 Shao-Lun Zhai1†
Shao-Lun Zhai1† Yi-Lun Xie1
Yi-Lun Xie1 Qi Zhai1Xiao-Hui Wen1Dian-Hong Lv1Qin-Ling Chen1Gang Wang1*Wen-Kang Wei1,2*
Qi Zhai1Xiao-Hui Wen1Dian-Hong Lv1Qin-Ling Chen1Gang Wang1*Wen-Kang Wei1,2*- 1Scientific Observation and Experiment Station of Veterinary Drugs and Diagnostic Techniques of Guangdong Province, Ministry of Agriculture of Rural Affairs, and Key Laboratory of Animal Disease Prevention of Guangdong Province, Institute of Animal Health, Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 2Agro-Biological Gene Research Center, Guangdong Academy of Agricultural Sciences, Guangzhou, China
Bovine rhinitis B virus (BRBV) is an emerging viral species in the genus Aphthovirus, family Picornaviridae. Studies suggested that BRBV was considered a potential etiological agent of bovine respiratory disease complex (BRDC). BRBV has been reported in the United States, Sweden, Canada, Japan, and Mexico. However, little information of BRBV was available in China. In this study, we performed viral metagenomic analysis in a calf with respiratory disease. The results showed high abundance (3.85) of BRBV nucleotide and 248 mapped reads in calf samples. Online BLASTn analysis showed that three contigs of those had the highest nucleotide similarity (95%) with one Swedish BRBV isolate (BRBV_SWE1, GenBank accession no. KY432299). To identify the genome characterization of the Chinese BRBV isolate (designated CHN1), six couples of overlapping RT-PCR primers were designed according to genome sequences of BRBV_SWE1. Through gene cloning and splicing, we obtained the genome information of CHN1, possessing 7,465 nucleotides (46.6% G+C). Although CHN1 had the highest nucleotide similarity (95.1%) with BRBV_SWE1, one 11-nucleotide (ACATTTGTTGT) deletion occurred in the 5′ untranslated region compared to SWE1. Phylogenetic analysis showed that CHN1 clustered together with BRBV_SWE1, and far from other BRBV isolates. This study recorded the first discovery of BRBV infection in China. Further investigation should be made in order to evaluate the infection status and epidemiological significance of BRBV in China.
Introduction
Bovine respiratory disease (BRD), an important multifactorial disease involving complex interactions between the animal, the pathogens, and the environment, severely affected the world bovine industry economically. In the Unites States alone, the economic impact due to BRD is estimated to exceed 1 billion dollars per year (1, 2). The BRD complex accounts for ~70–80% of morbidity in the Unites States and 84.5–99.9% of morbidity in Mexican feedlot cattle (3, 4). In Canada, over 80% of the vaccines licensed for cattle are applied for control and prevention of BRD (5, 6). BRD poses significant challenges to its prevention and control (7).
To date, major pathogens responsible for BRD include Histophilus somni, Pasteurella multocida, Mannheimia haemolytica, bovine herpesvirus 1 (BHV-1), bovine viral diarrhea virus (BVDV), bovine parainfluenza 3 virus (PI3V), and bovine respiratory syncytial virus (BRSV) (8). Despite the aggressive use of antibiotics and vaccines against the above pathogens, the morbidity and mortality rates of BRD among feedlot cattle have remained steady (3). This may be due to the etiological role of some new pathogens including bovine adenovirus 3 (BAdV3), influenza D virus (IDV), and bovine rhinitis virus (BRV) (9).
BRV, along with equine rhinitis virus (ERV) and foot-and-mouth disease virus (FMDV), is a species in the genus Aphthovirus, family Picornaviridae, according to the International Committee on Taxonomy of Viruses. BRV has two species, bovine rhinitis A virus (BRAV) and bovine rhinitis B virus (BRBV) (10). BRBV has been reported in bovine herds from the United States, United Kingdom, Mexico, Sweden, and Canada, and was considered to be associated with BRD (9, 11–14). However, little information of BRBV was available in China (15). In this study, we performed viral metagenomic analysis in a calf with respiratory disease. A nearly complete genome sequence of the first Chinese BRBV isolates was obtained and characterized.
Materials and Methods
Sample Information
On March 11, 2014, one dead Holstein calf was submitted to the Institute of Animal Health, Guangdong Academy of Agricultural Sciences. According to the calf owner, the calf before death mainly presented dyspnea and diarrhea. Autopsy result was consistent with the description of the calf owner. The pathological changes mainly focused on lung (pneumonia) and intestines (increased watery contents), and parasitic agents were not observed. To further identify the possible pathogens causing the death of the calf, the spleen and blood were collected to culture the bacteria, and the lung and intestine tissues were collected to perform viral metagenomic analysis.
Viral Metagenomic Analysis
Prior to viral RNA/DNA extraction, the lung and intestine tissues were ground and diluted one time using phosphate-buffered saline (PBS) (pH: 7.4). The supernatants were then collected by centrifugation at 5,000 × g for 5 min. Two hundred microliters of clarified supernatants was used to extract nucleotide following the manufacturer's recommendations (Axygen Scientific Inc., Union City, CA, USA). The nucleotide samples (100 μl) were mixed together and submitted to a sequencing company (Annoroad Gene Technology Inc., Beijing, China) to perform high-throughput sequencing. The results showed high abundance (3.85) of BRBV nucleotide and 248 mapped reads in the calf samples.
Genome Amplification of BRBV
From the three fragments of spliced sequences (446, 465, and 482 bp), the online BLASTn analysis revealed that they shared the highest nucleotide similarity (95%) with one Swedish BRBV isolate (GenBank no. KY432299). So, according to the genome sequence of the Swedish BRBV isolate, six overlapping pairs of primers (Table 1) were designed and used to amplify the genome sequence of the Chinese BRBV isolate. The 25-μl one-step RT-PCR reaction mixture included 12.5 μl of 2 × 1 Step Buffer, 1 μl of PrimeScript one Step Enzyme Mix, 1 μl of upstream primer (10 μM), 1 μl of downstream primer (10 μM), 2 μl of RNA sample, and 7.5 μl of RNase-Free ddH2O. RT-PCR protocol was designed as follows: reverse transcription at 50°C for 30 min, pre-denaturation at 95°C for 5 min, 45 cycles of (denaturation at 95°C for 30 s, annealing at optimized temperature listed in Table 1 for 30 s, extension at 72°C for 2 min), and final extension at 72°C for 5 min. The positive amplicons were cloned into the pMD19-T vector (Takara, Shiga, Japan), and all positive recombinant plasmids were submitted to a sequencing company (Sangon Biotech Co., Ltd., Shanghai, China) and sequenced for at least three times. One nearly complete genome sequence of BRBV (designated CHN1) was obtained (Table 2), and has been submitted to the GenBank database (accession number MT160419).
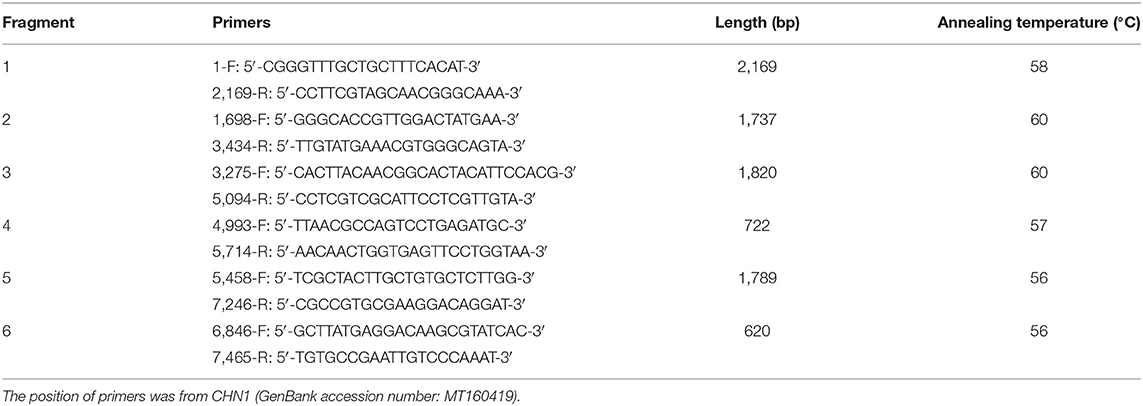
Table 1. RT-PCR primers used in this study.
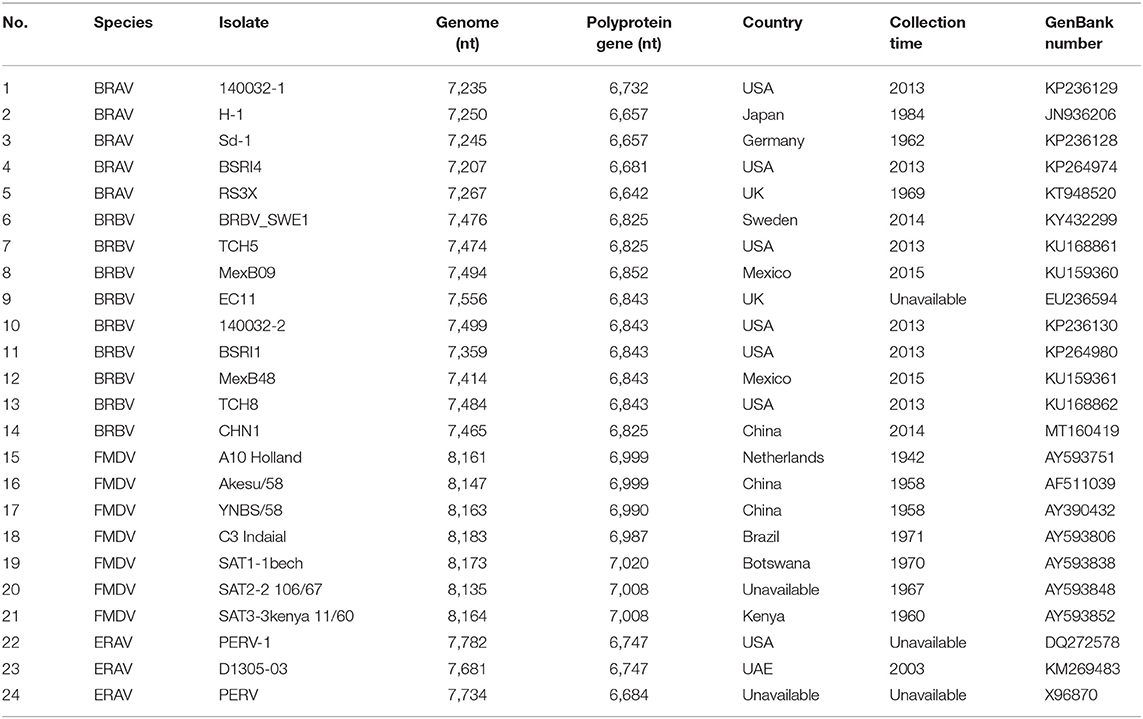
Table 2. Information of members in the genus Aphthovirus used in this study.
Sequence Analysis
Sequence alignment analysis based on CHN1 and reference BRAV, BRBV, FMDV, and ERV sequences (Table 2) was performed using the Clustal W program implemented in the Lasergene v7.1 sequence analysis software package (DNASTAR Inc., Madison, WI, USA). A phylogenetic tree was then constructed based on the polyprotein gene of three of viral species in the genus Aphthovirus by the Maximum Likelihood method using the Molecular Evolutionary Genetics Analysis (MEGA) software version 7.0 with bootstrap replication at 1,000 (16). Similarity plot for CHN1 against EC11, TCH5, and SWE1 was generated using the Kimura (2-parameter) distance model with a 200-bp window and the 20-bp step in the software SimPlot v3.5.1 (17).
Isolation of BRBV-CHN1
The supernatant of the homogenate of the lungs and intestine tissues from the calf in this study was filtered with a 0.22-μm filter, inoculated to a monolayer of Madin-Darby bovine kidney (MDBK) cells, and incubated at 33°C with concentration of CO2 at 5% for 120 h. The supernatant of the culture went on three passages. Each passage was observed for any CPE, and the supernatants of every passage were extracted (Axygen Scientific Inc.). RT-qPCR was run on the extracts to detect any viral replications.
Results and Discussion
Through RT-PCR amplification, a nearly full-length genome sequence (7,465 nucleotides, 46.6% G+C) of the first Chinese BRBV isolate (CHN1) was obtained, which shared the highest nucleotide similarity (95.1%) with BRBV_SWE1. Overall, CHN1 shared 77.9–95.1% similarity with the selected BRBV isolates, 55.5–56.2% with FMDV, 53.4–54.5% with BRAV, and 8.2–27.6% with ERAV on the level of genomics (Table 3).
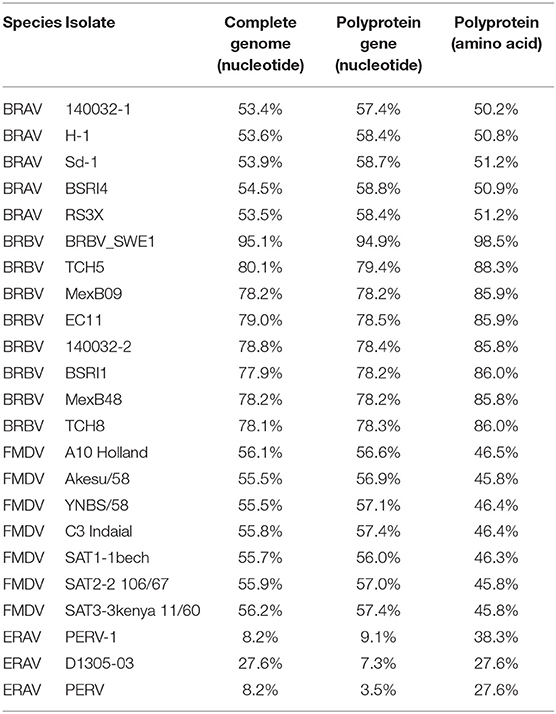
Table 3. Similarity of CHN1 with other member from the genus Aphthovirus.
It is noteworthy that CHN1 shared only 79.0% similarity on the level of complete genome and 85.9% on the level of polyprotein with the BRBV isolate EC11 that was first identified in England, for the reason that EC11 is the only isolate studied well on the level of genomics so far. Thus, the genomic regions of CHN1 genome in this study were mainly predicted based on the genomic characteristics of EC11 (18).
CHN1 had the closest relationship with SWE1. In the middle of CHN1 genome, there was a large complete open reading frame (ORF) (6,825 nt) (94.9% with SWE1) that encodes the polyprotein (2274 amino acid) (98.5% with SWE1). Meanwhile, the nearly complete 5′ and 3′ untranslated regions of CHN1 genome that had been successfully sequenced were 578 nucleotides (94.9% with SWE1) and 62 nucleotides (100% with SWE1) in length, respectively. However, compared with SWE1, one 11-nucleotide (ACATTTGTTGT) deletion occurred before the Leader region compared to SWE1 (Figure 1).
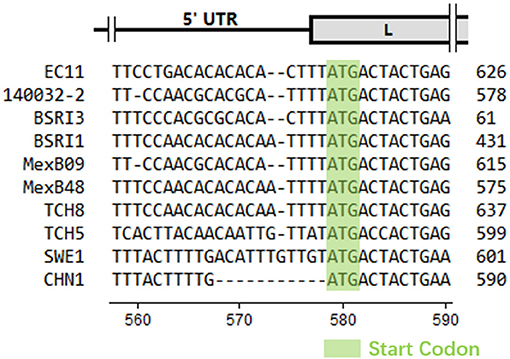
Figure 1. Partial nucleotide alignment of CHN1 with reference sequences. The absence of nucleotides was represented by a dot. The scale of genomic regions was illustrated according to EC11 predicted in a previous study (18) and the position scale was indicated for CHN1. Start codons were indicated with a light green background.
Phylogenetic analysis of the polyprotein genes showed that CHN1 clustered together with BRBV_SWE1, and was closer to the BRBV isolate TCH5 identified in United States but far from other BRBV isolates (including EC11) and related viral species (Figure 2A). Further investigation on the similarity between EC11, SWE1, TCH5, and CHN1 complete genome (Figure 2B) revealed that the 5′ NTR, L, VP4, 2BC, 3ABCD and 3′ NTR region of the selected isolates shared >60% of similarity, manifesting relatively high genomic conservation. On the contrary, VP2, VP3, VP1, and 2A region shared similarity ranging from 5 to 75%, which was more diverse compared to the rest of the regions mentioned above. For picornaviruses, generally, the continuous genomic region P1 encodes the structural proteins VP4, VP2, VP3, and VP1 that compose the viral capsid. In FMDV, VP1 is the most exposed protein on the viral capsid surface (19) while VP4 is an internal structural protein that is conversed in different serotypes (20), which accords with the similarity analysis where VP4 shared 65–80% similarity while VP2, VP3, and VP1 shared 5–70% among selected BRBV isolates. In addition, like EC11 and SWE1, the 2A/2B junction of CHN1 preserved the four-residue “NPGP” that involved a “cleavage” mechanism termed “ribosomal skipping” that was demonstrated in FMDV (21).
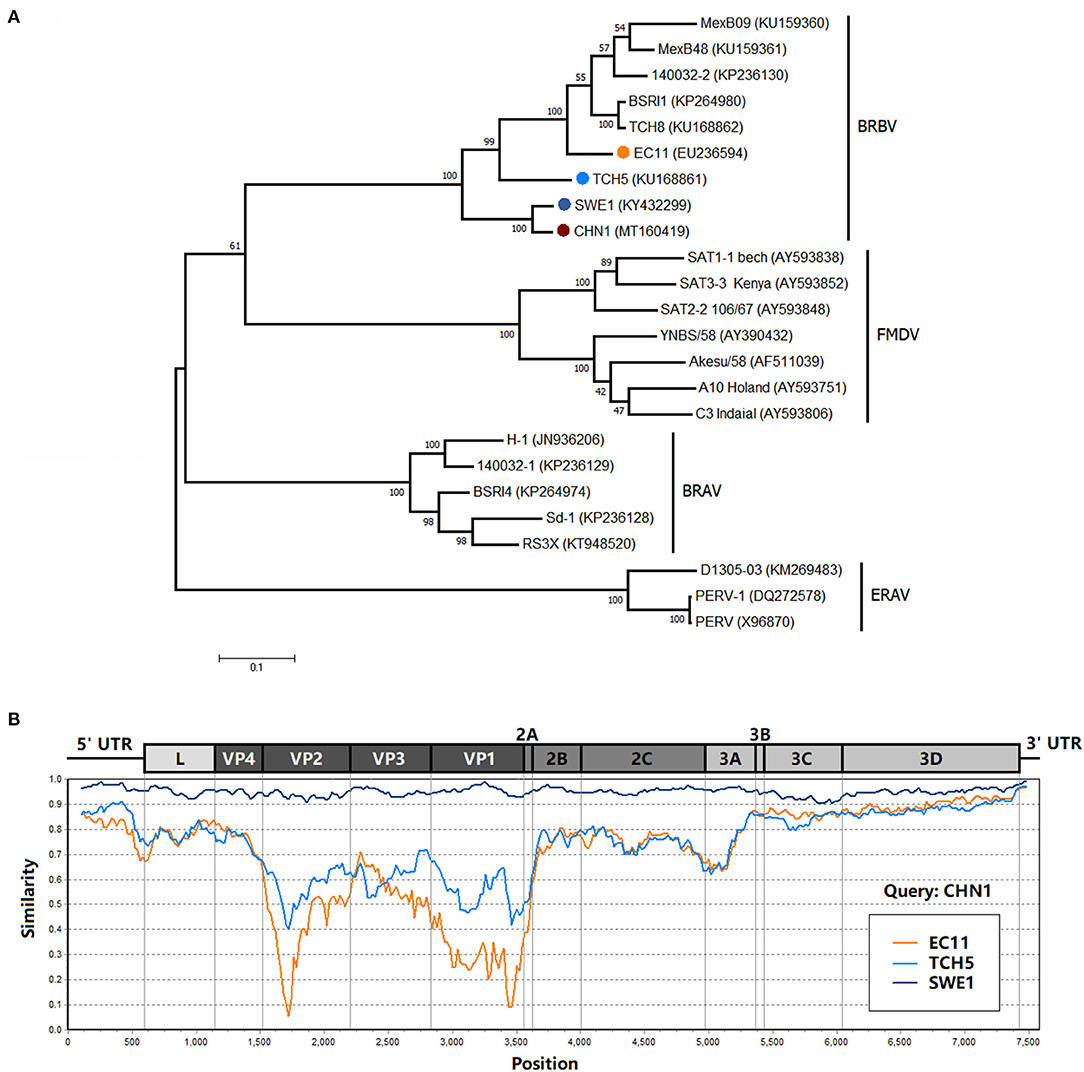
Figure 2. (A) Phylogenetic analysis of BRBV polyprotein sequence with other viruses from the genus Aphthovirus. The phylogenetic tree was drawn to scale using the Maximum Likelihood method and tested with the booststrap method at a replication of 1,000, and its frequency was marked at the nodes. The isolate CHN1 that was identified in this study was marked in bold and accompanied by a red circle. (B) Similarity analysis of isolate CHN1 with isolates EC11, TCH5, and SWE1 using the Kimura (2-parameter) distance model with a 200-bp window and 20-bp step. The genomic region scale was illustrated according to isolate EC11.
BRBV could exist worldwide. In a span of a relatively short period of time (2013–2015), with the next-generation sequencing technic thriving, reports of BRBV from different countries increased and most of the cases had connections with BRD. In an early study into the pathology of BRBV, increased body temperature, increased respiration rate, serious nasal discharge, and lesions in the lungs were observed (22). Similar conditions were noticed in the calf in this study and the calves in the studies mentioned above. In particular, the metagenomic analysis from this study confirmed no other common BRD-related virus presented, while the autopsy result showed that the infection in the calf was rather viral, and no bacteria had been isolated in vitro, indicating the potential pathogenicity of BRBV. However, although attempts were made to isolate the BRBV-CHN1, neither CPE- nor BRBV-positive RT-qPCR reaction was observed in all three passages. Therefore, further investigation and more evidence are needed to extrapolate the theory.
Generally, BRBV was frequently detected in those respiratory tract samples (such as nasal swabs, lungs) (9, 11, 13, 14). Little information was available in other samples. In this study, the nucleotide sample using viral metagenomic analysis was the mixture from lung and intestine tissues; this does not determine whether the virus comes from the lung or small intestine. However, further detection showed that BRBV was only detected in the lung sample rather than other samples (intestine tissue, spleen, and blood). Moreover, in our previous study, we detected a lower positive number of BRBV in stool samples than in the nasal swab (15). To a certain extent, it suggested that BRBV could spread via the respiratory tract and/or fecal–oral transmission.
Conclusions
This study recorded the first discovery of BRBV in China. Further investigation should be made in order to evaluate infection status and epidemiological significance of BRBV in China.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by Institute of Animal Health, Guangdong Academy of Agricultural Sciences. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
S-LZ: conceptualization. S-LZ and Y-LX: methodology and writing—original draft preparation. Y-LX: software. S-LZ, Y-LX, QZ, X-HW, D-HL, and Q-LC: writing—review and editing. W-KW: supervision. S-LZ, GW, and W-KW: project administration. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Jinying's Star Talent Program (Grant no. R2018PY-JX003) from Guangdong Academy of Agricultural Sciences, the grant (No. 201906040005) from Guangzhou Science and Technology Bureau, the grant (No. 2019B020217002) from the Guangdong Science and Technology Department, and the grant (Nos. 2020KJ114 and 2020KJ119) from the Department of Agriculture and Rural Affairs of Guangdong Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Griffin D. Antibiotic metaphylaxis to control respiratory disease. Cattle Prod Libr. (2006) 606:1–6.
2. Snowder GD, Van Vleck LD, Cundiff LV, Bennett GL, Koohmaraie M, Dikeman ME. Bovine respiratory disease in feedlot cattle: phenotypic, environmental, and genetic correlations with growth, carcass, and longissimus muscle palatability traits. J Anim Sci. (2007) 85:1885–92. doi: 10.2527/jas.2007-0008
3. Hilton WM. BRD in 2014: where have we been, where are we now, and where do we want to go? Anim Heal Res Rev. (2014) 15:120–2. doi: 10.1017/S1466252314000115
4. Villagómez-Cortés JA, Itzcoatl Martínez-Herrera D. Epidemiological evaluation of clinical bovine respiratory disease complex in a tropical Mexican feedlot. Res Opin Anim Vet Sci. (2013) 3:315–21.
5. Bowland SL, Shewen PE. Bovine respiratory disease: commercial vaccines currently available in Canada. Can Vet J. (2000) 41:31–48.
6. Edwards TA. Control methods for bovine respiratory disease for feedlot cattle. Vet Clin North Am Food Anim Pract. (2010) 26:273–84. doi: 10.1016/j.cvfa.2010.03.005
7. Murray GM, O'Neill RG, More SJ, McElroy MC, Earley B, Cassidy JP. Evolving views on bovine respiratory disease: an appraisal of selected key pathogens – Part 1. Vet J. (2016) 217:95–102. doi: 10.1016/j.tvjl.2016.09.012
8. Fulton RW. Bovine respiratory disease research (1983-2009). Anim Health Res Rev. (2009) 10:131–9. doi: 10.1017/S146625230999017X
9. Zhang M, Hill JE, Fernando C, Alexander TW, Timsit E, van der Meer F, et al. Respiratory viruses identified in western Canadian beef cattle by metagenomic sequencing and their association with bovine respiratory disease. Transbound Emerg Dis. (2019) 66:1379–86. doi: 10.1111/tbed.13172
10. Rai DK, Rieder E. Homology modeling and analysis of structure predictions of the bovine rhinitis B virus RNA dependent RNA polymerase (RdRp). Int J Mol Sci. (2012) 13:8998–9013. doi: 10.3390/ijms13078998
11. Ng TF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J Virol. (2015) 89:5340–9. doi: 10.1128/JVI.00064-15
12. Reed SE, Tyrrell DAJ. Studies on a Rhinovirus(EC11) Derived from a Calf I. Isolation in Calf tracheal organ cultures and characterization of the virus. J Comp Pathol. (1971) 81:33–40. doi: 10.1016/0021-9975(71)90052-1
13. Mitra N, Cernicchiaro N, Torres S, Li F, Hause BM. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J Gen Virol. (2016) 97:1771–84. doi: 10.1099/jgv.0.000492
14. Blomström AL, Oma V, Khatri M, Hansen HH, Stokstad M, Berg M, et al. Genome sequence of a bovine rhinitis B virus identified in cattle in Sweden. Genome Announc. (2017) 5:e00172–17. doi: 10.1128/genomeA.00172-17
15. Xie YL Lv DH, Wen XH, Zhai Q, Luo ML, Wei WK, Chen QL, et al. Development of a real-time quantitative RT-PCR assay for detection of bovine rhinitis B virus. Front Vet Sci. (2021) 8:680707. doi: 10.3389/fvets.2021.680707
16. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–1874. doi: 10.1093/molbev/msw054
17. Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. Full-Length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. (1999) 73:152–60. doi: 10.1128/JVI.73.1.152-160.1999
18. Hollister JR, Vagnozzi A, Knowles NJ, Rieder E. Molecular and phylogenetic analyses of bovine rhinovirus type 2 shows it is closely related to foot-and-mouth disease virus. Virology. (2008) 373:411–25. doi: 10.1016/j.virol.2007.12.019
19. Acharya R, Fry E, Stuart D, Fox G, Rowlands D, Brown F. The three-dimensional structure of foot-and-mouth disease virus at 2.9 Å resolution. Nature. (1989) 337:709–16. doi: 10.1038/337709a0
20. Carrillo C, Tulman ER, Delhon G, Lu Z, Carreno A, Vagnozzi A, et al. High throughput sequencing and comparative genomics of foot-and-mouth disease virus. Dev Biol. (2006) 126:23–30. doi: 10.1128/jvi.79.10.6487
21. Donnelly MLL, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, et al. Analysis of the aphthovirus 2A/2B polyprotein 'cleavage' mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip'. J Gen Virol. (2001) 82:1013–25. doi: 10.1099/0022-1317-82-5-1013
Keywords: bovine rhinitis B virus, bovine respiratory disease complex, high-throughput sequencing, complete genome, nucleotide deletion, genetic variation
Citation: Zhai S-L, Xie Y-L, Zhai Q, Wen X-H, Lv D-H, Chen Q-L, Wang G and Wei W-K (2021) Genome Characterization and Phylogenetic Analysis of the First Bovine Rhinitis B Virus Isolate in China. Front. Vet. Sci. 8:721284. doi: 10.3389/fvets.2021.721284
Received: 06 June 2021; Accepted: 20 August 2021;
Published: 22 September 2021.
Edited by:
Jesus Hernandez, Consejo Nacional de Ciencia y Tecnología (CONACYT), MexicoReviewed by:
Raquel Arruda Leme, State University of Londrina, BrazilMohanned Naif Alhussien, Technical University of Munich, Germany
Copyright © 2021 Zhai, Xie, Zhai, Wen, Lv, Chen, Wang and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Wang, d2FuZ2dhbmcmI3gwMDA0MDtnZGFhcy5jbg==; Wen-Kang Wei, d2Vpd2Vua2FuZyYjeDAwMDQwO2dkYWFzLmNu
†ORCID: Shao-Lun Zhai orcid.org/0000-0003-3217-2256