
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Vet. Sci., 23 March 2021
Sec. Comparative and Clinical Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fvets.2021.664718
This article is part of the Research TopicPrecision Medicine in Veterinary OncologyView all 11 articles
 Jason Chibuk1*
Jason Chibuk1* Andi Flory1*Kristina M. Kruglyak1Nicole Leibman2Alexis Nahama3
Andi Flory1*Kristina M. Kruglyak1Nicole Leibman2Alexis Nahama3 Nilesh Dharajiya4Dirk van den Boom5Taylor J. Jensen6
Nilesh Dharajiya4Dirk van den Boom5Taylor J. Jensen6 Jeffrey S. Friedman7
Jeffrey S. Friedman7 M. Richard Shen8
M. Richard Shen8 Francisco Clemente-Vicario9
Francisco Clemente-Vicario9 Ilya Chorny1John A. Tynan1
Ilya Chorny1John A. Tynan1 Katherine M. Lytle1
Katherine M. Lytle1 Lauren E. Holtvoigt1Muhammed Murtaza10Luis A. Diaz Jr.11
Lauren E. Holtvoigt1Muhammed Murtaza10Luis A. Diaz Jr.11 Dana W. Y. Tsui1*
Dana W. Y. Tsui1* Daniel S. Grosu1
Daniel S. Grosu1Cancer is the leading cause of death in dogs, in part because many cases are identified at an advanced stage when clinical signs have developed, and prognosis is poor. Increased understanding of cancer as a disease of the genome has led to the introduction of liquid biopsy testing, allowing for detection of genomic alterations in cell-free DNA fragments in blood to facilitate earlier detection, characterization, and management of cancer through non-invasive means. Recent discoveries in the areas of genomics and oncology have provided a deeper understanding of the molecular origins and evolution of cancer, and of the “one health” similarities between humans and dogs that underlie the field of comparative oncology. These discoveries, combined with technological advances in DNA profiling, are shifting the paradigm for cancer diagnosis toward earlier detection with the goal of improving outcomes. Liquid biopsy testing has already revolutionized the way cancer is managed in human medicine – and it is poised to make a similar impact in veterinary medicine. Multiple clinical use cases for liquid biopsy are emerging, including screening, aid in diagnosis, targeted treatment selection, treatment response monitoring, minimal residual disease detection, and recurrence monitoring. This review article highlights key scientific advances in genomics and their relevance for veterinary oncology, with the goal of providing a foundational introduction to this important topic for veterinarians. As these technologies migrate from human medicine into veterinary medicine, improved awareness and understanding will facilitate their rapid adoption, for the benefit of veterinary patients.
Cancer is frequent in dogs and is by far their most common cause of death (1–5). While dogs and humans have a similar lifetime risk of cancer (between 1:2 and 1:4), dogs have an annual incidence of cancer that is up to 10-fold higher than in humans, as their lifetime risk is compressed into a much-abbreviated lifespan (1, 2). Similar to humans, both genomic and environmental factors drive cancer incidence in dogs: cancer predisposition mutations are concentrated in many breeds as an inadvertent side effect of selective breeding; and dogs share the same environment as humans, including exposure to many carcinogens (6, 7). These considerations help explain why ~4–6 million dogs are newly diagnosed with cancer per year in the US in a population of under 90 million as compared to 1.8 million cancer diagnoses in humans in a population of ~330 million (8). Like humans, the burden of cancer in dogs increases with age: up to 50% of dogs over 10 years of age will develop cancer during the remainder of their lives (3, 9, 10).
Canine cancer also carries a significant mortality risk (3, 8, 11), since many canine cancers are diagnosed at advanced stages after there has been microscopic (12, 13) or macroscopic spread (12, 14–16) and a cure is no longer achievable. With rising pet ownership and increased emotional attachment to pets, the substantial burden of canine cancer goes well-beyond the immediate health implications for the dog, with significant emotional and financial impact on dog owners (17–21). Given the high incidence of cancer in dogs, all companion animal practices are exposed to oncology cases on a regular basis, and cancer care is an essential part of pet health care (13).
Over the past decade, genomic medicine has made great strides thanks to technological breakthroughs such as the introduction of next generation sequencing (NGS). In 2005, the National Institutes of Health (NIH) launched The Cancer Genome Atlas (TCGA), a landmark initiative aiming to molecularly characterize the genomic landscape of human cancer (22). By 2013, TCGA concluded enrollment with over 20,000 samples and built a knowledge base across all major human cancer types (22, 23). This effort, together with similar international initiatives such as the International Cancer Genome Consortium (24), enabled rapid cancer biology research and helped facilitate the development of new molecularly targeted therapeutic agents for cancer. As a result, tumor tissue-based molecular testing has become an integral part of the “precision medicine” trend in cancer care for humans (25). More recent innovations in the field have enabled non-invasive testing based on a simple blood draws; typically referred to as “liquid biopsy,” this type of testing most commonly relies on analysis of cell-free DNA (cfDNA) fragments released by the tumor cells into the bloodstream and known as circulating tumor DNA (ctDNA) (26–30).
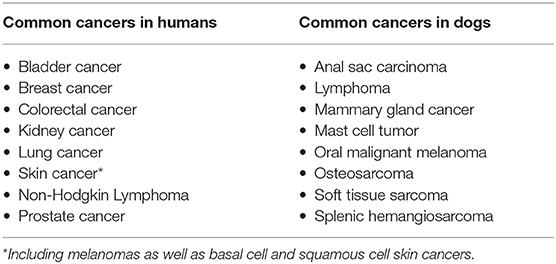
The first canine reference genome was published in 2005 (31), not long after the publication of the human reference genome (Figure 1) (32–34). However, progress in canine genomics has not been nearly as rapid as in humans, and most advances in genomic medicine have not yet been adopted in veterinary medicine. Certain areas of canine genetics have seen meaningful progress, including breed identification (35, 36), breed-specific disease predisposition (37–39), and genetic determinants of heritable disorders (40). Much of this accumulated knowledge is now available to veterinarians and pet owners through commercial testing options. However, only a small fraction of the scientific progress made in humans has been transferred into the arena of canine oncology. More research and development pertaining to the genetic predispositions underlying canine cancer syndromes, and to the detection, characterization, and management of cancer in dogs, is urgently needed to allow the standard of cancer care in veterinary medicine to catch up with human medicine standards.
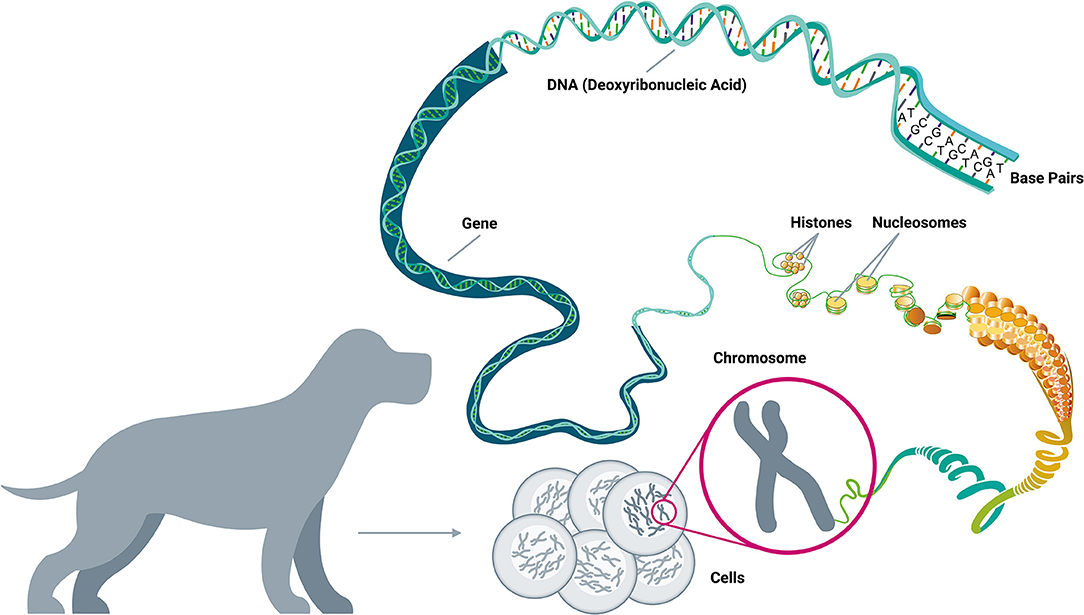
Figure 1. A brief guide to genomics. Cancer is a disease of the genome because DNA alterations provide the biological basis of cancer. Each body cell (except for mature red blood cells) contains a full copy of the organism's genome within a set of chromosomes packed in its nucleus. The DNA double-helix is formed by four nucleotides, or bases, assembled in complementary pairs via hydrogen bonds: adenine (A) is always paired with thymine (T), and cytosine (C) is always paired with guanine (G). The gene is the basic unit of heredity and consists of a long sequence of nucleotides that encodes for the synthesis of a protein by transcription to RNA (ribonucleic acid) in the cell's nucleus, followed by translation to a sequence of amino acids in the cytoplasm. The average gene comprises several thousand bases, with wide size variation. The DNA double-helix strand wraps around a set of histone proteins, forming structures known as “nucleosomes” at regular intervals along the length of the strand (Adapted from National Human Genome Research Institute, genome.gov).
With a few notable exceptions—such as BRAF testing in urine for detection of canine urinary tract cancer (41), and testing for c-kit mutations in mast cell tumors (42–44)—the field of veterinary oncology has yet to utilize the full power of genomics for its precision medicine benefits. However, the rapid adoption of genomics-based testing by the veterinary community could pose risks due to the current lack of regulatory oversight for high complexity molecular testing. Clinical genomic testing for veterinary applications can be currently marketed without any peer-reviewed clinical validation studies, or based on studies in small cohorts that may not be representative of the intended-use population (45). There is currently no established regulatory approval pathway in the United States for veterinary diagnostics, and while a form of accreditation is available through the American Association of Veterinary Laboratory Diagnosticians, this accreditation is limited to publicly funded, full-service laboratories and is not available to privately owned commercial labs (46, 47). In the United States, laboratories conducting high complexity molecular testing in humans must secure certification under CLIA (Clinical Laboratory Improvement Amendments) and may also pursue accreditation through CAP (College of American Pathologists); many laboratory-developed tests (LDTs) intended for oncology applications are also regulated by the US FDA (Food and Drug Administration) (48–50). Because no such standards exist for high complexity molecular testing in veterinary medicine, low-quality tests could easily find their way into clinical use, leading to poor outcomes for patients. The lack of external oversight in veterinary diagnostic testing means that it is critically important for highly complex, novel tests to undergo rigorous analytical and clinical validation, with detailed findings published in peer-reviewed journals for full transparency (51).
To develop reliable genomics-based testing solutions for veterinary applications, significant research and development efforts will be required. This is especially true for blood-based liquid biopsy tests since the proportion of ctDNA in the plasma can be very low and variable, requiring highly sensitive detection with minimal false positive results (52). Analytical validation of any such test must evaluate the entire process – from blood collection to shipping, accessioning, separation of plasma and buffy coat (white blood cell - WBC) components, DNA extraction and sequencing library preparation, data generation by NGS, and sophisticated bioinformatics analysis – through adequately designed and powered studies (53). Clinically, the test will need to be validated for each intended use. The unique, non-invasive nature of liquid biopsy allows it to be deployed in multiple clinical use cases across the full spectrum of cancer care in dogs, including: (1) screening for early detection in patients without any signs of cancer; (2) aid in diagnosis in patients with suspected cancer; (3) molecular profiling for targeted treatment selection; (4) detection of minimal residual disease after curative-intent interventions; (5) treatment response monitoring; and (6) recurrence monitoring in patients who achieve complete remission after initial treatment. Each of these use cases will require independent clinical validation in the corresponding intended-use population, with clinical utility ultimately determined by the test's demonstrated ability to inform clinical decision-making or improve clinical outcomes in each use case.
This article will review fundamental principles of cancer genomics for a contemporary understanding of cancer as a disease of the genome; describe key biological and technical considerations for developing and validating a liquid biopsy assay for veterinary cancer applications; and conclude with a review of the six clinical use cases for liquid biopsy described above. Armed with a well-informed appreciation for the validation requirements and the potential of liquid biopsy solutions to significantly improve care for their patients, veterinarians will be well-positioned to evaluate and employ validated liquid biopsy tests as they enter the clinic in the coming years. Once developed and commercialized, liquid biopsy solutions promise to usher in a new era for veterinary medicine, enabling personalized cancer care for pets at the same level of quality and sophistication already available to humans at major cancer centers today.
Historically, cancer has been defined by its organ or tissue of origin, or by its cellular characteristics, as the ability of clinicians to understand and describe it was limited to gross examination and/or microscopic evaluation. Advances in molecular medicine over the past two decades have revealed that normal cells accumulate random genomic alterations over time as a result of DNA replication errors, as well as exposure to endogenous factors (such as free radicals) and to environmental (exogenous) carcinogens such as various forms of radiation and mutagenic chemicals in food and air (54–57); and that cancer results when one or more of these alterations confer an uncontrolled growth advantage to a population of cells (58). These random alterations are called somatic alterations, as they are acquired “in the body” after birth; in some cases, cancer-predisposing alterations are already present at birth, having been inherited from parents as germline alterations.
Most somatic alterations are promptly corrected by intracellular DNA repair mechanisms or (if unrepaired) are severe enough to trigger death of the affected cell, with no ill consequences for the organism; however, when such alterations occur in specific locations in the genome, and are not corrected, a chain of events is set in motion that ultimately leads to the development of cancer. Such alterations confer a growth and/or survival advantage to the affected cells, either by triggering increased cell replication or by inhibiting the processes that keep cell division in check; these are analogous to pressing the gas pedal and cutting the brakes on a car, respectively. Tumor growth can be further accelerated by the accumulation of new somatic alterations with the passing of time; this causes cancer cells to replicate faster, invade surrounding tissues, travel to distant organs by lymphatic and vascular routes, and evade the immune system's surveillance and control mechanisms. When the number of cancer cells reaches around one billion, the malignant mass is ~1 cm in size and weighs about 1 g (59, 60); at this stage, the mass typically becomes detectable by physical and imaging examinations, and may have already started to cause clinical signs such as bleeding, lameness, weight loss, lethargy, etc. This clinical manifestation is called cancer, and is commonly described by its organ of origin, size, and appearance under the microscope (histological diagnosis and grading). The tumor spread is defined by the TNM (tumor, node, and metastasis) staging system. Fundamentally, however, cancer is a disease of the genome, as it is directly caused by genomic alterations and cannot develop in the absence of such alterations (61).
As malignant tumors grow, they develop the ability to invade adjacent areas and metastasize to distant locations in the body through the accumulation of DNA alterations in key genes (58). A primary “gatekeeping” alteration provides an initial growth advantage and allows the affected cell to replicate more quickly than the surrounding cells, becoming a microscopic clone (58); in time, a cell within this clone will randomly acquire a second alteration, typically in another gene, and initiate a subsequent round of clonal expansion with enhanced selective growth advantage for the cells containing both alterations. In this way, the process of novel mutation acquisition followed by clonal expansion continues, leading to the evolution of malignant subclones that can invade surrounding tissues, metastasize to lymph nodes, and spread to distant organs (58).
Genomic alterations that confer a selective growth advantage are termed driver mutations. The cumulative effect of this advantage, over many cell divisions, results in a mass of billions of malignant cells growing at an accelerating rate, with multiple subclonal populations emerging through the successive accumulation of additional mutations. In humans, this is a process that begins with a single driver mutation and ends with metastatic disease, and is estimated to take decades (58). On average, a human cancer genome contains 4–5 driver mutations, though there is wide variability across different cancer types (62). Cancer genomes also contain somatic alterations that do not confer a discernible growth advantage to the cell and are referred to as passenger mutations (58). Detection of either class (driver or passenger) can point to the presence of cancer, but only driver mutations can inform the selection of effective targeted therapies (58, 63).
Driver mutations are not randomly distributed across the genome; in fact, of the more than 20,000 human genes, fewer than 1,500 have been implicated in cancer development (58, 64–67). These cancer-related genes are implicated in 12 specific cellular pathways (Figure 2), which in turn relate to three main functions: (1) cell survival (ability to thrive in nutrient-poor conditions, dysregulation of apoptosis, angiogenic stimulation); (2) cell fate (division and differentiation); and (3) genome maintenance (ability to survive despite gross chromosomal abnormalities, acceleration of mutation acquisition, and DNA damage control) (58, 68, 69).
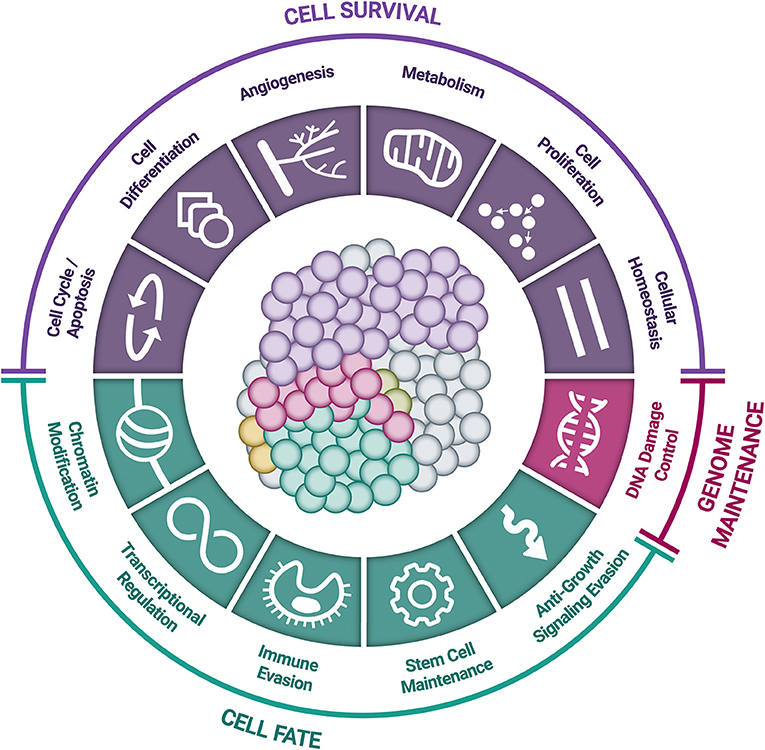
Figure 2. Cellular pathways and functional processes involved in cancer. Driver mutations in cancer-related genes are responsible for cancer development. These cancer-related genes are implicated in 12 cellular signaling pathways, which can be grouped into 3 core cellular functions: cell survival, cell fate, and genome maintenance [Inspired by Hanahan & Weinberg (2011) and Vogelstein et al. (2013)] (58, 68).
As noted previously, cancer-related alterations can be either somatic (acquired after birth, and present in only a subset of cells in the body) or germline (inherited, and present in every cell). Germline alterations resulting in cancer predisposition - for example BRCA1 and BRCA2 variants - increase the risk of breast cancer in humans, and alterations in these genes have also been documented in dogs with mammary tumors (70). In humans with cancer-predisposing germline alterations, the diagnosis is often made at a younger age than is typical for that cancer type, and therefore these patients benefit from proactive cancer screening that can detect such cancers at earlier stages (71). As researchers learn more about heritable canine cancer risk, proactive cancer screening in younger dogs, informed by the presence of germline alterations, will likely demonstrate increasing clinical utility and lead to better clinical outcomes.
Somatic driver mutations predominantly occur in two types of genes: oncogenes and tumor-suppressor genes (TSGs) (Figure 3) (58). Oncogenes typically acquire activating (or gain of function) mutations in very specific locations (known as “hotspots”); these activating mutations increase the rate of cell division, inhibit programmed cell death (apoptosis), or help the cell evade immune surveillance (58). TSGs, on the other hand, typically acquire inactivating (or loss of function) mutations, which can occur across the full length of the gene (58). As their name implies, TSGs serve as a built-in control mechanism to suppress the development and growth of tumors; inactivating mutations impair this critical protective function, leaving oncogene-driven cancers to grow unchecked (58).
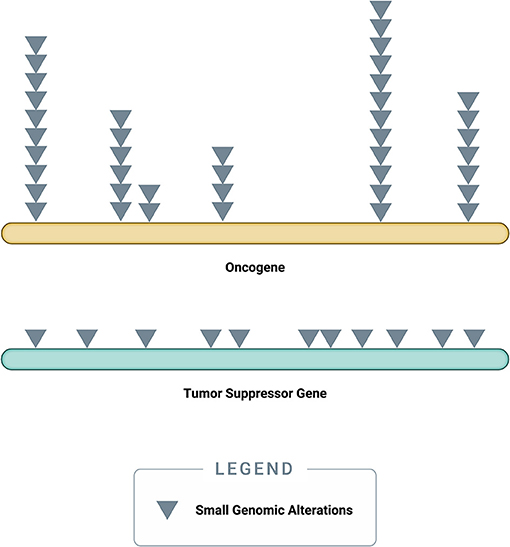
Figure 3. Accumulation of small genomic alterations in cancer-related genes. Small genomic alterations in oncogenes tend to be activating mutations, which cluster at very specific locations (“hotspots”), whereas small genomic alterations in tumor suppressor genes (TSGs) tend to be inactivating mutations and may occur across the full length of the gene. The design of a high-quality genomic assay needs to account for these characteristics in order to identify relevant alterations across cancer-related genes in an efficient manner.
Successive genomic alterations can accumulate in both oncogenes and TSGs, thereby accelerating the progression of the disease in advanced stages of cancer (72). Early in cancer formation, however, disease progression occurs at a relatively slow pace (72). In humans, many tumors grow over 10 to 30 years before clinical manifestation and remain confined to the organ of origin through most of this period (72). This timeframe represents a considerable window of opportunity for early detection that can allow for a cure to be achieved by simple surgical removal of the localized mass (72–74). This paradigm holds true in canine cancer as well: in some types of canine cancers, for example mast cell tumors and soft tissue sarcomas, clinical outcomes are often excellent with early detection and proper surgical excision (75, 76).
Each patient's cancer is characterized by a variety of genomic alterations, and even within a particular cancer type (breast, colon, etc.), no two cancers are the same (77). There is no established 1:1 correspondence between a given tumor type and a given genomic alteration. For example, the BRAF V600E mutation is most commonly seen in human melanoma but is also seen in other cancers (78); likewise, its canine ortholog V595E is common in transitional cell carcinoma but is also present in different canine cancer types (79). The presence of the same mutation in different cancer types may have different therapeutic implications. For example, in humans, targeting BRAF with the agent vemurafenib works more effectively in melanoma than in other cancer types (80). Significant amounts of focused research will be required to understand the efficacy of various targeted agents in specific canine cancers.
Cancer in adult humans typically has dozens to hundreds of mutations per case, while pediatric cancers usually have far fewer mutations per case (58). A commonly employed metric for describing the frequency of mutations in a given cancer case is the tumor mutational burden (TMB), represented by the number of mutations per Mb (megabase, i.e., one million DNA bases) (81). A recent review of over 100,000 human cancer cases across more than 500 cancer types revealed a wide TMB spectrum, ranging from 0 to over 1,000 mutations/Mb, with a median of 3.6 mutations/Mb and increasing with patient age (82). Although less extensively studied, canine cancer genomes have been shown to exhibit similar TMBs in published studies, with a median of 1.98 mutations/Mb in canine osteosarcoma (83), 2.04 mutations/Mb in primary canine lung cancer (84), and a range of 0.1–2.1 mutations/Mb in canine hemangiosarcoma (85, 86). TMB has been shown to be a marker for predicting response to immunotherapy in humans, with high-TMB tumors more likely to respond (87, 88). The ability to non-invasively measure TMB from a blood sample could gain clinical relevance as immunotherapies become increasingly utilized in the management of canine cancers (10).
The extreme diversity of genomic features across cancer types, coupled with the fundamental understanding of cancer as a “disease of the genome,” have opened the door to novel diagnostic approaches that go beyond the notion of a specific test for a specific type of cancer and favor a “pan-cancer” model where a single, highly complex diagnostic assay can be used to detect and characterize a broad range of cancer types (27, 28, 61).
To understand how genomics-based testing can characterize cancer accurately, it is important to first review the main classes of genomic alterations that drive cancer initiation and progression (Figure 4). Though counter-intuitive, many cancers are driven by single nucleotide variant (SNV) “hotspot” alterations that involve a change of just one letter out of several billion letters in the genome (58, 89–91). Another class of small genomic alterations are indels (insertions and deletions), in which one to several nucleotides are inserted into, or removed from, the normal DNA sequence (89–91). Larger genomic events, affecting thousands to millions of nucleotides and known as structural alterations, can also cause significant genomic disruption, leading to cancer (92). Cancer-related structural alterations include: (1) copy number variants (CNVs), in which large segments of DNA (thousands to millions of bases long, up to entire chromosomes) are either completely absent or are abnormally repeated, and (2) translocations, in which DNA strands from unrelated parts of the genome are joined together and result in “fusion genes” in the RNA transcript (92).
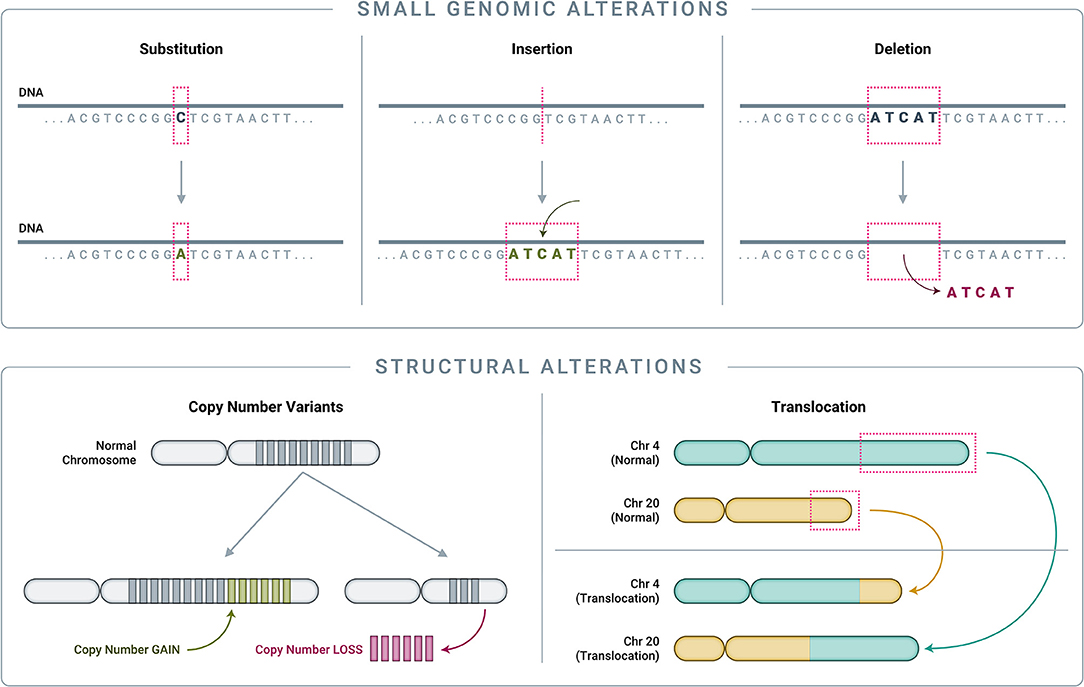
Figure 4. Classes of genomic alterations. Small genomic alterations include single nucleotide variants (SNVs) as well as small insertions and deletions (collectively known as “indels”). SNVs arise when one nucleotide is substituted for another, which can result in altered amino acid translation and an altered protein product. Indels involve the insertion or deletion of one or more nucleotides from the normal DNA sequence, resulting in an altered protein product. On a much larger scale, structural alterations typically involve thousands to millions of nucleotides. Copy number variants (CNVs) are a common type of structural alteration, involving gains or losses of large stretches of DNA. Translocations represent another type of structural alteration, whereby two distant, otherwise unrelated genomic regions are joined together, creating “gene fusions” that can drive tumor growth.
Numerous studies have revealed that the disease etiology of a given cancer is typically driven either by focal somatic alterations (SNVs, indels, and/or translocations) or by CNVs, but rarely by both categories (58, 93, 94). This association with specific classes of driver genomic alterations is often cancer type or subtype-specific, with cancers such as sarcomas—which are far more common in dogs than in humans (9)–being mostly CNV-driven while others, such as carcinomas of the lung or gastrointestinal tract, being mostly SNV and indel-driven (93).
By the time cancers are diagnosed, they are typically large—measuring centimeters in diameter—and thus comprised of billions of cells (59, 60). As described previously, cancer growth is characterized by the successive accumulation of somatic alterations, meaning that tumors are not static—they constantly evolve to include additional alterations beyond the original clonal (or “truncal”) alteration (Figure 5) (58). At the time of diagnosis, when the primary tumor is one or more centimeters in size, most patients do not in fact have “cancer”; rather, they have “cancers,” as the disease has already evolved to consist of multiple sub-populations of cells (subclones), each sharing the original clonal alteration but further evolved with its own additional unique mutational profile. This phenomenon is known as spatial heterogeneity, which can manifest as intratumor heterogeneity (within a single primary or metastatic tumor mass) and/or intrapatient heterogeneity (between different tumor masses within the same individual) (58, 95–98). Once seeded in a new location, metastatic deposits subsequently accumulate additional alterations, which can be distinct from those present in the primary tumor (58). New alterations, which are unique to a specific subclone within the primary tumor or at a metastatic site, are referred to as private mutations (95).
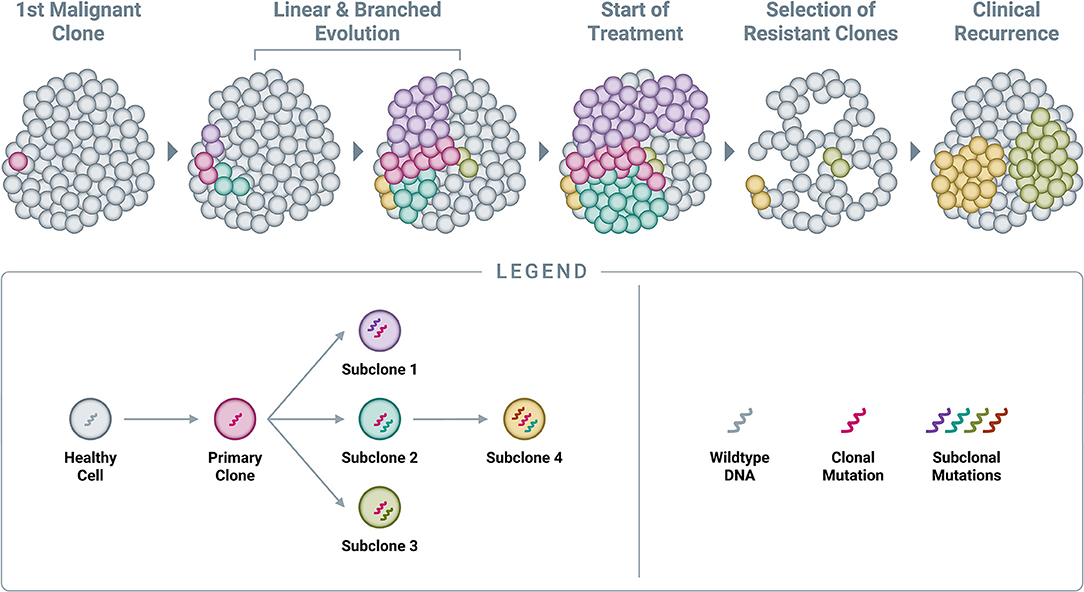
Figure 5. Accumulation of genomic alterations and emergence of resistance. Cancer begins with a single genomic alteration in a cancer-related gene, which provides a selective growth advantage that allows the cell with the original “clonal mutation” (also known as the “truncal mutation”) to grow and divide more quickly than neighboring healthy cells. Over time, additional genomic alterations accumulate in the DNA of these cancerous cells, leading to both linear and branched evolution from the original clonal population. This leads to a tumor comprised of various subclones, all of which share the original truncal mutation but also feature additional, unique mutations (known as “private mutations”). Administration of an efficacious treatment will typically eliminate many cells in the tumor, resulting in a reduction in tumor burden and clinical remission; however, certain subclones already harboring resistance mutations will often survive treatment at clinically undetectable levels and subsequently expand in the absence of competition. In time, this leads to the clinical observation of recurrence.
At the time of diagnosis under the current standard of care, a single biopsy of a single tumor will only reveal a set of mutations at one point in time for that one specific physical location in the tumor. However, it is likely that an adjacent area in the primary tumor, or a distant metastatic site, will have a different set of mutations (95, 99). As cancer therapeutics become increasingly guided by the tumor's molecular alterations, a representative and unbiased view of the mutational landscape across all subclones in the body will be essential (100).
Treatment success is currently determined by observing a reduction or apparent disappearance of the tumor mass on imaging or physical examination, but in many cases this is ultimately followed by reemergence of the cancer at the same anatomic site or elsewhere. From a molecular perspective, the treatment may have been successful in eliminating a large subset (perhaps the dominant clone) of cancer cells with a particular genomic signature but left behind other subclones that harbored private resistance mutations to the treatment (Figure 5) (101). The treatment-resistant cell populations (subclones) were likely already present in the tumor at the time of initial treatment, albeit in smaller numbers compared to the dominant clone; once the overall disease burden is reduced as a result of treatment pressure on the susceptible clone, these resistant subclones are allowed to prosper, with reduced competition for space and nutrients from the previously dominant clone (58, 102). This highlights an important benefit of detecting cancer earlier, before it accumulates a more diverse clonal composition that may increase its overall resistance to treatments.
In humans, this accumulation of additional somatic alterations is known to progress at relatively predictable rates. By the time a tumor reaches a clinically detectable size (typically 1 g, or 1 cm3, or 1 billion cells), it has undergone 30 volume doublings (103); the time that the tumor has been present in the body can be roughly estimated by back calculation via the tumor doubling time (TDT), if known. In human breast tumors across multiple subtypes, median tumor volume doubling times of 85–185 days have been reported (104). Assuming constant growth rates, the average breast cancer would need many years to reach a size at which it could be clinically detected. Currently recommended screening intervals in humans take these tumor growth estimates into account. For example, screening for breast cancer with mammography is recommended every 1–2 years beginning at age 45–50 (105, 106), while screening for colorectal cancer is recommended every 3–5 years beginning at age 50 (107). In effect, these recommendations reflect current understanding of the growth rates of specific cancers from early stage to late stage in humans. Routine screening at set intervals also provides the benefit of “cumulative detection”—the combination of detection rates compounded over time, such that after 2+ cycles of screening, the overall detection rate will be higher than if a single screening test were used at just one point in time (108, 109).
The rates of growth of various cancer types in dogs are not as well understood as in humans; however, given the shorter canine lifespan, it can be assumed that the time from a cancer's molecular inception to clinical manifestation is significantly compressed. Though TDTs have been rarely reported in veterinary medicine, those that have been reported support this assessment: for example, the mean TDT for induced canine lung adenocarcinomas was ~100 days, and for human pulmonary adenocarcinoma was greater than 1 year (110, 111). As in humans, TDT is important for informing the cadence of cancer screening in dogs; given these preliminary estimates, an annual or semiannual screening interval, when such testing becomes available, should allow for the detection of a significant proportion of canine cancers at the localized (resectable) stage.
Comparative oncology is typically described as the study of naturally occurring cancers in veterinary patients to benefit both humans and animals, through the study of cancer biology, pathogenesis, and treatment (112). Canine and human cancers share many histological, molecular, physiological, and even epidemiological features, and this commonality provides the rationale for the field of comparative oncology, wherein a deeper understanding of cancer in one species can drive corresponding insights in the other (7, 113–115). Dogs represent a powerful model system for the study of human cancers and vice versa, as cancers occur spontaneously in both species and are driven by orthologous genomic changes that impact corresponding biological pathways (114, 116, 117).
The human genome is ~3.1 billion nucleotides in length; the canine genome is ~20% smaller at ~2.4 billion nucleotides (31–33, 66, 118). Despite the size difference, the human and canine genomes have a high degree of homology (estimated at around 85%) (31); and among the top 100 human genes most frequently mutated in cancer, the extent of homology in the canine genome is likely even higher. Despite these commonalities, there are important differences between human and canine cancers, and these differences can be intelligently leveraged to drive faster translation of discoveries from one species to the other. For example, while dogs and humans are susceptible to cancers throughout the body, some cancers that are common in dogs are rare in humans (e.g., osteosarcoma, T-cell lymphoma) (83, 119, 120), and vice versa (Table 1). It is difficult to perform well-powered studies in rare cancer types; however, research efforts can progress faster in the species where the cancer is more common, and key insights can be translated back to the other species.
Liquid biopsy broadly refers to the sampling and analysis of analytes from various biological fluids (primarily blood, but in some cases also urine, cerebrospinal fluid, or other secretions) that can be sampled through minimally invasive or non-invasive methods (121). Blood-based liquid biopsy may include analysis of circulating nucleic acids (mainly cfDNA, which includes ctDNA in patients with cancer); circulating tumor cells (CTCs); and proteins (121). The ability to detect cancer-related analytes from blood has unique advantages, especially in cancer (or suspected cancer) cases where obtaining a tissue sample for traditional histological analysis might be particularly risky or difficult.
The conventional path to achieving cancer diagnosis in companion animals varies based on patient characteristics, tumor type, and tumor location (122). Fine needle aspiration (FNA) cytology is less invasive and lower risk compared to biopsy, and FNA is often used to make a preliminary or definitive diagnosis, develop a treatment plan, and predict prognosis (123, 124). However, inconclusive results or misdiagnoses can occur with FNA due to low cellularity, artifact, necrosis, minimal exfoliation of certain cell types, lack of tissue architecture, etc. (124, 125). Also, not all tumors are easily accessible by FNA (such as deep-seated abdominal tumors, many intrathoracic tumors, and tumors of the central nervous system); and some tumors with high vascularization, or those which might seed the body wall (e.g., urinary tract), are not amenable for sampling by FNA.
If FNA cytology is attempted and is non-diagnostic or equivocal, more invasive methods (such as a traditional biopsy or exploratory surgery) are often employed to obtain tissue for analysis prior to making a definitive diagnosis and initiating treatment (126). Compared with FNA, biopsies and surgeries entail higher risks of morbidity and mortality, which are dependent upon the site of the suspected mass and the characteristics of the procedure. Such risks include infection, internal bleeding, fracture after bone biopsy, intestinal perforation with endoscopic biopsy, pancreatitis after pancreatic biopsy, collapse of vertebra at spinal surgery sites, non-diagnostic results, and in the worst cases, death (122, 127–137).
The clinical and cost challenges of tissue analysis have stimulated the search for “non-invasive” methods that rely upon analysis of biomarkers found in easily accessible body fluids, such as blood, urine, and secretions. Despite decades of research, few such methods have entered broad clinical use, with the exception of testing for hematological malignancies where blood-based cytology is part of the standard of care. Solid tumors, which make up most malignancies in both humans and dogs, have seen limited benefits to date from methods that employ circulating biomarkers, with cfDNA-based approaches currently showing the greatest promise for the future.
In humans, blood-based testing has provided the opportunity to profile tumors to aid in the diagnosis of cancers, and to guide treatment decisions; and the earliest such tests have targeted protein markers (138, 139). A number of blood-based protein biomarkers have been used for human cancer screening and monitoring using immunohistochemical methods, including: CEA for colorectal cancer, PSA for prostate cancer, CA-125 for ovarian cancer, and alpha-fetoprotein (AFP) for hepatocellular carcinoma (140–143). Using similar ELISA (Enzyme-Linked ImmunoSorbent Assay) testing methods, recent attempts have been made to measure the concentrations of histone proteins that form the core of nucleosomes in order to detect the presence of cancer (144, 145). The nucleosome is the basic structural unit of DNA packaging, consisting of a segment of DNA wound around eight histone proteins. As cancer cells die, they release histone-bound DNA into circulation, whereupon the histone proteins can be separated and independently assayed (Figure 1).
However, biomarker assays based on circulating proteins suffer from high rates of false positives and false negatives, since the same proteins exist in circulation in healthy individuals and can be increased for reasons other than cancer, such as inflammation, sepsis, and trauma (146); also, these markers may not be significantly elevated in a significant proportion of individuals with even advanced stage cancer, reducing the potential for a highly sensitive test (146). Importantly in dogs, nucleosome concentration is also elevated in benign disease and in trauma (147–151), limiting its diagnostic utility for cancer (152). For these reasons, circulating protein markers have not been broadly adopted for cancer detection, and are more commonly used for monitoring cancer in cases where the level of the corresponding protein was shown to be already abnormal at the time of diagnosis.
In veterinary medicine, there has been interest in leveraging protein biomarkers such as thymidine kinase type 1 (TK1), canine C-reactive protein (cCRP), and alpha-fetoprotein receptor (RECAF) for canine cancer detection (153, 154); however, such protein biomarkers are not highly specific for canine cancer and can be elevated due to other reasons including immune-mediated hemolytic anemia, thrombocytopenia, and polyarthropathy (155). In human medicine, protein biomarkers such as RECAF and AFP have demonstrated limited sensitivity and specificity for cancer detection (143, 156).
CTCs are intact tumor cells originating in solid tumors that can sometimes be detected in circulation, a finding that has catalyzed a considerable body of research aimed at using CTCs for cancer detection. However, multiple studies in humans have demonstrated that even in metastatic disease, as many as 20% to over 50% of patients (depending on cancer type) have no detectable CTCs in the typical sample volume collected (157–162). Similar performance has been observed in metastatic canine cancer (163). As a result, CTCs have not seen broad clinical adoption, and remain primarily a research tool in both humans and dogs (164, 165).
Over the past decade, circulating nucleic acids – in particular cfDNA – have emerged as the most promising class of circulating biomarkers for non-invasive detection and characterization of cancer. cfDNA, which includes ctDNA in cancer subjects, is the focus of the remainder of this review.
As cells undergo programmed cell death (apoptosis) and necrosis, the membranes of cells and nuclei are broken down, and their contents are released into the circulation (Figure 6). Among these contents are fragments of DNA, known as “cell-free DNA” once they have left the confines of the cell and its nucleus. These cfDNA fragments are rapidly degraded by normal metabolic processes and have a very short half-life, estimated at 15 min to a few hours in both humans and dogs; as a result, they are usually cleared within a few days (168–170). The constant turnover of cells throughout the body provides a steady supply of cfDNA in the circulation, which is amenable to analysis with sophisticated technologies including NGS.
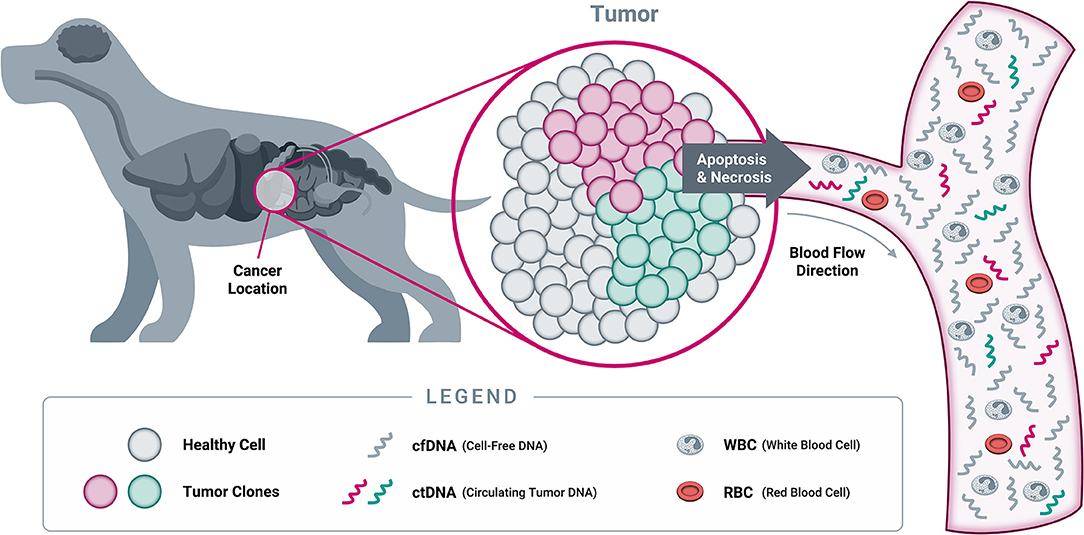
Figure 6. Origins of cell-free DNA. When a cell dies through either programmed cell death (apoptosis) or necrosis, its cellular contents (including DNA from the nucleus) are released into the bloodstream. At this point, the DNA becomes “cell-free DNA” and is rapidly degraded into small fragments through the action of circulating enzymes known as “DNAses.” As a result, most cfDNA fragments found in circulation are typically short, averaging 167 nucleotides in length in both humans and dogs (166, 167). While both healthy cells and tumor cells contain DNA that becomes cfDNA in circulation, only tumor cells will harbor somatic genomic alterations in cancer-related genes. Detection of such genomic alterations in the cfDNA of a patient is thus indicative of the presence of tumor cells in the body, providing the rationale for “liquid biopsy” testing approaches (Note: cfDNA exists as both single stranded DNA and double stranded DNA; only single stranded DNA is depicted here, for illustrative purposes).
The presence of cfDNA in humans was first reported in 1948, and while cfDNA was hypothesized to be linked to metastatic cancer in the mid-1960s, it took until 1977 for the first results evaluating cfDNA concentrations in patients with cancer compared to normal controls to be published, and neoplastic characteristics were reported in circulation in 1989 (171–174).
In 1996, two landmark publications reported the detection of cancer-derived alterations in plasma or serum of cancer patients as ctDNA (175, 176). Since then, significant efforts have been devoted to developing molecular tests to detect the presence of cancer-derived alterations in the blood (161, 177), and use the information for cancer detection, characterization, treatment, and monitoring (27, 28, 101, 178–183).
In parallel, fetal-derived cfDNA was discovered in maternal plasma in 1997 (184), leading to the first widely adopted clinical application for cfDNA testing: a screen for common fetal chromosomal abnormalities such as trisomy 21 (Down syndrome) using a sample of the pregnant woman's blood (185). Prior to this revolutionary advance, such fetal genetic information could only be derived from invasive diagnostic tests such as chorionic villus sampling (CVS) or amniocentesis, which carry a risk of miscarriage (186). As a result, the introduction of cfDNA-based non-invasive prenatal testing (NIPT) in 2011 (185) fundamentally changed the way prenatal care is delivered. Tens of millions of pregnancies have been screened with this cfDNA-based technology to date, leading to a marked decrease in the number of invasive diagnostic procedures for detection of fetal chromosomal abnormalities (187).
There are many documented instances of NIPT results incidentally identifying maternal cancer, highlighting plasma as a common repository for both fetal-derived and cancer-derived cfDNA fragments (188, 189), and suggesting the potential of using plasma cfDNA to screen for asymptomatic cancers. Indeed, a population-based study published in 2017 reported the performance of cfDNA-based liquid biopsy to detect nasopharyngeal cancer before symptoms develop (190), which marked the first demonstration of using a cfDNA-based blood test to screen for a specific type of cancer. Multiple commercial providers are currently offering or developing liquid biopsy tests for human cancer applications, and many clinical trials are underway to expand the clinical utility of this technology to additional use cases and/or cancer types.
Published research on canine cfDNA has covered a variety of clinical applications, including trauma, sepsis, thromboembolism, and neoplasia, and has focused primarily on determining the concentration of cfDNA in plasma as correlated to a particular clinical state or as a predictor for certain clinical outcomes (84, 148–151, 167, 169, 191–203). Studies that evaluated cfDNA concentrations in healthy canine subjects have reported median concentrations ranging from less than 1 ng/mL to greater than 500 ng/mL (148, 149, 167, 169, 194–203)—significantly wider than the range documented in healthy humans (typically 0–20 ng/mL) (204). These wide-ranging findings suggest that additional research employing well-controlled, large-scale studies is required to better understand the fundamental characteristics of cfDNA in dogs; they also point to the need for standardized, reproducible methods for blood collection, extraction, and measurement of canine cfDNA. Such standardization will be critical for the successful transfer of cfDNA-based technologies such as liquid biopsy—currently limited to the human space where such methods are well established—to routine clinical use in veterinary medicine.
To provide the highest clinical value, a liquid biopsy test should be able to detect multiple classes of cancer-associated genomic alterations (described above) in cfDNA with high accuracy, even at very low concentrations in the circulation. Furthermore, the biology of cfDNA uniquely facilitates the evaluation of certain genomic features in circulation that can provide additional information about the presence and the origin of cancer.
For example, it is well-known that the attachment of methyl (CH3) groups to the DNA strand at specific locations throughout the genome is associated with cancer; methylation of the promoter regions of tumor suppressor genes can inactivate the expression of these genes, allowing oncogene-driven cancers to proliferate unopposed (205). Furthermore, DNA in cells from specific organs have methylation profiles that are specific to that organ (206). When DNA from cancer cells in a particular organ is released into circulation as ctDNA, its methylation “signature” carries information about the presence of cancer and about the organ of origin of that cancer (27, 207). For this reason, NGS-based analysis of cfDNA methylation profiles has emerged as one of the most promising approaches for detecting cancer and assigning it to a specific organ of origin, which has obvious clinical benefits (27).
Another unique feature of cfDNA is the fact that it is highly fragmented according to specific patterns. In the nucleus of a cell, DNA is organized in chromosomes as an uninterrupted strand ranging in size from tens of millions to over 100 million nucleotides (or bases). However, by the time it enters circulation following cell death and nuclear DNA degradation, cfDNA has been biologically degraded into fragments that are typically less than 1,000 nucleotides in length. In both humans and dogs, much of the cfDNA exists in fragments that are ~167 bases in length, representing the length of the DNA strand between two nucleosomes plus one full wrap of DNA around the histone proteins that make up the core of the nucleosome (166, 167, 208). Furthermore, it has been shown that in humans with cancer, the fragment length of cfDNA tends to be shorter; one of the key observations that have led to fragment profile analysis becoming an emerging method to improve the sensitivity for cancer detection. In addition, fragmentation features in cfDNA can also encode information about the organ of origin (209–213). As a result, fragmentomics – like methylomics – has the potential to extract unique information from cfDNA that points to both the presence of cancer and its organ of origin (214).
Emerging methylomic and fragmentomic methods leverage features that are unique to circulating tumor DNA and offer additional possibilities for the detection and characterization of cancer in circulation. However, the canine methylome has not been comprehensively characterized, which means that significant research will have to be performed before methylomics-based liquid biopsy solutions can be offered for oncology applications in dogs. Likewise, the canine cfDNA fragmentome is poorly understood at this time, requiring a massive investment in research to fully understand its potential for clinical use.
Currently, the only technology that can simultaneously interrogate all the major classes of genomic alterations in cfDNA, as well as features such as methylation and fragmentation patterns, is next generation sequencing (NGS). Leading liquid biopsy assays currently in use or under development in human medicine use advanced NGS-based techniques to evaluate a broad range of alterations and features across the genome that are known to be associated with cancer. Most of these approaches do not target a particular cancer type; instead, they take a “pan-cancer” approach rooted in the premise that cancer is fundamentally a disease of the genome, and accurate analytical detection of somatic genomic alterations will lead to accurate clinical detection of a wide variety of cancer types. Assays that combine multiple classes of genomic alterations and/or orthogonal genomic features are likely to yield improved clinical performance (such as higher sensitivity and specificity) or provide additional useful information (such as organ of origin prediction and identification of molecular targets for personalized treatment) across a broad range of cancer types. The past few years have also seen the debut of multi-omic liquid biopsy approaches that combine (for example) genomic and proteomic methods, breathing new life into protein analysis as a valuable adjunct to cfDNA analysis (26, 28). Similar combinatorial strategies will likely be required for the successful development of a pan-cancer liquid biopsy test for dogs.
Liquid biopsy promises the convenience of a blood draw combined with the power of genomic technology. It is unlikely to fully replace the key role that traditional tissue biopsy plays in veterinary cancer diagnosis and management, but the non-invasive nature of liquid biopsy, coupled with its ability to detect tumor signal from any malignant mass in the body, should allow it to provide immediate value in several clinical scenarios once it becomes commercially available. In humans, liquid biopsy has demonstrated feasibility and great clinical potential across multiple use cases, spanning the entire continuum of cancer care; a similar spectrum of applications is in principle available for veterinary uses of the technology (Figure 7).
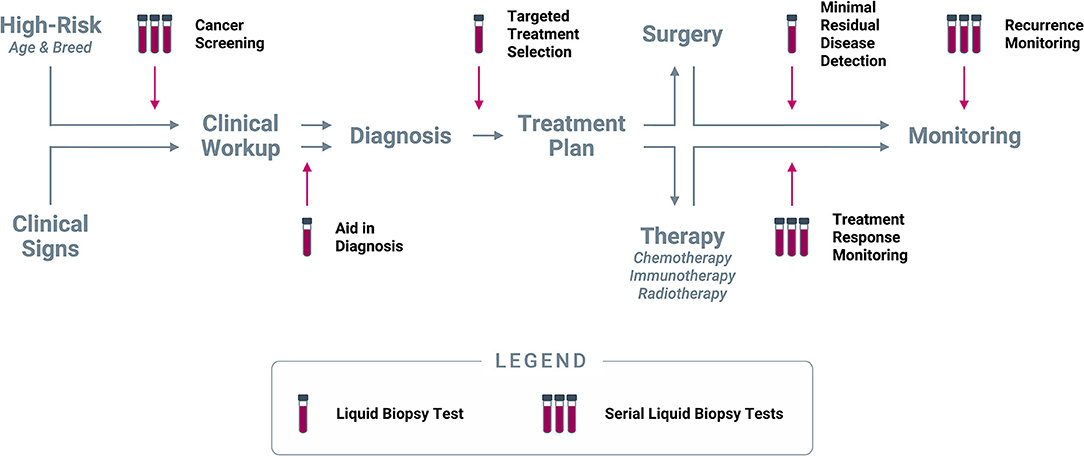
Figure 7. Clinical use cases for liquid biopsy in cancer. Liquid biopsy can be used to inform multiple decision points along the entire continuum of cancer care: (1) Cancer screening at regular intervals in patients deemed to be at higher risk for cancer based on age and/or breed; (2) Aid in diagnosis in patients who present with clinical signs (including incidental findings on imaging or laboratory tests) that are suspicious for cancer; (3) Targeted treatment selection based on the unique mutational profile of the tumor in patients diagnosed with cancer; (4) Minimal residual disease detection following a curative-intent intervention (such as surgery); (5) Treatment response monitoring at regular intervals during extended-duration therapeutic regimens; (6) Recurrence monitoring at regular intervals after complete remission or presumed cure.
Prior to a cancer diagnosis, liquid biopsy can provide valuable information in (1) presumably cancer-free patients as a screening test, and (2) in patients with clinical signs suspicious for cancer as an aid in diagnosis. Upon confirmation of a cancer diagnosis, liquid biopsy can be used to (3) identify a personalized treatment path based on the mutational profile of the tumor for targeted treatment selection; and (4) if the patient is to undergo a curative-intent intervention (such as a surgical procedure), a liquid biopsy immediately following the intervention can be used to test for minimal residual disease. After initiation of a longer-term therapy, such as chemotherapy or radiotherapy, liquid biopsy can be used (5) at regular intervals for treatment response monitoring. Finally, once a patient completes their course of treatment and is determined to be cured or in complete remission, liquid biopsy testing at longer intervals can be used for (6) recurrence monitoring. Each of these use cases, and their potential applicability in dogs, are described in more detail below.
Certain dog breeds are known to be more predisposed to cancer than others, presumably due to cancer-predisposing mutations that have become concentrated in the population over time as a result of the breeding process; however, the germline mutations responsible for most of these cancer predispositions are not as well-understood as in humans. It is also well-established that, just as in humans, cancer incidence in dogs increases with age (3). In a large fraction of cases, cancers in dogs are diagnosed at advanced stages after they have spread beyond the organ of origin, when prognosis is poor and the ability to extend life by treatment is limited (12–16). A liquid biopsy-based screening paradigm focused on high-risk populations, such as dogs from predisposed breeds or from geriatric populations, could help identify many of these cancers earlier. Early detection has been shown to drive better clinical outcomes in humans, such as increased life expectancy and higher rates of achieving complete remission following curative-intent interventions (e.g., surgery); historically, this has provided the rationale for well-established screening programs such as colonoscopy, mammograms, Pap smears, PSA screening, and low-dose CT scans (183, 215–217). Liquid biopsy solutions for universal cancer screening in humans are nearing commercialization (216, 218–220), and some of these assays have also shown potential for predicting the organ of origin of the tumor, facilitating the path to a definitive diagnosis (27, 28, 221–223).
State-of-the-art liquid biopsy assays currently in development for pan-cancer screening in humans have demonstrated detection rates (sensitivity) for early-stage cancer ranging from ~20 to 70% across multiple cancer types, at specificities of 98 to >99% (false positive rates of 2 to <1%) (27, 28, 211). High specificity is particularly important in cancer screening, given potential harms resulting from the diagnostic work-up of false positive screens, and from diagnosis and treatment of cancers that may never have become clinically apparent without screening (overdiagnosed and overtreated cases) (224). Screening results implying the possibility of a cancer diagnosis can also impose a considerable psychological burden on people who receive false positive results (225, 226), and it is reasonable to assume that pet owners would likewise experience distress as a result of false-positive cancer screening results in their companion animal.
A recent health economic modeling study revealed that adding an annual universal cancer screening test to the current standard of care in human medicine would reduce late-stage cancer incidence by 78% in those intercepted by the screening test, and result in an absolute reduction of 26% in all cancer deaths (227). The practice of screening at regular intervals relies on the concept of “cumulative detection” to improve the clinical sensitivity over time at the population level, as sequential testing holds the benefit of detecting cases missed on initial screening (108, 109, 228). Ultimately, this technology may support cancer screening in lower-risk canine populations as well, comparable to how NIPT technology expanded beyond high-risk cases to encompass all pregnancies in humans (229).
One of the most common scenarios in which liquid biopsy may add value in the veterinary clinic is as an aid in diagnosis, when cancer is suspected due to clinical signs (including incidental findings on imaging or laboratory tests) or clinical history. Due to the high-risk nature of this patient population, this scenario is likely to provide the initial opportunity for liquid biopsy to be deployed in veterinary medicine. In some cases, clinical signs may be non-specific and not localizing to a certain anatomic site; whereas in other cases an anatomic site may be evident, but the invasive procedures required to obtain tissue for diagnosis may carry a high risk of complications, or the suspected mass is inaccessible by biopsy or surgery. In such cases, liquid biopsy could significantly shorten the time to a definitive diagnosis and help avoid the challenges typically associated with a long diagnostic odyssey. Often, elucidation of such clinical cases requires additional appointments, time, and expense; and diagnosis may be delayed or missed completely. Many pet owners may decline biopsy or exploratory surgery due to the associated risks and cost, missing the opportunity to obtain an adequate diagnosis and select an appropriate treatment. A liquid biopsy can be conveniently performed from a routine blood collection drawn during the initial visit when cancer is first suspected, potentially saving time and money while increasing compliance.
In both the screening and the aid in diagnosis use cases, liquid biopsy can facilitate earlier detection of cancer compared to the current standards of care. In addition to improving outcomes, earlier diagnosis can mitigate the financial burden of treatment, making it a cost-effective paradigm both at the population level and at the level of individual patients. Health economic studies have shown that treatment costs for human cancer patients diagnosed early in the disease course to be 2 to 4 times less than for those diagnosed at later stages (216, 217). Treatment for early-stage cancer typically consists of localized resection, which is often curative and has a short recovery time (28); whereas treatment for late-stage disease involves repeat courses of chemotherapy or radiation therapy aimed at extending life rather than achieving a cure. Availability of an affordable and convenient liquid biopsy testing option for proactive serial screening of dogs at high risk of cancer, or for first-line evaluation of canine patients suspected of cancer, could reshape the clinical and economic landscapes of pre-diagnostic cancer management in veterinary medicine.
In situations where surgical interventions are not feasible, other therapeutic options may be utilized, either with curative intent or as a chronic treatment to extend life and/or improve quality of life. In such cases, selection of a specific therapy may be based on established clinical practice guidelines; however, an emerging area in human medicine, often designated by the terms “precision medicine” or “personalized medicine,” aims to utilize the genomic signature of an individual's cancer to select specific targeted therapies (230, 231). For humans, there are over 200 FDA-approved drugs for the treatment of cancer (232) including a subset of more than 50 drugs matched (or “targeted”) to specific genomic alterations in a tumor, with many additional targeted-treatment candidates in various phases of development (233–235). For dogs, there are only two drugs that are FDA approved at the time of this writing for the treatment of cancer - toceranib (Palladia™) and tigilanol tiglate (Stelfonta®), with two more drugs - rabacfosadine (Tanovea®-CA1) and verdinexor (Laverdia™-CA1) - available under a conditional FDA approval (236). In the EU, the European Medicines Agency (EMA) has approved toceranib, tigilanol tiglate and mastinib mesylate (Masivet®) (237). Of these approved or conditionally approved drugs, only toceranib (a multi-kinase inhibitor that inhibits c-kit, PDGFR, and VEGFR2), and mastinib (a c-kit inhibitor) can be used as a targeted drugs linked to specific genomic features of a tumor, as improvements in tumor response (43, 238) and outcome (239) have been demonstrated for tumors with an activating kit mutation; however, many targeted drugs used to treat human disease are currently used off-label in dogs (236, 240). Many compounds developed (and FDA-approved) for use in humans underwent preclinical safety testing in dogs; significant safety and dosing data are thus available to help inform the treatment of canine cancer patients with these agents (241).
State of the art liquid biopsy approaches have the potential to comprehensively evaluate the genomic signature of a patient's cancer directly from blood – the final common pathway for ctDNA derived from all tumor subclones in the patient's body; this unique capability makes therapy selection based on liquid biopsy results less susceptible to treatment selection bias resulting from tumor heterogeneity, a bias that is unavoidable when a tumor is only sampled by a single tissue biopsy. Liquid biopsy results could be used for targeted treatment selection, especially for treatments where the genomic alteration targeted in humans has a direct ortholog in the canine genome. This could lead to more rapid and widespread utilization in canine cancer patients of targeted therapies currently approved for human use. The availability of liquid biopsy assays, as subject selection tools during the drug development process and as companion diagnostics following regulatory approval, can also accelerate the development of canine-specific targeted therapeutics; ultimately this will likely be the preferred path to bringing targeted treatments into veterinary oncology, as human-oriented targeted treatments might not have the same efficacy in canine cancer even if the targeted genomic alteration is perfectly homologous across the two species (242).
After curative intent treatment (such as surgery) has been performed to remove the tumor, adjuvant therapy is often considered because of the risk of malignant deposits remaining in the body and resulting in relapse (or recurrence) in the future (243). MRD is defined as occult malignant disease that exists immediately after surgery and is undetectable by conventional methods; however, it can often be detected by the presence of ctDNA in the circulation (244, 245). The short half-life of cfDNA (minutes to hours in both humans and dogs) makes it an ideal analyte for MRD testing, as detection of any amount of ctDNA starting within a few days after surgery would point to the persistent presence of malignant disease in the body (168, 169). Many cancer types in humans have been studied in the context of MRD detection, including breast, pancreatic, lung, nasopharyngeal, and colorectal, as well as hematological malignancies (246, 247). In colorectal cancer for example, MRD detection has strong prognostic value, as patients with undetectable ctDNA post-operatively have significantly improved recurrence-free survival compared to those with detectable ctDNA in plasma (178, 244). In fact, detectable ctDNA post-operatively has a stronger prognostic association than many of the other traditional high-risk pathological and clinical features typically used by oncologists when considering adjuvant chemotherapy for patients with stage II colon cancer (246, 248). Similarly, the adoption of liquid biopsy-based MRD testing for canine patients could be used to inform the clinician about the relative risk of recurrence following curative-intent interventions, and thereby guide decisions regarding initiation of adjuvant treatment as soon as the patient has recovered from surgery.
Traditionally, treatment response monitoring has been performed by clinical observation and by imaging (mainly ultrasound and radiography, in the veterinary setting). Formalized procedures for documenting treatment response in dogs, such as the Canine Response Evaluation Criteria for Solid Tumors (cRECIST v1.0), have been published based on these methods (249, 250). However, reliance upon imaging alone for ascertaining treatment response has significant shortcomings. There are well-documented high inter-observer variabilities with imaging approaches in both dogs and humans, which can complicate the interpretation of imaging studies read by different radiologists (251–256).
In addition, hyperprogression (faster-than-expected tumor growth while under treatment) (257) and pseudoprogression (an initial apparent increase in tumor size or appearance of a new lesion on imaging during treatment, followed by tumor regression) (258, 259) can confound the interpretation of imaging for evaluation of treatment response. Lesion growth observed on imaging after treatment initiation may be due to advancing disease (secondary to ineffective treatment), an inflammatory response (resulting from tumor destruction by the treatment or from a direct side effect of the treatment), or simply ongoing tumor growth in the setting of a delayed treatment effect (257). Due to these complexities, real-time monitoring of tumor dynamics via serial liquid biopsy testing may help the clinician differentiate among these challenging scenarios and obtain more frequent updates on the patient's response to treatment than might be feasible with imaging alone.
The concentration of ctDNA in plasma can serve as a surrogate for the overall tumor burden (161), and patients with undetectable ctDNA after treatment are more likely to have had a complete response (178, 248, 260). Furthermore, the precise genomic variants in an individual's cancer can be used to follow the efficacy of the treatment in real time. This monitoring for treatment response may be useful regardless of the treatment modality (e.g., IV vs. oral chemotherapy, radiation, etc.). Since many chemotherapeutics are costly and typically require multiple clinic visits (20, 261), a ctDNA-based treatment response monitoring approach can offer significant value by detecting treatment response or treatment failure sooner than imaging or clinical observation would. This earlier detection may allow for early discontinuation of non-efficacious therapies in favor of alternate therapies that might have a better efficacy profile; or it may reassure the pet owner to continue a course of successful treatment even if clinical improvement is not readily apparent, or when a mixed clinical picture raises the question of disease progression vs. side effects of an otherwise efficacious treatment.
Monitoring for treatment response will also likely yield insights into the genomic evolution of tumor clones under the selective pressures of treatment—for example, the emergence of resistance mutations, or the emergence of new genomic variants potentially targetable by a different drug (262–264). Such molecular insights into tumor evolution are currently possible with standard tumor biopsy; however, even if molecular profiling of tumor tissue were widely available, longitudinal monitoring through repeat tissue biopsies would not be feasible in actual practice due to clinical, ethical, and financial considerations. Compared to current methods for monitoring treatment response, liquid biopsy would represent a complementary tool to better understand the evolution of the tumor, and its non-invasive nature could pave the way for liquid biopsy to become a routine monitoring test during cancer treatment in dogs.
Even in patients who are thought to have achieved complete remission or a cure following successful treatment, the possibility of disease recurrence remains an ever-present concern. Sequential cfDNA testing during the post-treatment period aims to detect residual disease at a pre-clinical stage and flag a “molecular relapse” well before clinical relapse becomes otherwise evident (246). Many recent studies have described the use of liquid biopsy to identify human patients with molecular relapse many months before clinical or radiological relapse (246). Early identification of cancer relapse may help guide treatment and management decisions in canine patients as well, with the goal of improving clinical outcomes through earlier adjuvant therapeutic intervention.
Development of high-quality liquid biopsy tests for dogs comparable to those currently available for human testing has the potential to revolutionize the detection, characterization, and management of cancer in pets. However, the challenges involved in such development are significant. To observe cancer-related genomic variants at low concentrations in blood, the assay must interrogate a large number of cfDNA fragments, the majority of which will not be tumor-derived. This drives the need to focus on genomic regions of known clinical relevance for cancer. Pending results from large-scale discovery efforts across all major canine cancer types, these clinically-relevant genomic regions can only be identified from the—limited—available literature describing genomic alterations in canine cancers, or by homology mapping from the much more substantial human knowledge base. Identifying high-confidence orthologous regions in dogs for the top cancer-related regions in humans is non-trivial and will require significant effort and expertise.
After defining the genomic regions and features of interest, the process of developing a robust assay to detect low ctDNA signal presents a number of challenges, including: (1) optimizing best practices for the collection and isolation of cfDNA from canine plasma; (2) optimizing enrichment of targeted genomic regions; (3) maximizing the signal-to-background ratio of tumor-derived ctDNA vs. non-cancer cfDNA during data analysis; and (4) establishing a normal reference baseline, so that a signal indicative of cancer can be confidently segregated from random signals in patients without cancer who may have other clinical conditions that also could present with cancer-like signatures. For example, a well-documented challenge in the human liquid biopsy field is posed by the presence of clonal hematopoiesis of indeterminate potential (CHIP), also known as age related clonal hematopoiesis (ARCH) and defined as the accumulation of somatic mutations in hematopoietic stem cells that are clonally propagated to their progeny, a process that is associated with aging (265, 266). This phenomenon has not yet been documented in dogs, but it is reasonable to expect that it could also be a confounder in canine liquid biopsy, requiring sophisticated approaches to mitigate the impact on the false positive rate of such tests.
An analytically robust and clinically accurate liquid biopsy assay for use in canine patients will be highly complex, potentially generating billions of data points (base reads) for each test from NGS data; and will require extensive analytical and clinical validation to demonstrate reliability and clinical performance. Although the veterinary diagnostics space is not subject to the extensive regulations that apply to human diagnostics, it is imperative that any candidate liquid biopsy solution undergo validation at a level similar to that expected for human use, to maximize benefit for veterinary patients and clinicians. Clinical validation should be performed in adequately sized cohorts of canine subjects with a variety of cancers as well as presumably cancer-free canine subjects, to demonstrate both high sensitivity (few missed cases of cancer) and high specificity (few false positives). The results of such studies should be published in peer-reviewed journals so that the veterinary community is able to review the full corpus of supporting data before starting to use liquid biopsy tests in routine practice.
As liquid biopsy solutions become available in veterinary medicine, the clinical paradigm can be expected to shift in order to accommodate the inclusion of additional information afforded by the new modality; over time, veterinarians will develop an informed appreciation for the clinical utility of liquid biopsy in each care setting and incorporate this new tool judiciously into their clinical algorithms. Specifically, screening and aid in diagnosis will likely show the most immediate clinical utility for liquid biopsy by shifting diagnosis to an earlier timepoint when clinical outcomes are superior. In addition, the use of liquid biopsy for detection of minimal residual disease and for recurrence monitoring promises to provide an earlier opportunity to determine if a curative-intent intervention (i.e., surgery) was successful – and to inform the timely use of adjuvant treatments if the disease has not been eradicated. Finally, as more treatment options become available in veterinary medicine in the form of targeted therapies aimed at specific genomic alterations, the standard of care may evolve to include liquid biopsy as a routine pre-treatment selection step, and as a complement to imaging for evaluating response to treatment.
Liquid biopsy solutions based on cfDNA analysis are well-positioned to revolutionize certain aspects of cancer care in veterinary medicine by enabling safe, non-invasive testing at frequent intervals as dictated by the needs of each clinical case. However, liquid biopsy is not a panacea for all the challenges facing veterinary cancer management, and limitations exist. Certain tumors may not shed sufficient ctDNA into circulation to allow for confident detection and characterization of the disease by liquid biopsy; this can happen with smaller sized tumors in early disease, or with certain malignancies that tend to release lower levels of ctDNA into the bloodstream (such as tumors of the central nervous system) (161, 267). Also, the novelty of liquid biopsy means that extensive education will be required before its use can become widespread in the veterinary community, presenting a practical limitation to the speed and extent of adoption. Finally, the economics of a liquid biopsy-based approach to veterinary cancer diagnostics are yet unknown, which can present challenges – especially in the early years. In some use cases, such as aid in diagnosis when cancer is already suspected on clinical grounds, liquid biopsy may offer obvious cost advantages over invasive diagnostic procedures; in other cases, the economic value of liquid biopsy may be less apparent, such as with annual screening of dogs who will never go on to develop cancer, or with testing for targeted treatment selection when the only available options are off-label human therapeutics that have not been directly shown to be efficacious in canine cancer. Pricing considerations will certainly play an important role in the overall economics of the emerging liquid biopsy paradigm; ongoing decreases in the cost of sequencing, rapid improvements in assay design and automation, volume-driven economies of scale, and competition among providers should all contribute to favorable developments in pricing, making liquid biopsy an increasingly affordable testing option for pet owners.
Tumor tissue analysis is likely to remain a core component of the standard of care, especially for cases where malignant masses can be easily sampled by biopsy or surgery. Traditional tissue histopathology can provide unique and highly valuable information, such as: establishing a definitive diagnosis of cancer; determining aggressiveness and prognosis; and selecting a treatment – this being especially useful in cases where genomic analysis of the tumor does not provide any obvious targeted treatment options. As experience with liquid biopsy builds within the veterinary community, this new testing method may prove to be a replacement for older methods in some cases but is more likely to establish itself as a complementary or backup method alongside existing approaches, expanding the overall ability of the clinician to provide the most personalized care to each patient.
The genomic revolution has already had a marked impact on cancer care for human patients and is poised to revolutionize veterinary medicine in a similar manner in the coming years. As genomics becomes a routine part of veterinary care, expansion into multi-omic liquid biopsy approaches is likely to follow, including epigenomics (methylation and histone-modification analyses), transcriptomics (gene expression, micro RNAs), proteomics (tumor markers, other peptides), metabolomics, fragmentomics, etc. (121, 214). When combined, these orthogonal datasets will enable a multidimensional view of the cancer in real-time, enabling delivery of the highest quality of care. The introduction of high-quality, clinically validated pan-cancer liquid biopsy tests into the realm of veterinary medicine has the potential to substantially impact every step along the clinical journey of a canine cancer patient, from early detection to recurrence monitoring.
Long known as “man's best friends,” dogs live much shorter lives than humans, yet they form exceptionally close bonds with their human companions as well as with other pet dogs in the family; the loss of a pet dog often has a devastating emotional impact on the surviving family members, whether humans or other pet dogs (268–270). Cancer is by far the single most common cause of death in dogs, and having a pet companion that is fighting a losing battle with late-stage cancer is particularly difficult for families because of the financial strain of managing the disease in its final stages, and because the process is often drawn out over weeks or months and may involve considerable physical pain for the patient (271–273). The decision to euthanize a pet family member is one of the most difficult decisions a family will make. As veterinary medicine stands on the threshold of the new era of genomic medicine, novel tools – convenient, affordable, non-invasive, and widely available – will enable veterinarians to routinely screen for cancer and detect it early, when it can be cured; pursue rapid diagnosis of cancer as soon as the disease is suspected; and select targeted treatments and monitor for response and recurrence after a diagnosis has been made. These new tools will allow countless families to spend more time with their beloved pet family members and will further empower veterinarians to honor their professional oath to use their “scientific knowledge and skills for the benefit of society through the protection of animal health and welfare. [and] the prevention and relief of animal suffering” (274).
Humans have benefited extensively from medical advances that were first trialed in our canine sidekicks. By implementing lessons learned from recent genomic advances in cancer care for humans, we can now raise the level of cancer care for our canine companions as well. It is fitting to consider that widespread adoption of liquid biopsies in veterinary medicine may represent an upcoming historic opportunity to repay our “best friends” for their many prior contributions to our well-being.
JC and DG contributed to conception and design of the manuscript and co-wrote the first draft. All authors contributed to the manuscript and provided critical revisions, read, and approved the submitted version.
This article funded by PetDx.
JC, AF, KK, IC, JT, KL, LH, DT, and DG are employed by or affiliated with PetDx. JC, AF, KK, NL, AN, ND, DB, TJ, JF, MS, IC, JT, KL, LH, MM, LD, DT, and DG hold vested or unvested equity in PetDx. TJ is employed by Laboratory Corporation of America. JF is Managing Partner at Friedman Bioventure, Inc. MS is Managing Director at RS Technology Ventures LLC. KK is an inventor on multiple patent applications related to bioinformatics methods for cancer diagnostics and holds equity in Illumina. MM is an inventor on multiple patent applications covering technologies for canine and human cancer diagnostics, and has licensing or consulting relationships with PetDx, Exact Sciences, AstraZeneca, Bristol Myers Squibb, and TGen. LD is a member of the board of directors of Personal Genome Diagnostics (PGDx) and Jounce Therapeutics. LD is a compensated consultant to PGDx, 4Paws (PetDx), Innovatus CP, Se'er, Kinnate and Neophore. LD is an uncompensated consultant for Merck but has received research support for clinical trials from Merck. LD is an inventor of multiple licensed patents related to technology for circulating tumor DNA analyses and mismatch repair deficiency for diagnosis and therapy from Johns Hopkins University. Some of these licenses and relationships are associated with equity or royalty payments directly to Johns Hopkins and LD. LD holds equity in PGDx, Jounce Therapeutics, Thrive Earlier Detection, Se'er, Kinnate and Neophore. LD's spouse holds equity in Amgen. The terms of all these arrangements for LD are being managed by Johns Hopkins and Memorial Sloan Kettering in accordance with their conflict of interest policies.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Leonid Kruglyak for his advice and critical review of the manuscript. We also thank Jason Loftis and Chuba Oyolu for their contributions to the illustrations.
cfDNA, Cell-Free DNA; CNV, Copy Number Variant; ctDNA, Circulating Tumor DNA; CTC, Circulating Tumor Cell; DNA, Deoxyribonucleic Acid; FNA, Fine Needle Aspiration; MRD, Minimal Residual Disease; NGS, Next Generation Sequencing; SNV, Single Nucleotide Variant; TDT, Tumor Doubling Time; TMB, Tumor Mutational Burden.
1. Pang LY, Argyle DJ. Using naturally occurring tumours in dogs and cats to study telomerase and cancer stem cell biology. Biochim Biophys Acta. (2009) 1792:380–91. doi: 10.1016/j.bbadis.2009.02.010
2. Pang LY, Argyle DJ. Veterinary oncology: biology, big data and precision medicine. Vet J. (2016) 213:38–45. doi: 10.1016/j.tvjl.2016.03.009
3. Fleming JM, Creevy KE, Promislow DEL. Mortality in North American dogs from 1984 to 2004: an investigation into age-, size-, and breed-related causes of death. J Vet Intern Med. (2011) 25:187–98. doi: 10.1111/j.1939-1676.2011.0695.x
4. LeBlanc AK, Mazcko CN. Improving human cancer therapy through the evaluation of pet dogs. Nat Rev Cancer. (2020) 20:727–42. doi: 10.1038/s41568-020-0297-3
5. Baioni E, Scanziani E, Vincenti MC, Leschiera M, Bozzetta E, Pezzolato M, et al. Estimating canine cancer incidence: findings from a population-based tumour registry in northwestern Italy. BMC Vet Res. (2017) 13:203. doi: 10.1186/s12917-017-1126-0
6. Dobson JM. Breed-predispositions to cancer in pedigree dogs. Isrn Vet Sci. (2013) 2013:1–23. doi: 10.1155/2013/941275
7. Pinello KC, Niza-Ribeiro J, Fonseca L, Matos AJ. Incidence, characteristics and geographical distributions of canine and human non-Hodgkin's lymphoma in the Porto region (North West Portugal). Vet J. (2019) 245:70–6. doi: 10.1016/j.tvjl.2019.01.003
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
9. Schiffman JD, Breen M. Comparative oncology: what dogs and other species can teach us about humans with cancer. Philos Trans R Soc B Biol Sci. (2015) 370:20140231. doi: 10.1098/rstb.2014.0231
10. Klingemann H. Immunotherapy for dogs: running behind humans. Front Immunol. (2018) 9:133. doi: 10.3389/fimmu.2018.00133
11. Cancer Statistics. National Cancer Institute. NCI (2020) Available online at: https://www.cancer.gov/about-cancer/understanding/statistics (accessed October 5, 2020)
12. Williams LE, Packer RA. Association between lymph node size and metastasis in dogs with oral malignant melanoma: 100 cases (1987-2001). J Am Vet Med Assoc. (2003) 222:1234–6. doi: 10.2460/javma.2003.222.1234
13. Biller B, Berg J, Garrett L, Ruslander D, Wearing R, Abbott B, et al. 2016 AAHA oncology guidelines for dogs and cats*. J Am Anim Hosp Assoc. (2016) 52:181–204. doi: 10.5326/jaaha-ms-6570
14. Polton GA, Brearley MJ. Clinical stage, therapy, and prognosis in canine anal sac gland carcinoma. J Vet Intern Med. (2007) 21:274–80. doi: 10.1111/j.1939-1676.2007.tb02960.x
15. Flory AB, Rassnick KM, Stokol T, Scrivani PV, Erb HN. Stage migration in dogs with lymphoma. J Vet Intern Med. (2007) 21:1041–7. doi: 10.1111/j.1939-1676.2007.tb03062.x
16. Wendelburg KM, Price LL, Burgess KE, Lyons JA, Lew FH, Berg J. Survival time of dogs with splenic hemangiosarcoma treated by splenectomy with or without adjuvant chemotherapy: 208 cases (2001-2012). J Am Vet Med Assoc. (2015) 247:393–403. doi: 10.2460/javma.247.4.393
17. Nakano Y, Matsushima M, Nakamori A, Hiroma J, Matsuo E, Wakabayashi H, et al. Depression and anxiety in pet owners after a diagnosis of cancer in their pets: a cross-sectional study in Japan. BMJ Open. (2019) 9:e024512. doi: 10.1136/bmjopen-2018-024512
18. Spitznagel MB, Jacobson DM, Cox MD, Carlson MD. Caregiver burden in owners of a sick companion animal: a cross-sectional observational study. Vet Rec. (2017) 181:321. doi: 10.1136/vr.104295
19. Bowen J, García E, Darder P, Argüelles J, Fatjó J. The effects of the Spanish COVID-19 lockdown on people, their pets and the human-animal bond. J Vet Behav. (2020) 40:75–91. doi: 10.1016/j.jveb.2020.05.013
20. Rebhun RB, Kent MS, Borrofka SAEB, Frazier S, Skorupski K, Rodriguez CO. CHOP chemotherapy for the treatment of canine multicentric T-cell lymphoma. Vet Comp Oncol. (2011) 9:38–44. doi: 10.1111/j.1476-5829.2010.00230.x
21. Shaevitz MH, Tullius JA, Callahan RT, Fulkerson CM, Spitznagel MB. Early caregiver burden in owners of pets with suspected cancer: owner psychosocial outcomes, communication behavior, and treatment factors. J Vet Intern Med. (2020) 34:2636–44. doi: 10.1111/jvim.15905
22. The Cancer Genome Atlas (TCGA) Timeline & Milestones. National Cancer Institute. NCI. Available online at: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/history/timeline (accessed October 5, 2020)
23. National Institutes of Health National Cancer Institute. TCGA Research Network Publications. NCI (2020) Available online at: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/publications (accessed October 5, 2020)
24. International Cancer Genome Consortium (ICGC). ICGC (2020. Available online at: https://icgc.org/ (accessed November 20, 2020)
25. National Institutes of Health (NIH). The Promise of Precision Medicine. NIH (2020). Available online at: https://www.nih.gov/about-nih/what-we-do/nih-turning-discovery-into-health/promise-precision-medicine (accessed November 20, 2020)
26. Lennon AM, Buchanan AH, Kinde I, Warren A, Honushefsky A, Cohain AT, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. (2020) 369:eabb9601. doi: 10.1126/science.abb9601
27. Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Consortium C, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. (2020) 31:745–59. doi: 10.1016/j.annonc.2020.02.011
28. Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. (2018) 359:eaar3247. doi: 10.1126/science.aar3247
29. Plagnol V, Woodhouse S, Howarth K, Lensing S, Smith M, Epstein M, et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE. (2018) 13:e0193802. doi: 10.1371/journal.pone.0193802
30. Gale D, Lawson ARJ, Howarth K, Madi M, Durham B, Smalley S, et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE. (2018) 13:e0194630. doi: 10.1371/journal.pone.0194630
31. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. (2005) 438:803–19. doi: 10.1038/nature04338
32. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. (2001) 409:860. doi: 10.1038/35057062
33. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. (2001) 291:1304–51. doi: 10.1126/science.1058040
34. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. (2004) 431:931. doi: 10.1038/nature03001
35. Koskinen MT. Individual assignment using microsatellite DNA reveals unambiguous breed identification in the domestic dog. Anim Genet. (2003) 34:297–301. doi: 10.1046/j.1365-2052.2003.01005.x
36. Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, et al. Genetic structure of the purebred domestic dog. Science. (2004) 304:1160–4. doi: 10.1126/science.1097406
37. Grall A, Guaguère E, Planchais S, Grond S, Bourrat E, Hausser I, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet. (2012) 44:140–7. doi: 10.1038/ng.1056
38. Plassais J, Guaguère E, Lagoutte L, Guillory A-S, Citres de CD, Degorce-Rubiales F, et al. A spontaneous KRT16 mutation in a dog breed: a model for human focal non-epidermolytic palmoplantar keratoderma (FNEPPK). J Invest Dermatol. (2015) 135:1187–90. doi: 10.1038/jid.2014.526
39. Donner J, Anderson H, Davison S, Hughes AM, Bouirmane J, Lindqvist J, et al. Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs. PLoS Genet. (2018) 14:e1007361. doi: 10.1371/journal.pgen.1007361
40. Nicholas FW. Online Mendelian Inheritance in Animals (OMIA): a comparative knowledgebase of genetic disorders and other familial traits in non-laboratory animals. Nucleic Acids Res. (2003) 31:275–7. doi: 10.1093/nar/gkg074
41. Mochizuki H, Kennedy K, Shapiro SG, Breen M. BRAF mutations in canine cancers. PLoS ONE. (2015) 10:e0129534. doi: 10.1371/journal.pone.0129534
42. London CA, Galli SJ, Yuuki T, Hu Z-Q, Helfand SC, Geissler EN. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp Hematol. (1999) 27:689–97. doi: 10.1016/s0301-472x(98)00075-7
43. London CA, Malpas PB, Wood-Follis SL, Boucher JF, Rusk AW, Rosenberg MP, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. (2009) 15:3856–65. doi: 10.1158/1078-0432.ccr-08-1860
44. Kurita S, Miyamoto R, Tani H, Kobayashi M, Sasaki T, Tamura K, et al. Genetic alterations of KIT during clonal expansion and subsequent acquisition of resistance to toceranib in a canine mast cell tumor cell line. J Vet Pharmacol Ther. (2019) 42:673–81. doi: 10.1111/jvp.12816
45. Moses L, Niemi S, Karlsson E. Pet genomics medicine runs wild. Nature. (2018) 559:470–2. doi: 10.1038/d41586-018-05771-0
46. American Association of Veterinary Laboratory Diagnosticians (AAVLD). AAVLD Accreditation Program. AAVLD (2017). Available online at: https://www.aavld.org/accreditation-explained (accessed January 29, 2021)
47. American Association of Veterinary Laboratory Diagnosticians (AAVLD). Accreditation Requirements. AAVLD (2018). Available online at: https://www.aavld.org/accreditation-requirements-page (accessed October 5, 2020)
48. College of American Pathologists (CAP). Accreditation. CAP. Available online at: https://www.cap.org/laboratory-improvement/accreditation (accessed October 5, 2020)
49. Centers for Medicare & Medicaid Services (CMS). Clinical Laboratory Improvement Amendments (CLIA). CMS (2020). Available online at: https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index (accessed October 5, 2020)
50. US Food & Drug Administration (FDA). List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). FDA (2020). Available online at: https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools (accessed January 29, 2021)
51. Cawley J, Khanna C. The challenge of cognitive dissonance in the delivery of precision medicine in veterinary oncology. Arch Cancer Biol Ther. (2020) 1:37–41. doi: 10.33696/cancerbiology.1.008
52. Stetson D, Ahmed A, Xu X, Nuttall BRB, Lubinski TJ, Johnson JH, et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. Jco Precis Oncol. (2019) 3:1–9. doi: 10.1200/po.18.00191
53. Tsui DWY, Blumenthal GM, Philip R, Barrett JC, Montagut C, Bramlett K, et al. Development, validation, and regulatory considerations for a liquid biopsy test. Clin Chem. (2020) 66:408–14. doi: 10.1093/clinchem/hvaa010
54. Morley AA, Turner DR. The contribution of exogenous and endogenous mutagens to in vivo mutations. Mutat Res Fundam Mol Mech Mutagen. (1999) 428:11–5. doi: 10.1016/s1383-5742(99)00026-5
55. Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. (2015) 347:78–81. doi: 10.1126/science.1260825
56. Wu S, Powers S, Zhu W, Hannun YA. Substantial contribution of extrinsic risk factors to cancer development. Nature. (2016) 529:43. doi: 10.1038/nature16166
57. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. (2017) 355:1330–4. doi: 10.1126/science.aaf9011
58. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
59. Fiala C, Diamandis EP. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. (2018) 16:166. doi: 10.1186/s12916-018-1157-9
60. DelMonte U. Does the cell number 10 9 still really fit one gram of tumor tissue? Cell Cycle. (2009) 8:505–6. doi: 10.4161/cc.8.3.7608
61. The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. (2013) 45:1113. doi: 10.1038/ng.2764
62. Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature. (2020) 578:82–93. doi: 10.1038/s41586-020-1969-6
63. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. (2018) 173:371–85.e18. doi: 10.1016/j.cell.2018.02.060
64. Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, et al. Ensembl 2020. Nucleic Acids Res. (2019) 48:D682–8. doi: 10.1093/nar/gkz966
65. Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. (2018) 18:696–705. doi: 10.1038/s41568-018-0060-1
66. Ensembl. Human (GRCh38.p13). Ensembl (2020). Available online at: https://uswest.ensembl.org/Homo_sapiens/Info/Annotation (accessed October 5, 2020)
67. Catalogue of Somatic Mutations in Cancer (COSMIC). COSMIC Census. COSMIC. Available online at: https://cancer.sanger.ac.uk/census (accessed October 5, 2020)
68. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
69. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/s0092-8674(00)81683-9
70. Rivera P, Melin M, Biagi T, Fall T, Häggström J, Lindblad-Toh K, et al. Mammary tumor development in dogs is associated with BRCA1 and BRCA2. Cancer Res. (2009) 69:8770–4. doi: 10.1158/0008-5472.can-09-1725
71. Forbes C, Fayter D, de Kock S, Quek RG. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manage Res. (2019) 11:2321–37. doi: 10.2147/cmar.s189627
72. Jones S, Chen W, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. (2008) 105:4283–8. doi: 10.1073/pnas.0712345105
73. Chenam A, Lau C. Genitourinary cancers. Canc Treat. (2018) 175:105–26. doi: 10.1007/978-3-319-93339-9_5
74. Zigras T, Lennox G, Willows K, Covens A. Early cervical cancer: current dilemmas of staging and surgery. Curr Oncol Rep. (2017) 19:51. doi: 10.1007/s11912-017-0614-5
75. Horta RS, Lavalle GE, Monteiro LN, Souza MCC, Cassali GD, Araújo RB. Assessment of canine mast cell tumor mortality risk based on clinical, histologic, immunohistochemical, and molecular features. Vet Pathol. (2018) 55:212–23. doi: 10.1177/0300985817747325
76. Milovancev M, Tuohy JL, Townsend KL, Irvin VL. Influence of surgical margin completeness on risk of local tumour recurrence in canine cutaneous and subcutaneous soft tissue sarcoma: a systematic review and meta-analysis. Vet Comp Oncol. (2019) 17:354–64. doi: 10.1111/vco.12479
77. Stephens PJ, Tarpey PS, Davies H, Loo PV, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. (2012) 486:400–4. doi: 10.1038/nature11017
78. Luu L-J, Price TJ. BRAF mutation and its importance in colorectal cancer. Advances in the Molecular Understanding of Colorectal Cancer. Intech Open. (2019) doi: 10.5772/intechopen.82571
79. Mochizuki H, Breen M. Comparative aspects of BRAF mutations in canine cancers. Vet Sci. (2015) 2:231–45. doi: 10.3390/vetsci2030231
80. Sclafani F, Gullo G, Sheahan K, Crown J. BRAF mutations in melanoma and colorectal cancer: a single oncogenic mutation with different tumour phenotypes and clinical implications. Crit Rev Oncol Hemat. (2013) 87:55–68. doi: 10.1016/j.critrevonc.2012.11.003
81. Shao C, Li G, Huang L, Pruitt S, Castellanos E, Frampton G, et al. Prevalence of high tumor mutational burden and association with survival in patients with less common solid tumors. JAMA Netw Open. (2020) 3:e2025109. doi: 10.1001/jamanetworkopen.2020.25109
82. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
83. Gardner HL, Sivaprakasam K, Briones N, Zismann V, Perdigones N, Drenner K, et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun Biol. (2019) 2:266. doi: 10.1038/s42003-019-0487-2
84. Lorch G, Sivaprakasam K, Zismann V, Perdigones N, Contente-Cuomo T, Nazareno A, et al. Identification of recurrent activating HER2 mutations in primary canine pulmonary adenocarcinoma. Clin Cancer Res. (2019) 25:5866–77. doi: 10.1158/1078-0432.ccr-19-1145
85. Wang G, Wu M, Maloneyhuss MA, Wojcik J, Durham AC, Mason NJ, et al. Actionable mutations in canine hemangiosarcoma. PLoS ONE. (2017) 12:e0188667. doi: 10.1371/journal.pone.0188667
86. Megquier K, Turner-Maier J, Swofford R, Kim J-H, Sarver AL, Wang C, et al. Comparative genomics reveals shared mutational landscape in canine hemangiosarcoma and human angiosarcoma. Mol Cancer Res. (2019) 17:2410–21. doi: 10.1158/1541-7786.mcr-19-0221
87. Büttner R, Longshore JW, López-Ríos F, Merkelbach-Bruse S, Normanno N, Rouleau E, et al. Implementing TMB measurement in clinical practice: considerations on assay requirements. Esmo Open. (2019) 4:e000442. doi: 10.1136/esmoopen-2018-000442
88. Klempner SJ, Fabrizio D, Bane S, Reinhart M, Peoples T, Ali SM, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncology. (2020) 25:e147–59. doi: 10.1634/theoncologist.2019-0244
89. Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. (2014) 32:246–51. doi: 10.1038/nbt.2835
90. Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. (2016) 37:564–9. doi: 10.1002/humu.22981
91. Wagner J, Olson ND, Harris L, Khan Z, Farek J, Mahmoud M, et al. Benchmarking challenging small variants with linked and long reads. BioRxiv. (2020)2020.07.24.212712. doi: 10.1101/2020.07.24.212712
92. Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature. (2020) 578:112–21. doi: 10.1038/s41586-019-1913-9
93. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. (2013) 45:1127–33. doi: 10.1038/ng.2762
94. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415. doi: 10.1038/nature12477
95. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/nejmoa1113205
96. McQuerry JA, Chang JT, Bowtell DDL, Cohen A, Bild AH. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J Mol Med. (2017) 95:1167–78. doi: 10.1007/s00109-017-1587-4
97. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. (2017) 168:613–28. doi: 10.1016/j.cell.2017.01.018
98. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. Omics J Integr Biol. (2018) 22:17–34. doi: 10.1089/omi.2017.0174
99. Crowley E, Nicolantonio FD, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. (2013) 10:472. doi: 10.1038/nrclinonc.2013.110
100. Murtaza M, Dawson S-J, Pogrebniak K, Rueda OM, Provenzano E, Grant J, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. (2015) 6:8760. doi: 10.1038/ncomms9760
101. Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. (2012) 486:537. doi: 10.1038/nature11219
102. Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. (2015) 525:261–4. doi: 10.1038/nature14971
103. Tubiana M. Tumor cell proliferation kinetics and tumor growth rate. Acta Oncol. (2009) 28:113–121. doi: 10.3109/02841868909111193
104. Nakashima K, Uematsu T, Takahashi K, Nishimura S, Tadokoro Y, Hayashi T, et al. Does breast cancer growth rate really depend on tumor subtype? Measurement of tumor doubling time using serial ultrasonography between diagnosis and surgery. Breast Cancer. (2019) 26:206–14. doi: 10.1007/s12282-018-0914-0
105. Siu AL, Force USPST. Screening for breast cancer: U.S. preventive services task force recommendation statement. Ann Intern Med. (2016) 164:279. doi: 10.7326/m15-2886
106. American Cancer Society (ACS). American Cancer Society Recommendations for the Early Detection of Breast Cancer. ACS (2020). Available online at: https://www.cancer.org/cancer/breast-cancer/screening-tests-and-early-detection/american-cancer-society-recommendations-for-the-early-detection-of-breast-cancer.html (accessed November 20, 2020)
107. US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, Curry SJ, Davidson KW, Epling JW, et al. Screening for colorectal cancer: US preventive services task force recommendation statement. JAMA. (2016) 315:2564. doi: 10.1001/jama.2016.5989
108. Melnikow J, Henderson JT, Burda BU, Senger CA, Durbin S, Weyrich MS. Screening for cervical cancer with high-risk human papillomavirus testing: updated evidence report and systematic review for the US preventive services task force. JAMA. (2018) 320:687–705. doi: 10.1001/jama.2018.10400
109. Zorzi M, Hassan C, Capodaglio G, Fedato C, Montaguti A, Turrin A, et al. Long-term performance of colorectal cancer screening programmes based on the faecal immunochemical test. Gut. (2018) 67:2124. doi: 10.1136/gutjnl-2017-314753
110. Perry RE, Weller RE, Buschbom RL, Dagle GE, Park JF. Radiographically determined growth dynamics of primary lung tumors induced in dogs by inhalation of plutonium. Am J Vet Res. (1992) 53:1740–3.
111. Aoki T, Nakata H, Watanabe H, Nakamura K, Kasai T, Hashimoto H, et al. Evolution of peripheral lung adenocarcinomas: CT findings correlated with histology and tumor doubling time. Am J Roentgenol. (2000) 174:763–68. doi: 10.2214/ajr.174.3.1740763
112. Paoloni MC, Khanna C. Comparative oncology today. Vet Clin North Am Small Anim Pract. (2007) 37:1023–32. doi: 10.1016/j.cvsm.2007.08.003
113. Rowell JL, McCarthy DO, Alvarez CE. Dog models of naturally occurring cancer. Trends Mol Med. (2011) 17:380–8. doi: 10.1016/j.molmed.2011.02.004
114. LeBlanc AK, Mazcko CN, Khanna C. Defining the value of a comparative approach to cancer drug development. Am Assoc Cancer Res. (2016) 22:2133–8. doi: 10.1158/1078-0432.ccr-15-2347
115. Sakthikumar S. Characterizing the Spectrum of Somatic Alterations in Canine and Human Cancers. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1624. Uppsala: Acta Universitatis Upsaliensis (2020). p. 64.
116. Alsaihati BA, Ho K-L, Watson J, Feng Y, Wang T, Zhao S. Canine tumor mutation rate is positively correlated with TP53 mutation across cancer types and breeds. Biorxiv. (2020)2020.07.15.205286. doi: 10.1101/2020.07.15.205286
117. LeBlanc AK, Breen M, Choyke P, Dewhirst M, Fan TM, Gustafson DL, et al. Perspectives from man's best friend: National Academy of Medicine's Workshop on Comparative Oncology. Sci Transl Med. (2016) 8:324ps5. doi: 10.1126/scitranslmed.aaf0746
118. Ensembl. Dog Assembly and Gene Annotation. Ensembl (2020). Available online at: https://uswest.ensembl.org/Canis_lupus_familiaris/Info/Annotation (accessed October 5, 2020)
119. McDonald JT, Kritharis A, Beheshti A, Pilichowska M, Burgess K, Ricks-Santi L, et al. Comparative oncology DNA sequencing of canine T cell lymphoma via human hotspot panel. Oncotarget. (2018) 9:22693–702. doi: 10.18632/oncotarget.25209
120. Villarnovo D, McCleary-Wheeler AL, Richards KL. Barking up the right tree. Curr Opin Hematol. (2017) 24:359–66. doi: 10.1097/moh.0000000000000357
121. Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. (2019) 20:71–88. doi: 10.1038/s41576-018-0071-5
122. Ehrhart N, Culp W. Chapter 1: Principles of surgical oncology. In: Kudnig S, Séguin B, editors. Veterinary Surgical Oncology. Wiley Blackwell (2012). Retrieved from: www.mazon.com
123. Friedrichs K, Young K. Diagnostic cytopathology in clinical oncology. In: Vail D, Thamm D, Liptak J, editors. Withrow & MacEwen's Small Animal Clinical Oncology. 6th ed. St. Louis, MO: Elsevier (2020). p. 126–45.
124. Sharkey LC, Dial SM, Matz ME. Maximizing the diagnostic value of cytology in small animal practice. Vet Clin North Am Small Animal Pract. (2007) 37:351–72. doi: 10.1016/j.cvsm.2006.11.004
125. DeBerry JD, Norris CR, Samii VF, Griffey SM, Almy FS. Correlation between fine-needle aspiration cytopathology and histopathology of the lung in dogs and cats. J Am Anim Hosp Assoc. (2014) 38:327–36. doi: 10.5326/0380327
126. Ehrhart N. Biopsy and sentinel lymph node mapping principles. In: Vail D, Thamm D, Liptak J, editors. Withrow & MacEwen's Small Animal Clinical Oncology. 6th ed. St. Louis, MO: Elsevier (2020). p. 164–74.
127. Simon MA. Biopsy of musculoskeletal tumors. J Bone Jt Surg. (1982) 64:1253–57. doi: 10.2106/00004623-198264080-00020
128. Guilford W. Gastrointestinal endoscopy. In: Guilford W, Center S, Strombeck D, editors. Strombeck's Small Animal Gastroenterology. 3rd ed. Philadelphia, PA: Saunders. (1995). p. 114–29.
129. Willard M, Schuz K, Hayashi K. Chapter 14: Principles of minimally invasive surgery and imaging of the surgical patient. In: Fossum T, Duprey LP, editors. Small Animal Surgery. 5th ed. Elsevier. Retrieved from: www.Amazon.com
130. Pratschke KM, Ryan J, McAlinden A, McLauchlan G. Pancreatic surgical biopsy in 24 dogs and 19 cats: postoperative complications and clinical relevance of histological findings. J Small Anim Pract. (2015) 56:60–6. doi: 10.1111/jsap.12262
131. de Rycke LMJH, van Bree HJJ, Simoens PJM. Ultrasound-guided tissue-core biopsy of liver, spleen and kidney in normal dogs. Vet Radiol Ultrasound. (1999) 40:294–9. doi: 10.1111/j.1740-8261.1999.tb00364.x
132. Bagley RS. Spinal neoplasms in small animals. Vet Clin North Am Small Animal Pract. (2010) 40:915–27. doi: 10.1016/j.cvsm.2010.05.010
133. Shales CJ, Warren J, Anderson DM, Baines SJ, White RAS. Complications following full-thickness small intestinal biopsy in 66 dogs: a retrospective study. J Small Anim Pract. (2005) 46:317–21. doi: 10.1111/j.1748-5827.2005.tb00326.x
134. Weisse C, Soares N, Beal MW, Steffey MA, Drobatz KJ, Henry CJ. Survival times in dogs with right atrial hemangiosarcoma treated by means of surgical resection with or without adjuvant chemotherapy: 23 cases (1986-2000). J Am Vet Med Assoc. (2005) 226:575–9. doi: 10.2460/javma.2005.226.575
135. Mamelak AN, Owen TJ, Bruyette D. Transsphenoidal surgery using a high definition video telescope for pituitary adenomas in dogs with pituitary dependent hypercortisolism: methods and results. Vet Surg. (2014) 43:369–79. doi: 10.1111/j.1532-950x.2014.12146.x
136. van Rijn SJ, Galac S, Tryfonidou MA, Hesselink JW, Penning LC, Kooistra HS, et al. The influence of pituitary size on outcome after transsphenoidal hypophysectomy in a large cohort of dogs with pituitary-dependent hypercortisolism. J Vet Intern Med. (2016) 30:989–95. doi: 10.1111/jvim.14367
137. Harris BJ, Lourenço BN, Dobson JM, Herrtage ME. Diagnostic accuracy of three biopsy techniques in 117 dogs with intra-nasal neoplasia. J Small Anim Pract. (2014) 55:219–24. doi: 10.1111/jsap.12187
138. Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. (2014) 32:579–86. doi: 10.1200/jco.2012.45.2011
139. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. (2017) 7:805–17. doi: 10.1158/2159-8290.cd-17-0343
140. Nicholson BD, Shinkins B, Pathiraja I, Roberts NW, James TJ, Mallett S, et al. Blood CEA levels for detecting recurrent colorectal cancer. Cochrane Database Syst Rev. (2015) 12:CD011134. doi: 10.1002/14651858.cd011134.pub2
141. Fenton JJ, Weyrich MS, Durbin S, Liu Y, Bang H, Melnikow J. Prostate-specific antigen–based screening for prostate cancer: evidence report and systematic review for the US preventive services task force. JAMA. (2018) 319:1914–31. doi: 10.1001/jama.2018.3712
142. Henderson J, Webber E, Sawaya G. Evidence Synthesis Number 157 Screening for Ovarian Cancer: An Updated Evidence Review for the U.S. Preventive Services Task Force. USPSTF (2018).
143. Zhang J, Chen G, Zhang P, Zhang J, Li X, Gan D, et al. The threshold of alpha-fetoprotein (AFP) for the diagnosis of hepatocellular carcinoma: a systematic review and meta-analysis. PLoS ONE. (2020) 15:e0228857. doi: 10.1371/journal.pone.0228857
144. Bauden M, Pamart D, Ansari D, Herzog M, Eccleston M, Micallef J, et al. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin Epigenetics. (2015) 7:106. doi: 10.1186/s13148-015-0139-4
145. Rahier J-F, Druez A, Faugeras L, Martinet J-P, Géhénot M, Josseaux E, et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin Epigenetics. (2017) 9:53. doi: 10.1186/s13148-017-0351-5
146. Fiala C, Kulasingam V, Diamandis EP. Circulating tumor DNA for early cancer detection. J Appl Lab Med. (2018) 3:300–13. doi: 10.1373/jalm.2018.026393
147. Lawson C, Smith SA, O'Brien M, McMichael M. Neutrophil extracellular traps in plasma from dogs with immune-mediated hemolytic anemia. J Vet Intern Med. (2018) 32:128–34. doi: 10.1111/jvim.14881
148. Letendre J-A, Goggs R. Concentrations of plasma nucleosomes but not cell-free DNA are prognostic in dogs following trauma. Front Vet Sci. (2018) 5:180. doi: 10.3389/fvets.2018.00180
149. Letendre J-A, Goggs R. Determining prognosis in canine sepsis by bedside measurement of cell-free DNA and nucleosomes: cell-free DNA and nucleosomes in canine sepsis. J Vet Emerg Crit Care. (2018) 28:503–11. doi: 10.1111/vec.12773
150. Goggs R. Effect of sample type on plasma concentrations of cell-free DNA and nucleosomes in dogs. Vet Rec Open. (2019) 6:e000357. doi: 10.1136/vetreco-2019-000357
151. Martiny P, Goggs R. Biomarker guided diagnosis of septic peritonitis in dogs. Front Vet Sci. (2019) 6:208. doi: 10.3389/fvets.2019.00208
152. McAnena P, Brown J, Kerin M. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers. (2017) 9:5. doi: 10.3390/cancers9010005
153. Lechowski R, Jagielski D, Hoffmann-Jagielska M, Zmudzka M, Winnicka A. Alpha-fetoprotein in Canine Multicentric Lymphoma. Vet Res Commun. (2002) 26:285–96. doi: 10.1023/a:1016086508286
154. Selting KA, Ringold R, Husbands B, Pithua PO. Thymidine kinase type 1 and C-reactive protein concentrations in dogs with spontaneously occurring cancer. J Vet Intern Med. (2016) 30:1159–66. doi: 10.1111/jvim.13954
155. Grobman M, Outi H, Rindt H, Reinero C. Serum thymidine kinase 1, canine-C-reactive protein, haptoglobin, and vitamin D concentrations in dogs with immune-mediated hemolytic anemia, thrombocytopenia, and polyarthropathy. J Vet Intern Med. (2017) 31:1430–40. doi: 10.1111/jvim.14787
156. Smrkolj T, Gubina B, Bizjak J, Kumer K, Fabjan T, Osredkar J. Tumor marker α-fetoprotein receptor does not discriminate between benign prostatic disease and prostate cancer. Adv Clin Exp Med. (2017) 26:1085–90. doi: 10.17219/acem/65432
157. Bidard F-C, Proudhon C, Pierga J-Y. Circulating tumor cells in breast cancer. Mol Oncol. (2016) 10:418–30. doi: 10.1016/j.molonc.2016.01.001
158. Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BGM, et al. Evaluation of circulating tumor cells and circulating tumor DNA in non–small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin Cancer Res. (2012) 18:2391–401. doi: 10.1158/1078-0432.ccr-11-3148
159. Beinse G, Berger F, Cottu P, Dujaric M -E, Kriegel I, Guilhaume M -N, et al. Circulating tumor cell count and thrombosis in metastatic breast cancer. J Thromb Haemost. (2017) 15:1981–8. doi: 10.1111/jth.13792
160. Politaki E, Agelaki S, Apostolaki S, Hatzidaki D, Strati A, Koinis F, et al. A comparison of three methods for the detection of circulating tumor cells in patients with early and metastatic breast cancer. Cell Physiol Biochem. (2017) 44:594–606. doi: 10.1159/000485115
161. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. (2014) 6:224ra24. doi: 10.1126/scitranslmed.3007094
162. Menarini Silicon Biosystems Inc. CellSearch Circulating Tumor Cell Kit (Epithelial) [Package Insert]. Available online at: https://documents.cellsearchctc.com/pdf/e631600006/e631600006_EN.pdf (accessed January 2, 2021)
163. Marconato L, Facchinetti A, Zanardello C, Rossi E, Vidotto R, Capello K, et al. Detection and prognostic relevance of circulating and disseminated tumour cell in dogs with metastatic mammary carcinoma: a pilot study. Cancers. (2019) 11:163. doi: 10.3390/cancers11020163
164. da Costa A, Oliveira JT, Gärtner F, Kohn B, Gruber AD, Klopfleisch R. Potential markers for detection of circulating canine mammary tumor cells in the peripheral blood. Vet J. (2011) 190:165–8. doi: 10.1016/j.tvjl.2010.09.027
165. Wright T, Brisson BA, Wood GA, Oblak M, Mutsaers AJ, Sabine V, et al. Flow cytometric detection of circulating osteosarcoma cells in dogs. Cytom Part A. (2019) 95:997–1007. doi: 10.1002/cyto.a.23847
166. Sanchez C, Snyder MW, Tanos R, Shendure J, Thierry AR. New insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. Npj Genome Med. (2018) 3:31. doi: 10.1038/s41525-018-0069-0
167. Akter S, Nakao R, Imasato Y, Alam MZ, Katakura K. Potential of cell-free DNA as a screening marker for parasite infections in dog. Genomics. (2018) 111:906–12. doi: 10.1016/j.ygeno.2018.05.020
168. Kustanovich A, Schwartz R, Peretz T, Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol Ther. (2019) 20:1057–67. doi: 10.1080/15384047.2019.1598759
169. Wilson IJ, Burchell RK, Worth AJ, Burton SE, Gedye KR, Clark KJ, et al. Kinetics of plasma cell-free DNA and creatine kinase in a canine model of tissue injury. J Vet Intern Med. (2018) 32:157–64. doi: 10.1111/jvim.14901
170. Chen K, Zhao H, Shi Y, Yang F, Wang LT, Kang G, et al. Perioperative dynamic changes in circulating tumor DNA in patients with lung cancer (DYNAMIC). Clin Cancer Res. (2019) 25:7058–67. doi: 10.1158/1078-0432.ccr-19-1213
171. Mandel P, Metais P. [Nuclear acids in human blood plasma]. Comptes Rendus Séances Soc Biol Fil. (1948) 142:241–3.
172. Bendich A, Wilczok T, Borenfreund E. Circulating DNA as a possible factor in oncogenesis. Science. (1965) 148:374–6. doi: 10.1126/science.148.3668.374
173. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. (1977) 37:646–50.
174. Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. (1989) 46:318–22. doi: 10.1159/000226740
175. Chen XQ, Stroun M, Magnenat J-L, Nicod LP, Kurt A-M, Lyautey J, et al. Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nat Med. (1996) 2:1033–5. doi: 10.1038/nm0996-1033
176. Nawroz H, Koch W, Anker P, Stroun M, Sidransky D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat Med. (1996) 2:1035–7. doi: 10.1038/nm0996-1035
177. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. (2005) 102:16368–73. doi: 10.1073/pnas.0507904102
178. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. (2007) 14:985–90. doi: 10.1038/nm.1789
179. Yung TKF, Chan KCA, Mok TSK, Tong J, To K-F, Lo YMD. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non–small cell lung cancer patients. Clin Cancer Res. (2009) 15:2076–84. doi: 10.1158/1078-0432.ccr-08-2622
180. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. (2012) 486:532–6. doi: 10.1038/nature11156
181. Wan N, Weinberg D, Liu T-Y, Niehaus K, Ariazi EA, Delubac D, et al. Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA. BMC Cancer. (2019) 19:832. doi: 10.1186/s12885-019-6003-8
182. Ulz P, Perakis S, Zhou Q, Moser T, Belic J, Lazzeri I, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun. (2019) 10:4666. doi: 10.1038/s41467-019-12714-4
183. Aravanis AM, Lee M, Klausner RD. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell. (2017) 168:571–4. doi: 10.1016/j.cell.2017.01.030
184. Lo YMD, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. (1997) 350:485–7. doi: 10.1016/s0140-6736(97)02174-0
185. Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. (2011) 13:913–20. doi: 10.1097/gim.0b013e3182368a0e
186. Akolekar R, Beta J, Picciarelli G, Ogilvie C, D'Antonio F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta-analysis. Ultrasound Obst Gyn. (2015) 45:16–26. doi: 10.1002/uog.14636
187. Samura O. Update on noninvasive prenatal testing: a review based on current worldwide research. J Obstet Gynaecol Res. (2020) 46:1246–54. doi: 10.1111/jog.14268
188. Bianchi DW, Chudova D, Sehnert AJ, Bhatt S, Murray K, Prosen TL, et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA. (2015) 314:162–9. doi: 10.1001/jama.2015.7120
189. Dharajiya NG, Grosu DS, Farkas DH, McCullough RM, Almasri E, Sun Y, et al. Incidental detection of maternal neoplasia in noninvasive prenatal testing. Clin Chem. (2018) 64:329–35. doi: 10.1373/clinchem.2017.277517
190. Chan KCA, Woo JKS, King A, Zee BCY, Lam WKJ, Chan SL, et al. Analysis of plasma epstein–barr virus DNA to screen for nasopharyngeal cancer. N Engl J Med. (2017) 377:513–22. doi: 10.1056/nejmoa1701717
191. Beck J, Hennecke S, Bornemann-Kolatzki K, Urnovitz HB, Neumann S, Ströbel P, et al. Genome aberrations in canine mammary carcinomas and their detection in cell-free plasma DNA. PLoS ONE. (2013) 8:e75485. doi: 10.1371/journal.pone.0075485
192. Devall VC, Goggs R, Hansen C, Frye CW, Letendre J, Wakshlag JJ. Serum myoglobin, creatine kinase, and cell-free DNA in endurance sled dogs and sled dogs with clinical rhabdomyolysis. J Vet Emerg Crit Care. (2018) 28:310–6. doi: 10.1111/vec.12731
193. Tagawa M, Tambo N, Maezawa M, Tomihari M, Watanabe K-I, Inokuma H, et al. Quantitative analysis of the BRAF V595E mutation in plasma cell-free DNA from dogs with urothelial carcinoma. PLoS ONE. (2020) 15:e0232365. doi: 10.1371/journal.pone.0232365
194. Schaefer DMW, Forman MA, Kisseberth WC, Lehman AM, Kelbick NT, Harper P, et al. Quantification of plasma DNA as a prognostic indicator in canine lymphoid neoplasia. Vet Comp Oncol. (2007) 5:145–55. doi: 10.1111/j.1476-5829.2007.00122.x
195. Uzuelli JA, Dias-Junior CAC, Izidoro-Toledo TC, Gerlach RF, Tanus-Santos JE. Circulating cell-free DNA levels in plasma increase with severity in experimental acute pulmonary thromboembolism. Clin Chim Acta. (2009) 409:112–6. doi: 10.1016/j.cca.2009.09.011
196. Burnett DL, Cave NJ, Gedye KR, Bridges JP. Investigation of cell-free DNA in canine plasma and its relation to disease. Vet Q. (2016) 36:1–8. doi: 10.1080/01652176.2016.1182230
197. Jeffery U, Ruterbories L, Hanel R, LeVine DN. Cell-free DNA and DNase activity in dogs with immune-mediated hemolytic anemia. J Vet Intern Med. (2017) 31:1441–50. doi: 10.1111/jvim.14808
198. Beffagna G, Sammarco A, Bedin C, Romualdi C, Mainenti M, Mollo A, et al. Circulating cell-free DNA in dogs with mammary tumors: short and long fragments and integrity index. PLoS ONE. (2017) 12:e0169454. doi: 10.1371/journal.pone.0169454
199. Letendre J, Goggs R. Measurement of plasma cell-free DNA concentrations in dogs with sepsis, trauma, and neoplasia. J Vet Emerg Crit Care. (2017) 27:307–14. doi: 10.1111/vec.12592
200. Lee K-H, Shin T-J, Kim W-H, Lee S-Y, Cho J-Y. Methylation of LINE-1 in cell-free DNA serves as a liquid biopsy biomarker for human breast cancers and dog mammary tumors. Sci Rep. (2019) 9:175. doi: 10.1038/s41598-018-36470-5
201. Tagawa M, Shimbo G, Inokuma H, Miyahara K. Quantification of plasma cell-free DNA levels in dogs with various tumors. J Vet Diag Invest. (2019) 31:836–43. doi: 10.1177/1040638719880245
202. Favaro PF, Stewart SD, McDonald BR, Cawley J, Contente-Cuomo T, Wong S, et al. Feasibility and promise of circulating tumor DNA analysis in dogs with naturally-occurring sarcoma. BioRxiv. (2020) 2020.08.20.260349. doi: 10.1101/2020.08.20.260349
203. Prouteau A, Denis JA, Fornel PD, Cadieu E, Derrien T, Kergal C, et al. Circulating tumor DNA is detectable in canine histiocytic sarcoma, oral malignant melanoma, and multicentric lymphoma. Sci Rep. (2021) 11:877. doi: 10.1038/s41598-020-80332-y
204. Chang H-W, Lee SM, Goodman SN, Singer G, Cho SKR, Sokoll LJ, et al. Assessment of plasma DNA levels, allelic imbalance, and CA 125 as diagnostic tests for cancer. J Natl Cancer Inst. (2002) 94:1697–703. doi: 10.1093/jnci/94.22.1697
205. Baylin SB, Jones PA. A decade of exploring the cancer epigenome — biological and translational implications. Nat Rev Cancer. (2011) 11:726–34. doi: 10.1038/nrc3130
206. Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. (2012) 22:407–19. doi: 10.1101/gr.119867.110
207. Sun K, Jiang P, Chan KCA, Wong J, Cheng YKY, Liang RHS, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA. (2015) 112:E5503–12. doi: 10.1073/pnas.1508736112
208. Lo YMD, Chan KCA, Sun H, Chen EZ, Jiang P, Lun FMF, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. (2010) 2:61ra91. doi: 10.1126/scitranslmed.3001720
209. Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA. (2015) 112:E1317–25. doi: 10.1073/pnas.1500076112
210. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. (2016) 164:57–68. doi: 10.1016/j.cell.2015.11.050
211. Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. (2019) 570:385–9. doi: 10.1038/s41586-019-1272-6
212. Jiang P, Sun K, Peng W, Cheng SH, Ni M, Yeung PC, et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov. (2020) 10:664–73. doi: 10.1158/2159-8290.cd-19-0622
213. Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. (2019) 29:418–27. doi: 10.1101/gr.242719.118
214. Chiu RWK, Heitzer E, Lo YMD, Mouliere F, Tsui DWY. Cell-free DNA fragmentomics: the new “omics” on the block. Clin Chem. (2020) 66:1480–4. doi: 10.1093/clinchem/hvaa258
215. Cho H, Mariotto AB, Schwartz LM, Luo J, Woloshin S. When do changes in cancer survival mean progress? The insight from population incidence and mortality. Jnci Monogr. (2014) 2014:187–97. doi: 10.1093/jncimonographs/lgu014
216. Beer TM. Novel blood-based early cancer detection: diagnostics in development. Am J Manage Care. (2020) 26:S292–9. doi: 10.37765/ajmc.2020.88533
217. Brill JV. Screening for cancer: the economic, medical, and psychosocial issues. Am J Manage Care. (2020) 26:S300–6. doi: 10.37765/ajmc.2020.88534
218. Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic — implementation issues and future challenges. Nat Rev Clin Oncol. (2021). doi: 10.1038/s41571-020-00457-x. [Epub ahead of print].
219. Oncology TL. Cancer detection: the quest for a single liquid biopsy for all. Lancet Oncol. (2020) 21:733. doi: 10.1016/s1470-2045(20)30033-4
220. Bradley SH, Barclay ME. “Liquid biopsy” for cancer screening. Bmj. (2021) 372:m4933. doi: 10.1136/bmj.m4933
221. Kalinich M, Haber DA. Cancer detection: seeking signals in blood. Science. (2018) 359:866–7. doi: 10.1126/science.aas9102
222. Penson A, Camacho N, Zheng Y, Varghese AM, Al-Ahmadie H, Razavi P, et al. Development of genome-derived tumor type prediction to inform clinical cancer care. JAMA Oncol. (2020) 6:84–91. doi: 10.1001/jamaoncol.2019.3985
223. Liu ET, Mockus SM. Tumor origins through genomic profiles. JAMA Oncol. (2020) 6:33–4. doi: 10.1001/jamaoncol.2019.3981
224. Pinsky PF. Principles of cancer screening. Surg Clin N Am. (2015) 95:953–66. doi: 10.1016/j.suc.2015.05.009
225. Wu GX, Raz DJ, Brown L, Sun V. Psychological burden associated with lung cancer screening: a systematic review. Clin Lung Cancer. (2016) 17:315–24. doi: 10.1016/j.cllc.2016.03.007
226. Vermeer NCA, Valk MJM, Snijders HS, Vasen HFA, Hoop AG, van der Guicherit OR. Psychological distress and quality of life following positive fecal occult blood testing in colorectal cancer screening. Psycho Oncol. (2020) 29:1084–91. doi: 10.1002/pon.5381
227. Hubbell E, Clarke CA, Aravanis AM, Berg CD. Modeled reductions in late-stage cancer with a multi-cancer early detection test. Cancer Epidemiol Prev Biomarkers. (2020). doi: 10.1158/1055-9965.epi-20-1134. [Epub ahead of print].
228. Kooyker AI, Toes-Zoutendijk E, Winden AWJO, Spaander MCW, Buskermolen M, Vuuren HJ, et al. The second round of the Dutch colorectal cancer screening program: impact of an increased fecal immunochemical test cut-off level on yield of screening. Int J Cancer. (2020) 147:1098–106. doi: 10.1002/ijc.32839
229. Rose NC, Kaimal AJ, Dugoff L, Norton ME, Bulletins—Obstetrics AC of O and GC on P Genetics C on. Screening for fetal chromosomal abnormalities: ACOG practice bulletin summary, number 226. Obstetr Gynecol. (2020) 136:859–67. doi: 10.1097/aog.0000000000004107
230. Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. (2017) 168:584–99. doi: 10.1016/j.cell.2016.12.015
231. Wise HC, Solit DB. Precision oncology: three small steps forward. Cancer Cell. (2019) 35:825–6. doi: 10.1016/j.ccell.2019.05.009
232. American Association for Cancer Research (AACR). AACR Cancer Progress Report 2020. (2020). Available online at: http://www.cancerprogressreportr.org (accessed October 5, 2020)
233. National Cancer Institute (NCI). Targeted Cancer Therapies. NCI (2020). Available online at: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet (acessed October 5, 2020)
234. Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. Jco Precis Oncol. (2017) 2017:1–16. doi: 10.1200/po.17.00011
235. OncoKB. OncoKB Level 1 FDA-Approved Drugs. OncoKB (2020). Available online at: https://www.oncokb.org/actionableGenes#levels=1 (accessed October 5, 2020)
236. US Food & Drug Administration (FDA). Animal Drugs at FDA. FDA. Available online at: https://animaldrugsatfda.fda.gov/ (accessed October 5, 2020)
237. European Medicines Agency (EMA). European Medicines Agency (EMA). EMA (2021). Available online at: https://www.ema.europa.eu/en/medicines (accessed January 29, 2021)
238. London CA, Hannah AL, Zadovoskaya R, Chien MB, Kollias-Baker C, Rosenberg M, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res. (2003) 9:2755–68.
239. Thamm DH, Weishaar KM, Charles JB, Ehrhart EJ. Phosphorylated KIT as a predictor of outcome in canine mast cell tumours treated with toceranib phosphate or vinblastine. Vet Comp Oncol. (2020) 18:169–175. doi: 10.1111/vco.12525
240. US Food & Drug Administration (FDA). The Ins and Outs of Extra-Label Drug Use in Animals: A Resource for Veterinarians. FDA (2020). Available online at: https://www.fda.gov/animal-veterinary/resources-you/ins-and-outs-extra-label-drug-use-animals-resource-veterinarians (accessed October 5, 2020)
241. Londhe P, Gutwillig M, London C. Targeted therapies in veterinary oncology. Vet Clin North Am Small Anim Pract. (2019) 49:917–31. doi: 10.1016/j.cvsm.2019.04.005
242. Cronise KE, Hernandez BG, Gustafson DL, Duval DL. Identifying the ErbB/MAPK signaling cascade as a therapeutic target in canine bladder cancer. Mol Pharmacol. (2019) 96:36–46. doi: 10.1124/mol.119.115808
243. Murray NP. Biomarkers detecting minimal residual disease in solid tumors: what do they mean in the clinical management of patients? Biomark Med. (2019) 13:1535–8. doi: 10.2217/bmm-2019-0401
244. Bork U, Grutzmann R, Rahbari NN, Scholch S, Distler M, Reissfelder C, et al. Prognostic relevance of minimal residual disease in colorectal cancer. World J Gastroenterol. (2014) 20:10296–304. doi: 10.3748/wjg.v20.i30.10296
245. Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. (2019) 16:409–24. doi: 10.1038/s41571-019-0187-3
246. Coakley M, Garcia-Murillas I, Turner NC. Molecular residual disease and adjuvant trial design in solid tumors. Clin Cancer Res. (2019) 25:6026–34. doi: 10.1158/1078-0432.ccr-19-0152
247. Sánchez R, Ayala R, Martínez-López J. Minimal residual disease monitoring with next-generation sequencing methodologies in hematological malignancies. Int J Mol Sci. (2019) 20:2832. doi: 10.3390/ijms20112832
248. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. (2016) 8:346ra92. doi: 10.1126/scitranslmed.aaf6219
249. Nguyen SM, Thamm DH, Vail DM, London CA. Response evaluation criteria for solid tumours in dogs (v1.0): a Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet Comp Oncol. (2015) 13:176–83. doi: 10.1111/vco.12032
250. Vail DM, Michels GM, Khanna C, Selting KA, London CA, Group VCO. Response evaluation criteria for peripheral nodal lymphoma in dogs (v1.0)–a veterinary cooperative oncology group (VCOG) consensus document. Vet Comp Oncol. (2010) 8:28–37. doi: 10.1111/j.1476-5829.2009.00200.x
251. Erasmus JJ, Gladish GW, Broemeling L, Sabloff BS, Truong MT, Herbst RS, et al. Interobserver and intraobserver variability in measurement of non–small-cell carcinoma lung lesions: implications for assessment of tumor response. J Clin Oncol. (2003) 21:2574–82. doi: 10.1200/jco.2003.01.144
252. Alexander K, Joly H, Blond L, D'Anjou M, Nadeau M, Olive J, et al. A comparison of computed tomography, computed radiography, and film-screen radiography for the detection of canine pulmonary nodules. Vet Radiol Ultrasound. (2012) 53:258–65. doi: 10.1111/j.1740-8261.2012.01924.x
253. McErlean A, Panicek DM, Zabor EC, Moskowitz CS, Bitar R, Motzer RJ, et al. Intra- and interobserver variability in CT measurements in oncology. Radiology. (2013) 269:451–9. doi: 10.1148/radiol.13122665
254. Guglielmini C, Toaldo MB, Quinci M, Romito G, Luciani A, Cipone M, et al. Sensitivity, specificity, and interobserver variability of survey thoracic radiography for the detection of heart base masses in dogs. J Am Vet Med Assoc. (2016) 248:1391–8. doi: 10.2460/javma.248.12.1391
255. Leffler AJ, Hostnik ET, Warry EE, Habing GG, Auld DM, Green EM, et al. Canine urinary bladder transitional cell carcinoma tumor volume is dependent on imaging modality and measurement technique. Vet Radiol Ultrasound. (2018) 59:767–76. doi: 10.1111/vru.12652
256. Woo M, Lowe SL, Devane AM, Gimbel RW. Intervention to reduce inter-observer variability in CT measurement of cancer lesions among experienced radiologists. Curr Probl Diagn Radiol. (2020). doi: 10.1067/j.cpradiol.2020.01.008. [Epub ahead of print].
257. Zhou L, Zhang M, Li R, Xue J, Lu Y. Pseudoprogression and hyperprogression in lung cancer: a comprehensive review of literature. J Cancer Res Clin. (2020) 146:3269–79. doi: 10.1007/s00432-020-03360-1.
258. Yekula A, Muralidharan K, Rosh ZS, Youngkin AE, Kang KM, Balaj L, et al. Liquid biopsy strategies to distinguish progression from pseudoprogression and radiation necrosis in glioblastomas. Adv Biosyst. (2020) 4:2000029. doi: 10.1002/adbi.202000029
259. Ma Y, Wang Q, Dong Q, Zhan L, Zhang J. How to differentiate pseudoprogression from true progression in cancer patients treated with immunotherapy. Am J Cancer Res. (2019) 9:1546–53.
260. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. (2015) 7:302ra133. doi: 10.1126/scitranslmed.aab0021
261. Hosoya K, Kisseberth WC, Lord LK, Alvarez FJ, Lara-Garcia A, Kosarek CE, et al. Comparison of COAP and UW-19 protocols for dogs with multicentric lymphoma. J Vet Intern Med. (2007) 21:1355–63. doi: 10.1111/j.1939-1676.2007.tb01959.x
262. Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. (2017) 7:999–1005. doi: 10.1158/2159-8290.cd-17-0146
263. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov. (2017) 7:137–55. doi: 10.1158/2159-8290.cd-16-1123
264. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
265. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
266. Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. (2019) 25:1928–37. doi: 10.1038/s41591-019-0652-7
267. Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA. (2015) 112:9704–9. doi: 10.1073/pnas.1511694112
268. Field NP, Orsini L, Gavish R, Packman W. Role of attachment in response to pet loss. Death Stud. (2009) 33:334–55. doi: 10.1080/07481180802705783
269. Uccheddu S, Cataldo LD, Albertini M, Coren S, Pereira GDG, Haverbeke A, et al. Pet humanisation and related grief: development and validation of a structured questionnaire instrument to evaluate grief in people who have lost a companion dog. Animals. (2019) 9:933. doi: 10.3390/ani9110933
270. Winch G. Why We Need to Take Pet Loss Seriously. Scientific American. (2018). Available online at: https://www.scientificamerican.com/article/why-we-need-to-take-pet-loss-seriously/ (accessed January 29, 2021)
271. Bishop G, Cooney K, Cox S, Downing R, Mitchener K, Shanan A, et al. 2016 AAHA/IAAHPC End-of-Life Care Guidelines*. J Am Anim Hosp Assoc. (2016) 52:341–56. doi: 10.5326/jaaha-ms-6637
272. Hetts S, Lagoni L. The owner of the pet with cancer. Vet Clin North Am Small Animal Pract. (1990) 20:879–96. doi: 10.1016/s0195-5616(90)50076-5
273. Butler CL, Lagoni L, Dickinson KL, Withrow SJ. Animal illness and human emotion. Probl Vet Med. (1991) 3:21–37.
274. American Veterinary Medical Association (AVMA). Veterinarian's Oath. AVMA Policies. (2021) Available online at: https://www.avma.org/resources-tools/avma-policies/veterinarians-oath (accessed January 29, 2021)
Keywords: dog, cfDNA, cell-free DNA, circulating tumor DNA, cancer, genomic, liquid biopsy, one health
Citation: Chibuk J, Flory A, Kruglyak KM, Leibman N, Nahama A, Dharajiya N, van den Boom D, Jensen TJ, Friedman JS, Shen MR, Clemente-Vicario F, Chorny I, Tynan JA, Lytle KM, Holtvoigt LE, Murtaza M, Diaz Jr. LA, Tsui DWY and Grosu DS (2021) Horizons in Veterinary Precision Oncology: Fundamentals of Cancer Genomics and Applications of Liquid Biopsy for the Detection, Characterization, and Management of Cancer in Dogs. Front. Vet. Sci. 8:664718. doi: 10.3389/fvets.2021.664718
Received: 05 February 2021; Accepted: 23 February 2021;
Published: 23 March 2021.
Edited by:
Carlos Eduardo Fonseca-Alves, Paulista University, BrazilReviewed by:
David John Argyle, University of Edinburgh, United KingdomCopyright © 2021 Chibuk, Flory, Kruglyak, Leibman, Nahama, Dharajiya, van den Boom, Jensen, Friedman, Shen, Clemente-Vicario, Chorny, Tynan, Lytle, Holtvoigt, Murtaza, Diaz Jr., Tsui and Grosu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason Chibuk, amNoaWJ1a0BwZXRkeC5jb20=; Andi Flory, YWZsb3J5QHBldGR4LmNvbQ==; Dana W. Y. Tsui, ZHRzdWlAcGV0ZHguY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.