncover the molecular network mechanism of VEGFA regulation of seasonal breeding in Tan sheep.
In addition, we discovered that the DEGs contained in the PPI network formed by Cluster 24 and 35 were primarily located in the pituitary and ovaries. Both CDC20 and ASPM are associated with meiosis, but interestingly, CDC20 inhibits meiosis while ASPM promotes it. CDC20 is a co-activator of the anaphase-promotion complex/cyclosome (APC/C) during mitosis and can maintain the genome by controlling the spindle assembly checkpoint (53). The activation of APC/C by CDC20 is a critical stage in the transition from meiosis I to meiosis II and homolog segregation in oocyte meiosis (54). This shows that CDC20 plays an important role in regulating mammalian follicle development and oocyte maturation. It has been demonstrated that CDC20 is able to maintain oocyte meiosis arrest in pig (55), mice (56), and cattle (57). ASPM is found in the mammalian cell intermediate (58) and is localized at both poles of the spindle during cell mitosis (59). According to the research, ASPM is essential for meiotic spindle assembly and meiotic progression in mouse oocytes (60). Additionally, it has also been found that estrogen has a conserved depressed effect on ASPM expression in primates (61). Therefore, we speculate that it is possible that CDC20 and ASPM may form e a dynamic balance in the oocyte division cycle, and hence may play an important role in seasonal reproduction of Tan sheep by regulating follicle development.
In Cluster 8, PLCG2 (the gene encoding phospholipase Cγ2) was found to be located at the core of the PPI network. It was found that PLCG2 may be involved in the regulation of placental function in goats (62). Additionally, PLCG2 has also been discovered to control uterine muscle activity in laying hens (63). Meanwhile, Környei et al. (64) discovered that PLCG2 is also expressed in human myometrium, which may be involved in the regulation of myometrial cell differentiation, maturation and uterine tissue growth. In addition, it was also demonstrated that PLCG2 can regulate the development of epididymis in mice, and that male mice lacking PLCG2 are sterile (65). Accordingly, we supposed that PLCG2 may affect animal reproduction by regulating the growth and development of animal reproductive organs. In present study, we discovered that the DEGs in the PPI network, which consisted of Cluster 8 with PLCG2 as the core gene, were all upregulated between the two controls and were dispersed cross all four tissues. Therefore, we speculate that PLCG2 may similarly play a role controlling seasonal reproduction of Tan sheep along the PHPO axis, however this remains to be proven in future studies.
In this study, RNA-Seq was used to identify genes and signaling pathways associated with the regulation of seasonal reproduction in the PHPO axis of Tan sheep. We discovered that two pathways, bile acid secretion and neuroactive ligand-receptor interaction, may be related to the regulation of seasonal reproduction in Tan sheep. We also identified four core genes, VEGFA, CDC20, ASPM, and PLCG2, which are closely associated with the regulation of seasonal reproduction in Tan sheep. Our findings can provide the foundation for further research in the molecular regulation mechanisms of seasonal reproduction, as well as certain references for molecular breeding of high-fertility in Tan sheep. Although we have discovered core genes and key signaling pathways that regulate seasonal reproduction throughout the PHPO axis, we still do not fully understand the specific molecular mechanisms by which these key genes and the key signaling pathways regulating seasonal reproduction in Tan sheep. Therefore, it is necessary to conduct functional and mechanistic studies on these genes and signaling pathways in the future.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
All experimental animal studies were reviewed and approved by the Animal Welfare Committee of Ningxia University (permit number NXUC20180306).
SW analyzed the data and wrote the paper. XK, CY, FW, and TD performed the experiment, provided text and data for the methods, and contributed comments on the manuscript. XG, ZM, and CL participated in analysis of the data and writing of the manuscript. HZ designed and supervised the experiment. XD supervised the project. All authors read and approved the final manuscript.
This research was funded by the National Natural Science Foundation of China (31760665) and the Ningxia Natural Science Foundation (2022AAC03057).
We thank all the authors of this paper for their hard work. We would also like to thank the reviewers for their insightful comments on this paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.1068882/full#supplementary-material
1. Jaiswal S, Nandi S, Iquebal MA, Jasrotia RS, Patra S, Mishra G, et al. Revelation of candidate genes and molecular mechanism of reproductive seasonality in female rohu (Labeo rohita Ham) by RNA sequencing. BMC Genomics. (2021) 22:685. doi: 10.1186/s12864-021-08001-6
2. Yang Y, Zhou R, Li W, Liu Y, Zhang Y, Ao H, et al. Dynamic transcriptome analysis reveals potential long non-coding RNAs governing postnatal pineal development in pig. Front Genet. (2019) 10:409. doi: 10.3389/fgene.2019.00409
3. Wurtman RJ. The pineal gland in relation to reproduction. Am J Obstet Gynecol. (1969) 104:320–6. doi: 10.1016/S0002-9378(16)34186-2
4. Herbison AE. Control of puberty onset and fertility by gonadotropin-releasing hormone neurons. Nat Rev Endocrinol. (2016) 12:452–66. doi: 10.1038/nrendo.2016.70
5. Alatzoglou KS, Gregory LC, Dattani MT. Development of the pituitary gland. Compr Physiol. (2020) 10:389–413. doi: 10.1002/cphy.c150043
6. Capra E, Lazzari B, Frattini S, Chessa S, Coizet B, Talenti A, et al. Distribution of ncRNAs expression across hypothalamic-pituitary-gonadal axis in Capra hircus. BMC Genomics. (2018) 19:417. doi: 10.1186/s12864-018-4767-x
7. Zhang X, Zhang L, Shang J, Tao Q, Tian M, Ma Y, et al. Combined microRNAome and transcriptome analysis of follicular phase and luteal phase in porcine ovaries. Reprod Domest Anim. (2019) 54:1018–25. doi: 10.1111/rda.13457
8. Kim JM, Park JE, Yoo I, Han J, Kim N, Lim WJ, et al. Integrated transcriptomes throughout swine oestrous cycle reveal dynamic changes in reproductive tissues interacting networks. Sci Rep. (2018) 8:5436. doi: 10.1038/s41598-018-23655-1
9. Li J, Li C, Li Q, Li G, Li W, Li H, et al. Novel regulatory factors in the hypothalamic-pituitary-ovarian axis of hens at four developmental stages. Front Genet. (2020) 11:591672. doi: 10.3389/fgene.2020.591672
10. Zhang C, Wang G, Wang J, Ji Z, Dong F, Chao T. Analysis of differential gene expression and novel transcript units of ovine muscle transcriptomes. PLoS ONE. (2014) 9:e89817. doi: 10.1371/journal.pone.0089817
11. Wang X, Zhou G, Xu X, Geng R, Zhou J, Yang Y, et al. Transcriptome profile analysis of adipose tissues from fat and short-tailed sheep. Gene. (2014) 549:252–7. doi: 10.1016/j.gene.2014.07.072
12. Yao L, Bao A, Hong W, Hou C, Zhang Z, Liang X, et al. Transcriptome profiling analysis reveals key genes of different coat color in sheep skin. PeerJ. (2019) 7:e8077. doi: 10.7717/peerj.8077
13. Ullah Y, Li C, Li X, Ni W, Yao R, Xu Y, et al. Identification and profiling of pituitary microRNAs of sheep during anestrus and estrus stages. Animals. (2020) 10:402. doi: 10.3390/ani10030402
14. Di R, Liu QY, Song SH, Tian DM, He JN, Ge Y, et al. Expression characteristics of pineal miRNAs at ovine different reproductive stages and the identification of miRNAs targeting the AANAT gene. BMC Genomics. (2021) 22:217. doi: 10.1186/s12864-021-07536-y
15. Tao Z, Song W, Zhu C, Xu W, Liu H, Zhang S, et al. Comparative transcriptomic analysis of high and low egg-producing duck ovaries. Poult Sci. (2017) 96:4378–88. doi: 10.3382/ps/pex229
16. Zheng J, Wang Z, Yang H, Yao X, Yang P, Ren C, et al. Pituitary transcriptomic study reveals the differential regulation of lncRNAs and mRNAs related to prolificacy in different FecB genotyping sheep. Genes. (2019) 10:157. doi: 10.3390/genes10020157
17. Zhang H, He L, Cai L. Transcriptome sequencing: RNA-Seq. Methods Mol Biol. (2018) 1754:15–27. doi: 10.1007/978-1-4939-7717-8_2
18. Li B, Qiao L, An L, Wang W, Liu J, Ren Y, et al. Transcriptome analysis of adipose tissues from two fat-tailed sheep breeds reveals key genes involved in fat deposition. BMC Genomics. (2018) 19:338. doi: 10.1186/s12864-018-4747-1
19. Fan R, Xie J, Bai J, Wang H, Tian X, Bai R, et al. Skin transcriptome profiles associated with coat color in sheep. BMC Genomics. (2013) 14:389. doi: 10.1186/1471-2164-14-389
20. Di R, He J, Song S, Tian D, Liu Q, Liang X, et al. Characterization and comparative profiling of ovarian microRNAs during ovine anestrus and the breeding season. BMC Genomics. (2014) 15:899. doi: 10.1186/1471-2164-15-899
21. Shin DJ, Wang L. Bile acid-activated receptors: a review on FXR and other nuclear receptors. Handb Exp Pharmacol. (2019) 256:51–72. doi: 10.1007/164_2019_236
22. Martinot E, Sedes L, Baptissart M, Holota H, Rouaisnel B, Damon-Soubeyrand C, et al. The bile acid nuclear receptor FXR alpha is a critical regulator of mouse germ cell fate. Stem Cell Reports. (2017) 9:315–28. doi: 10.1016/j.stemcr.2017.05.036
23. Takae K, Nakata M, Watanabe T, Sasada H, Fujii H, Tomioka I. Evidence for the involvement of FXR signaling in ovarian granulosa cell function. J Reprod Dev. (2019) 65:47–55. doi: 10.1262/jrd.2018-054
24. Nagy RA, Hollema H, Andrei D, Jurdzinski A, Kuipers F, Hoek A, et al. The origin of follicular bile acids in the human ovary. Am J Pathol. (2019) 189:2036–45. doi: 10.1016/j.ajpath.2019.06.011
25. Schuermann Y, Welsford GE, Nitschmann E, Wykes L, Duggavathi R. Association between pre-breeding metabolic profiles and reproductive performance in heifers and lactating dairy cows. Theriogenology. (2019) 131:79–88. doi: 10.1016/j.theriogenology.2019.03.018
26. Nagy RA, van Montfoort AP, Dikkers A, van Echten-Arends J, Homminga I, Land JA, et al. Presence of bile acids in human follicular fluid and their relation with embryo development in modified natural cycle IVF. Hum Reprod. (2015) 30:1102–9. doi: 10.1093/humrep/dev034
27. Sanchez R, Schuermann Y, Gagnon-Duval L, Baldassarre H, Murphy BD, Gevry N, et al. Differential abundance of IGF1, bile acids, and the genes involved in their signaling in the dominant follicle microenvironment of lactating cows and nulliparous heifers. Theriogenology. (2014) 81:771–9. doi: 10.1016/j.theriogenology.2014.01.005
28. Yang X, Wu R, Qi D, Fu L, Song T, Wang Y, et al. Profile of bile acid metabolomics in the follicular fluid of PCOS patients. Metabolites. (2021) 11:845. doi: 10.3390/metabo11120845
29. Yan X, Liu H, Hu J, Han X, Qi J, Ouyang Q, et al. Transcriptomic analyzes of the HPG axis-related tissues reveals potential candidate genes and regulatory pathways associated with egg production in ducks. BMC Genomics. (2022) 23:281. doi: 10.1186/s12864-022-08483-y
30. Zhang T, Chen L, Han K, Zhang X, Zhang G, Dai G, et al. Transcriptome analysis of ovary in relatively greater and lesser egg producing Jinghai yellow chicken. Anim Reprod Sci. (2019) 208:106114. doi: 10.1016/j.anireprosci.2019.106114
31. Chen H, Feng W, Chen K, Qiu X, Xu H, Mao G, et al. Transcriptomic analysis reveals potential mechanisms of toxicity in a combined exposure to dibutyl phthalate and diisobutyl phthalate in zebrafish (Danio rerio) ovary. Aquat Toxicol. (2019) 216:105290. doi: 10.1016/j.aquatox.2019.105290
32. Jia YY, Chi ML, Jiang WP, Liu SL, Cheng S, Zheng JB, et al. Identification of reproduction-related genes and pathways in the Culter alburnus H-P-G axis and characterization of their expression differences in malformed and normal gynogenetic ovaries. Fish Physiol Biochem. (2021) 47:1–20. doi: 10.1007/s10695-020-00859-9
33. Xu S, Wang D, Zhou D, Lin Y, Che L, Fang Z, et al. Reproductive hormone and transcriptomic responses of pituitary tissue in anestrus gilts induced by nutrient restriction. PLoS ONE. (2015) 10:e0143219. doi: 10.1371/journal.pone.0143219
34. Su F, Guo X, Wang Y, Wang Y, Cao G, Jiang Y. Genome-wide analysis on the landscape of transcriptomes and their relationship with DNA methylomes in the hypothalamus reveals genes related to sexual precocity in Jining gray goats. Front Endocrinol. (2018) 9:501. doi: 10.3389/fendo.2018.00501
35. Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. (2005) 21 Suppl 1:i159–68. doi: 10.1093/bioinformatics/bti1022
36. Pizzi C, Bortoluzzi S, Bisognin A, Coppe A, Danieli GA. Detecting seeded motifs in DNA sequences. Nucleic Acids Res. (2005) 33:e135. doi: 10.1093/nar/gni131
37. Fan Y, Han Z, Lu X, Arbab AAI, Nazar M, Yang Y, et al. Short time-series expression transcriptome data reveal the gene expression patterns of dairy cow mammary gland as milk yield decreased process. Genes. (2021) 12:942. doi: 10.3390/genes12060942
38. Zhang J, Zhang J, Gao B, Xu Y, Liu H, Pan Z. Detection of the effects and potential interactions of FSH, VEGFA, and 2-methoxyestradiol in follicular angiogenesis, growth, and atresia in mouse ovaries. Mol Reprod Dev. (2019) 86:566–75. doi: 10.1002/mrd.23133
39. Robinson RS, Nicklin LT, Hammond AJ, Schams D, Hunter MG, Mann GE. Fibroblast growth factor 2 is more dynamic than vascular endothelial growth factor A during the follicle-luteal transition in the cow. Biol Reprod. (2007) 77:28–36. doi: 10.1095/biolreprod.106.055434
40. McFee RM, Rozell TG, Cupp AS. The balance of proangiogenic and antiangiogenic VEGFA isoforms regulate follicle development. Cell Tissue Res. (2012) 349:635–47. doi: 10.1007/s00441-012-1330-y
41. Abedal-Majed MA, Kurz SG, Springman SA, McNeel AK, Freetly HC, Largen V, et al. Vascular endothelial growth factor A isoforms modulate follicle development in peripubertal heifers independent of diet through diverse signal transduction pathways. Biol Reprod. (2020) 102:680–92. doi: 10.1093/biolre/ioz211
42. Ortega HH, Veiga-Lopez A, Sreedharan S, del Luján Velázquez MM, Salvetti NR, Padmanabhan V. Developmental programming: does prenatal steroid excess disrupt the ovarian VEGF system in sheep? Biol Reprod. (2015) 93:58. doi: 10.1095/biolreprod.115.131607
43. Sagsoz H, Saruhan BG. The expression of vascular endothelial growth factor and its receptors (flt1/fms, flk1/KDR, flt4) and vascular endothelial growth inhibitor in the bovine uterus during the sexual cycle and their correlation with serum sex steroids. Theriogenology. (2011) 75:1720–34. doi: 10.1016/j.theriogenology.2011.01.012
44. Castle-Miller J, Bates DO, Tortonese DJ. Mechanisms regulating angiogenesis underlie seasonal control of pituitary function. Proc Natl Acad Sci USA. (2017) 114:E2514–23. doi: 10.1073/pnas.1618917114
45. Sagsoz H, Liman N, Kucukaslan I, Saruhan BG. Immunolocalization of vascular endothelial growth factor, its receptors (flt1/fms, flk1/KDR, flt4) and vascular endothelial growth inhibitor in the bitch uterus during the sexual cycle. Anim Reprod Sci. (2013) 140:241–54. doi: 10.1016/j.anireprosci.2013.05.014
46. Chowdhury MW, Scaramuzzi RJ, Wheeler-Jones CP, Khalid M. The expression of angiogenic growth factors and their receptors in ovarian follicles throughout the estrous cycle in the ewe. Theriogenology. (2010) 73:856–72. doi: 10.1016/j.theriogenology.2009.10.011
47. Patergnani S, Danese A, Bouhamida E, Aguiari G, Previati M, Pinton P, et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int J Mol Sci. (2020) 21:8323. doi: 10.3390/ijms21218323
48. Abdou HS, Villeneuve G, Tremblay JJ. The calcium signaling pathway regulates leydig cell steroidogenesis through a transcriptional cascade involving the nuclear receptor NR4A1 and the steroidogenic acute regulatory protein. Endocrinology. (2013) 154:511–20. doi: 10.1210/en.2012-1767
49. Motola S, Cao X, Popliker M, Tsafriri A. Involvement of mitogen-activated protein kinase (MAPK) pathway in LH- and meiosis-activating sterol (MAS)-induced maturation in rat and mouse oocytes. Mol Reprod Dev. (2008) 75:1533–41. doi: 10.1002/mrd.20899
50. Sha QQ, Dai XX, Dang Y, Tang F, Liu J, Zhang YL, et al. A MAPK cascade couples maternal mRNA translation and degradation to meiotic cell cycle progression in mouse oocytes. Development. (2017) 144:452–63. doi: 10.1242/dev.144410
51. Macedo TJS, Menezes VG, Barberino RS, Silva RLS, Gouveia BB, Monte APO, et al. Leptin decreases apoptosis and promotes the activation of primordial follicles through the phosphatidylinositol-3-kinase/protein kinase B pathway in cultured ovine ovarian tissue. Zygote. (2021) 29:445–51. doi: 10.1017/S0967199421000034
52. Lins T, Barberino RS, Monte APO, Pinto JGC, Campinho DSP, Palheta RC Jr, et al. Rutin promotes activation and reduces apoptosis of primordial follicles by regulating Akt phosphorylation after in vitro culture of ovine ovarian tissue. Theriogenology. (2021) 173:64–72. doi: 10.1016/j.theriogenology.2021.07.018
53. Musacchio A, Hardwick KG. The spindle checkpoint: structural insights into dynamic signaling. Nat Rev Mol Cell Biol. (2002) 3:731–41. doi: 10.1038/nrm929
54. Jones KT. Anaphase-promoting complex control in female mouse meiosis. Results Probl Cell Differ. (2011) 53:343–63. doi: 10.1007/978-3-642-19065-0_15
55. Yamamuro T, Kano K, Naito K. Functions of FZR1 and CDC20, activators of the anaphase-promoting complex, during meiotic maturation of swine oocytes. Biol Reprod. (2008) 79:1202–9. doi: 10.1095/biolreprod.108.070326
56. Jin F, Hamada M, Malureanu L, Jeganathan KB, Zhou W, Morbeck DE, et al. Cdc20 is critical for meiosis I and fertility of female mice. PLoS Genet. (2010) 6:e1001147. doi: 10.1371/journal.pgen.1001147
57. Yang WL, Li J, An P, Lei AM. CDC20 downregulation impairs spindle morphology and causes reduced first polar body emission during bovine oocyte maturation. Theriogenology. (2014) 81:535–44. doi: 10.1016/j.theriogenology.2013.11.005
58. Paramasivam M, Chang YJ, LoTurco JJ. ASPM and citron kinase co-localize to the midbody ring during cytokinesis. Cell Cycle. (2007) 6:1605–12. doi: 10.4161/cc.6.13.4356
59. Kouprina N, Pavlicek A, Collins NK, Nakano M, Noskov VN, Ohzeki J, et al. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum Mol Genet. (2005) 14:2155–65. doi: 10.1093/hmg/ddi220
60. Xu XL, Ma W, Zhu YB, Wang C, Wang BY, An N, et al. The microtubule-associated protein ASPM regulates spindle assembly and meiotic progression in mouse oocytes. PLoS ONE. (2012) 7:e49303. doi: 10.1371/journal.pone.0049303
61. Shi L, Lin Q, Su B. Estrogen regulation of microcephaly genes and evolution of brain sexual dimorphism in primates. BMC Evol Biol. (2015) 15:127. doi: 10.1186/s12862-015-0398-x
62. Yan Q, Xu J, Wu X, Su D, Tan Z. Stage-specific feed intake restriction differentially regulates placental traits and proteome of goats. Br J Nutr. (2018) 119:1119–32. doi: 10.1017/S0007114518000727
63. Chen X, He Z, Li X, Song J, Huang M, Shi X, et al. Cuticle deposition duration in the uterus is correlated with eggshell cuticle quality in white leghorn laying hens. Sci Rep. (2021) 11:22100. doi: 10.1038/s41598-021-01718-0
64. Kornyei JL, Li X, Lei ZM, Rao CV. Analysis of epidermal growth factor action in human myometrial smooth muscle cells. J Endocrinol. (1995) 146:261–70. doi: 10.1677/joe.0.1460261
Keywords: Tan sheep, seasonal reproduction, transcriptome, PHPO axis, estrus, anestrus
Citation: Wei S, Kang X, Yang C, Wang F, Dai T, Guo X, Ma Z, Li C, Zhao H and Dan X (2022) Analysis of reproduction-related transcriptomes on pineal-hypothalamic-pituitary-ovarian tissues during estrus and anestrus in Tan sheep. Front. Vet. Sci. 9:1068882. doi: 10.3389/fvets.2022.1068882
Received: 13 October 2022; Accepted: 08 November 2022;
Published: 24 November 2022.
Edited by:
Yalan Yang, Agricultural Genomics Institute at Shenzhen (CAAS), ChinaReviewed by:
Wenting Li, Henan Agricultural University, ChinaCopyright © 2022 Wei, Kang, Yang, Wang, Dai, Guo, Ma, Li, Zhao and Dan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxi Zhao, emhhb2hvbmd4aTIwMDZAMTYzLmNvbQ==; Xingang Dan, eGdkYW5Abnh1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.