 Tong Qin1,2
Tong Qin1,2 Mingjie Shi
Mingjie Shi Yi Sun
Yi Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 13 January 2023
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.1057977
This article is part of the Research Topic Tick-borne Viruses of Domestic Livestock: Epidemiology, Evolutionary Trends, Biology and Climate Change Impact View all 5 articles
Background: A wide range of bacterial pathogens have been identified in ticks, yet the diversity of viruses in ticks is largely unexplored.
Methods: Here, we used metagenomic sequencing to characterize the diverse viromes in three principal tick species associated with pathogens, Haemaphysalis concinna, Dermacentor silvarum, and Ixodes persulcatus, in North China.
Results: A total of 28 RNA viruses were identified and belonged to more than 12 viral families, including single-stranded positive-sense RNA viruses (Flaviviridae, Picornaviridae, Luteoviridae, Solemoviridae, and Tetraviridae), negative-sense RNA viruses (Mononegavirales, Bunyavirales, and others) and double-stranded RNA viruses (Totiviridae and Partitiviridae). Of these, Dermacentor pestivirus-likevirus, Chimay-like rhabdovirus, taiga tick nigecruvirus, and Mukawa virus are presented as novel viral species, while Nuomin virus, Scapularis ixovirus, Sara tick-borne phlebovirus, Tacheng uukuvirus, and Beiji orthonairovirus had been established as human pathogens with undetermined natural circulation and pathogenicity. Other viruses include Norway mononegavirus 1, Jilin partitivirus, tick-borne tetravirus, Pico-like virus, Luteo-like virus 2, Luteo-likevirus 3, Vovk virus, Levivirus, Toti-like virus, and Solemo-like virus as well as others with unknown pathogenicity to humans and wild animals.
Conclusion: In conclusion, extensive virus diversity frequently occurs in Mononegavirales and Bunyavirales among the three tick species. Comparatively, I. persulcatus ticks had been demonstrated as such a kind of host with a significantly higher diversity of viral species than those of H. concinna and D. silvarum ticks. Our analysis supported that ticks are reservoirs for a wide range of viruses and suggested that the discovery and characterization of tick-borne viruses would have implications for viral taxonomy and provide insights into tick-transmitted viral zoonotic diseases.
Ticks (Arachnida: Ixodida) are common hematophagous arthropods that have been implicated as vectors of human and animal diseases worldwide (1). Approximately 900 species of ticks have been described and taxonomically classified into four families Argasidae (soft ticks), Ixodidae (hard ticks), Nuttalliae, and the newly proposed Deinotheriidae (2, 3). Their propensity for feeding on multiple hosts, expansive range, and long life cycle underlines the importance of active surveillance of ticks for the presence of potential pathogens threatening both human and animal health (4). Argasid and ixodid ticks combined transmit a greater diversity of viral, bacterial, and protozoan pathogens than any other arthropod vectors (5). The increasing incidence of tick-borne disease worldwide is caused partly by the increased frequency of human exposure to ticks or their endemic habitats, burgeoning tick populations, and the discoveries of new tick-associated agents (5, 6). The significance of tick-borne viral diseases (TBVDs) in human health has raised global health concerns in recent decades due to current suboptimal diagnostics, treatment options for emerging viruses, and a scarcity of vaccines (7). Attention from clinicians and researchers had alerted the public to the frequent emergences of novel viral pathogens causing febrile human diseases, such as Dabie bandavirus, also known as severe fever with thrombocytopenia syndrome virus (SFTSV) (8, 9), Alongshan virus (10), Bourbon virus (11), most recently Jingmen tick virus (12), Yezo virus (13), Toyo virus (14), and Songlin virus (15), which had been identified in China and neighboring countries. In addition, the re-emergence and continuous spread of known tick-borne viral diseases (TBVDs), such as Crimean–Congo hemorrhagic fever virus (CCHFV) (16), Heartland virus (17), tick-borne encephalitis virus (18), Powassan virus (19), and African swine fever virus (20), positively correlate with the increasing incidence of TBVDs in humans and animals. The emergence of some undefined TBVDs pathogens as well as the dearth of data on tick virome highlights an urgent requirement for active viral surveillance and discovery in ticks, which greatly promotes our knowledge of the biodiversity and evolution of viruses transmitted by ticks, although it seems plausible that traditional isolation via tissue culture in cell lines has been proven a prerequisite gold standard to characterize a novel virus or insight into a known one (5). However, not all tick-borne viruses are amenable to being isolated due to technological limitations, extensive culture-independent studies, such as transcriptomics analysis following the high-throughput sequences (HTSs), have been attempted to examine tick virome (21, 22). Such studies might not only identify viruses associated with acute diseases but also could provide insights into the pathogenesis of more controversial chronic illnesses associated with tick bites (23). Therefore, profiling viruses in ticks and investigating the substantial contact between ticks and human or animal hosts are necessary to better understand the viral sphere harbored by ticks and identify the potential viral pathogens in China. In this study, the virome of tick pools was profiled via metagenomic sequencing aimed at identifying the baseline viruses carried by ticks and characterizing their biodiversity and evolution. Then, the viral transmission patterns between ticks and humans were discussed based on the active surveillance results of molecular prevalence and potential exposure of humans to these ticks. These findings may shed light on the tick virome endemic to China and reveal the potential risks of TBVDs in the human population, which helps to guide tick-borne disease prevention and control in China.
Questing Ixodes persulcatus, Haemaphysalis concinna, and Dermacentor silvarum were collected by sweeping flags on the vegetation in Zhalantun county, Inner Mongolia Autonomous Region, and Mudanjiang city, Heilongjiang province, respectively (Supplementary Figure 1). Morphological identification of ticks was conducted using the standard key for Chinese ticks (24) under stereomicroscopes. We further assigned these ticks into eight pools by species, sex/nymph, and origin location, each with 30 individuals (the nymphal pool of H. concinna was abandoned for inadequate individuals). All tick specimens were thoroughly surface sterilized to remove the possible contaminants before DNA/RNA isolation. Surface sterilization included sequential 1 mL washes with a vortex for 1 min in 3% hydrogen peroxide (H2O2), two 30 s washes in 70% ethanol (w/v), 2 min in phosphate-buffered saline (PBS, pH = 7.4), and finally drying on clean tissue paper to avoid cross contaminations. Extraction of total DNA and RNA from ticks was performed using E.Z.N.A. DNA/RNA Isolation Kit (OMEGA) with modifications. In brief, ticks were homogenized in RLT solution under liquid nitrogen and then incubated at 55°C for 10 min with proteinase K (Qiagen) and centrifuged for 30 s at 15,000 × g. We took a proportion (~30 μL) of the homogenates for genomic DNA extraction with RNAase A treatment (15 min) for later DNA purification. Simultaneously, another 300 μL of tick homogenates was utilized to enrich viral particles and total RNA extraction. Before RNA extraction, the homogenates were filtered (0.45 μM) at room temperature and then treated with RNase A (15 min), followed by Turbo DNase and Benzonase (MilliporeSigma, Burlington, MA, USA) (30 min) (22). The enrichment of viral particles had been achieved due to the degradations of unprotected nucleic acid absence of a viral capsid (25).
To verify tick species identification, genomic DNA was extracted from each specimen pool using E.Z.N.A. DNA/RNA Isolation Kit (OMEGA). Two genes were used for tick identification: the partial 18S rRNA gene (~1,100 nt) which was amplified using primer pairs 18S-1 (5′-CTGGTGCCAGCGAGCCGCGGYAA-3′) and 18S-2 (5′-TCCGTCAATTYCTTTAAGTT-3′) and partial COI gene (~680 nt) using primer pairs LCO1490 (5′-GGTCAACAAATCATAAAGATA TTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′). PCRs were performed as previously described (26, 27). For taxonomic determination, the resulting sequences were compared against the non-redundant nucleotide (nt) database and with all COI barcode records on the Barcode of Life Data (BOLD) Systems.
The enriched total nucleic acid (11 μL) from each tick pool was then subjected to first- and second-strand cDNA synthesis with Super Script IV reverse transcriptase (Invitrogen, Waltham, MA, USA) and exo-Klenow fragment (New England Biolabs, Ipswich, MA, USA), respectively. For all libraries, the Ribo-Zero Gold Kit (Illumina) was used to remove ribosomal RNA under the manufacturer's guidance. Subsequently, all rRNA-depleted RNA samples were resuspended to construct libraries using the KAPA Stranded RNA-Seq Kit (KAPA biosystems, Roche) with TruSeq Index PCR Primer barcodes (Illumina, SanDiego, CA, USA) following the manufacturer's instructions. Qubit high-sensitive RNA assays (Thermo Fisher Scientific) were performed to quantify cDNA levels before, during, and after library preparation, and the fragment sizes were simultaneously determined with an Agilent Bioanalyzer. Subsequently, equimolar amounts of nucleic acids were pooled and submitted for sequencing in each library. All libraries were sequenced on a single lane (paired-end, 125 bp read-length) on an Illumina HiSeq 2,500 platform at the BGI Sequencing Center (www.genomics.cn).
Raw sequencing reads were first subjected to adapter removal and quality trimmed with TrimGalore (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) (28, 29). Clean reads were de novo assembled using the Trinity v2.8.5 program (30, 31).
Trinity assemblies with a length above 200 bp were subjected to BLASTN against all nt databases using a local BLAST tool and BLASTX against all non-redundant protein (nr) databases (available as early as May 2022) of reference RNA viruses as well as those recently published, with hits at an e-value of 1 × 10−5 or better collated. All potential virus assemblies were screened against the Conserved Doman Database (www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) with an expected value threshold of 1 × 10−3 to identify viral gene segments. Putative viral contigs were further merged by high-identity overlaps with a threshold value of 95% similarity using the SeqMan program of the Lasergene package v7.1 (32). To complete the remaining gaps, original reads were aligned to the viral contigs again using the Bowtie2 program (33), and the resultant assembly was verified in the Integrated Genomics Viewer (34). A novel viral species should be satisfied with one of the following conditions described before (35), namely (i) <80% nucleotides identity across the complete genome or (ii) <90% amino acids identity of RNA-dependent RNA polymerase (RdRp) domain with the known viruses. To eliminate possible endogenous viruses, all viral assemblies were blasted against the reference genomes of I. persulcatus (GCA_013339685.1), D. silvarum (GCA_013339745.1), and whole-genome shotgun database of Ixodida (Taxonomy ID: 6935, accession data:1/1/2022), respectively. If aligned bases of the query contigs covered more than 50% and the nucleotide similarity exhibited higher than 85% from any comparison with the earlier databases, they were discarded from the downstream analysis. Transcript abundance containing RNA-Seq fragment counts for each transcript (or gene) across each sample was estimated using the alignment-based abundance estimation method RSEM (36). The trimmed mean of M-values normalization method (TMM) was employed to normalize the transcript abundance.
Reverse transcription-PCR and nested PCR were performed to confirm the existence of virus on RNA samples from the tick pools which were originally subjected to RNA-seq with primer sets (Supplementary Table 2) targeting the RdRp contigs of virus hit. Then, these primers were utilized to really exclude the presence of endogenous viral elements by PCR amplifications on genomic DNA templates from the same tick pools. Only positively detected potential virus would undergo a further epidemiological survey. The contigs which hit the viral genome were used as templates for overlapping primers design. The overlapping primers for amplifying the complete genome of the representative viruses (Taiga tick Nigecruvirus and Mukawa virus) were provided for the nested RT-PCR, genome walking, and rapid amplification of complementary DNA ends (5′-, 3′-RACE) with a commercial kit (Takara) according to the manufacturer's protocol (Supplementary Tables 3, 4). All the PCR products were separated on 0.8% agarose gel and purified through an agarose gel DNA extraction kit for Sanger sequencing.
To infer the evolutionary relationships of the viruses discovered, the protein-translated RdRp open reading frame segments produced in this study were combined with representative complete proteomes and (or) RdRp-segments of the Bunyavirales, Mononegavirales, Flaviviridae, Luteoviridae, Partitiviridae, Totiviridae, and Picornaviridae were retrieved from NCBI GenBank (www.ncbi.nlm.nih.gov/genbank) and aligned using MAFFT v.7.266, employing the E-INS-i algorithm (37). Ambiguous regions in the alignments were removed with TrimAl v.1.2 (38). Following sequence alignment, ProtTest v.3.4 was employed to select the best-fit model of amino acid substitution (39). Finally, maximum likelihood trees for all alignments were inferred using the best-fit model of amino acid substitution (LG + I + Γ + F for all alignments) with 1,000 bootstrap replicates with the PhyML v.3 program (40). Phylogenetic trees were edited and visualized with FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree). All phylogenetic trees were mid-point rooted for purposes of clarity only. The flowchart of the study design diagram was illustrated with different types of treatment and analysis (Supplementary Figure 2).
After morphological classification, there were 216 I. persulcatus (129 females, 52 males, and 35 nymphs) and 147 H. concinna (87 females, 49 males, and 11 nymphs) achieved alive from Mudanjiang city, Heilongjiang province, which were then randomly assigned into five groups of 30 individuals each species, sexes, and development stage. Among them, I. persulcatus groups included female, male, and nymphal ones, respectively, while only two groups (female and male) of H. concinna were arranged for inadequate nymphs. An average of 10.96 Gb data, including 7.1–7.6 × 107 or so 150-base paired-end reads, were generated from male, female, and nymph groups of I. persulcatus, while the male and female groups of H. concinna generated ~11.18 Gb of data on average, including 6.8 × 107 and 8.0 × 107 or so 150-base paired-end reads. While in Zhalantun city, Hulunbuir, Inner Mongolia, a total of 274 D. silvarum (58 females, 170 males, and 46 nymphs) and 40 I. persulcatus (28 females and 12 males) were harvested alive. The D. silvarum ticks were also divided into three groups (female, male, and nymphal ones), each with 30 individuals, which generated ~12.30 Gb data on average. After quality filtration and host subtraction, a total of 614,213,814 reads remained which were assembled into 815,457 contigs. Assembled contigs were compared to the NCBI Viral Genomes database, resulting in 1,311, 1,033, and 709 viral contigs from I. persulcatus, D. silvarum, and H. concinna as viral origin through BLASTN and BLASTX, which were kept for a subsequent manual inspection (Supplementary Table 1). A prediction of ORFs was also implemented and compared to the viral protein database through BLASTP. As a result, sequences of 28 putative viruses were identified (Table 1), with five representing novel species. Seventeen viruses were identified within the I. persulcatus pool, 11 in D. silvarum, while only six were discovered in H. concinna. Of them, single-stranded positive-sense RNA viruses (Flaviviridae, Luteoviridae, Picornaviridae, Solemoviridae, and Tetraviridae), negative-stranded RNA viruses (Mononegavirales and Bunyavirales), and double-stranded RNA viruses (Totiviridae and Partitiviridae) were involved (Table 1). The presence of the representative viral strains in the corresponding pool was further verified by nested RT-PCR and Sanger sequencing (Supplementary Tables 2, 3, Supplementary Figures 3, 4). In total, 41 viral sequences from the 28 identified RNA viruses were verified and submitted to NCBI under the accession numbers OP863261–OP863295 and OP863301–OP863305.
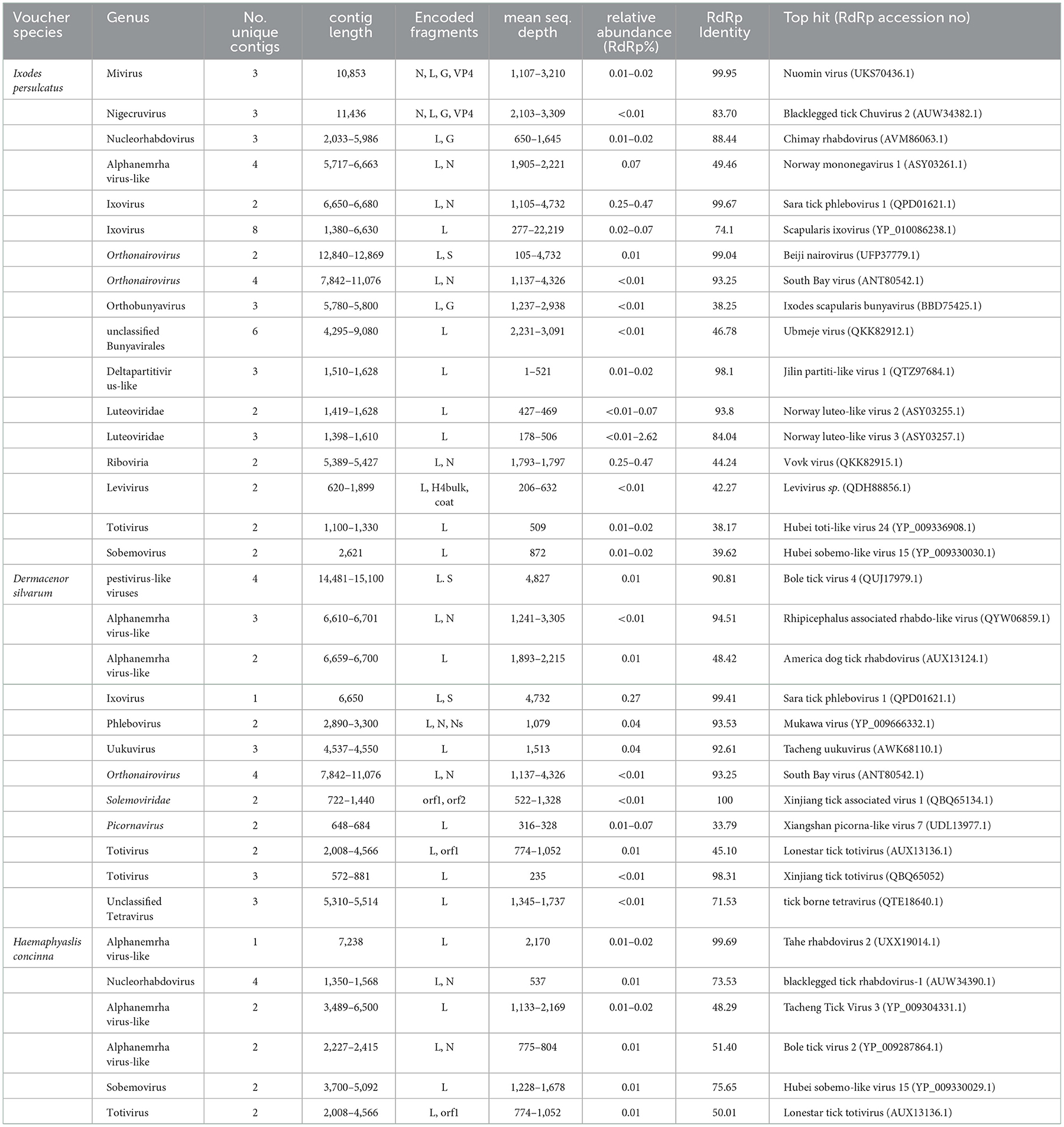
Table 1. Virus identified in three dominant tick species in North China.
Sequences for a novel viral species belonging to the family Flaviviridae, tentatively named Dermacentor silvarum pestivirus-like virus 1 (DSPV), were identified in all the three D. silvarum pools (Table 1, Figures 1A, B). DSPV comprises a single polyprotein that shares the closest homology within the NS3 and NS5 of viruses within the genus Pestivirus (41), which clustered DSPV with other recently identified Pestivirus-like viruses, Bole tick virus 4 and Trinbago virus (42). Additional segmented Flavivirus, Jingmen tick virus, was also discovered in I. persulcatus with 97.16 and 96.90% amino acid similarities with RdRp (QFR36159.1) and VP3 protein (QHW66956.1), respectively (Table 1). These contigs were shown positioned on corresponding sites in our phylogenetic tree of these Flavivirus-associated viruses (43) constructed based on their RdRp genes (Figures 1A, C).
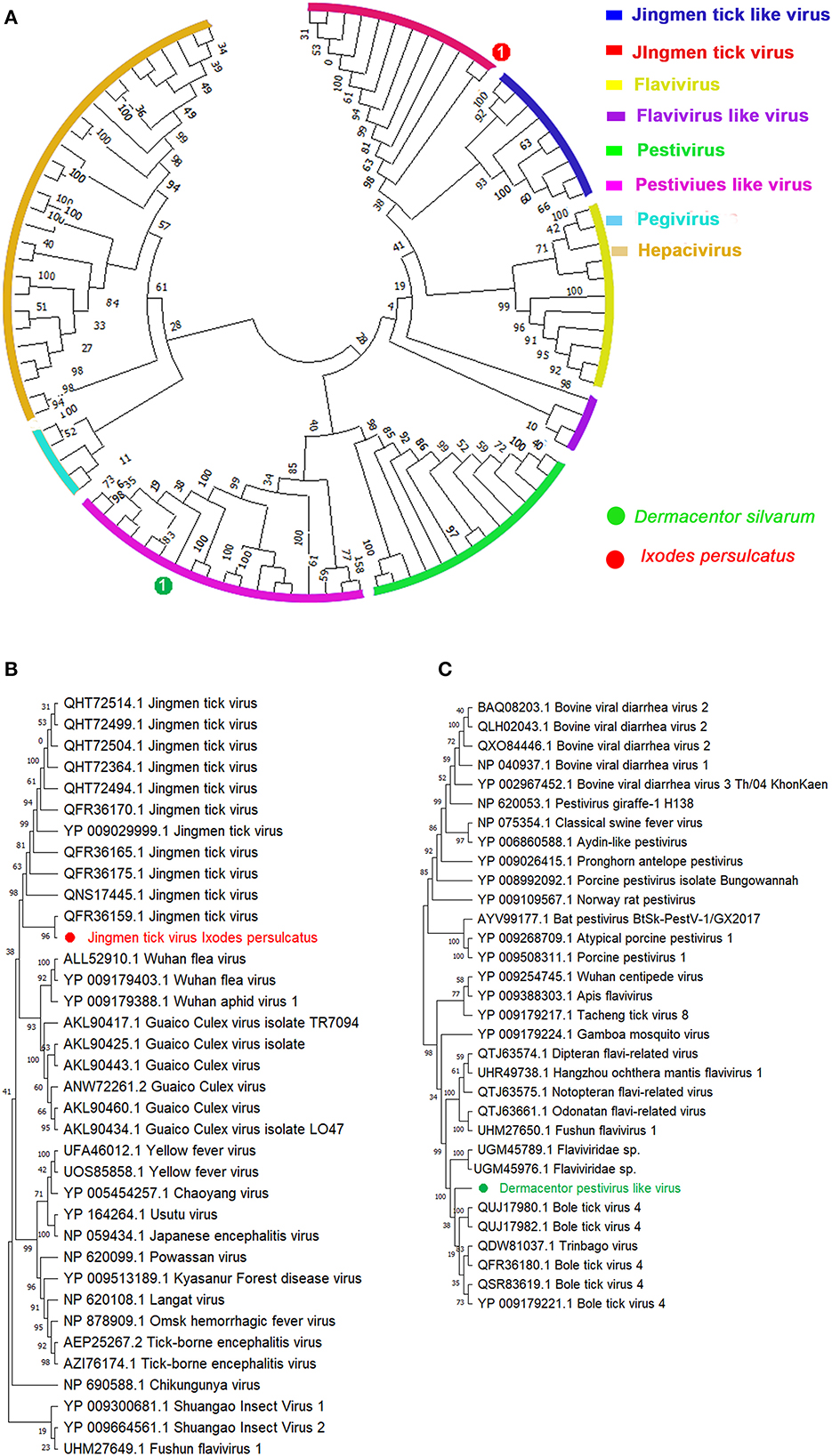
Figure 1. Phylogenetic analysis of representative branches in the family Flaviviridae based on the RdRp sequences. (A) Enhanced region showing the relationship of the unclassified Pestivirus-like group in relation to Pestivirus and Flavivirus. (B) Enhanced region showing the relationship of Jingmen tick virus in relation to the family Flaviviridae. (C) Enhanced region showing the relationship of Dermacentor pestivirus-like virus in relation to genus Pestivirus. Red dot marked with 1 represents Jingmen tick virus from Ixodes persulcatus, and green dot marked with 1 represents Dermacentor pestivirus-like virus from Dermacentor silvarum. Maximum likelihood tree inferred using the best-fit model of amino acid substitution (LG + I + Γ + F for all alignments) with 1,000 bootstrap replicates.
Luteovirus, Picornavirus, Tetravirus, and Solemovirus are single-stranded positive-sense RNA viruses that are endowed with important and potential viral pathogen parasites on humans and animals (44). In the present study, taiga tick luteo-like virus was identified with shared amino acid sequences of Norway luteo-virus 2 (93.80%, ASY03255.1) and Norway luteo-virus 3 (84.04%, ASY03257.1), respectively, across 99% RdRp gene (Table 1). The two luteo-like viruses grouped with Ixodes scapularis-associated viruses in our ML phylogeny tree, although low abundant Norway luteo-virus 2 was present in one library (Supplementary Figure 7). Moreover, a total of three Solemoviruses were discovered in the present study as well (Supplementary Figure S8). Of which, a novel Solemoviridae member, Xinjiang tick-associated virus 1 from D. silvarum, was shown 100% identical to those sequences on orf1 and orf2, respectively (Table 1). Sobemovirus-like viruses from H. concinna and I. persulcatus were found to be identical to Hubei sobemo-like virus 15 (YP_009330030.1) with 75.65 and 39.62% identities on RdRp coverage over 2,620 amino acid sequences, respectively (Table 1). In addition, another positive-sense ssRNA viruses, tick-borne tetravirus and Picorna-like virus from D. silvarum, exhibited 71.53 and 33.79% amino acid identities of RNA polymerase genes with tick-borne tetravirus-like virus (QTE18640.1) (45) (Table 1, Supplementary Figure 9) and Xiangshan picorna-like virus 7 (UDL13977.1) over 95% coverage spaced across L segments, respectively (Table 1, Supplementary Figure 10). Further classification of the two ss (+) RNA viruses was challenging due to high sequence divergence in combination with a limited number of available Tetravirus and Picornavirus sequences.
Assembly of the viral contigs from I. persulcatus yields two viruses belonging to Chuviridae (Figures 2A, B). Among them, a Mivirus was discovered from I. persulcatus with shared viral RdRp amino acid (aa) sequences at 99.95% on the 100% query coverage of Nuomin virus (UKS70432.1) (Table 1). Nuomin virus was reported as a negative-strand circular RNA virus, whose genome at a length of 10,863 nucleotides, coding open reading frames (ORFs) of RNA-dependent RNA polymerase (RdRp), glycoprotein (G), nucleoprotein (NP), and VP4 protein (Supplementary Figure 11, left), shows high similarity to RdRp of Lesone mivirus (QPD01622.1) and Suffolk virus (YP 009177218.1) with distinct amino acids sequences on VP4. Nuomin virus had been recognized as pathogenic to humans in Tahe, Heilongjiang province, China (46), and then assigned to the recently proposed Mivirus genus of Chuviridae (21). Fortunately, we also achieved a novel Nigecruvirus, tentatively named Taiga tick nigecruvirus, which was shown at a length of 11,436 nucleotides and identical to blacklegged tick chuvirus 2 in L (AUW34382.1), G (AUW34383.1), NP (AUW34384.1), and VP4 (AUW34385.1) (Table 1, Supplementary Figure 11, right) based on the amino acids' similarities at 83.07, 79.94, 66.22, and 67.31%, respectively, over the 100% coverage. Homology searches revealed this virus to be very distant from genus Mivirus. The similarities of the four ORFs of the novel taiga tick nigecruvirus and Nuomin virus ensure the prevalence of these two Chuviridae members in I. persulcatus in China (47).
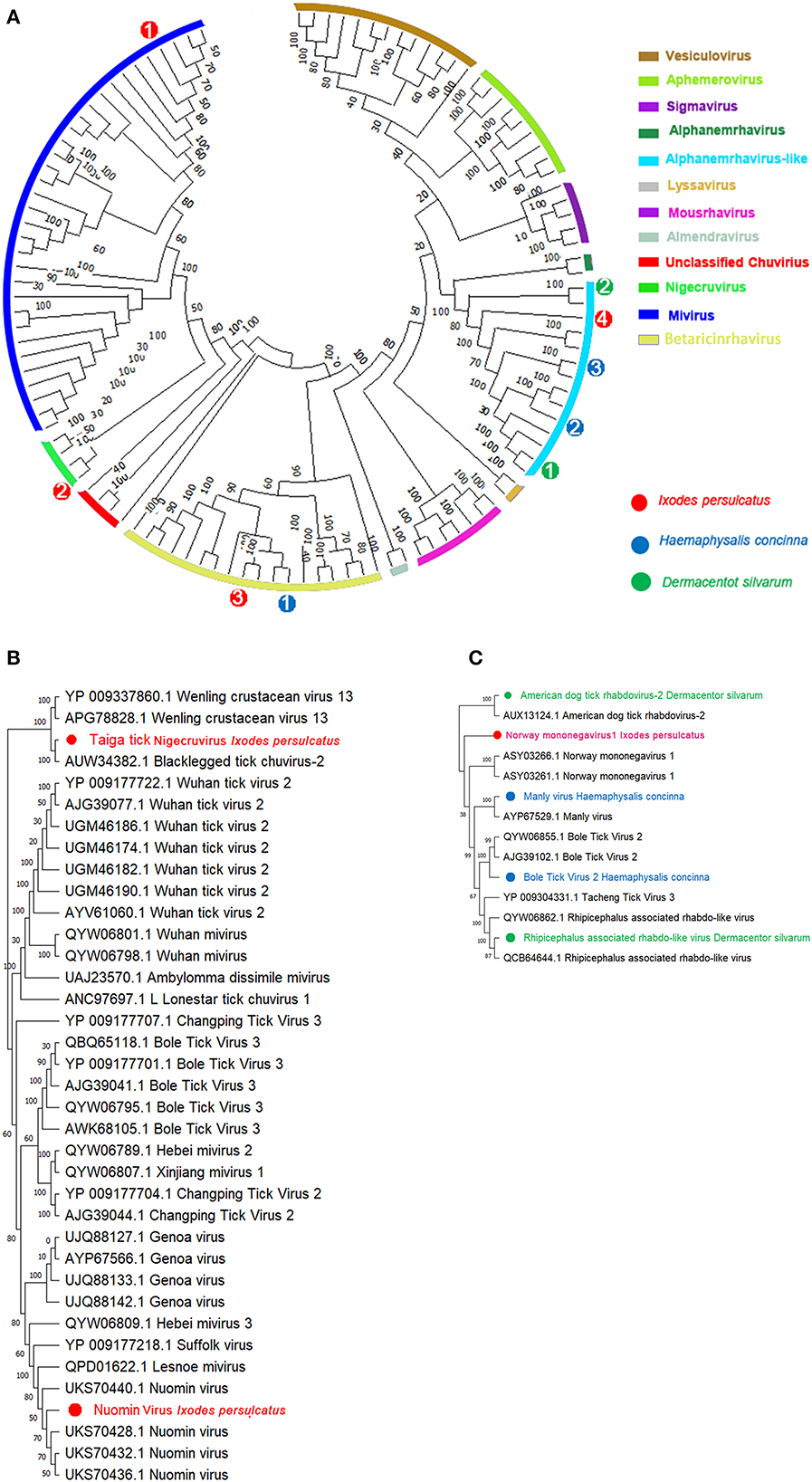
Figure 2. Phylogenetic analysis of representative branches in the order Mononegavirales. (A) Alignment of all branches belonging to the order Mononegavirales. (B) Enhanced region showing the genera relationship of Mivirus and Nigecruvirus. (C) Enhanced region showing the relationships of Mononegavirus and Rhabdovirus. Red dots marked with 1 to 4 represent Nuomin chuvirus, taiga tick nigecruvirus, Chimay rhabdovirus, and Norway mononegavirus-like virus 1 from Ixodes persulcatus, respectively. Green dots marked with 1 and 2 represent Rhipicephalus-associated rhabdo-like virus and America dog tick rhabdovirus from Dermacentor silvarum, respectively. Blue dots marked with 1 to 3 represents blacklegged tick rhabdovirus-1, Bole tick virus 2, and Manly virus from Haemaphysalis concinna, respectively. Maximum likelihood tree inferred using the best-fit model of amino acid substitution (LG + I + Γ + F for all alignments) with 1,000 bootstrap replicates.
Except for Chuviridae, we also identified several viral contigs belonging to Mononegavirales (48), among which one Nucleorhabdovirus and two Alphanemrhavirus-like viruses (Tacheng tick virus 3 and Norway mononegavirus 1) were discovered from I. persulcatus with 88.44, 47.32, and 49.46% identities to those aligned RdRp sequences of Chimay-like rhabdovirus (62.52% AVM86063.1), Tacheng tick virus 3 (YP_009304331.1), and Norway mononegavirus 1 (ASY03261.1) (Table 1) (Figures 2A, C, Supplementary Figure 5). Moreover, similar glycoprotein (G) and nucleoprotein (NP) sequences were also obtained from I. persulcatus and assigned as glycoprotein of Chimay rhabdovirus (AVM86062.1) and nucleoprotein of Tacheng tick virus 3 (62.17%, AJG39137.1) and Norway mononegavirus 1 (57.43%, KAG0427517.1), respectively (Table 1). As a case of H. concinna, assembly contigs obtained were shown with a high degree of diversity in Mononegavirales involving three Alphanemrhavirus-like rhabdoviruses and one Nucleorhabdovirus. Of Alphanemrhavirus-like rhabdovirus, Bole tick virus 2, Tacheng tick virus 3, and Tahe rhabdovirus 2 were involved, and their RdRp sequence accession numbers of reference strains as YP_009287864.1 (51.40%, 804 aa), YP_009304331.1 (48.29%, 2,179aa), and UXX19014.1 (99.69%, 2,168aa) in L segments, respectively (Table 1) (Figures 2A, C, Supplementary Figure 5). Bole tick virus 2 was also identified with nucleoprotein segment (73.02%, YP_009287860.1) over 100% coverage. The nucleorhabdovirus shared 73.53% similarities as (1,568aa) in RdRp genes of blacklegged tick rhabdovirus-1 (AUW34390.1), whereas D. silvarum from Zhaluntun, Inner Monogolia, yield two Alphanemrhavirus-like viruses identical to Rhipicephalus associated rhabdo-like virus (QYW06859.1) and American dog tick rhabdovirus (AUX13124.1) with 94.51% and 48.42% similarities over the 99.0% coverage, respectively (Figures 2A, C). The details of other related protein fragments in the viruses described earlier are shown in Table 1.
Bunyaviruses are segmented negative-stranded viruses that include at least nine families and 13 genera, many of which were involved as pathogens to humans and animals (49). In our study, 11 species in five genera of three families in the order Bunyavirales were detected. Assembly of these contigs revealed the presence of two Orthonairovirus (Nairoviridae), three Phlebovirus, two Ixovirus, and one Uukuvirus (Phenuiviridae), one Orthobunyavirus (Peribunyaviridae), and two unclassified Bunyaviruses from I. persulcatus and D. silvarum, whereas no Bunyavirales contigs detected in H. concinna (Table 1). The phylogenetic tree of Bunyavirales constructed based on the RdRp genes indicated that these phleboviruses, orthonairoviruses, and unclassified bunyaviruses clustered with referenced strains and formed well-supported monophyletic groups as the “classic” schematic diagram involved with Orthohantanviridae, Nairoviridae, Phenuiviridae, Peribunyaviridae, and others (Figure 3A).
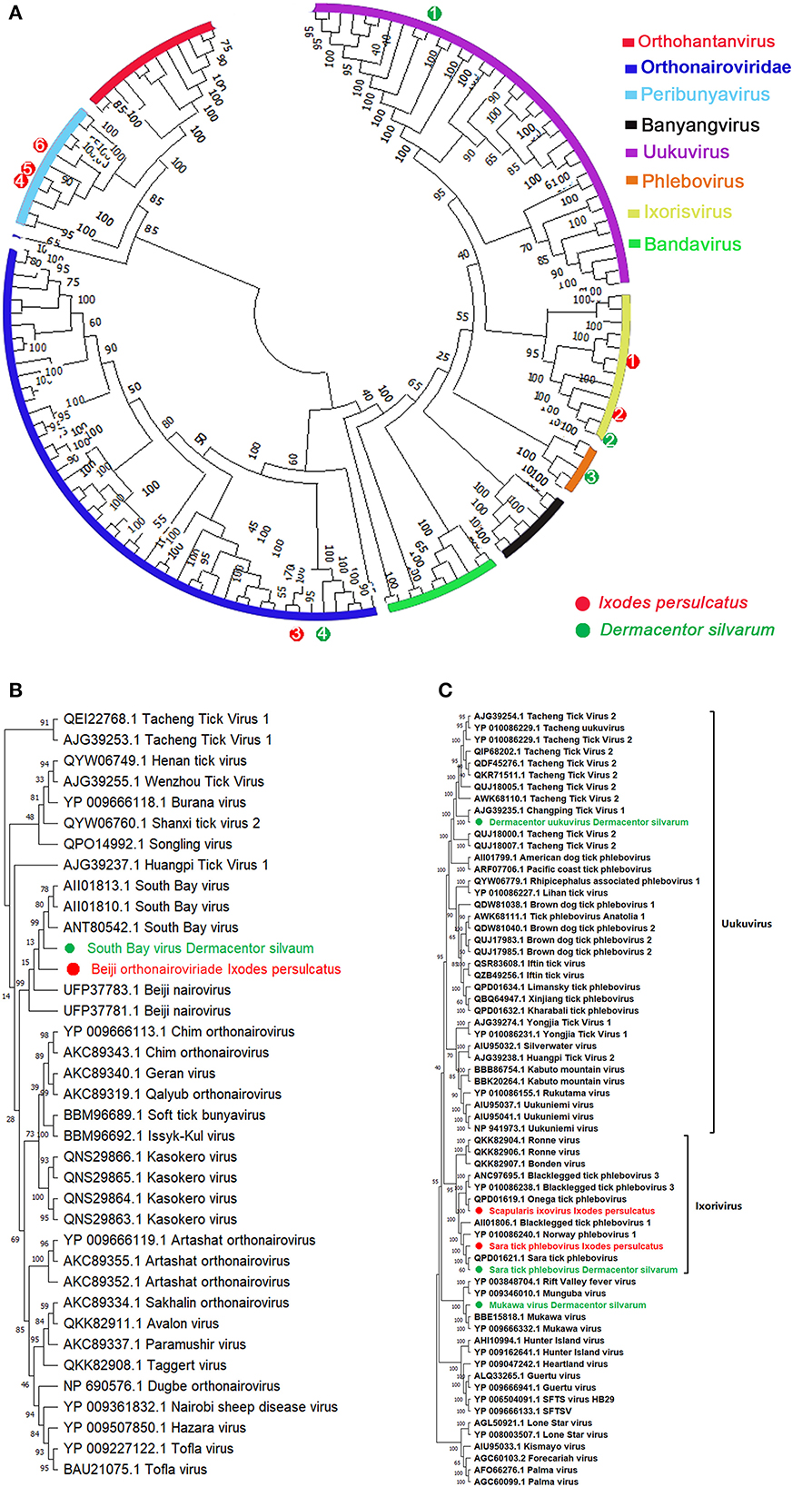
Figure 3. Phylogenetic analysis of representative branches in the order Bunyavirales. (A) Alignment of all branches belonging to the order Bunyavirales. (B) Enhanced region showing Phenuiviridae members. (C) Enhanced region showing Orthonairoviridae members. Red dots marked with 1 to 6 represent Scapularis ixovirus, Sara tick phlebovirus 1, Beiji orthonairovirus, unclassified Bunyavirales 1, unclassified Bunyavirales 2, and Ixodes scapularis orthobunyavirus viruses from Ixodes persulcatus, respectively. Green dots marked with 1 to 4 represent Tacheng uukuvirus, Sara tick phlebovirus 2, Mukawa virus, and South Bay virus from Dermacentor silvarum, respectively. Maximum likelihood tree inferred using the best-fit model of amino acid substitution (LG + I + Γ + F for all alignments) with 1,000 bootstrap replicates.
Assembly of multiple contigs from I. persulcatus and D. silvarum pools contained with coding sequences is similar to viruses in the genus Orthonairovirus by BLASTx. With CCHFV as a reference genome, assembly of these Orthonairovirus-like contigs provided 100% coverage of L and S segments. The assembled sequences from I. persulcatus showed high identity (99.81%) to Beiji orthonairovirus (BJNV) (Table 1, Figures 3A, B), a novel species initially discovered from I. persulcatus (50) and human patients (51). BJNV reads were the dominant viral reads obtained from all I. persulcatus pools and accounted for 5–7% of filtered reads. Moreover, South Bay virus was also revealed from D. silvarum with 97.12% similarity in RdRp (AII01810.1) and 77.61% in nucleocapsid protein (AII01798.1) over the 100% coverage (Table 1, Figures 3A, B). However, despite an exhaustive bioinformatics analysis, we were unable to identify any contigs or reads with any similarity to M segments of Orthonairovirus.
In the present study, a total of six species of three genera in the family Phenuiviridae are present in I. persulcatus and D. silvarum. The most diversity appeared in genus Phlebovirus, which is currently comprised of over 70 phleboviruses isolated from ticks, mosquitoes, and sand flies (52). We identified multiple contigs with homology to phleboviruses by BLASTx in pools of I. persulcatus and D. silvarum (Table 1, Figures 3A, C). The contigs derived from the pools of each tick species were assembled separately to a reference phlebovirus genome. We obtained phlebovirus contigs from D. silvarum, which were identical to Mukawa virus with shared amino acids similarities in L segments (98.60%, YP 009666332.1) and non-structural protein (93.53%, YP_009666334.1) and nucleoprotein (99.60%, YP_009666333.1) over the 98% coverage (Table 1, Figures 3A, C). Moreover, an unclassified Phlebovirus, Changping tick virus 2 (AJG39235.1), was harvested from D. silvarum with 85.46% similarity on the 100% sequence coverage of RdRp gene (Table 1, Figures 3A, B). In addition, two members in genus Ixovirus were also achieved, one is Sara tick phlebovirus from both D. silvarum and I. persulcatus with their RdRp identities up to 99.32 and 99.41%, respectively, across the 2,210 amino acids sequences (Table 1, Figures 3A, C). Another Ixovirus in Phenuiviridae, Scapularis ixovirus (previously cited as blacklegged tick Phlebovirus 3, YP_010086238.1), was also documented in I. persulcatus with 74.1% identity in L segments (Table 1, Figures 3A, C). Fortunately, the third genus Uukuvirus in the family Phenuiviridae was harvested from D. silvarum and identical to Tacheng Uukuvirus (Tacheng tick virus 2, AWK68110.1) with 92.61% similarity on the amino acids' sequences on polymerase gene (Table 1, Figures 3A, C). All sequences assigned to the Phenuiviridae family were presented with missed M segment coding for the viral glycoprotein.
Except for Nairoviridae and Phenuiviridae, we also obtained another viral contig belonging to Bunyavirales from I. persulcatus (Figure 3A, Supplementary Figure 6), which exhibited a relatively lower identity with Volzhskoe tick virus 1 (72.41%, QPD01626.1) (53) and Ixodes scapularis bunyavirus (38.25%, BBD75425.1) (Orthobunyavirus, Peribunyaviridae) based on over 90% sequence coverage of RdRp gene (Table 1). Moreover, Ubmeje-like virus (QKK82912.1) from D. silvarum was presented with a lower RdRp identity of 46.78%, which was also known as an unclassified Bunyavirales (Table 1).
Both Partitiviridae and Totiviridae are double-stranded RNA viruses mostly known as abundant and diverse in arthropods, some of which are pathogens to animals and humans. Notably, several lineages of Partitivirus appear related to endogenous virus elements in the genomes of these arthropod hosts (54). We identified three highly divergent Partiti-like viruses, Jilin partiti-like virus 1 from three Ixodes persulcatus pools (Table 1, Supplementary Figure 12). Interestingly, this virus formed a cluster with other arthropod-associated partiti-like viruses, which shared a close relationship with those found in host animals associated with arthropods. Whether Jilin partiti-like virus 1 is a truly tick-borne virus will need to be examined in detail. In addition to partiti-like virus, another dsRNA virus close to the family Totiviridae was also discovered from H. concinna with relatively lower amino acid identities with over 95% coverages of RdRp genes (50.01%, AUX13136.1) and ORF1 (67.01%, AUX13135.1) of Lonestar tick totivirus (Table 1). Meanwhile, I. persulcatus yields contigs similar to Hubei toti-like virus 24 based on over 95% sequences L segment (38.17%, YP_009336908.1) (Table 1). As a case of D. silvarum, both Lonestar tick totivirus and Hubei toti-like virus 24 were found with RdRp genes (Table 1, Supplementary Figure 13).
Other more distantly related viruses including unclassified Vovk virus (44.24% RdRp aa identity with QKK82915.1) and Levivirus (42.27% RdRp aa identity QDH88856.1, 31.75% H4bulk (QDH87337.1) and 35.34% coat (UJQ85258.1) in the family of Leviviridae were also detected from I. persulcatus (Table 1).
This study focused on the characterization of virome diversity of questing D. silvarum, I. persulcatus, and H. concinna ticks from North China. The outcome from our sampling efforts supports the results throughout various Chinese regions, showing that I. persulcatus, D. silvarum, and H. concinna are three of the most abundant questing ticks within the regions (55). All three tick species have been heavily implicated in the transmission of TBDs throughout the regions. I. persulcatus is the principal vector of the agents of Lyme borreliosis, Rickettsioses, and Babesioses along with A. phagocytophilum and clinically relevant human pathogen Jingmen tick virus (Alongshan virus) and Far-Eastern strain of TBEV (56). D. silvarum and H. concinna had also been linked with several tick-borne pathogens found throughout China, including Francisella tularensis, Coxiella burnetii, Rickettsia spp., Babesia spp., Anaplasma spp., TBEV, and SFTSV/Dabie bandavirus (50, 57). Despite examining more pools of D. silvarum and H. concinna, we identified a greater number of viral contigs in I. persulcatus, while no Bunyavirales viral sequences were detected within H. concinna pools. Combined, these studies support a hypothesis that different tick species can harbor diverse viruses at different levels. Interestingly, no viral sequence of TBEV was identified in the known endemic region, in addition to a low abundance of Jingmen tick virus identified. This most likely is attributed to the low prevalence of these viruses within the tick populations along with sampling bias. For example, data show that the prevalence of TBEV maintained within I. persulcatus populations in China is <0.1% (58). Since we only examined 90 I. persulcatus ticks, it is unlikely we would identify a positive tick.
More than 350 viruses are classified in the order Bunyavirales, which were assigned as nine families and over 13 genera, with various species only recently discovered and characterized (50). Our studies have demonstrated molecular evidence of the emergence of over 10 bunyaviruses present in ixodid ticks in China. Based on the phylogenetic analysis of the polymerase coding regions, Beiji orthonairovirus and South Bay virus form a distinct phylogenetic lineage within currently described orthonairoviruses, which indicate that the genus Orthonairovirus is significantly more diverse than previously appreciated. Based on a very short sequence fragment, the earlier phylogenetic analysis of tick-associated orthonairoviruses yielded two main phylogenetic clades, with which one clade being isolated exclusively from ixodid ticks and the other from argasid ticks (59). The phylogeny has been proven complex and confounded as some previously unidentified genetically distinct or emerged Nairovirus presents. A more accurate Orthonairovirus classification scheme is urgent to be updated with oncoming contributions from genome sequencing of unclassified Nairovirus, especially those isolated from ticks of Argasid or Ixodid. In addition to the genus Orthonairovirus, we also identified several Phenuiviridae viruses from I. persulcatus and D. silvarum. Phenuiviridae was the latest update representing the newly combined Phlebovirus and Tenuivirus along with the newly erected genera Goukovirus and Phasivirus (49). Historically, phleboviruses were classified into at least three phylogenetic clusters, each comprised of several potential species: the Uukuniemi group, the Bhanja group, and the STFS group based on vector, genomic, and serological relationships (49). Of them, Tacheng uukuvirus in D. reticulatus has been proven phylogenetically close to a human clinical isolate with a history of tick bite in northwestern China (60), which raised widespread concerns about the health risk of Dermacetor ticks to transmit Uukuvirus in China. As one of the predominant human-bitten species, the potential transmission ability of Uukuvirus in D. silvarum remains to be determined in detail. Furthermore, the Mukawa virus, another novel Phlebovirus outsides the described three clades earlier, was first detected from D. silvarum. Together with the evidence of Mukawa virus proliferation in the Huh-7 cell line and the innate immune responses caused by the viral NSs during mice infection (61), the immense medical significance of D. silvarum to human victims should be addressed and more detailed surveys on the epidemiological characters of Mukawa virus should not be ignored (62). Similarly, although no clinical manifestations were documented to elucidate the pathogenicity of some phleboviruses in humans and animals, the discovery of Sara tick phlebovirus, Scapularis ixovirus, and Changping tick virus 2 in our studies has deepened our knowledge about diverse Phenuiviridae viruses widely prevalent in China and adjacent areas. However, the relative abundance of these Bunyavirales viruses was shown as quite lower among the viral transcripts obtained. The expansive infectious ticks, for example, up to 3% infection rate of Beiji orthonairovirus in I. persulcatus (9), might cause a series of human victims suffering from bunyavirus infections. Considering the unknown pathogenicity of Volzhskoe tick virus, Ubmeje virus, and Ixodes scapularis-orthobunyavirus discovered in the present article, the findings of Bunyavirus-like contigs from I. persulcatus and D. silvarum suggest that these viruses may have much medical relevance and broader geographical distributions, perhaps throughout the range of the two tick species, comparable to bacterial and protozoan pathogens transmitted by them. Enhanced by the spreading of ticks through the movements of the viremic host reservoir, the real geographical ranges should be further extended. We do not know whether these bunyaviruses are specific to their vector tick species. As a matter of fact, some tick-borne viruses can usually be isolated from more than one tick species. CCHFV, for example, has been isolated from over 30 tick species and multiple genera throughout Europe, Africa, and Asia, and it is unlikely that all of the 30 species represent competent vectors (16, 63). Although the HTS analysis suggests vector specificity for tick species, we cannot rule out the possibility that other tick species may serve as vectors of these viruses.
One particularly notable characteristic of these Bunyaviruses is the lack of a recognizable glycoprotein-coding segment. Although we recovered ~90% of the L and S segments for Sara tick phlebovirus, Mukava virus, and other Bunyavirus by HTS and dispelled the possibility of viral integration, we were unable to identify any scenario with similarity to Bunyavirales M segments. We have considered various explanations for this confounding result. First, these viruses encode a more complicated secondary structure of the M segments, which may inhibit efficient cDNA synthesis, subsequently resulting in the failure to detect by amplifications (23). Alternatively, these viruses may employ special pathways in the process of cellular attachment and entry other than the well-known glycoprotein precursor (GPC) depending manner (64). Finally, we also cannot exclude the possibility that an episome-like form is coded by S and L segments of these viruses. As such, they may not assemble an infectious virion but instead use other vehicles, for instance, extracellular vesicles, for their dissemination to new hosts (65).
In our analysis, a novel Chuvirus, Taiga tick nigecruvirus, was also revealed in I. persulcatus other than Nuomin virus. Phylogenic analysis based on L, G, NP, and VP4 fragments suggested that Taiga tick nigecruvirus and blacklegged tick chuvirus 2 formed a separate monophyletic clade outside genus Mivirus under the Chuviridae branch of Jingchuvirales in the order Mononegavirales, although a complete genetic characterization will be required to fully determine its taxonomy. The taiga tick nigecruvirus was found to have high identity in RdRp and relatively lower identities in N and G genes with blacklegged tick chuvirus 2, with no pathogenicity in humans and other animals. However, the reported human pathogenicity of Nuomin virus in genus Mivirus from Tahe, Heilongjiang province, suggested the potential threats of the Taiga tick nigecruvirus on humans and animals via tick bites or other transmission routes. Based on the phylogeny relationship, we considered the virus closely related to blacklegged tick chuvirus 2 as a novel Chuvirus named Taiga tick nigecruvirus tentative, while Nuomin virus as a member of Mivirus, called Mivirus nuomin.
Rhabdoviridae are another diverse set of single-stranded negative-sense RNA viruses with frequent host-switching features during their evolution history (66) and widely found to naturally infect both animals and plants. In the present study, almost all the pools of the three tick species yield the positive contains of Rhabdovirus-like contigs, which demonstrates relatively high divergence of RdRp sequences of the rhabdoviruses. Among these rhabdoviruses, only two Nucleorhabdovirus, Chimay-like rhabdovirus and blacklegged tick rhabdovirus-1, were harvested from I. persulcatus and H. concinna, respectively. While, in the Alphanemrha virus-like group, extensive diversity was observed with three virus species in H. concinna, two species in D. silvarum, and one species in I. persulcatus. Together with two Nucleorhabdovirus detected, diverse Rhabdovirus in the three tick species might be associated with high frequencies of host switches within their comparable broader host spectrums. It should be noted that more diversity of Rhabdovirus is still unexplored due to limited tick samples or geographical sites.
Although our surveys have demonstrated a wide array of highly diverse viruses harbored in the three tick species sampled, however, the limitations of the study were also prominent due to our sample bias. One constraint of our study was caused by the limited geographical regions and aggregated distributions of these ticks, as our HTS analysis merely focused on specimens collected from limited locations. We could not determine the pathogenicity and transmission capability of the viruses carried by these ticks. Necessary investigations should be carried out in detail to characterize their epidemiological features and determine their potential threats to public health. Meanwhile, in our sequence-based query strategies, the presence of “dark contigs” which had no BLAST hits, may include novel, highly divergent viruses that are unrecognizable (Supplementary Table 1). We still anticipate that many of the viruses uncovered by our HTS analysis would be along the distribution ranges of their presumed natural cycle between vector ticks and host animals, and we also speculate that analysis of ticks from diverse geographical areas would reveal greater viral diversity with more emerged agents to be discovered in future. Thus, accurate viral taxonomy would allow more precise elucidation of tick-associated virus evolutionary relationships (67).
In summary, we metagenomically analyzed the virome of I. persulcatus, D. silvarum, and H. concinna, three predominant tick species associated with pathogens transmitted in China. In addition to the exploration of viral diversity, our study also identified several tick-borne viruses with potential relevance for diseases of humans and livestock, for example, Tacheng uukuvirus, Sara tick-borne phlebovirus, Beiji orthonairovirus, Nuomin virus, and Jingmen tick virus. The novel viruses found in the present study, such as Dermacentor pestivirus-like virus, taiga tick nigecruvirus, Mukawa virus, South Bay virus, and Scapularis ixovirus, do not allow reliable conclusions on the transmissibility issues merely depending on available genetic similarities to human pathogens. Our discoveries have paved a cornerstone for more detailed investigations and rational control strategies for tick-borne viral diseases in China.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
YS contributed to the concept, design, and statistical analysis of the manuscript. TQ and MS were involved in the field survey. YS and HF were involved in the taxonomic identification of ticks. TQ and ZL were involved in the sequencing and molecular analysis of viruses. YS, TQ, MS, and MZ were involved in the analysis, interpretation, and drafting of the manuscript. All authors contributed to revising it critically for important intellectual content, final approval of the version to be published, and agreement to be accountable for all aspects of the manuscript in ensuring that questions related to the accuracy or integrity of any part of the manuscript are appropriately investigated and resolved.
This study was supported by the National Key Program for Research and Development (2022YFC2305001), the Science & Technology Fundamental Resource Investigation Program (2022FY100902), and the State Key Program of Infectious Diseases of China (2017ZX10303404).
We thank all the staff in the tick-borne disease department at Mudanjiang Forestry Central Hospital and Balin town Clinics of Zhanlantun City for their help with sample collections.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.1057977/full#supplementary-material
1. Guglielmone AA, Robbins RG, Apanaskevitch DA, Petney TN, Estrada-Pena A, Horak IG. The hard ticks of the world (Acari: Ixodida: Ixodidae). Heidelberg: Springer (2014). p. xiii + 738. doi: 10.1007/978-94-007-7497-1
2. Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG, et al. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: a list of valid species names. Zootaxa. (2010) 2528:1–28. doi: 10.11646/zootaxa.2528.1.1
3. Peñalver E, Arillo A, Delclòs X, Peris D, Grimaldi DA, Anderson SR, et al. And De La Fuente RP. Ticks parasitised feathered dinosaurs as revealed by Cretaceous amber assemblages Nat Commun. (2017) 8:1924. doi: 10.1038/s41467-017-01550-z
4. Jongejan, F, Uilenberg G. The global importance of ticks. Parasitology. (2004) 129:S3–14. doi: 10.1017/S0031182004005967
5. Madison-Antenucci S, Kramer LD, Gebhardt LL, Kauffman EB. Emerging tick-borne diseases. Clin Microbiol Rev. (2020) 33:e00083–18. doi: 10.1128/CMR.00083-18
6. Fang LQ, Liu K, Li X-L, Liang S, Yang Y, Yao H-W, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. (2015) 15:1467–79. doi: 10.1016/S1473-3099(15)00177-2
7. Ortiz DI, Piche-Ovares M, Romero-Vega LM, Wagman JM, Troyo A. The impact of deforestation, urbanization, and changing land use patterns on the ecology of mosquito and tick-borne diseases in central America. J Med Entomol. (2021) 58:1546–64. doi: 10.3390/insects13010020
8. Yu X, Liang M, Zhang S, Liu Y, Li J, Sun Y, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. (2011) 364:1523–32. doi: 10.1056/NEJMoa1010095
9. Liu Q, He B, Huang S, Wei F, Zhu X. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect Dis. (2014) 148:763–72. doi: 10.1016/S1473-3099(14)70718-2
10. Wang Z, Wang B, Wei F, Han S, Zhang L, Yang Z, et al. A new segmented virus associated with human febrile illness in China. N Engl J Med. (2019) 380:2116–25. doi: 10.1056/NEJMoa1805068
11. Ejiri H, Lim C, Isawa H, Fujita R, Murota K, Sato T, et al. Characterization of a novel thogotovirus isolated from Amblyomma testudinarium ticks in Ehime, Japan: a significant phylogenetic relationship to Bourbon virus. Virus Res. (2018) 249:57–65. doi: 10.1016/j.virusres.2018.03.004
12. Jia N, Wang JF, Shi WQ, Du LF, Sun Y, Zhan W, et al. Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell. (2022) 182:1–13. doi: 10.1016/j.cell.2020.07.023
13. Kodama F, Yamaguchi H, Park E, Tatemoto K, Sashika M, Nakao R, et al. A novel nairovirus associated with acute febrile illness in Hokkaido, Japan. Nat Commun. (2021) 12:5539. doi: 10.1038/s41467-021-25857-0
14. Kobayashi D, Kuwata R, Kimura T, Faizah AN, Higa Y, Hayashi T, et al. Toyo virus, a novel member of the Kaisodi group in the genus Uukuvirus (family Phenuiviridae) found in Haemaphysalis formosensis ticks in Japan. Arch Virol. (2021) 166:2751–62. doi: 10.1007/s00705-021-05193-w
15. Ma J, Lv X, Zhang X, Han S, Wang Z, Li L, et al. Identification of a new orthonairovirus associated with human febrile illness in China. Nat Med. (2021) 27:434–39. doi: 10.1038/s41591-020-01228-y
16. Monsalve-Arteagaid L, Alonso-Sardon M, Bellido JLM, Santiago MBV, Lista MCV, Aban JL, et al. Seroprevalence of Crimean-Congo hemorrhagic fever in humans in the world health organization European region: a systematic review. PLoS Negl Trop Dis. (2020) 14:e0008094. doi: 10.1371/journal.pntd.0008094
17. McMullan L, Folk SM, Kelly AJ, MacNeil A, Goldsmith CS, Metcalfe MG, et al. A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med. (2012) 367:834–41. doi: 10.1056/NEJMoa1203378
18. Grard G, Moureau G, Charrel RN, Lemasson JJ, Gonzalez JP, Gallian P, et al. Genetic characterization of tick-borne Flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology. (2007) 361:80–92. doi: 10.1016/j.virol.2006.09.015
19. Sharma R, Cozens DW, Armstrong PM, Brackney DE. Vector competence of human-biting ticks Ixodes scapularis, Amblyomma americanum and Dermacentor variabilis for Powassan virus. Parasit Vectors. (2021) 14:466. doi: 10.1186/s13071-021-04974-1
20. Gaudreault NN, Madden DW, Wilson WC, Trujillo JD, Richt JA. African swine fever virus: an emerging DNA arbovirus. Front Vet Sci. (2020) 7:215. doi: 10.3389/fvets.2020.00215
21. Li C, Shi M, Tian J, Lin X, Kang Y, Chen L, et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife. (2015) 4:e05378. doi: 10.7554/eLife.05378
22. Pettersson JH, Shi M, Bohlin J, Eldholm V, Brynildsrud OB, Paulsen KM, et al. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci Rep. (2017) 7:1–7. doi: 10.1038/s41598-017-11439-y
23. Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol. (2017) 15:183–92. doi: 10.1038/nrmicro.2016.182
24. Teng KF, Jiang ZJ. Economic insect fauna of China. Fasc 39. Acarina: Ixodidae. Beijing: Science Press, Academia Sinica. (1991). p. 1–359.
25. Loens K, Bergs K, Ursi D, Goossens H, Ieven M. Evaluation of NucliSens easyMAG for automated nucleic acid extraction from various clinical specimens. J Clin Microbiol. (2007) 45:421–5. doi: 10.1128/JCM.00894-06
26. Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek RDNA. primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. (1994) 3:294–9.
27. Machida RJ, Knowlton NPCR. primers for metazoan nuclear 18S and 28S ribosomal DNA sequences. PLoS ONE. (2012) 7:e46180. doi: 10.1371/journal.pone.0046180
28. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatic. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
29. Krueger F, James FO, Ewels PA, Afyounian E, Schuster-Boeckler B. FelixKrueger /TrimGalore: v0, 6.7. (2021).
30. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. (1883) 29:644–52. doi: 10.1038/nbt.1883
31. Haas BJ, Papanicolaou A, Yassour M, Grabherr MG, Blood PD, Bowden JC, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. (2013) 8:1494–512. doi: 10.1038/nprot.2013.084
32. Burland TG. DNASTAR's Lasergene sequence analysis software. Methods Mol Biol. (2000) 132:71–91. doi: 10.1385/1-59259-192-2:71
33. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
34. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
35. Xu L, Guo MJ, Hu B, Zhou H, Yang W, Hui LX, et al. Tick virome diversity in Hubei Province, China, and the influence of host ecology. Virus Evolut. (2021) 7:1–12. doi: 10.1093/ve/veab089
36. Xia LC, Cram JA, Chen T, Fuhrman JA, Sun F. Accurate genome relative abundance estimation based on shotgun metagenomic reads. PLoS ONE. (2011) 6:e2799. doi: 10.1371/journal.pone.0027992
37. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol. (2013) E30:772–80. doi: 10.1093/molbev/mst010
38. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. (2009) 25:1972–3. doi: 10.1093/bioinformatics/btp348
39. Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. (2011) 27:1164–5. doi: 10.1093/bioinformatics/btr088
40. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. (2010) 59:307–21. doi: 10.1093/sysbio/syq010
41. Sameroff S, Tokarz R, Vucelja M, Jain K, Oleynik A, Boljfetić M„ et al. Virome of Ixodes ricinus, Dermacentor reticulatus, and Haemaphysalis concinna ticks from Croatia. Viruses. (2022) 14:929. doi: 10.3390/v14050929
42. Sameroff S, Tokarz R, Charles RA, Jain K, Oleynik A, Che X, et al. Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci Rep. (2019) 9:10421. doi: 10.1038/s41598-019-46914-1
43. Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, Chen LJ, et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J Virol. (2016) 90:659–69. doi: 10.1128/JVI.02036-15
44. Ali M, Hameed S, Tahir M. Luteovirus: insights into pathogenicity. Arch Virol. (2014) 159:2853–60. doi: 10.1007/s00705-014-2172-6
45. Tokarz R, Williams SH, Sameroff S, Sanchez Leon M, Jain K, Lipkin WI, et al. (2014). Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J Virol. 88:11480–92. doi: 10.1128/JVI.01858-14
46. Quan L, Wang ZD, Gao Y, Lv X, Han S, Zhang X, et al. Identification of a new chuvirus associated with febrile illness in China. Nat Med. (2021) 27:434–9. doi: 10.21203/rs.3.rs-104938/v1
47. Shi J, Shen S, Wu H, Zhang Y, Deng F. Metagenomic profiling of viruses associated with Rhipicephalus microplus ticks in Yunnan province, China. Virol Sin. (2021) 36:623–35. doi: 10.1007/s12250-020-00319-x
48. Afonso CL, Amarasinghe GK, Bányai K, Bào Y, Basler CF, Bavari S, et al. Taxonomy of the order Mononegavirales: update 2016. Arch Virol. (2016) 161:2351–60. doi: 10.1007/s00705-016-2880-1
49. Kuhn JH, Adkins S, Alioto D, Alkhovsky SV, Amarasinghe GK, Anthony SJ, et al. Taxonomic update of phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch Virol. (2021) 166:3513–66. doi: 10.1007/s00705-021-05143-6
50. Meng F, Ding M, Tan Z, Zhao Z, Xu L, Wu J, et al. Virome analysis of tick-borne viruses in Heilongjiang province, China. Ticks Tick Borne Dis. (2019) 10:412–20. doi: 10.1016/j.ttbdis.2018.12.002
51. Wang Y, Wei Z, Lv X, Han S, Wang Z, Fan C, et al. A new nairo-like virus associated with human febrile illness in China. Emerg Microbes Infect. (2021) 10:1200–08. doi: 10.1080/22221751.2021.1936197
52. Elliott RM, Brennan B. Emerging phleboviruses. Current Opinion Virol. (2014) 5:50–7. doi: 10.1016/j.coviro.2014.01.011
53. Pettersson JH, Ellström P, Ling J, Nilsson I, Bergström S, González-Acuña DA, et al. Circumpolar diversification of the Ixodes uriae tick virome. PLoS Pathog. (2020) 16:e1008759. doi: 10.1371/journal.ppat.1008759
54. Nibert ML, Ghabrial SA, Maiss E, Lesker TR, Vainio EJ, Jiāng D, et al. Taxonomic reorganization of family Partitiviridae and other recent progress in Partitivirus research. Virus Res. (2014) 188:128–41. doi: 10.1016/j.virusres.2014.04.007
55. Jia N, Liu H, Ni X, Bell-Sakyi L, Zheng Y, Song J, et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine. (2019) 43:317–24. doi: 10.1016/j.ebiom.2019.04.004
56. Li LJ, Ning NZ, Zheng YC, Chu YL, Cui XM, Zhang MZ, et al. Virome and blood meal-associated host responses in Ixodes persulcatus naturally fed on patients. Front Microbiol. (2022) 12:728996. doi: 10.3389/fmicb.2021.728996
57. Liu Z, Li L, Xu W, Yuan Y, Liang X, Zhang L, et al. Extensive diversity of RNA viruses in ticks revealed by metagenomics in northeastern China. PLoS Negl Trop Dis. (2022) 16:e0011017. doi: 10.1101/2022.04.27.489762
58. Jiao J, Lu Z, Yu Y, Ou Y, Fu M, Zhao Y, et al. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasit Vectors. (2021) 14:287. doi: 10.1186/s13071-021-04740-3
59. Kuhn JH, Wiley MR, Rodriguez SE, Bào Y, Prieto K, Travassos da Rosa AP, et al. Genomic characterization of the genus Nairovirus (Family Bunyaviridae). Viruses. (2016) 8:164. doi: 10.3390/v8060164
60. Dong Z, Yang M, Wang Z, Zhao S, Xie S, Yang Y„ et al. (2021). Human Tacheng tick virus 2 infection, China, 2019. Emerg Infect Dis. 27:594. doi: 10.3201/eid3201/191486
61. Matsuno K, Kajihara M, Nakao R, Nao N, Mori-Kajihara A, Muramatsu M, et al. (2018). The unique phylogenetic position of a novel tick-borne phlebovirus ensures an ixodid origin of the genus Phlebovirus. mSphere. 3:e00239–18. doi: 10.1128/mSphere.00239-18
62. Wang YN, Jiang RR, Ding H, Zhang XL, Wang N, Zhang YF, et al. First Detection of Mukawa Virus in Ixodes persulcatus and Haemaphysalis concinna in China. Front. Microbiol. (2022) 13:791563. doi: 10.3389/fmicb.2022.791563
63. Bente DA, Forrester NL, Watts DM, McAuley AJ, Whitehouse CA, Bray M, et al. Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antiviral Res. (2013) 100:159–89. doi: 10.1016/j.antiviral.2013.07.006
64. Welch SR, Scholte FE, Spengler JR, Ritter JM, Coleman-McCray JD, Harmon JR, et al. The Crimean-Congo hemorrhagic fever virus nsm protein is dispensable for growth in vitro and disease in Ifnar-/- mice. Microorganisms. (2020) 8:775. doi: 10.3390/microorganisms8050775
65. Zhou W, Woodson M, Neupane B, Bai F, Sherman MB, Choi KH, et al. Exosomes serve as novel modes of tick-borne flavivirus transmission from arthropod to human cells and facilitates dissemination of viral RNA and proteins to the vertebrate neuronal cells. PLoS Pathog. (2018) 14:e1006764. doi: 10.1371/journal.ppat.1006764
66. Longdon B, Murray GG, Palmer WJ, Day JP, Parker DJ, Welch JJ, et al. The evolution, diversity, and host associations of rhabdoviruses. Virus Evolut. (2015) 1:1. doi: 10.1093/ve/vev014
Keywords: virome, diversity, high-throughput sequencing, Ixodidae, China
Citation: Qin T, Shi M, Zhang M, Liu Z, Feng H and Sun Y (2023) Diversity of RNA viruses of three dominant tick species in North China. Front. Vet. Sci. 9:1057977. doi: 10.3389/fvets.2022.1057977
Received: 30 September 2022; Accepted: 16 December 2022;
Published: 13 January 2023.
Edited by:
Daniel Oladimeji Oluwayelu, University of Ibadan, NigeriaReviewed by:
Shahid Karim, University of Southern Mississippi, United StatesCopyright © 2023 Qin, Shi, Zhang, Liu, Feng and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Sun,  Mzc4MTc2OTM4QHFxLmNvbQ==
Mzc4MTc2OTM4QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.