 Xin-yan Lu1†
Xin-yan Lu1† Quan-fu Zhang2†Dan-dan Jiang3Ya-fang Liu1Bin Chen1Shuang-ping Yang3Zong-ti Shao4Hang Jiang4Jian Wang4
Quan-fu Zhang2†Dan-dan Jiang3Ya-fang Liu1Bin Chen1Shuang-ping Yang3Zong-ti Shao4Hang Jiang4Jian Wang4 Yi-hao Fang5*Chun-hong Du4*
Yi-hao Fang5*Chun-hong Du4* Xing Yang1*
Xing Yang1*- 1Integrated Laboratory of Pathogenic Biology, College of Preclinical Medicine, Dali University, Dali, China
- 2Department of Digestion, First Affiliated Hospital of Chengdu Medical College, Chengdu, China
- 3School of Public Health, Dali University, Dali, China
- 4Yunnan Provincial Key Laboratory of Natural Epidemic Disease Prevention and Control Technology, Puer, China
- 5Fu-gong Administration Bureau, Gaoligong Mountain National Nature Reserve, Yunnan, China
The mitochondrial genome may include crucial data for understanding phylogenetic and molecular evolution. We sequenced the complete mitogenome of Haemaphysalis nepalensis and Haemaphysalis yeni for the first time. H. nepalensis and H. yeni's complete mitogenomes were 14,720 and 14,895 bp in size, respectively, and both contained two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and 13 protein-coding genes (PCG). Haemaphysalis nepalensis have one control region (D-loop). The adenine + thymine concentration of the genomes of H. nepalensis and H. yeni was 77.75 and 78.41%, respectively. The codon use pattern and amino acid content of proteins were both observed to be affected by the AT bias. Genes in the mitogenome were organized and located in a comparable manner to previously known genes from Haemaphysalis ticks. Mitochondrial PCGs were used to perform phylogenetic relationships based on the Minimum Evolution (ME) approach using MEGA 7.0 software, the results reveal that H. nepalensis has tight links with H. tibetensis, H. yeni and H. kolonini share a sister group relationship, and that H. nepalensis and H. yeni belong to Haemaphysalis. The results of this study include the following: (i) discovered and supplied new tick records (H. nepalensis) for China, (ii) provided the first complete mitochondrial genome for H. nepalensis and H. yeni and revealed their phylogenetic relationships, and (iii) the features of the mitochondrial genome of H. nepalensis and H. yeni provided more genetic reference for Phylogeography, systematics, and population genetics of the Haemaphysalis species.
Introduction
Ticks are obligatory ectoparasites of all vertebrate species and are blood-sucking arachnids. They can spread the broadest spectrum of zoonotic pathogens that lead to animal and human diseases, causing substantial financial damage to animal productivity (1). Tick populations are also growing as a result of climate change (2). Recently, numerous major tick-borne pathogens have been identified in ticks, such as Babesia ovata, Chlamydiaceae bacteria, Rickettsia japonica, Anaplasma bovis, and Severe Fever with Thrombocytopenia Syndrome Bunyavirus, have sparked increased interest in the field of public health (3). Despite the medical importance of ticks in the spread of Lyme disease, spotted fever group rickettsiosis, and other human diseases, the details of the entire mitochondrial genome are not well recognized, and the phylogenetic links are not established (4).
Haemaphysalis nepalensis (5) (Ixodidae) is an important tick that belongs to the Ixodidae family, Metastriata group, Herpetobia Canestrini subgenus. H. nepalensis has previously been discovered in India and Nepal. H. nepalensis, a parasite that affects people, sheep, and dzo, is most widely distributed in Tibet in China (5). India, Japan, the Philippines, Indonesia, Ceylon, Borneo, and China are the main distribution areas for Haemaphysalis yeni (6) (Ixodidae). In China, Fujian, Guangdong, Hainan, Hunan, Hubei, Shanxi, and Yunnan account for the majority of the records (7). Although the morphological characteristics of H. nepalensis and H. yeni have been illustrated and documented, it is unknown what their complete mitochondrial genome looks like (8). In recent years, tick bite reports on people have increased. The quality of human health continues to decline as a result of relapsing and persistent illness, long-term effects linked to tick-borne diseases, and even fatalities brought on by delayed or incorrect diagnosis. It is crucial to verify tick classification in order to combat disease (9).
The mitochondrion, a vital organelle in eukaryotes, contains a distinct genome from the cell nucleus. The mitogenome typically contains minimal levels of recombination, a simple structure, maternal inheritance, and fast evolution. It is therefore acknowledged as one of the most trustworthy and effective molecular tools for research on tick phylogenetic studies, species identification, and population structure (10). Complete mitochondrial genomes are normally double-stranded, with the length of circular nucleotides ranging between 14 and 19 kb, and consist of 13 PCGs: cox1-cox3, nad1-nad6, atp8, atp6, nad4l, and cytb, 22 tRNAs, two rRNAs, and D-loop. Researchers have used mitochondrial genomes as instructive molecular labels to investigate numerous evolutionary studies among animals (11). Recent advances in sequencing technology have made it easier than ever to reconstruct phylogenies using animal mitochondrial genomes in their entirety rather than just partial DNA sequences.
Using Illumina sequencing technology, we first sequenced and annotated the entire mitochondrial genomes of H. nepalensis and H. yeni in the current work, then we compared them to other Haemaphysalis mitochondrial genomes. Further study of phylogenetics, mitochondrial genomes, nuclear rRNA genes, and taxonomy revision of related Haemaphysalis species and the Ixodidae family may benefit from the information provided here (12).
Materials and methods
Samples and DNA extraction
The H. nepalensis adult specimens used in this study were procured in October 2020 from Deqin in the Yunnan Province of China (27°33′N, 98°3′E) (n = 2, female; n = 1, male), Diqing Tibetan Autonomous Prefecture. Adult samples of H. yeni were collected in March 2021 from High Li Gong Shan in the Nujiang Lisu Autonomous Prefecture of the Chinese Yunnan Province (26°34′N, 98°48′E) (n = 2, female; n = 1, male). Professor Chunhong Du examined the species' morphological identification using the essential diagnostic features (13). After collection, one adult female specimen was used for DNA extraction, and the remainder of the ticks were held as voucher specimens. The collected tick specimens were deposited at the Parasitological Museum of Dali University under the voucher numbers DLUP2010 and DLUP2103, respectively. The samples were maintained at −80°C and preserved in 95% alcohol before being utilized for DNA extraction. Single tick genomic DNA was extracted using DNAzol (Life Technologies, USA) following the manufacturer's instructions and stored for further processing (13).
DNA amplification and sequencing
Two overlapping sets of primers were used to amplify the mitogenomes of the H. nepalensis and H. yeni species. The long-PCR primers were created using the 12S rRNA and cox1 genes of Haemaphysalis bancrofti (NC041076) and Haemaphysalis japonica (MG253031). The following PCR primers were employed:
HN1 (5′-CTCYAATTAAATTCTTTATRGAAT-3′, 5′-ATTAGGCTTGGTTGTATGAAWAA-3′);
HN2 (5′-TCTGTATTAAYTACAGCAATTTTAC-3′, 5′-CAAATWTTAAATTTAACACCCCAATTTTA-3′).
HY1 (5′-CTCTAGTTAATYTTGTGCCAGCAA-3′, 5′-AGCAACAGCGGTTATACAAWAAG-3′);
HY2 (5′-TCCGTATTAATTACTGCAATTCTAC-3′, 5′-CAACTTTAWAATGTAACACTCCAATCTTA-3′) (14).
The PCR was implemented in a 50 μl reaction mixture including 10 μl of 5X PrimeSTAR GXL Buffer (Takara, Japan), 1 μl of PrimeSTAR GXL DNA Polymerase (Takara, Japan), 4 μl of each primer, 4 μl of dNTPs, 4 μl of DNA template, and 23 μl of nuclease-free water. The PCR conditions used in the amplification procedure were as follows: initial denaturation at 95°C for 5 mins, followed by 45 cycles of denaturation (98°C for 10 s), annealing (68°C for 30 s), and extension (68°C for 10 mins), and a final extension was subjected to 68°C for 10 mins. The findings of the PCR were examined using 1.2 percent agarose gel electrophoresis stained with ethidium bromide (15). Libraries were sequenced on the Illumina HiSeq 2500 platform at Shanghai Biotechnology Co. Ltd. after the amplified PCR products had been purified.
Gene annotation and sequence analysis
Data quality was evaluated from four indicators: single base quality of sequencing data, base content distribution, GC content distribution, and sequence base quality. The software used is FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). AdapterRemoval software (v2.0) was used to remove the contamination of original data, and SOAPec software (v2.01) was used to correct the quality of data based on k-mer distribution. With the mitochondrial genome sequence of H. kolonini as a reference, the whole mitogenomes of H. nepalensis and H. yeni were extracted using online BLAST tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi). SPAdesv3.9.0 (http://cab.spbu.ru/software/spades/) and A5-miseqv20150522 were utilized to compile high-quality next-generation sequencing data for de novo mitogenome construction (16). BLASTn (BLAST v2.2.31+) alignment was performed between the sequences with high sequencing depth and the NT Library in NCBI to pick out the mitochondrial sequences of each spliced result. The mitochondrial stitching results obtained by the above different software were analyzed by using MUMmer (v3.1) software for collinearity analysis to determine the position relationship between conting and to fill the gap of contigs. Pilon software (v1.18) was used to correct the results to obtain the final mitochondrial sequence. Using the MITOS web server (http://mitos.bioinf.uni-leipzig.de/), the mitochondrial genomes of H. nepalensis and H. yeni were annotated (17). The program tRNAscan-SE was used to forecast the secondary cloverleaf architectures of tRNAs (18). The skewness of the composition was estimated using the formulas GC-skew = [G – C]/[G + C] and AT-skew = [A – T]/[A + T]. The analysis of the nucleotide composition and relative synonymous codon usage was done using the software MEGA v7.0 and Geneious Prime (RSCU). The whole mitogenome annotation findings were submitted to NCBI in table (.tbl) format (19).
Phylogenetic analysis
Based on the concatenated datasets of 13 PCGs from 40 ticks, including 34 Metastriata and six species of Prostriata, the phylogenetic connection was examined. The concatenated nucleotide sequence of the mitochondrial 13 PCGs was used to determine the evolutionary relationship using the ME approach in the software MEGA v.7.0 based on 1,000 bootstrapped datasets. The PCGs' multiple sequence alignments were conducted using the MUSCLE nucleotide mode. All places with incomplete or blank information were eliminated. Based on the Akaike information criterion (AIC), the GTR + G + I best model was selected as the best-fit replacement model for nucleotide phylogenetic connectivity (20). To examine the RSCU and nucleotide composition, MEGA program was utilized. A chronogram made with FigTree v1.4.2 was used to demonstrate the evolutionary relationships that emerged.
Genome organization and nucleotide composition
The entire mitogenomes of H. nepalensis and H. yeni used in this study were closed circular molecules with a size of 14,720 and 14,895 bp, respectively (Figure 1). The complete mitochondrial genome had 37 distinguishing genes, including 22 tRNAs, two rRNAs (rrnL and rrnS), 13 PCGs (cox1-3, nad1-6, atp8, atp6, nad4l, and cob), and H. nepalensis has one D-loop (Table 1). Under the accession numbers NC064124 and ON853615, the whole mitochondrial genomes of H. nepalensis and H. yeni, respectively, had been uploaded to GenBank. The mitochondrial genomes of H. nepalensis and H. yeni included the following nucleotide compositions: adenines = 38.46% (38.66%), thymines = 39.29% (39.74%), guanines = 9.51% (9.5%), and cytosines = 12.74% (12.28%). The complete mitochondrial genome of H. nepalensis and H. yeni were biased toward AT nucleotides (77.75 and 78.41%) (Table 3). Fourteen genes from both tick species are encoded on the majority (J) strand (Table 1). The H. nepalensis mitogenome includes 15 overlapping areas and intergenic nucleotides with a total length of 116 bp (range from 1 to 41 bp) and 464 bp (ranging from 2 to 334 bp). There were 14 overlapping areas in the mitochondrial genome of H. yeni, and the intergenic nucleotides were 49 base pairs long (ranging from 1 to 13 bp).
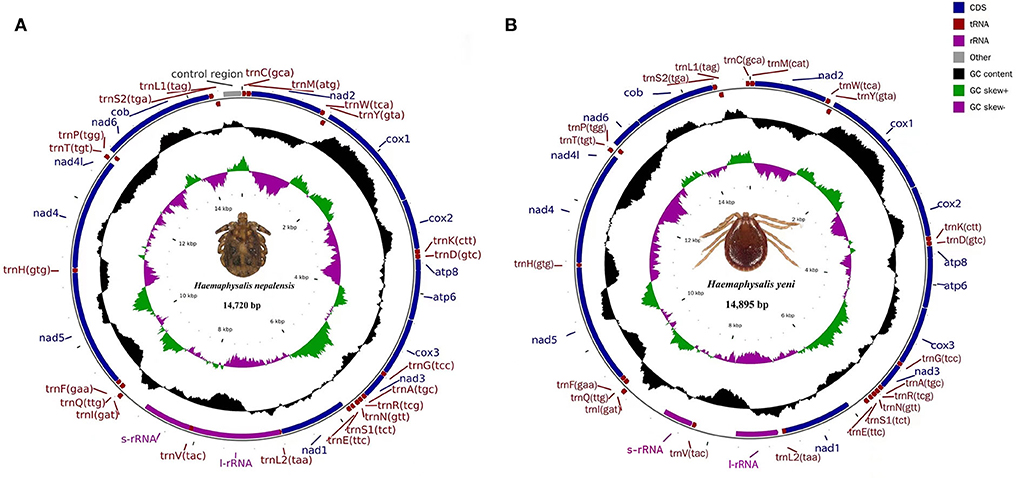
Figure 1. Mitochondrial genome map of (A) H. nepalensis and (B) H. yeni. Genes encoded in the forward direction are located on the outside of the ring, while those encoded in the reverse direction are located on the inside of the ring.
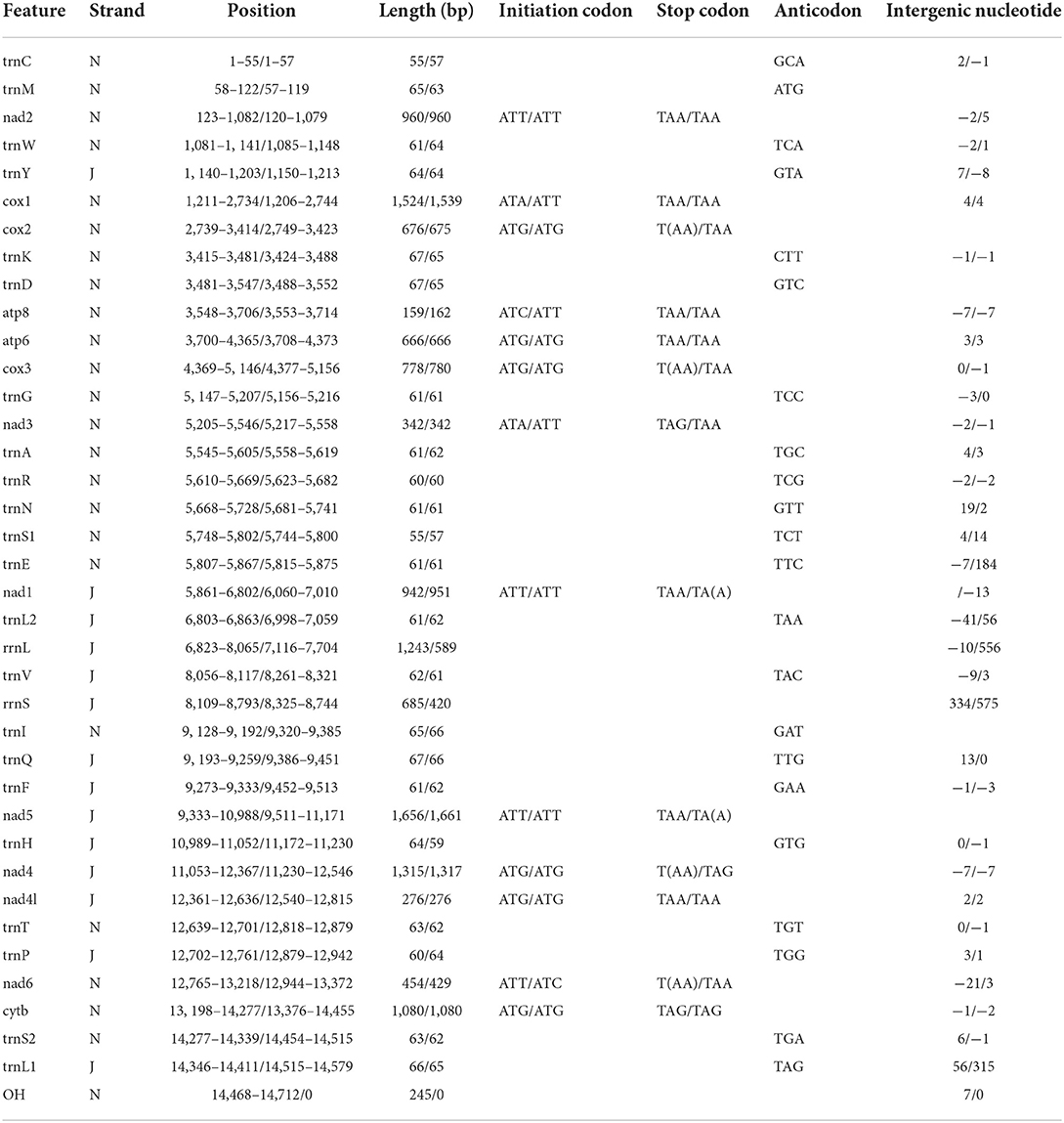
Table 1. Organization of the H. nepalensis and H. yeni mitochondrial genomes.
PCGs and codon usage
Both H. nepalensis and H. yeni possessed 13 typical PCGs in their whole mitogenomes, including four cytochrome genes (cytb and cox1-3), two ATP genes (atp6 and atp8), seven NADP genes (nad4l and nad1-6) (Figure 1). The PCGs' respective regions were 10,828 bp and 10,837 bp in size. The PCGs of H. nepalensis began with ATA (cox1 and nad3), ATT (nad1, nad2, nad5, nad6), ATG (cox3, atp6, cox2, nad4l, and nad4), and ATC (atp8), also seven PCGs that were terminated by TAA (cox1, nad2, atp8, atp6, nad1, and5, and nad4l). The stop codon employed by the cox2, cox3, nad4, and nad6 was a single T, whereas the nad3 and cytb were terminated with TAG. The PCGs of H. yeni begin with ATT (cox1, nad2, nad3, atp8, nad1, and nad5), ATG (cox3, atp6, cox2, nad4l, nad4, and cytb), and only nad6 with ATC. Nad1 and nad5 used incomplete termination codons that consisted of TA, while the majority of PCGs terminated with TAA (Table 2).
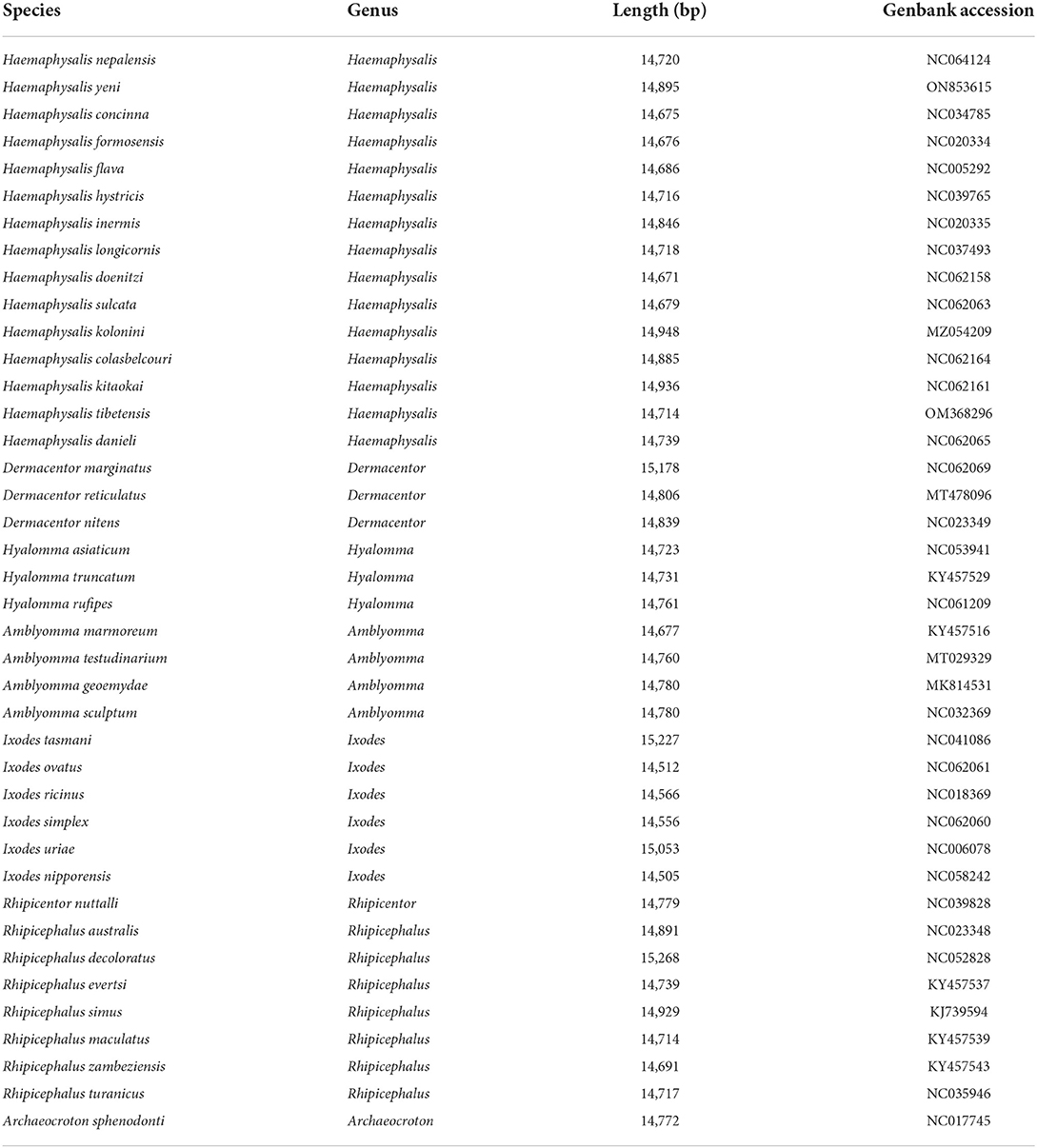
Table 2. List of the 40 tick species analyzed in this paper with their GenBank numbers.
We investigated the RSCU and codon use patterns in the mitochondrial genomes of H. nepalensis and H. yeni. 3,405 amino acids in total were encoded by the mitochondrial genome of H. nepalensis. Leucine (16.18%) was the amino acid that was used the most, followed by phenylalanine (13.71%) and isoleucine (9.36%). The PCGs encoded a total of 3,474 amino acids of the H. yeni mitogenome; phenylalanine (14.42%), leucine (12.95%), and isoleucine (12.14%) were the most often utilized amino acids; arginine (0.9%) was the least frequently used amino acid, indicating the popularity of biasness toward AT content among the PCGs (Table 3; Figure 2).
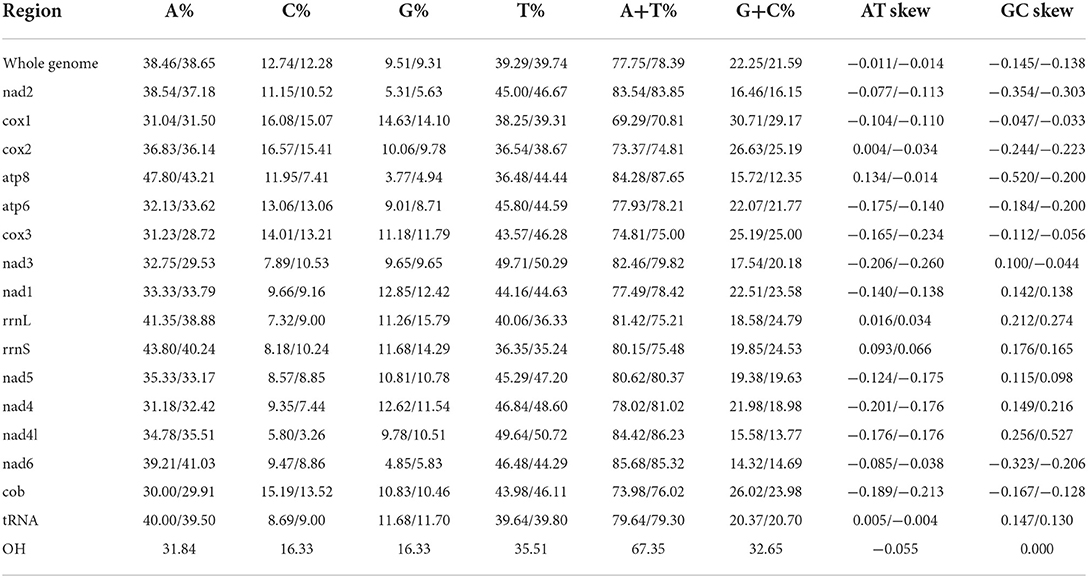
Table 3. Composition and skewness of H. nepalensis and H. yeni mitogenome.
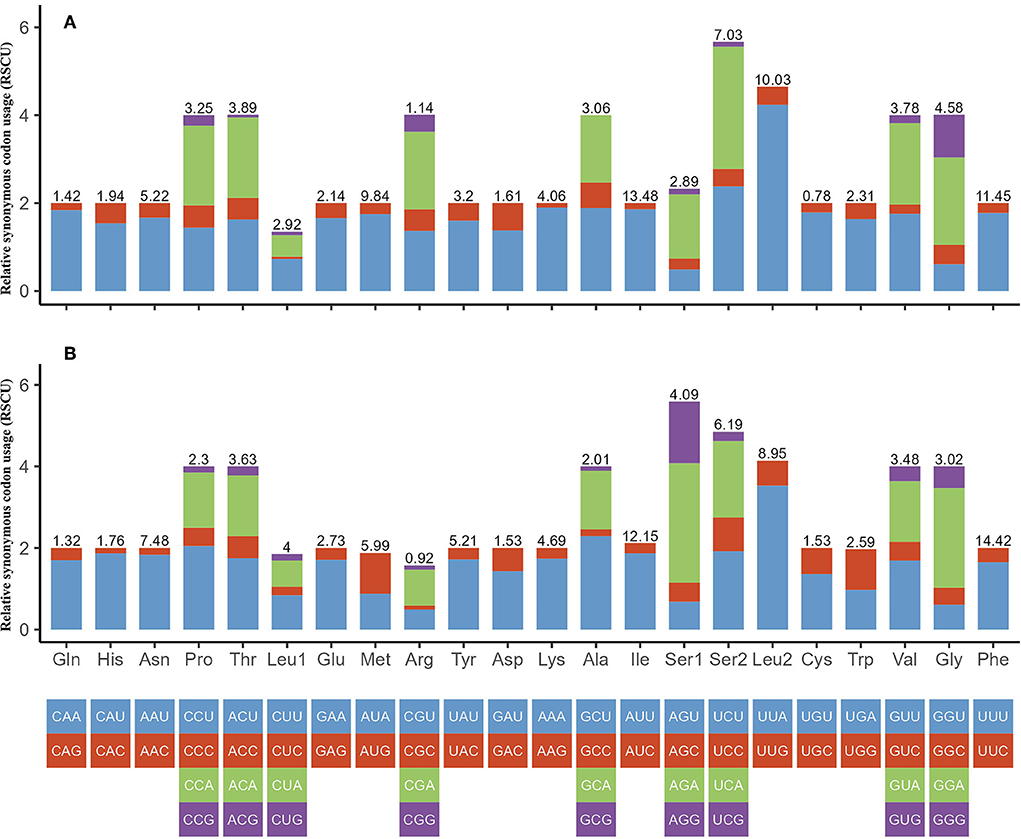
Figure 2. Relative synonymous codon usage (RSCU) of codons. (A) H. nepalensis and (B) H. yeni. The box below the bar chart represents all codons encoding each amino acid, and the height of the column above represents the sum of all RSCU values.
A+T skewness and transfer RNAs
Positive AT and GC skew in the whole mitochondrial genomes of H. nepalensis and H. yeni indicates that bases A and G are less frequent than their comparable bases (Table 3). The mitogenomes of H. nepalensis and H. yeni had a set of 22 tRNAs, like the majority of mitochondrial genome DNA. The tRNAs of H. nepalensis are 1,370 bp long and range in size from 55 nucleotides (trnC and trnS1) to 67 nucleotides (trnK, trnD, and trnQ). H. yeni's tRNAs were between 57 and 66 bp in length. The tRNA-C, tRNA-F, and tRNA-S1 genes, as well as the 14 tRNAs encoded on the majority (J) strand, did not exhibit the normal cloverleaf structure.
Phylogenetic analysis
To determine the phylogenetic tree of 40 Ixodida, the whole mitogenomes of H. nepalensis and H. yeni were further examined (Table 2). Using ME investigations in the context of the Maximum Composite Likelihood model, the topologies of the phylogenetic tree were examined based on the concatenated nucleotide sequences of 13 PCGs (Figure 3). The majority of the genera Ixodes, Amblyomma, Dermacentor, Hyalomma, and Rhipicephalus in the tree formed a monophyletic branch in the phylogenetic analyses, according to the ML analyses. Haemaphysalis species were paraphyletic, despite this. The sequences in the tree are divided into the Metastriata and Prostriata main branches. There is only one explicit Ixodes species in the Prostriata group. H. nepalensis and H. tibetensis are clustered together on one branch of the phylogenetic tree with a high nodal support value, and H. yeni and H. kolonini have a close phylogenetic relationship, indicating a sister group link between them. Furthermore, the phylogenetic relationship revealed that H. nepalensis and H. yeni were divided into various clades, yet they belonged to Haemaphysalis within Ixodida, correlating with other studies.
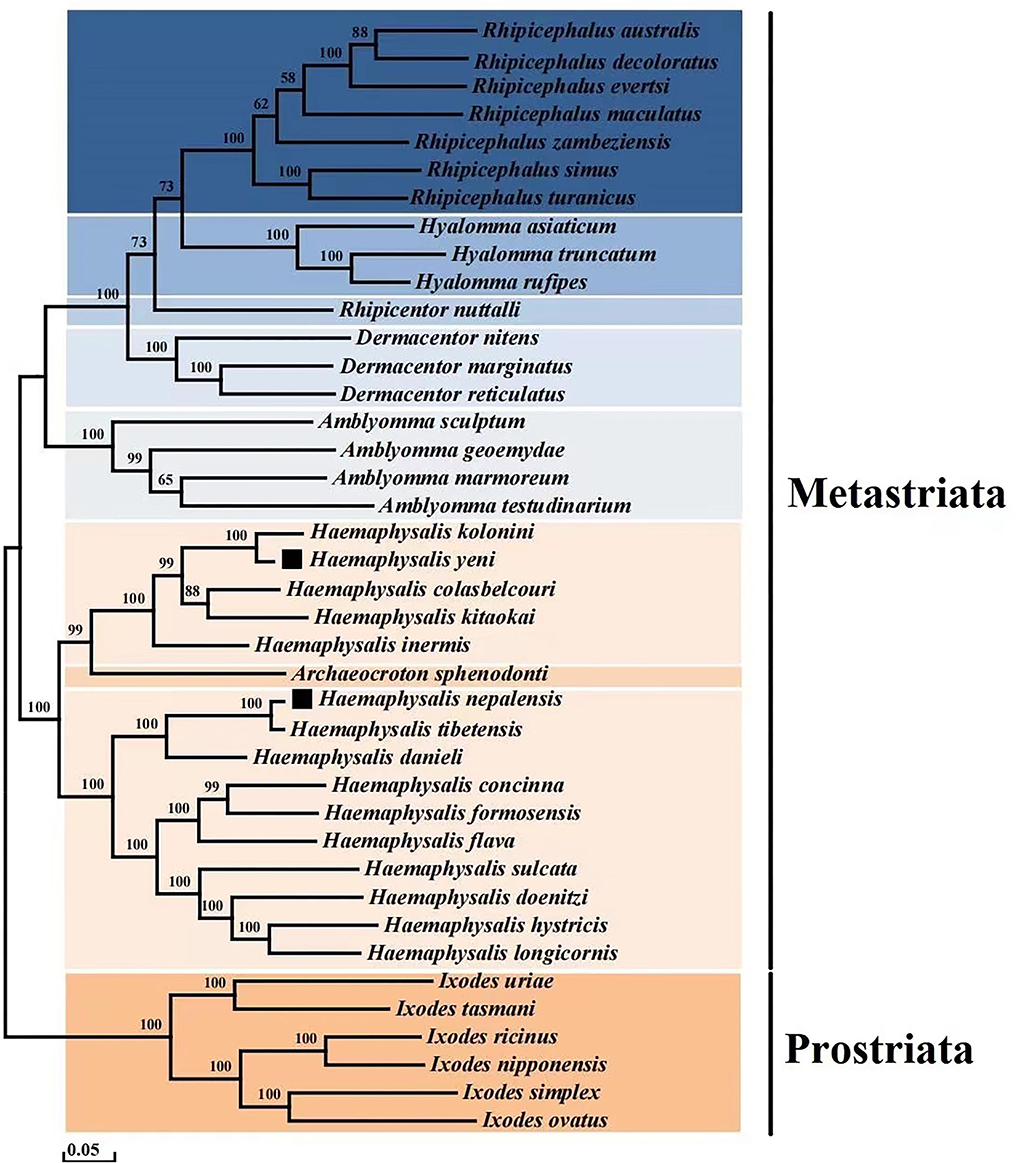
Figure 3. Phylogenetic analysis based on the nucleotide sequences of the 13 PCGs in the mitogenome. Each genus is represented by different colors.
Discussion
Similar to previously known tick mitochondrial genomes, a comparison of the mitochondrial genome sequences in the genus Haemaphysalis suggests that the D-loop is where the size shift is most prominent. The D-loop is the largest non-coding segment of the mitogenome and contains the major regulatory elements for its replication and expression. Furthermore, the high-level of intraspecific genetic variation found in the D-loop favors its use in population genetic studies of all kinds of organisms and phylogeographic analysis. Differences in the size of the complete mitogenome between species were driven by variation in the size of the control area, which in turn differed in both the size of different short repeat nucleotides and those replicated within it. In this study, most ticks in the Haemaphysalis genus have two D-loops. Nevertheless, H. yeni and H. longicornis have no D-loop, H. nepalensis and H. hystricis have one D-loop, and H. colasbelcouri and H. kitaokai have three D-loops. Because the D-loop lacks characteristic coding constraints, it accumulates indels, a variable number of tandem repeats, and base substitutions which are responsible for the widespread length differentiation found in the mtDNA molecule. Most of the tRNA secondary structures, the genome's codon use, and the gene makeup and quantity of Haemaphysalis are identical to other mitochondrial species that have been observed (21). The tRNA-F gene of H. nepalensis, however, differs from those of most of the genus Haemaphysalis in that it did not reveal the usual cloverleaf structure. The mitochondrial genomes' gene arrangement is comparable with that of other Haemaphysalis.
Through the creation of two new primer nucleotide sequences, next-generation sequencing technologies, and a brand-new long-range PCR amplification, we explored the mitogenomes. This will open a new way for study into H. nepalensis and H. yeni in the future. Population biology, behavior, phylogenetics studies, and tick ecology are all made easier by genetic information. Only the 16s rDNA partial sequences for H. nepalensis and H. yeni are currently available in the database. The mitochondrial partial sequencing can only offer relevant data, though. The entire mitochondrial genome can provide more sensitivity and resolution for analysis of the evolutionary relationships between closely related species as compared to partial mitochondrial sequence.
The current study shows that whereas Haemaphysalis species are paraphyletic, the majority of genera analyzed are monophyletic. According to the results of the phylogenetic analysis, there are two branches within the eight genera: one major clade contains a branch of Ixodes species that is quite explicit and monophyletic, and the other clade is made up of the genera Haemaphysalis, Archaeocroton, Amblyomma, Dermacentor, Rhipicentor, Hyalomma, and Rhipicephalus. Within the Metastriata, they are split into two branches, one of which has a sister-taxon (Haemaphysalis + Archaeocroton), and the other of which contains the genera Amblyomma, Dermacentor, and Rhipicentor, which are sisters to the sister-group genera Hyalomma and Rhipicephalus.
In addition, the clade (H. nepalensis + H. tibetensis + H. danieli) that includes our target species H. nepalensis grouped into one branch, and H. yeni and H. kolonini have strong links and a high nodal bootstrap support value.
The H. nepalensis and H. tibetensis, the H. yeni and H. kolonini genes are arranged in the same order and the PCG encoding of H. nepalensis is 11 bp less than the PCG encoding of H. tibetensis, and the PCG encoding of H. yeni is 35 bp more than the PCG encoding of H. kolonini. The percent identity of the cox1 gene of these two groups species were 98.37 and 90.16%, respectively. The percent identity of the complete mitochondrial sequences were 98.14 and 92.75%, respectively. Species identification mainly depends on morphology and molecular studies. Although the two groups (H. nepalensis and H. tibetensis, H. yeni and H. kolonini) were phylogenetically very closely related in phylogenetic tree analysis, the morphological differences and percent identity are < 99%, which may lead to the formation of different species. The H. tibetensis and H. nepalensis species are now only known to have been found in Tibet alone. The phylogenetic analysis result demonstrates a close relationship between H. nepalensis and H. tibetensis, demonstrating the correctness of our sequencing findings and demonstrating the first distribution of H. nepalensis in Yunnan Province.
Conclusion
In this study, we sequenced the entire mitochondrial genomes of H. nepalensis and H. yeni, measuring 14,720 base pairs (bp) and 14,895 base pairs (bp), respectively. These genomes contained 37 genes (13 PCGs, 22 tRNAs, and two rRNAs), which are typical of the Haemaphysalis mitochondrial genome. H. nepalensis had an extra D-loop. All PCGs began with the ATN codon, with ATT and ATG being the most frequent initiation codons. The cox2, cox3, nad4, and nad6 of H. nepalensis and nad1 and nad5 of H. yeni had incomplete termination codons consisting of T or TA, and the other PCGs stop with the canonical TAG or TAA. The whole mitochondrial genomes of H. nepalensis and H. yeni had negative AT-skew and GC-skew, which is consistent with the majority of sequenced Haemaphysalis. Higher-level phylogenies might be provided by the whole mitochondrial genome. This study provides a crucial resource for better understanding the phylogenetics, molecular evolution, and population dynamics of these significant tick species.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number NC064124 and ON853615.
Ethics statement
This study was approved by the Administration Committee of Experimental Animals, Dali University, First Affiliated Hospital of Chengdu Medical College, and Yunnan Provincial Key Laboratory of Natural Epidemic Disease Prevention and Control Technology.
Author contributions
X-yL and Q-fZ conceived the study and wrote the manuscript. D-dJ, Y-fL, BC, and S-pY carried out the experiments and analyzed the data. Z-tS, HJ, and JW contributed to the collection of Haemaphysalis nepalensis and Haemaphysalis yeni and discussions. Y-hF, C-hD, and XY are responsible for the interpretation of experimental data, critical revision of important knowledge content, and final approval of the version to be published.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81760607 and U2002219), Yunnan Natural Science Foundation (2017FD139), and Scientific Research Fund of Yunnan Education Department (2022J0687).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tao M, You C, Zhao R, Liu S, Zhang Z, Zhang C, et al. Animal mitochondria: evolution, function, and disease. Curr Mol Med. (2014) 14:115. doi: 10.1038/nature18902
2. Yang X, Gao Z, Zhou T, Zhang J, Wang L, Xiao L, et al. Mapping the potential distribution of major tick species in China. Int J Environ Res Public Health. (2020) 17:5145. doi: 10.3390/ijerph17145145
3. Kelava S, Mans BJ, Shao R, Moustafa M, Matsuno K, Takano A, et al. Phylogenies from mitochondrial genomes of 120 species of ticks: insights into the evolution of the families of ticks and of the genus Amblyomma. Ticks Tick Borne Dis. (2021) 12:101577. doi: 10.1016/j.ttbdis.2020.101577
4. Waterhouse RM, Sonenshine DE, Roe RM, Ribeiro JM, Sattelle DB, et al. Tick genome assembled: new opportunities for research on tick-host-pathogen interactions. Front Cell Infect Microbiol. (2016) 6:103. doi: 10.3389/fcimb.2016.00103
5. Dhanda V. Haemaphysalis nepalensis Hoogstraal, 1962 (Ixodoidea: Ixodidae), systematic position based on description of the nymph, and host and locality records. J Parasitol. (1964) 50:783–5. doi: 10.2307/3276201
6. Kwak ML. A checklist of the ticks (Acari: Argasidae, Ixodidae) of Japan. Exp Appl Acarol. (2018) 75:263–7. doi: 10.1007/s10493-018-0259-6
7. Saito Y, Hoogstraal H. Haemaphysalis (kaiseriana) yeni toumanoff (ixodoidea: ixodidae): discovery in Japan, description of female and immature stages, environment, and life cycle. J Parasitol. (1972) 58:950–9.
8. Hoogstraal H, Trapido H. Redescription of the type materials of Haemaphysalis (kaiseriana) bispinosa neumann (India), h. (K) Neumanni donitz (Japan), h (K) Lagrangei larrousse (Vietnam), and h (K) Yeni toumanoff (vietnam) (Ixodoidea, Ixodidae). J Parasitol. (1966) 52:1188–98.
9. Jia N, Wang J, Shi W, Du L, Sun Y, Zhan W, et al. Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell. (2020) 182:1328–40. doi: 10.1016/j.cell.2020.07.023
10. Xin ZZ, Liu Y, Zhang DZ, Wang ZF, Tang BP, Zhang HB, et al. Comparative mitochondrial genome analysis of Spilarctia subcarnea and other noctuid insects. Int J Biol Macromol. (2018) 107:121–8. doi: 10.1016/j.ijbiomac.2017.08.153
11. Ciloglu A, Ibis O, Yildirim A, Aktas M, Duzlu O, Onder Z, et al. Complete mitochondrial genome characterization and phylogenetic analyses of the main vector of crimean-congo haemorrhagic fever virus: Hyalomma marginatum koch, 1844. Ticks Tick-Borne Dis. (2021) 12:101736. doi: 10.1016/j.ttbdis.2021.101736
12. Lu X, Zhang Q, Jiang D, Wang T, Sun Y, Du C, et al. Characterization of the complete mitochondrial genome of Haemaphysalis (alloceraea) kolonini (ixodidae) and its phylogenetic implications. Parasitol Res. (2022) 121:1951–62. doi: 10.1007/s00436-022-07535-2
13. Lu X, Zuo X, Jiang D, Yang X. The complete mitochondrial genome of Ixodes vespertilionis (acari: ixodidae). Mitochon DNA Part B Resour. (2021) 6:3001–3. doi: 10.1080/23802359.2021.1976686
14. Rozen S, Skaletsky H. Primer3 on the www for general users and for biologist programmers. Methods Mol Biol (Clifton, NJ). (2000) 132:365–86.
15. Lu X, Zhang Q, Jiang D, Du C, Xu R, Guo X, et al. Characterization of the complete mitochondrial genome of Ixodes granulatus (ixodidae) and its phylogenetic implications. Parasitol Res. (2022) 121:2347–58. doi: 10.1007/s00436-022-07561-0
16. Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from illumina miseq data. Bioinformatics. (2015) 31:587–9. doi: 10.1093/bioinformatics/btu661
17. Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. Mitos: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. (2013) 69:313–9. doi: 10.1016/j.ympev.2012.08.023
18. Lowe TM, Chan PP. Trnascan-se on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. (2016) 44:W54–7. doi: 10.1093/nar/gkw413
19. Kumar S, Stecher G, Tamura K. Mega7: molecular evolutionary genetics analysis version 70 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
20. Yamaoka K, Nakagawa T, Uno T. Application of akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. (1978) 6:165–75. doi: 10.1007/BF01117450
Keywords: ticks, Haemaphysalis nepalensis, Haemaphysalis yeni, mitogenome, genome annotation
Citation: Lu X-y, Zhang Q-f, Jiang D-d, Liu Y-f, Chen B, Yang S-p, Shao Z-t, Jiang H, Wang J, Fang Y-h, Du C-h and Yang X (2022) Complete mitogenomes and phylogenetic relationships of Haemaphysalis nepalensis and Haemaphysalis yeni. Front. Vet. Sci. 9:1007631. doi: 10.3389/fvets.2022.1007631
Received: 30 July 2022; Accepted: 01 September 2022;
Published: 02 November 2022.
Edited by:
Nicola Pugliese, University of Bari Aldo Moro, ItalyReviewed by:
Sapto Andriyono, Airlangga University, IndonesiaOlusesan Adeyemi Adelabu, University of the Free State, South Africa
Arunee Ahantarig, Mahidol University, Thailand
Copyright © 2022 Lu, Zhang, Jiang, Liu, Chen, Yang, Shao, Jiang, Wang, Fang, Du and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-hao Fang, ZmFuZ3loQGVhc3Rlcm4taGltYWxheWEuY24=; Chun-hong Du, ZGNoNjg5MDcyOEAxNjMuY29t; Xing Yang, eWFuZzA4MjIwMDEzQDE2My5jb20=
†These authors have contributed equally to this work