 Herbert F. Jelinek1,2,3
Herbert F. Jelinek1,2,3 Mira Mousa4
Mira Mousa4 Eman Alefishat1,5,6Wael Osman7
Eman Alefishat1,5,6Wael Osman7 Ian Spence8
Ian Spence8 Dengpan Bu9
Dengpan Bu9 Samuel F. Feng1,10Jason Byrd11Paola A. Magni12,13
Samuel F. Feng1,10Jason Byrd11Paola A. Magni12,13 Shafi Sahibzada14Guan K. Tay1,15,16
Shafi Sahibzada14Guan K. Tay1,15,16 Habiba S. Alsafar1,2,17*
Habiba S. Alsafar1,2,17*- 1Center for Biotechnology, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 2Department of Biomedical Engineering, College of Engineering, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 3Center of Heath Engineering Innovation, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 4Nuffield Department of Women's and Reproduction Health, Oxford University, Oxford, United Kingdom
- 5Department of Pharmacology, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 6Department of Biopharmaceutics and Clinical Pharmacy, School of Pharmacy, The University of Jordan, Amman, Jordan
- 7Department of Chemistry, College of Arts and Sciences, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 8Discipline of Pharmacology, University of Sydney, Sydney, NSW, Australia
- 9State Key Laboratory of Animal Nutrition, Institute of Animal Science, Chinese Academy of Agricultural Science, Beijing, China
- 10Department of Mathematics, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 11Department of Pathology, Immunology and Laboratory Medicine, University of Florida, Gainesville, FL, United States
- 12Discipline of Medical, Molecular and Forensic Sciences, Murdoch University, Murdoch, WA, Australia
- 13Murdoch University Singapore, King's Centre, Singapore, Singapore
- 14Antimicrobial Resistance and Infectious Diseases Laboratory, College of Science, Health, Engineering and Education, Murdoch University, Murdoch, WA, Australia
- 15Division of Psychiatry, Faculty of Health and Medical Sciences, The University of Western Australia, Crawley, WA, Australia
- 16School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia
- 17Department of Genetics and Molecular Biology, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
Coronavirus infections have been a part of the animal kingdom for millennia. The difference emerging in the twenty-first century is that a greater number of novel coronaviruses are being discovered primarily due to more advanced technology and that a greater number can be transmitted to humans, either directly or via an intermediate host. This has a range of effects from annual infections that are mild to full-blown pandemics. This review compares the zoonotic potential and relationship between MERS, SARS-CoV, and SARS-CoV-2. The role of bats as possible host species and possible intermediate hosts including pangolins, civets, mink, birds, and other mammals are discussed with reference to mutations of the viral genome affecting zoonosis. Ecological, social, cultural, and environmental factors that may play a role in zoonotic transmission are considered with reference to SARS-CoV, MERS, and SARS-CoV-2 and possible future zoonotic events.
Introduction
The emergence of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the virus responsible for the COVID-19 pandemic, has focused attention on the phenomenon of zoonosis. Zoonoses, as defined by the World Health Organization (WHO), are diseases and infections which are naturally transmitted between vertebrate animals and humans. The challenge with emerging zoonoses, such as SARS-CoV-2, is to establish the origin and mechanism(s) of transmission of the new disease. Viral mutation and recombination, viral host physiology and immune response, ecogeography, and human factors including ACE2 receptor structure and immune function have all been proposed as possible mechanisms (1, 2) (Figure 1).
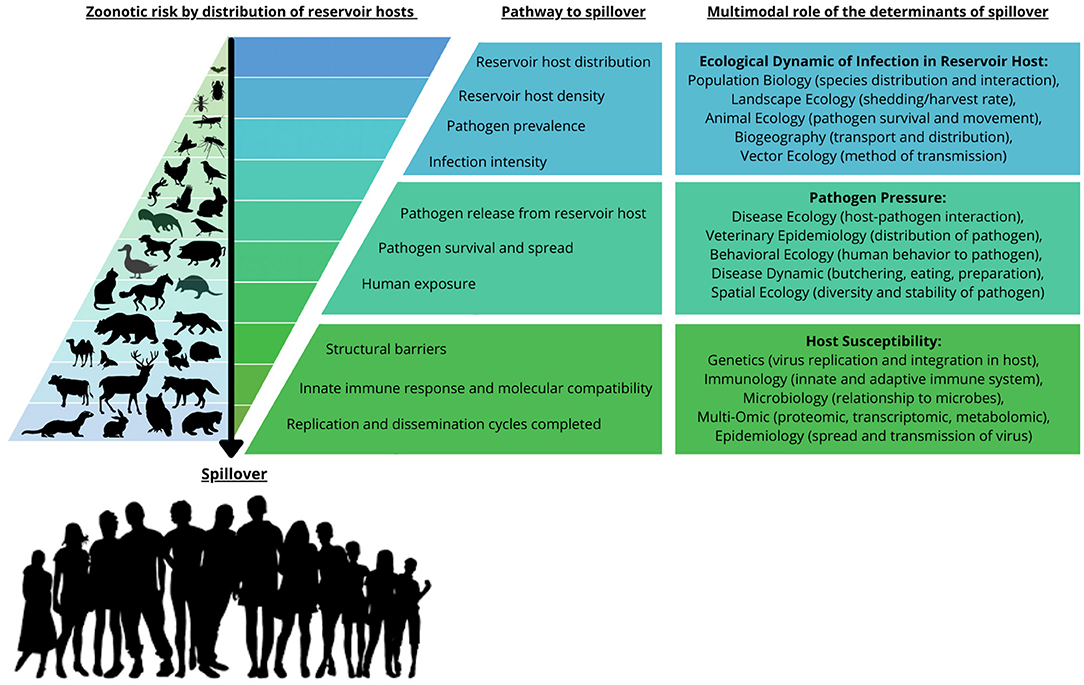
Figure 1. Zoonotic risk distribution, pathway to spillover, and the multimodal role of the determinants of spillover. The zoonotic risk is demonstrated by the accumulated distribution of reservoir hosts and vectors that play a role in the pathway to spillover. The risk of spillover is determined by a series of processes from the ecological dynamics of reservoir host distribution and density, to the susceptibility, replication and dissemination of the biological factors in the recipient host. This is also reflected in the multimodal role of the determinants of spillover, demonstrating the disciplines that are being used to study zoonotic transmission and the determinants of spillover. This figure was adapted from Figure 1 in Plowright et al. (2). Copyright permission was obtained with license number 55218980848529.
Viruses account for ~25–44% of all emerging infectious diseases and present an increased risk of zoonotic transmission (3–5). Zoonotic viral transmission has been well established in the links between human immunodeficiency virus-1 (HIV-1) and simian immunodeficiency viruses (SIVs) from chimpanzees and gorillas in West Central Africa, and HIV-2 and SIVs from sooty mangabeys (6). Zoonotic virus to human transmission and the number of viral-based infections has been steadily increasing, prompting a worldwide investigation of potential zoonotic pathogens, not only to understand current zoonoses but, more importantly, to also identify potential future transmissions of viral pathogens between animals and humans (4, 5). The emergence of SARS-CoV, Middle East Respiratory Syndrome (MERS-CoV), and SARS-CoV-2, which are all highly pathogenic, is the end-point in a sequence of events involving viral evolution and ecology (7). Mutation, recombination, genetic drift, and other evolutionary mechanisms, combined with ecological pressures and opportunities, allow the viruses to cross several species barriers and overcome immune responses in the new hosts. However, the origin and the evolutionary changes of the SARS-CoV-2 virus in humans are currently unknown.
Evidence from other viral zoonoses suggests an origin in bats via possibly an intermediate host (8, 9). This review will examine the origin of SARS-CoV-2 in the context of possible zoonotic transmission, evolution, ecological factors, and the role of reservoir species in other coronaviruses (10).
Coronavirus Characteristics
Coronaviruses are members of the family Coronaviridae, order Nidovirales. The Coronaviridae family is further subdivided into the Torovirinae and Coronavirinae subfamilies. Coronaviridae have a well-conserved genomic organization and a substantial number of nonstructural genes, as well as unique enzymatic activities (3, 11). The sub-family of Coronavirinae contains four genera: alphacoronavirus (α-CoV) and betacoronavirus (β-CoV), which infect mammals, and gammacoronavirus (γ-CoV) and deltacoronavirus (δ-CoV), which principally infect birds (12, 13). Coronaviruses (CoVs) are large enveloped positive-sense single-stranded (+)ssRNA viruses. Genomes of CoVs range from 25 to 32 kilobases (kb), and reproduction of this genome involves a unique transcription mechanism (14). CoV genomes contain five open reading frames (ORFs) encoding for the replicase protein 1a/1b nucleocapsid (N), spike (S), membrane (M), and envelope (E) proteins essential for the replication of the virus. CoVs also contain a variable number of ORFs coding for accessory proteins, not essential for replication but with possible roles in pathogenesis (15, 16).
SARS-CoV-2 belongs to the genus betacoronavirus (β-CoV). GenBank sequences of β-CoVs from the first patients in the Chinese city of Wuhan, who visited the seafood market, indicated that bat-SL-CoVZC45 and bat-SL-CoVZXC21 collected in 2018 in Zhoushan, eastern China, have the highest sequence identities (~90% similarity) with SARS-CoV-2. When gene regions were analyzed individually, the S gene (Spike protein) had the lowest sequence identity (75%), and the E gene the highest (98.7%). Significantly, the SARS-CoV-2 strains in the initial Chinese study of nine patients had a sequence identity of only 79% with SARS-CoV and ~50% with MERS-CoV (17). These findings suggest that SARS-CoV-2 is a distinct β-CoV within the Coronaviridae family (18). However, SARS-CoV-2 does share a similar ACE2-binding domain with only a few amino acid differences from SARS-CoV (19, 20). Later sequencing of SARS-CoV-2 from Taiwan samples indicated further differences from SARS-CoV, especially in the ORF8 loci (21).
Within the Coronavirinae subfamily, different host species are involved with different transmission profiles, and not all have demonstrated zoonotic transmission to humans at this time. Of the seven CoVs which have transferred to humans, two (HCoV-229E and HCoV-NL63) are α-CoVs (22, 23). HCoV-OC43, HCoV-HKU1 (24, 25), SARS-CoV, MERS-CoV (26–29), and SARS-CoV-2 are β-CoVs (30, 31). Infections with HCoV-229E, HCoV-OC43, HCoV-HKU1, and HCoV-NL63 are usually self-limiting with mild symptoms (5, 25, 32–36). Although HCoV-OC43 infection is usually mild, more severe pathologies, including fatal pulmonary infections and lethal encephalitis, have been reported (37). SARS-CoV-2 does not have any close sequence similarity to HCoV-229E, HCoV-OC43, HCoV-HKU1, or HCoV-NL63 but is antigenically related to SARS-CoV (38, 39).
Entry to host cells by CoVs, including SARS-CoV, MERS, and SARS-CoV-2, is mediated via the spike protein (40). The S1 subunit of this protein binds to a receptor on the host cell surface, and the S2 subunit fuses the viral and host membranes (41). An important property of CoVs is the diversity in receptors recognized by the S1 protein in different viruses. These include zinc peptidases angiotensin-converting enzyme 2 (ACE2) (SARS-CoV; SARS-CoV-2; HCoV-NL63), aminopeptidase N (APN) (HCoV-229E), the serine peptidase dipeptidyl peptidase 4 (DPP4) (MERS-CoV), 9-O-acetylated sialic acid (9-O-Ac-Sia) (HCoV-HKU1; HCoV-OC43), a cell adhesion molecule carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) as is the case with mouse hepatitis virus (MHV), and an angiotensin II receptor type 2 (AGTR2) possibly with SARS-CoV-2 (42–45). Other species use sugars as receptors or co-receptors (41). Apart from the ACE2 receptor-binding site for virus entry, TMPRSS2 is another key protein required for viral entry into human tissue (46–50).
Some spike proteins may also have different access routes to cells of possible host species. PDF2180-CoV, a MERS-like virus found in a Ugandan bat, does not use the DPP4 receptor for cell entry but requires proteolytic cleavage using possibly gastrointestinal tract (GIT) released trypsin; it, then, may enter via the digestive tract rather than via the respiratory system (51). The MERS DPP4-binding site has also been shown to have natural polymorphisms (K267E, K267N, A291P, and Δ346-348) that reduce cell entry capacity of the virus and two polymorphisms (K267E and A291P) that reduce viral replication (52). SARS-CoV-2, HCoV-OC43, MERS-CoV, and MHV-A59 are characterized by a furin-like cleavage site that may be important in virus cell entry since furin is found in large amounts in the lung (53, 54). In addition, potential routes of transmission of SARS-CoV-2, including entry via oral, sexual, and ocular pathways, cannot be discounted due to the wide spread of the ACE2 receptor in different tissues and organs (55, 56). MERS-CoV has already been shown to have multiple transmission and infection options by entering cells within endosomes or at the plasma membrane, depending on the location of the host proteases that cleave the viral spike protein to allow membrane fusion (57).
Coronavirus Evolution
Evolution-associated genotypic and phenotypic changes are a function of complex regulatory cellular, organismic, environmental, and geographical interactions (58). Coronaviruses and other zoonotic viruses are genetically heterogeneous while in prehuman hosts, such as bats, but are often poorly adapted for zoonotic transmission from their initial reservoir pool. This is due to physiological differences between host species, and hence requires significant evolutionary adaptation, which is often a co-evolutionary process (59, 60). Evolutionary changes associated with mutations and recombination, as well as genetic drift or pleiotropy, play an essential role in the adaptation of viruses to new hosts. These changes may keep viruses not only in the host population but also determine their virulence as seen in the adaptive evolution of the S glycoprotein of bovine coronavirus and human coronavirus (HCoV) OC43 using phylogenetic and phylodynamic analysis (61–63).
For successful transmission to a new species, viruses must adapt to cell surface receptors to bind successfully to the host receptor, escape host immune surveillance and response, and ensure further transmission by the host (64). Successful viral cross-species transmission and infection depend on rapid evolution accomplished by high mutation rates, recombination, and assortment and how closely related possible host species are (17, 65–68). Mutations that favor more efficient host entry, optimize virus replication, and decrease susceptibility to host immune responses, will increase virulence and maximize transmission potential either within the host species or between hosts (69–71). Findings of previous cross-species transmission has identified the importance of tracking viral mutations in the new host, in SARS-CoV-2 mutations to the receptor-binding domain changes virus virulence, and either natural immune response or vaccine efficacy (72).
Modification of the CoV genome and associated structural and non-structural proteins contribute to successful zoonoses (73–76). These genomic changes are further enhanced by geographical spread (77). Mutation rates in viruses are generally much higher (by a factor of ~106) (78) than those of their hosts but vary for different viruses by several orders of magnitude (79). Estimated mutation rates in CoVs are moderately higher than others in the ssRNA virus category (78). In addition, SARS-CoV and SARS-CoV-2 may have evolved rapidly due to different parts of the genome mutating at different rates leading to a possible increase in phenotypic diversity (80, 81). Mutations may also lead to viral resistance to host immune reactions and resistance to antiviral drugs (82, 83). Forty-seven SNPs (point mutations) that may affect virulence have been identified in samples of SARS-CoV-2 from 12 countries from which complete genome sequencing data were available (84). Mutations in the sequence of the spike protein have led to the great versatility of viral receptor binding strategies, which characterizes the SARS-CoV and SARS-CoV-2 spike proteins. This versatility may underlie their high, yet different, affinities to the human ACE2 receptor compared with that of the spike protein in Bat-CoV and affect virulence and transmission (82, 85–87). However, mutations also lead to disparity in fatality rates in different countries following SARS-CoV-2 infection, which has been linked to the D614G substitution in the SARS-CoV-2 spike (S) protein suggesting changes in pathogenicity and transmission associated with specific mutations (21, 88–90).
Major drivers of CoV evolution also include mutation rate, viral mutation capacity, and recombination, influenced by environmental changes, as well as surface glycoprotein plasticity often promoted by host-parasite interaction without reducing virulence (91, 92). Polymorphisms of specific genes, however, do not only change the possible phenotypic expression of the gene but may also influence other genes and their expression. Genetic polymorphisms that affect host phenotype upon infection have been reported for HIV-1, hepatitis C, dengue, influenza A virus infections, and other types of parasites (93, 94). These studies, as well as plant studies, suggest that viruses may have both virulence and avirulence genes that map to susceptibility and resistance genes in the host (94). Improved immunity, for instance, has been found for the cellular co-receptor CCR5, which prevents HIV-1 from entering the cell, leading to resistance against HIV-1 in some individuals (95). Similarly, the human leukocyte antigen (HLA) polymorphisms may be associated with resistance or susceptibility to viral infection (96). SARS-CoV and SARS-CoV-2 both enter cells via the ACE2 receptor and access to or ability to bind to ACE2 may be associated with zoonotic transmission potential to humans (60, 97). This is particularly interesting as the ACE2 gene is located on the X-chromosome; hence, females may potentially be heterozygous with different susceptibility to males who are hemizygous (98). ACE1 is a similar receptor with a similar function; it is characterized by an insertion (I)/deletion (D) polymorphism where the D allele is associated with reduced expression of ACE2 and therefore may confer some resistance to SARS-type viruses by changes in the ACE1/ACE2 balance (98–101). Similarly, two mutations affecting the non-structural protein 6 (NSP6) and the ORF 10 may lead to a lower virulence and pathogenicity of SARS-CoV-2 due to a decrease in protein stability with similar mutations within the receptor-binding domain (RBD) required for retaining viral strength and virulence (102, 103). A recent study also identified a novel SARS-CoV-2 mutation in the RBD furin-associated cleavage site that may lead to a decrease in virulence and transmission and is associated with the sequence of ZJ01 (BataCov/Zhejiang/ZJ01/2019) in a mild case of COVID-19 reported in China (104). Novel CoVs have been identified in bats in Vietnam (105) and Mexico, where 13 distinct CoVs (nine α-CoVs; four β-CoVs) were identified, 12 of which were novel (106). These observations highlighted the fact that bats may not only carry multiple viruses but harbor distinct CoVs across different geographical regions. Sequence homology between the novel and known viruses underscores the importance of identifying the viral reservoirs in different bat families (42).
The evolution of the virus does not occur in isolation but is accompanied by alterations in the host. Host evolution and ecology associated with genetic mutations and recombination events provide the necessary host factors that need to occur in parallel with viral evolution (107–109). For effective zoonoses and the ability to establish in a new host, several factors need to come together, including the emergence of a founder virus that possesses a modification and allows more efficient infection/transmission in the new host, as well as host alterations. For example, bats have to have a specific immune response that allows them to survive following viral infection, and not affect virus survival, viral load, or viral spill over. In bats, this may be associated with the type I interferon and interferon-associated antiviral activity (5, 110, 111). These evolutionary adaptations are linked to damping of cytoplasmic DNA through the loss of PYHIN genes and a regulatory site mutation in STING, which leads to changes in the inflammatory response in association with a change in TNFα, IL-10, IL-1β, and IL-18, and IFN-α levels (112, 113). Recent studies have also shown that frequency and synchronization of the reproduction cycle of bats can influence the prevalence and persistence of viruses. The Egyptian fruit bat, Rousettus aegyptiacus E. Geoffroy, tested in Africa indicated two peak periods of horizontal transmission throughout the year and coincidental increase in human infections of Marburg virus, which is carried by these bats (114).
Persistence in the new host requires further genetic modifications. Mechanisms including repressor-operon function in gene action, the role of viruses incorporated as extrachromosomal elements, recombination, reassortment, codon bias, hypermutations, and epigenetics in host evolution all play a role in either resistance or susceptibility in viral infections including zoonoses (115, 116). In particular, epigenetic modifications by viruses, including CoVs, such as histone modifications, may influence host viral susceptibility (117). In this regard, following the zoonotic introduction of HCoV-OC43 to humans, the hemagglutinin-esterase protein required for binding to the 9-O-acetylated sialic acid as receptor was selected against contributing to the persistence of the virus in the human population (118). In addition, rapid recombination and mutations have led to a diversification of HCoV-OC43, with seven genotypes identified. In contrast, bovine coronaviruses (BoCoV) exhibit relative genetic stability that may be similar to porcine hemagglutinating encephalomyelitis virus (PHEV) (119–126).
In the case of SARS-CoV-2, the large ssRNA genome provides the virus with a multitude of pathways for adaptation-associated base substitutions and deletions and contributes to altering host gene expression and counteracting the host immune responses (127, 128). The previous pandemic associated with SARS-CoV did not identify a direct evolutionary pathway toward zoonotic transmission as no evidence has been found for mutations from a known coronavirus, nor does it seem to be a recombination or mutation of any known CoVs suggesting that SARS-CoV possibly spilled over into an intermediary, unidentified animal, bird, or reptile host prior to infecting humans (129, 130). A similar lack of identification of an intermediary host for SARS-CoV-2 is the case.
Ecological Factors And The Role Of Reservoir Species
Zoonoses depend on the relationship between the virus and a reservoir host or hosts. Both the ecology of the virus and the ecology of the host(s) play roles in creating the potential for zoonotic transmission. Viral ecology is concerned with the interactions between the virus and its host or hosts but broader ecological factors affecting the host(s) directly play a role in the ecology of viruses. Viral ecology is an important link in the process of zoonoses as ecological factors can facilitate or inhibit virus spillover (131, 132). Comparisons of viral sequence data with demographic and geographic sampling location data can assist in our understanding of current and potential zoonoses (133). Animal to human transmission opportunities have been increasing in line with ecological changes worldwide (134–139). Zoonotic transmission risk is higher where population growth has had the greatest effect on land-use, and biodiversity is high (77, 140, 141). A global heat map from 2017 indicated that China, the Indian subcontinent, and the Himalayan region are hot spots with an increased risk of future zoonotic events (77). Although viruses play a role in broader environmental ecology, this discussion will focus on examining interactions of CoVs with identified host species and the types of transmission cycles that have evolved with respect to zoonoses.
Recent studies have indicated that viral diversity is reflective of the number of natural host species. Rodentia (rodents) and Chiroptera (bats) contain the most species among mammals (142). With over 1,400 bat species, bats are one of the most common mammals on the planet and have been identified as being likely associated with zoonosis (143). Bats form an important part of many ecosystems by regulating possible crop pest outbreaks, seed dispersal, and pollination, as well as soil fertilization, and as a food source are a major intermediate host of coronavirus species (144). The islands of the West Indian Ocean, which are home to 50 of the 1,411 known bat species provide a suitable environment for the co-evolution of hosts and viruses (145). Approximately 9% of the bat species in the West Indies tested positive for coronavirus of which nearly half were members of the α- and β-CoVs subfamilies. Evidence of co-evolution was shown with limited species switching, possibly due to habitat separation. The potential for spillover in this environment, however, remains to be investigated (144).
Viral transmission to humans may occur through the consumption of reservoir species for food or medicinal use of animal products from reservoir species as well as other commercial enterprises. Transmission of MERS-CoV, where the only identified host species are Dromedary camels, has been attributed to consumption of unpasteurized camel milk, raw meat, or medicinal use of camel urine, as well as proximity to the animals (146). Ecological factors thus facilitate the persistence and the frequent transmission of a virus in the case of MERS and dromedary camels (147). Consumption of wild-caught animals may not be necessary for transmission to humans as simple proximity to the host species appears to be sufficient for transmission in the case of SARS-CoV from palm civets and mink (148, 149). Other potential sources of contact between humans and wild animals occur when these animals are used for entertainment, as is the case for performing monkeys in Indonesia or zoos (150, 151). Thus, behavioral and cultural factors, which alter the probability of interactions between humans and animals have increased the possibility of cross-species infections, which may lead to more zoonotic events (66, 131, 142, 152–154).
Dispersal and host spillover dynamics are a function of animal ecology. Bats, birds, and especially migrating birds, which are hosts of CoVs, provide an opportunity for dispersal and spillover across large distances, continents, and host species. Feeding habits in airborne animals are constrained by the aerodynamics of flight and associated energy requirements and location of food sources as well as geography. Bats may either be frugivorous or insectivorous, and the fecal contents of bats may be ingested by ground-foraging animals, leading to transmission of the virus (155). An interesting corollary is that flight also leads to greater energy expenditure and higher body temperatures in bats, which has required viruses to adapt and led to a greater diversity of zoonotic viruses in bats (156, 157). The adaptation as part of a co-evolution may then have led to better resistance to inflammatory reactions, as part of the innate immune response in spillover species such as humans (158).
Warm-blooded flying vertebrates appear to be ideal hosts for coronaviruses, bats for alpha and beta CoVs and birds for delta and gamma CoVs (159). Migratory birds are known to have led to transfection of the West Nile virus, tick-borne encephalitis virus, influenza A virus (IAV), Newcastle disease virus (NDV), and H5Nx avian influenza viruses to humans (160, 161).
Bat ecogeographical characteristics, including food choices, population dynamics, territorial range, migration patterns, a long-life span, virus immunity, and the human-led changes in habitat that brings bats in more contact with humans, lead to bats being an ideal candidate to spread viruses to humans. Viruses with a diverse host range and host plasticity are also more likely to amplify viral spillover by secondary human-to-human transmission (31, 162–164). Shifts in the migratory behavior of species can alter the risk of infection for wildlife, domestic animals, and humans. These shifts in migratory behavior occur in conjunction with changes in food availability due to natural ecological change, agriculture, or feed supplementation (165). An increased risk of infection may occur in smaller regional areas if animals become sedentary and long migrations do not cull infected hosts but reduce geographical infection spread. Climate change and deforestation have the potential to cause many such ecological changes in the immediate future with increased risk of zoonotic events. Habitat deterioration, which may have detrimental effects on the health of the reservoir species, is an important adjunct to viral spillover probability, leading to higher susceptibility to pathogens and increases in shedding of virus (166). The extent that habitat deterioration or human activity affects bat species health is quite specific and depends on the roost type and human activity with respect to the roost type (167).
Reservoir Species
A factor permitting viruses to persist in one host and, possibly, to cross to another is the presence in different hosts of common receptors. For movement across species, host evolution and ecology associated with genetic mutations and recombination events are the necessary host factors that need to occur in parallel with viral evolution (107–109). The cross-species infection can take several forms, but a common model is the spillover of the virus from bats through undigested food. Bats mainly break down food to obtain the high-energy sugars and spit out the remaining fruit which is consumed by other species including insects, which are a potential intermediary transmission vector depending on the bat species (168). Direct human transmission may also occur through eating bats and the use of bat feces for medicinal purposes (66, 169).
Studies from China in the last 15 years have shown that bats are the natural reservoir of a range of CoVs, including SARS-CoV and SARS-like CoVs, found in Rhinolophus macrotis Blythe, Rhinolophus ferrumequinum Schreber, Rhinolophus pearsoni Horsfield, Rhinolophus sinicus Andersen, Pipistrellus abramus Temminck, Pipistrellus pipistrellus Schreber, Tylonycteris pachypus Temminck, Myotis ricketti Thomas, and Scotophilus kuhlii Leach (136, 170–173). However, despite the high numbers of bat species harboring diverse CoVs, any direct link between these viruses and their bat hosts with viruses isolated from human samples is lacking. Several possibilities exist including host shifts between closely related hosts and/or viruses found in host species. Preadaptation of a virus strain to overcome the immune response of a new host is a mechanism that allows spill-over. In addition, a previrulent virus entry into a new host can allow mutations in the new host that eventually lead to increased virulence. These mechanisms are further outlined here with respect to reported studies of coronaviruses. From phylogenetic association studies, results show that for instance there is no strict match between Rhinolophid bats and their association with CoVs, suggesting that interspecies transmission followed by establishment and long-term persistence in the new host species have occurred during the evolutionary history of the virus rather than the current virus genome establishing in the human population (31, 119, 120). Host shifts within Rhinolophid bats have been identified in all bats harboring two distinct lineages of CoVs, except for R. macrotis (119, 135). This indicates that there is a divergence in behavior and ecogeography of the close phylogenetic relationship between viruses harbored by M. ricketti and S. kuhlii and reflects the similar behavior and ecology of the R. ferrumequinum, R. pearsoni, and R. sinicus hosts. Novel bat species have also been recently reported harboring CoVs being Myotis (Myotis pequinius Thomas) and Fringed long-footed Myotis (Myotis fimbriatus Peters). In addition, the genotypes of lineage C, β-CoV in Fringed long-footed Myotis (M. fimbriatus), and common serotine (Eptesicus serotinus Schreber) in China have a nucleotide similarity of 85–88% with MERS-CoV (124). Since viruses have probably evolved more recently than their host bats, the apparent co-evolutionary patterns could be due to a host shift that would allow viruses to undergo preadaptation against the immune responses of the host. This mechanism may explain the movement of SARS-like CoVs between hosts (121, 174).
Interspecies spill-over and viral recombination may lead to eventual zoonotic events that can also be viewed from the receptor binding domain of the virus with the ACE2 receptor perspective. Recent data suggests that the RBD for ACE2 may be an ancestral trait of SARS-CoV-2 and not a result of a recent recombinant event. Thus, SARS-CoV-2 may have diverged from its ancestral bat sarbecovirus reservoir sometime between 1948 and 1982 and circulated in bats for some time (175). Intriguing is the proposition that RaTG13 CoV with a high nucleotide sequence identity with the Spike protein of SARS-CoV-2 may not be the historic ancestor, as suggested by some reports but that the RBD was acquired from the MP789 pangolin CoV by recombination (176).
MERS-CoV
MERS-CoV, which causes respiratory-type illness but differing from that caused by SARS-related coronavirus (SARSr-CoV) was isolated in 2012 (27, 177). MERS-CoV replicates in cell lines from Rousettus, Rhinolophus, Pipistrellus, Myotis, and Carollia bats as well as pigs, substantiating that MERS-CoV uses a different receptor to ACE2 that is conserved in bats, pigs, and humans, demonstrating a continued strong zoonotic potential (178, 179). Phylogenetic characterization has placed MERS-CoV into the β-CoV group (180, 181). No animal reservoir for MERS-CoV has been identified. The closest relatives of MERS-CoV occur in a bat species Neoromicia zuluensis Roberts (182–184). It is closely related to the Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5 (27, 159). The closest viral relative to MERS-CoV sequence identity with MERS-CoV EMC/2012 (MERS coronavirus Erasmus Medical Center/2012) isolated in the infected person in Saudi Arabia has been reported for Taphozous perforatus E. Geoffroy (Egyptian Tomb bats), suggesting a possible regional host reservoir (182, 185). Studies of the evolutionary origins of MERS-CoV link Pipistrellus cf. hesperidus (strain PREDICT/PDF-2180) from the bat family Vespertilionidae as a possible evolutionary ancestor for MERS-like CoV, indicating a 30% homology between the MERS-CoV S protein antigenic sites and HKU4, but a 70% homology with HKU5 bat-CoV. A 100% homology between MERS-CoV and both HKU4 and HKU5 was found for the E, M, and N protein antigenic sites (157, 186, 187). Cross-reactive antibodies to MERS-CoV have been detected in dromedary camels, which have been suggested as an intermediary host, in Oman and the Canary Islands, but no virus was detected in these animals (188, 189). Another possibility has been suggested by Zhang et al. that at least in Kenya, co-infection with MERS-CoV and a HKU8r-CoV in camels occurred, leading to the possibility of recombination events between a precursor virus of MERS-CoV and HKU8r-CoV (190). Apart from dromedary camels, other new-world camelids, including alpacas, llamas, and Bactrian camels, are also at risk of MERS-CoV infection. The DPP4 receptor in Bactrian camels has a 98.8% sequence identity with the dromedary camel DPP4 receptor (191).
The precise mode of transmission for MERS-CoV, therefore, remains uncertain due to differences in the viral genome reported from different geographic regions (110, 192–198), including differences between the original African MERS-CoV and the current Saudi Arabian MERS-CoV clade (29, 199–201). MERS-CoV differs from the closely related bat viruses in binding to the DPP4 receptor, also referred to as CD26 cellular receptor (202, 203) found in humans, dromedaries, and pigs (204–206). The potential of MERS-CoV to undergo rapid alterations in the surface charge of the spike protein enhances viral entry and demonstrated that MERS-CoV utilizes multiple paths to adapt to changes in the receptor sites of host species (207–209). A study of naturally occurring DPP4 protein variants indicated that four (K267E, K267N, A291P, and Δ346-348) reduced binding and cell entry of MERS-CoV and two polymorphisms (K267E and A291P) attenuated viral replication (52). Whether human DPP4 variants differ in geographical location or between ethnic groups remains to be investigated and may shed a better light on why MERS-CoV infection in the African continent is less than in the Middle East. An alternative explanation of the low zoonotic potential of MERS-CoV to humans may be related to the level of human DPP4 receptor expression (210). In contrast, viral escape mutations as an adaptive mechanism toward neutralizing antibodies in humans have been shown to most often lead to a reduction in MERS-CoV fitness and DPP4 binding (211). Similar mutations and recombination events are also characteristic of SARS-CoV and SARS-CoV-2 (1, 11, 30, 212). The mechanisms that lead to a balance between enhanced transmissibility associated with the rapid mutations observed in the Spike protein gene to DPP4 variants and adaptive mechanisms, for neutralizing host immune response that lead to a reduction in viral fitness, has yet to be elucidated (213).
SARS-CoV
SARS-CoV, discovered in 2003 in association with patients being admitted to hospitals with severe acute respiratory syndrome obtains human cell entry through the ACE2 receptor (214). SARS-CoV shows a close relationship to murine hepatitis virus (MHV) based on phylogeny but MHV binds to the murine receptor CEACAM1 (215). The structure of the RBD of SARS that binds to ACE2 reveals possible sites of importance for crossing to humans (216–218). The pathway of zoonotic transmission however remains unknown despite intensive investigations and the discovery of closely related viruses that have the capacity to infect multiple hosts using the ACE2 receptor (219). Interspecies similarities have been reported with the RBDs of civet and humans differing only in four positions. Amino acids at these positions are responsible for the imbalanced salt bridges at the hydrophobic virus/receptor interface leading to a 1,000-fold difference in affinity of the respective spike protein for human ACE2 that creates the observed species barrier for zoonosis (220, 221). This makes the civet an unlikely candidate for intermediary host. Arguments for civets as an intermediary host therefore hinge on the rapid stepwise mutation rate of SARS-related CoV that contributed to SARS-CoV adapting to the human RBD within a short period (222, 223).
Any viral mutations must then be non-virulent to the reservoir or host species. Of interest is that civets showed clinical signs when infected with two strains of human SARS-CoV and suggesting a complex relationship between virus and civets as possible intermediate hosts (219). More recent research has identified two viruses, RsSHC014 and Rs3367, which are closely related to SARS-CoV in horseshoe bats (R. sinicus) from Yunnan, China that also have a close similarity in the RBD of the spike protein. One, Rs3367 demonstrated that it was equally able to infect HeLa cells expressing ACE2 from human, civet and Chinese horseshoe bats. Rs3367 has 99.9% genome identity with CoV-WIV-1 (224). This suggests that horseshoe bats may not only be a natural reservoir for SARS-CoVs and SARS-like CoVs but future virus mutations, may be able to directly cross to humans without spilling over into intermediate species such as palm civets (Paguma Larvata C.E.H. Smith) or raccoon dogs (Nyctereutes procyonoides Gray) (222, 225). Continued research by Ge and co-researchers have also published a newer WIV mutation, SARS-like CoV-WIV16 (SL-CoV-WIV16), which has an identical gene organization to SL-CoV-WIV1 but a slightly different organization to that of civet SARS-CoV and other bat SL-CoVs. SL-CoV-WIV16 has a 96% nucleotide sequence identity with human (SARS-CoV GZ0 human isolate from 2003 SARS outbreak) and civet SARS-CoVs compared with SL-CoV-WIV1 with 83% nucleotide sequence identity (226). The large genetic diversity of ACE2 among bats and humans and the rapid mutation rate seen in coronaviruses and the often close similarity of the virus RBD with human ACE2 receptors suggests possible future zoonotic events either directly from bats as the reservoir species or via intermediary species including cat (Felis catus Linnaeus), red fox (Vulpes vulpes Linnaeus), and the Chinese ferret badger (Melogale moschata Gray), all found in market places (12, 227–230).
SARS-CoV-2
SARS-CoV-2 is a novel recombinant virus for which the ancestral host species or intermediate spillover species is as yet unidentified, although horseshoe bats (R. sinicus) across China, Europe, and Africa harbor SARS-CoVs (163, 231). Two SARS-CoV-2-related bat coronaviruses (BatCoV RmYN02 and BatCoV RaTG13) have been identified in horseshoe bats (Rhinolophus malayanus Bonhote and Rhinolophus affinis Horsfield). BatCoV RaTG13 sampled from R. affinis in Yunnan in 2013 displayed an approximate 95% similarity to SARS-CoV-2, suggesting that SARS-CoV-2 may have crossed from bats in this area of China (51). Several studies, including one investigating the polyprotein 1ab (pp1ab) of CoVs that is involved in replication and transcription of the viral genome and virus sequence similarity, has identified bat SARS-CoV RaTG13 as being the closest to SARS-CoV-2, rather than CoVZXC21 and CoVZXC45, which also show some similarity to SARS-CoV-2 (51, 81, 232, 233). RaTG13, which has the most closely related CoV sequence to SARS-CoV-2 (96.2%), still has a distinctive RBD which is less efficient in binding to ACE2 (45, 51) and phylogenetic tree and haplotype network analysis suggest historic divergence between SARS-CoV-2 and RaTG13 CoV between 40 and 70 years ago (176, 234).
The animal origin of SARS-CoV-2 is possibly quite diverse, with several SARSr-CoVs having been detected in horseshoe bats co-located in a single cave in China (224). Several sequential recombination events may then have led to changes in the coding sequences of the S-protein, ORF3, and ORF8 of SARSr-CoVs in these bat species prior to spillover to the civet as a possible intermediary host and the emergence of SARS-CoV variants including SARS-CoV and SARS-CoV-2 (45, 235). In the same cave, further SARSr-CoVs with different S protein sequences that were able to recognize ACE2 were identified that have the potential to spillover to humans, including SARS-Like CoV-WIV1 (SL-CoV-WIV1) and β-GX210 found in Cynopterus sphinx in Guangxi (224, 236–238). Human ACE2 is one of the most polymorphic genes, with 317 missense single-nucleotide variations (SNVs) identified that play an important role in susceptibility to SARS-CoV-2 due to changes in binding affinity of the virus to the receptor (230). The diversity of the ACE2 receptor may lead to a greater likelihood of possible infection with future SARS-CoV-2 variants that currently may not be as infectious in the majority of the population. SARS-CoV-2 mutations are identified in South Africa and UK that may be spreading faster and with different infectivity (239). India has also revealed dispersion of SARS-CoV-2 into at least three clades with mutations found in the spike, RdRP, and nucleocapsid coding genes (89). A further possibility is the T265I mutation in the USA which aids SARS-CoV-2 survival in the host cells and may play a role in virulence, transmissibility, or infectivity (240). Whether the viral spread and infectivity is solely due to the change in the spike protein structure or a combination of mutations in other viral proteins, as well as differences in the ACE2 receptors, is currently being investigated (241).
The CoVs from bats in Sichuan and Yunnan province in China also had sequence similarities with SARSr-CoV including β-YN2018A, β-YN2018B, β-YN2018C, β-YN2018D, and β-SC2018 and between 92 and 97% sequence identity with the human SARS-CoV SZ3 (238). These findings indicate the capacity of the coronavirus to undergo recombination between different parts of the genome in different hosts. Such analysis helps to elucidate the possible role of Malayan pangolins (Meloidogyne javanica Treub) as an intermediate host, between bats and humans (242, 243). CoV sequence similarities have identified a virus cluster, including 6 pangolin CoVs (MP789, PCoV-GX-P5L, P5E, P1E, P4L, and P2V) and two bat CoVs (RaTG13 and RmYN02 from R. affinis and R. malayanus, respectively). Pangolins, sampled from Guangdong province in China harbor the Pan-SL-CoV-GD with a genetic similarity of 91.2% to SARS-CoV-2 and others sampled in Guangxi province harbor the Pan-SL-CoV-GX with an 85.4% similarity to SARS-CoV-2 (244–247). Moderate similarity was reported on a genome level between SARS-CoV-2 and pangolin CoV samples using phylogenetic analysis, and the ACE2 receptor binding domain of current SARS-CoV-2 resembles most closely that of the pangolin CoVs from Guangdong. However, it is unlikely that SARS-CoV-2 has emerged directly from the virus isolates in pangolins but rather from an ancestral virus that may have had sequence similarity with RaTG13 CoV that may have obtained the S protein from the MP789 CoV (232, 233, 245, 248, 249).
Coronavirus can infect many animal species other than humans any of which could, conceivably act as an intermediate host species. Possibilities include snakes (Serpentes), hedgehogs (Erinaceidae), bats (Chiroptera), marmots (Marmota), turtles (Chrysemys picta bellii Gray, Chelonia mydas Linnaeus, and Pelodiscus sinensis Wiegmann) (250–253), as well as domesticated species (254–256). Farmed mink has been shown both to harbor SARS-CoV-2 and to transmit the virus to humans. Infections in farms have been reported with an estimated mutation rate of 1.16*10−3 substitutions/site/year, suggesting virus circulation (including the D614G mutation) within the mink farms for some time prior to identification (257, 258). Other animals that have been identified with SARS-CoV-2 or SARS-CoV2-like virus include tigers, lions, pigs, ferrets, golden hamsters, chickens (Gallus), and rabbits (RbCoV HKU14). Of these, pigs, cats, ferrets, and primates may have increased susceptibility to SARS-CoV-2 but possibly lesser transmission potential to humans (259–261). This lower transmission potential may be due to lower amplification potential associated with the presence of virus neutralizing antibodies or conversely a higher virulence in these animals (262, 263). The Savanna Monkeys (Chlorocebus spp.) are an example where a high level of human interaction occurs throughout sub-Saharan Africa and the Caribbean. Savannah Monkeys carry the ACE2 receptor for SARS-CoV-2 binding, which increases the risk of bi-directional cross-species transmission and potential of viral mutations to optimize transmission and virulence (50, 264). Ecological and environmental factors, however, may lead to natural host species that carry different coronaviruses and were initially separated geographically to come in contact in the same new host. This type of host switching and dual infection can lead to a novel recombinant virus with higher transmissibility and virulence (265).
Possible ectoparasite vectors including ticks, fleas, and mites have also been reported as transmission factors (234, 260, 266–271). However, for effective transmission and virulence, sufficient adaptation and amplification within the new host of the viral variant are required regardless of the transmission vector (272).
Conclusion
Although direct evidence of zoonotic transfer of SARS-CoV-2 has not been demonstrated, zoonotic transfer to humans remains an ongoing concern. Recent increases in infections and the emergence of new zoonotic transmissions associated with climate change, habitat changes due to agriculture, and human-animal interaction increases the complexity of identifying possible zoonotic pathways (152, 273, 274). Research into receptor utilization has provided critical information on possible zoonotic transmission but at this stage has not identified a possible host or intermediate species for SARS-CoV, MERS, or SARS-CoV-2.
Diverse concepts aimed at explaining zoonotic transmission including phylogenetic similarities, and environmental effects, have highlighted the potential of zoonotic transmission. As such, animal hosts most closely related to humans harbor zoonoses of lower impact in terms of morbidity and mortality, while more distantly related hosts such as Chiroptera (bats) may carry highly virulent zoonoses but with a lower capacity for endemic establishment in human hosts (275). Similar environments, sharing immune response mechanisms, or cell receptors, increase the likelihood that a virus is preadapted to a novel host (66, 121, 276). The plasticity of CoVs, especially β-CoVs, allows these viruses to adapt to diverse host species en route to human spillover (277). This has been shown to be achieved through stepwise adaptive evolution of different functional proteins of SARS-CoVs at different epidemic stages and in different hosts (263, 267, 278–284). However, evidence of zoonotic transmission of specific animal CoVs has not been proven for many of the animals studied, although there is evidence that SARS-CoV may be transmitted from domestic hosts (285, 286).
Co-habitation of hosts carrying different (+)ssRNA viruses within the Coronaviridae will continue to lead to the emergence of new virulent strains by mutation, recombination, or reassortment. HCoV-NL63, an α-CoV, which leads to common cold symptoms, may be essential to follow as, in contrast to other α-CoVs, which rely on the CD13 (aminopeptidase) receptor for entry into host cells, it has been shown to lead to frequent infection in humans and utilizes the ACE2 receptor, and so may only require minor alterations to become more virulent (227, 287, 288). S-protein from HCoV-NL63 and respiratory syndrome coronavirus bind overlapping regions in ACE2. Although the factors responsible for the difference in response between SARS-CoV-2 and HCoV-NL63 are not known, several possibilities have been suggested including accessory protein incorporation into HCoV-NL63 or differences in interaction with the ACE2 receptor. The HCoV-NL63 S protein may only require minor modification to become more virulent, and this is possible due to the frequent HCoV-NL63 infections in humans and the high mutation rates in these viruses (287).
The potential for further β-CoV transmission has been highlighted by the discovery of replication-competent viruses such as bat SL-Cov-WIV1 and SL-Cov-WIV16. SL-Cov-WIV1 discovered between 2012 and 2013 has shown low-level replication in human alveolar basal epithelial (A549) cells and other mammalian cell lines. SL-Cov-WIV1 can also use ACE2 from humans and other species for cell entry (224, 226, 289).
Future mutations or recombination events of SL-CoV-WIV1 could increase virulence and the likelihood of spillover of this viral strain as a related SL-CoV-WIV16 viral mutation has already been identified with greater nucleotide similarity with SARS-CoV GZ0 human isolate from 2003 SARS outbreak. Similarly, HKU4-CoV and NL140422-CoV are capable of binding to human DPP4 without adaptation of the spike protein and pose a zoonotic risk due to possible accumulation of adaptive mutations with persistent infections (290–293). Another SARS-like virus, SHC014-CoV carrying the SHC014 spike mutation, and also found in Chinese horseshoe bats, has the potential to enter via ACE2 and, importantly, showed that immune-therapeutic and prophylactic modalities failed to stop the infection in a mouse model (294). The Hong Kong University 9 (HKU9) virus also has a close similarity with sections of the SARS-CoV and the Wuhan SARS-CoV-2 within the receptor-binding domain for ACE2 and is also widely distributed in diverse species including Rousettus leschenaultii Desmarest, Hypostomus commersonii Valenciennes, Eidolon helvum Kerr, and R. aegyptiacus, from Asia to Africa (20, 292, 295). Whether diversity of human ACE2 increases the potential for new zoonotic transmission of viruses with similarity to SARS-CoV and SARS-CoV-2, or as yet unidentified ACE2-binding viruses, needs to be further investigated.
The factors determining the potential for spillover have been more generally investigated from different perspectives, including phylogenetic analysis and ecogeographical perspectives. These two concepts are intricately linked as viral diversity tends to increase as ecogeographical factors become more favorable for virus to host transmission. Hence, the proportion of potentially zoonotic viruses per species can be predicted by phylogenetic relatedness to humans, host taxonomy, and extent of human population within a species range. Associated with this is the concept that ecosystems that have a large biodiversity of animals, tend to have a greater diversity of viruses. Ecogeographical factors such as clearing of land allows diverse animals that are usually isolated to interact. This is highlighted by Olival et al. (42), who reported that of their mammalian virus dataset consisting of 2,805 mammal–virus associations, and 586 viruses listed, 44.9% were found in humans, and 71.5% of human viruses had a zoonotic origin associated with virus diversity, phylogenetic proximity to humans, and opportunity for human contact (42). The viral richness or diversity in animals including bats and other mammals is most pronounced in Flaviviruses, Bunyaviruses, and Rhabdoviruses in bats (42). Marburg virus found in R. aegyptiacus bats and Lassa virus (LASV) found in the Natal multimammate mouse, Mastomys natalensis Smith (1834), across parts of Africa, further illustrate the role of geoecology of these mammals and the role this plays in the risk of zoonosis (296, 297). A similar database and extraction protocol to that discussed by Olival et al. (42) has also been developed for MERS-CoV virus host species distribution and risk of zoonotic infection, which can be used for future mapping efforts for MERS-CoV and other infectious diseases (298). In the case of a more species-restricted virus, the Epstein-Barr virus (EBV), found in Hypsignathus monstrosus Allen, Eucalyptus torquate Luehm, and Mecynorhina torquata Drury, fruit bats of Pteropodidae family and mainly located in Africa, modeling of possible zoonotic transmission has identified regional spread in Africa and India as a possible future outbreak risk due to bat distribution and bat-human contact probabilities (299, 300). The example of the frequent human infection associated with HCoV-NL63 suggests that highly pathogenic variants have ample opportunity to evolve different strategies for binding to the ACE2 receptor or other pathways of zoonoses (287). MERS-CoV, but not HKU4, has adapted to use human DPP4 and human cellular proteases for efficient human cell entry, contributing to the enhanced pathogenesis of MERS-CoV in humans. A similar risk of future zoonosis is associated with the capability of the S protein to adapt to humans for cross-species transmissions, as may be the case with the HKU4 virus, which binds to DPP4 receptors but with current affinity to bat DPP4 rather than the human DPP4 (291). To understand the zoonotic potential of coronaviruses, further research is required into the role of the ORF genes as well as viral non-structural proteins responsible for the replication and transcription of the viral genome (15). ACE2 receptor variants may also contribute to identifying potential future zoonotic transmission and spillover to other mammals (301).
Determining zoonotic niches for viral host species and continued surveillance of cases of viral infection also play an essential role in predicting the risk and potential spread of zoonotic transmission with reference to virus evolution and ecology. Reverse zoonoses followed by secondary infection may play an important part in SARS-CoV-2 viral transmission and virulence (8, 302). Human to animal host leads to further mutations of the virus in humans which may, in some cases, lead to an improved adaptability of the virus and hence enhanced spread and infection back to humans. Genetic sequence homology between coronaviruses including MERS and SARS-CoV2 may lead to combined infection in human hosts as a function of dual transmission, especially in North Africa and in the Middle East and North Africa (MENA) region in general. Dual infection can then lead to viral mutations in human hosts that may spillover back to animals and be more virulent than the original. This possibility needs to be closely monitored (265). Viral mutations in spillover species such as mink and domesticated animals also needs to be monitored as SARS-CoV-2 mutations in some animal species have already been observed that may change the virulence of SARS-CoV-2 and reduce the likelihood of SARS-CoV-2 losing either its virulence and/or transmissibility. Future pandemics may not only come from α-CoV or β-CoV but also from γ-CoV that are found in domestic birds such as turkeys, guinea fowls, or quails and more recently in beluga whales and bottleneck dolphins (303–305). δ-CoVs have potential to spillover to humans and are present in different mammalian (Asian leopard cat CoV, Chinese ferret badger CoV, porcine CoV HKU15) and avian (bulbul CoV HKU11, thrush CoV HKU12, munia CoV HKU13, white-eye CoV HKU16, sparrow CoV HKU17, magpie-robin CoV HKU18, night heron CoV HKU19, wigeon CoV HKU20, and common moorhen CoV HKU21) species (159, 306–308).
Author Contributions
HJ, MM, EA, WO, IS, DB, GT, and HA contributed to conception and design of the study. HJ took the lead in writing the manuscript. A comprehensive search using Ovid software on Global Health, Medline, PubMed, Zoological Record was performed by MM. GT and HA supervised the project. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This review was commissioned as part of a project to study the host cell receptors of coronaviruses funded by Khalifa University's CPRA grant (Reference number 2020-004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AV declared a shared affiliation with one of the authors JB, to the handling Editor at time of review.
References
1. Yadav T, Saxena SK. “Transmission Cycle of SARS-CoV and SARS-CoV-2,” in Coronavirus disease 2019 (COVID-19): Epidemiology, pathogenesis, diagnosis, and therapeutics., ed. S. K. Saxena (Singapore: Springer Science and Business Media, LLC.) (2020). p. 33–42. doi: 10.1007/978-981-15-4814-7_4
2. Plowright RK, Parrish CR, McCallum H, Hudson PJ, Ko AI, Graham AL, et al. Pathways to zoonotic spillover. Nat Rev Microbiol. (2017) 15:502–10. doi: 10.1038/nrmicro.2017.45
3. Millar BC, Moore JE. Emerging pathogens in infectious diseases: definitions, causes and trends. Rev Med Microbiol. (2006) 17:101–6. doi: 10.1097/MRM.0b013e32818a6b9e
4. Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, et al. Global trends in emerging infectious diseases. Nature. (2008) 451:990–3. doi: 10.1038/nature06536
5. Baker ML, Schountz T, Wang LF. Antiviral immune responses of bats: a review. Zoonoses Publ Hlth. (2013) 60:104–16. doi: 10.1111/j.1863-2378.2012.01528.x
6. Peeters M, Jung M, Ayouba A. The origin and molecular epidemiology of HIV. Expert Rev Anti Infect Ther. (2013) 11:885–96. doi: 10.1586/14787210.2013.825443
7. Abdullahi IN, Emeribe AU, Mustapha JO, Fasogbon SA, Ofor IB, Opeyemi IS. Exploring the genetics, ecology of SARS-COV-2 and climatic factors as possible control strategies against COVID-19. Infez Med. (2020) 28:166–73.
8. Anderson LJ, Baric RS. Emerging human coronaviruses–disease potential and preparedness. N Engl J Med. (2012) 367:1850–2. doi: 10.1056/NEJMe1212300
9. Lau SKP, Wong ACP, Luk HKH, Li KSM, Fung J, He Z, et al. Differential tropism of SARS-CoV and SARS-CoV-2 in bat cells. Emerg Infect Dis. (2020) 26:2961–5. doi: 10.3201/eid2612.202308
10. Haider N, Rothman-Ostrow P, Osman A, Arruda L, Macfarlane-Berry L, Elton L, et al. COVID-19—zoonosis or emerging infectious disease? Front Public Health. (2020) 8:596944. doi: 10.3389/fpubh.2020.596944
11. Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. (2003) 331:991–1004. doi: 10.1016/S0022-2836(03)00865-9
12. Woo PC, Lau SK, Yuen, K.-Y. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr Opin Infect Dis. (2006) 19:401–7. doi: 10.1097/01.qco.0000244043.08264.fc
13. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Meth Mol Biol. (2015) 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1
14. Weiss SR, Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev. (2005) 69:635–64. doi: 10.1128/MMBR.69.4.635-664.2005
15. Tan YJ, Teng E, Shen S, Tan TH, Goh PY, Fielding BC, et al. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J Virol. (2004) 78:6723–34. doi: 10.1128/JVI.78.13.6723-6734.2004
16. Alam I, Kamau AA, Kulmanov M, Jaremko Ł, Arold ST, Pain A, et al. Functional pangenome analysis shows key features of e protein are preserved in SARS and SARS-CoV-2. Front Cell Infect Microbiol. (2020) 10:405. doi: 10.3389/fcimb.2020.00405
17. Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining 'host jump' of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. (2015) 23:468–78. doi: 10.1016/j.tim.2015.06.003
18. Wang L, Wang Y, Ye D, Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents. (2020) 55:105948. doi: 10.1016/j.ijantimicag.2020.105948
19. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
20. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. (2020) 63:457–60. doi: 10.1007/s11427-020-1637-5
21. Gong YN, Tsao KC, Hsiao MJ, Huang CG, Huang PN, Huang PW, et al. SARS-CoV-2 genomic surveillance in Taiwan revealed novel ORF8-deletion mutant and clade possibly associated with infections in Middle East. Emerg Microb Infect. (2020) 9:1457–66. doi: 10.1080/22221751.2020.1782271
22. Hamre D, Procknow JJ. A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med. (1966) 121:190–3. doi: 10.3181/00379727-121-30734
23. Mcintosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci USA. (1967) 57:933–40. doi: 10.1073/pnas.57.4.933
24. Van Der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, et al. Identification of a new human coronavirus. Nat Med. (2004) 10:368–73. doi: 10.1038/nm1024
25. Woo PC, Lau SK, Tsoi HW, Huang Y, Poon RW, Chu CM, et al. Clinical and molecular epidemiological features of coronavirus HKU1–associated community-acquired pneumonia. J Infect Dis. (2005) 192:1898–907. doi: 10.1086/497151
26. Drosten C, Günther S, Preiser W, Van Der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. (2003) 348:1967–76. doi: 10.1056/NEJMoa030747
27. Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. (2012) 367:1814–20. doi: 10.1056/NEJMoa1211721
28. Xu D, Sun H, Su H, Zhang L, Zhang J, Wang B, et al. SARS coronavirus without reservoir originated from an unnatural evolution, experienced the reverse evolution, and finally disappeared in the world. Chin Med J. (2014) 127:2537–42.
29. Drosten C, Muth D, Corman VM, Hussain R, Al Masri M, Hajomar W, et al. An observational, laboratory-based study of outbreaks of Middle East Respiratory Syndrome coronavirus in Jeddah and Riyadh, Kingdom of Saudi Arabia, 2014. Clin Infect Dis. (2015) 60:369–77. doi: 10.1093/cid/ciu812
30. Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. (2016) 24:490–502. doi: 10.1016/j.tim.2016.03.003
31. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
32. Bucknall RA, King LM, Kapikian AZ, Chanock RM. Studies with human coronaviruses. II. Some properties of strains 229E and OC43. Proc Soc Exp Biol Med. (1972) 139:722–7. doi: 10.3181/00379727-139-36224
33. Salkeld DJ, Stapp P, Tripp DW, Gage KL, Lowell J, Webb CT, et al. Ecological traits driving the outbreaks and emergence of zoonotic pathogens. Bioscience. (2016) 66:118–29. doi: 10.1093/biosci/biv179
34. Kindler E, Gil-Cruz C, Spanier J, Li Y, Wilhelm J, Rabouw HH, et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. (2017) 13:e1006195. doi: 10.1371/journal.ppat.1006195
35. Kiyuka PK, Agoti CN, Munywoki PK, Njeru R, Bett A, Otieno JR, et al. Human coronavirus NL63 molecular epidemiology and evolutionary patterns in rural coastal kenya. J Infect Dis. (2018) 217:1728–39. doi: 10.1093/infdis/jiy098
36. Gussow AB, Auslander N, Faure G, Wolf YI, Zhang F, Koonin EV. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc Nat Acad Sci USA. (2020) 117:15193–9. doi: 10.1073/pnas.2008176117
37. Hulswit RJG, Lang Y, Bakkers MJG, Li W, Li Z, Schouten A, et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc Nat Acad Sci USA. (2019) 116:2681–90. doi: 10.1073/pnas.1809667116
38. Che XY, Qiu LW, Liao ZY, Wang YD, Wen K, Pan YX, et al. Antigenic cross-reactivity between severe acute respiratory syndrome-associated coronavirus and human coronaviruses 229E and OC43. J Infect Dis. (2005) 191:2033–7. doi: 10.1086/430355
39. Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. J Med Virol. (2020) 92:522–8. doi: 10.1002/jmv.25700
40. Rabaan AA, Al-Ahmed SH, Haque S, Sah R, Tiwari R, Malik YS, et al. SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Infez Med. (2020) 28:174–84.
41. Li F. Structure, function, and evolution of coronavirus spike proteins. Ann Rev Virol. (2016) 3:237–61. doi: 10.1146/annurev-virology-110615-042301
42. Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature. (2017) 546:646–50. doi: 10.1038/nature22975
43. Serrano I, Gomes D, Ramilo D, Rebelo MT, Da Fonseca IP, Moreira A, et al. An overview of zoonotic disease outbreaks and its forensic management over time. J Forensic Sci. (2019) 64:1304–11. doi: 10.1111/1556-4029.14034
44. Cui Q, Cui C, Huang C, Zhou W, Ji X, Zhang F, et al. AGTR2, One possible novel key gene for the entry of 2019-nCoV into human cells. Preprints. (2020) 2020020194. doi: 10.20944/preprints202002.0194.v1
45. Li X, Giorgi EE, Marichannegowda MH, Foley B, Xiao C, Kong P, et al. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv. (2020) 6:eabb9153. doi: 10.1126/sciadv.abb9153
46. Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. (2011) 85:4122–34. doi: 10.1128/JVI.02232-10
47. Zmora P, Blazejewska P, Moldenhauer AS, Welsch K, Nehlmeier I, Wu Q, et al. DESC1 and MSPL activate influenza A viruses and emerging coronaviruses for host cell entry. J Virol. (2014) 88:12087–97. doi: 10.1128/JVI.01427-14
48. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e278. doi: 10.1016/j.cell.2020.02.052
49. Hou Y, Zhao J, Martin W, Kallianpur A, Chung MK, Jehi L, et al. New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med. (2020) 18:216. doi: 10.1186/s12916-020-01673-z
50. Schmitt CA, Bergey CM, Jasinska AJ, Ramensky V, Burt F, Svardal H, et al. ACE2 and TMPRSS2 variation in savanna monkeys (Chlorocebus spp.): Potential risk for zoonotic/anthroponotic transmission of SARS-CoV-2 and a potential model for functional studies. PLoS ONE. (2020) 15:e0235106. doi: 10.1371/journal.pone.0235106
51. Zhou H, Chen X, Hu T, Li J, Song H, Liu Y, et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol. (2020) 30:2196–203.e2193. doi: 10.1016/j.cub.2020.09.030
52. Kleine-Weber H, Schroeder S, Krüger N, Prokscha A, Naim HY, Müller MA, et al. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg Microbes Infect. (2020) 9:155–68. doi: 10.1080/22221751.2020.1713705
53. Coutard B, Valle C, De Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. (2020) 176:104742. doi: 10.1016/j.antiviral.2020.104742
54. Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol. (2020) 27:763–7. doi: 10.1038/s41594-020-0468-7
55. Meyerowitz EA, Richterman A, Gandhi RT, Sax PE. Transmission of SARS-CoV-2: a review of viral, host, environmental factors. Ann Intern Med. (2020) 17:M20-5008. doi: 10.7326/M20-5008
56. Patel KP, Vunnam SR, Patel PA, Krill KL, Korbitz PM, Gallagher JP, et al. Transmission of SARS-CoV-2: an update of current literature. Eur J Clin Microbiol Infect Dis. (2020) 7:1–7. doi: 10.1007/s10096-020-03961-1
57. Rothan HA, Acharya A, Reid SP, Kumar M, Byrareddy SN. Molecular aspects of COVID-19 differential pathogenesis. Pathogens. (2020) 9:538. doi: 10.3390/pathogens9070538
58. Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol. (2020) 11:1800. doi: 10.3389/fmicb.2020.01800
59. Retel C, Märkle H, Becks L, Feulner PGD. Ecological and evolutionary processes shaping viral genetic diversity. Viruses. (2019) 11:220. doi: 10.3390/v11030220
60. Fam BSO, Vargas-Pinilla P, Amorim CEG, Sortica VA, Bortolini MC. ACE2 diversity in placental mammals reveals the evolutionary strategy of SARS-CoV-2. Gen Mol Biol. (2020) 43:e20200104. doi: 10.1590/1678-4685-gmb-2020-0104
61. Miller CS. Pleiotropic mechanisms of virus survival and persistence. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2005) 100:S27–36. doi: 10.1016/j.tripleo.2005.03.017
62. Ren X, Glende J, Al-Falah M, De Vries V, Schwegmann-Wessels C, Qu X, et al. Analysis of ACE2 in polarized epithelial cells: surface expression and function as receptor for severe acute respiratory syndrome-associated coronavirus. J Gene Virol. (2006) 87:1691–5. doi: 10.1099/vir.0.81749-0
63. Bidokhti MRM, Tråvén M, Krishna NK, Munir M, Belák S, Alenius S, et al. Evolutionary dynamics of bovine coronaviruses: natural selection pattern of the spike gene implies adaptive evolution of the strains. J Gen Virol. (2013) 94:2036–49. doi: 10.1099/vir.0.054940-0
64. Oberemok VV, Laikova KV, Yurchenko KA, Fomochkina II, Kubyshkin AV. SARS-CoV-2 will continue to circulate in the human population: an opinion from the point of view of the virus-host relationship. Inflamm Res. (2020) 69:635–40. doi: 10.1007/s00011-020-01352-y
65. Wu L, Kewalramani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nature Rev Immunol. (2006) 6:859–68. doi: 10.1038/nri1960
66. Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, Mccracken GF, Rupprecht CE. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science. (2010) 329:676–9. doi: 10.1126/science.1188836
67. Abdullahi IN, Emeribe AU, Ajayi OA, Oderinde BS, Amadu DO, Osuji AI. Implications of SARS-CoV-2 genetic diversity and mutations on pathogenicity of the COVID-19 and biomedical interventions. J Taibah Univ Med Sci. (2020) 15:258–64. doi: 10.1016/j.jtumed.2020.06.005
68. Wu A, Niu P, Wang L, Zhou H, Zhao X, Wang W, et al. Mutations, recombination and insertion in the evolution of 2019-nCoV. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.02.29.971101
69. Longdon B, Brockhurst MA, Russell CA, Welch JJ, Jiggins FM. The evolution and genetics of virus host shifts. PLoS Pathog. (2014) 10:e1004395. doi: 10.1371/journal.ppat.1004395
70. Ahmed SSSJ, Paramasivam P, Raj K, Kumar V, Murugesan R, Ramakrishnan V. Regulatory cross talk between SARS-CoV-2 receptor binding and replication machinery in the human host. Front Physiol. (2020) 11:802. doi: 10.3389/fphys.2020.00802
71. Brook CE, Boots M, Chandran K, Dobson AP, Drosten C, Graham AL, et al. Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. Elife. (2020) 9:e48401. doi: 10.7554/eLife.48401.sa2
72. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182:812–27.e819. doi: 10.1016/j.cell.2020.06.043
73. Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. (2010) 84:3134–46. doi: 10.1128/JVI.01394-09
74. Taylor JK, Coleman CM, Postel S, Sisk JM, Bernbaum JG, Venkataraman T, et al. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol. (2015) 89:11820–33. doi: 10.1128/JVI.02274-15
75. Giotis ES, Carnell G, Young EF, Ghanny S, Soteropoulos P, Wang F, et al. Entry of the bat influenza H17N10 virus into mammalian cells is enabled by the MHC class II HLA-DR receptor. Nat Microbiol. (2019) 4:2035–8. doi: 10.1038/s41564-019-0517-3
76. Kirtipal N, Bharadwaj S, Kang SG. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect Genet Evol. (2020) 85:104502. doi: 10.1016/j.meegid.2020.104502
77. Allen T, Murray KA, Zambrana-Torrelio C, Morse SS, Rondinini C, Di Marco M, et al. Global hotspots and correlates of emerging zoonotic diseases. Nat Commun. (2017) 8:1124. doi: 10.1038/s41467-017-00923-8
78. Fitzsimmons WJ, Woods RJ, Mccrone JT, Woodman A, Arnold JJ, Yennawar M, et al. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. (2018) 16:e2006459. doi: 10.1371/journal.pbio.2006459
79. Zhao Z, Li H, Wu X, Zhong Y, Zhang K, Zhang P, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. (2004) 4:21. doi: 10.1186/1471-2148-4-21
80. Sheikh JA, Singh J, Singh H, Jamal S, Khubaib M, Kohli S, et al. Emerging genetic diversity among clinical isolates of SARS-CoV-2: Lessons for today. Infect Genet Evol. (2020) 84:104330. doi: 10.1016/j.meegid.2020.104330
81. Tan Y, Schneider T, Leong M, Aravind L, Zhang D. Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS-CoV-2-related viruses. mBio. (2020) 11:e00760–e00720. doi: 10.1128/mBio.00760-20
82. Ortega JT, Serrano ML, Pujol FH, Rangel HR. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. Excli J. (2020) 19:410–7. doi: 10.17179/excli2020-1167
83. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Trans Med. (2020) 18:179–9. doi: 10.1186/s12967-020-02344-6
84. Vankadari N. Overwhelming mutations or SNPs of SARS-CoV-2: a point of caution. Gene. (2020) 752:144792. doi: 10.1016/j.gene.2020.144792
85. Guruprasad L. Evolutionary relationships and sequence-structure determinants in human SARS coronavirus-2 spike proteins for host receptor recognition. Proteins. (2020) 88:1387–93. doi: 10.1002/prot.25967
86. Brielle ES, Schneidman-Duhovny D, Linial M. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses. (2020) 12:497. doi: 10.3390/v12050497
87. Jaimes JA, André NM, Chappie JS, Millet JK, Whittaker GR. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J Mol Biol. (2020) 432:3309–25. doi: 10.1016/j.jmb.2020.04.009
88. Eaaswarkhanth M, Al Madhoun A, Al-Mulla F. Could the D614G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int J Infect Dis. (2020) 96:459–60. doi: 10.1016/j.ijid.2020.05.071
89. Maitra A, Sarkar MC, Raheja H, Biswas NK, Chakraborti S, Singh AK, et al. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci. (2020) 45:76. doi: 10.1007/s12038-020-00046-1
90. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike mutation D614G alters SARS-CoV-2 fitness and neutralization susceptibility. Nature. (2021) 592:116–121. doi: 10.1038/s41586-020-2895-3
91. Alexander HK, Day T. Risk factors for the evolutionary emergence of pathogens. J R Soc Interface. (2010) 7:1455–74. doi: 10.1098/rsif.2010.0123
92. Alnazawi M, Altaher A, Kandeel M. Comparative genomic analysis MERS CoV isolated from humans and camels with special reference to virus encoded helicase. Biol Pharm Bull. (2017) 40:1289–98. doi: 10.1248/bpb.b17-00241
93. Bang FB. Genetics of resistance of animals to viruses: Introduction I, and studies in mice. Adv Virus Res. (1978) 23:269–348. doi: 10.1016/S0065-3527(08)60102-5
94. Van Sluijs L, Pijlman GP, Kammenga JE. Why do individuals differ in viral susceptibility? A story told by model organisms. Viruses. (2017) 9:284. doi: 10.3390/v9100284
95. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. (1996) 382:722–5. doi: 10.1038/382722a0
96. Pittman KJ, Glover LC, Wang L, Ko DC. The legacy of past pandemics: common human mutations that protect against infectious disease. PLoS Pathog. (2016) 12:e1005680. doi: 10.1371/journal.ppat.1005680
97. Walls AC, Park YJ, Tortorici MA, Wall A, Mcguire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92.e286. doi: 10.1016/j.cell.2020.02.058
98. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation might the double X-chromosome in females be protective against SARS-CoV-2 compared to the single X-chromosome in males? Int J Mol Sci. (2020) 21:3474. doi: 10.3390/ijms21103474
99. Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. (2006) 6:271–6. doi: 10.1016/j.coph.2006.03.001
100. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
101. Delanghe JR, Speeckaert MM, De Buyzere ML. The host's angiotensin-converting enzyme polymorphism may explain epidemiological findings in COVID-19 infections. Clin Chim Acta. (2020) 505:192–3. doi: 10.1016/j.cca.2020.03.031
102. Armijos-Jaramillo V, Yeager J, Muslin C, Perez-Castillo Y. SARS-CoV-2, an evolutionary perspective of interaction with human ACE2 reveals undiscovered amino acids necessary for complex stability. Evol Appl. (2020) 13:2169–78. doi: 10.1101/2020.03.21.001933
103. Benvenuto D, Angeletti S, Giovanetti M, Bianchi M, Pascarella S, Cauda R, et al. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J Infect. (2020) 81:e24–7. doi: 10.1016/j.jinf.2020.03.058
104. Jin X, Xu K, Jiang P, Lian J, Hao S, Yao H, et al. Virus strain from a mild COVID-19 patient in Hangzhou represents a new trend in SARS-CoV-2 evolution potentially related to Furin cleavage site. J Emerging Microbes Infect. (2020) 9:1474–88. doi: 10.1080/22221751.2020.1781551
105. Anthony SJ, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, et al. Coronaviruses in bats from Mexico. J Gen Virol. (2013) 94:1028–38. doi: 10.1099/vir.0.049759-0
106. Berto A, Anh PH, Carrique-Mas JJ, Simmonds P, Van Cuong N, Tue NT, et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of southern Viet Nam. Zoo Public health. (2018) 65:30–42. doi: 10.1111/zph.12362
107. Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Bresolin N, Clerici M, et al. Genome-wide identification of susceptibility alleles for viral infections through a population genetics approach. PLoS Genet. (2010) 6:e1000849. doi: 10.1371/journal.pgen.1000849
108. Cui J, Eden JS, Holmes EC, Wang LF. Adaptive evolution of bat dipeptidyl peptidase 4 (dpp4): implications for the origin and emergence of Middle East respiratory syndrome coronavirus. Virol J. (2013) 10:304. doi: 10.1186/1743-422X-10-304
109. Nasir A, Sun FJ, Kim KM, Caetano-Anollés G. Untangling the origin of viruses and their impact on cellular evolution. Ann N Y Acad Sci. (2015) 1341:61–74. doi: 10.1111/nyas.12735
110. Adney DR, Bielefeldt-Ohmann H, Hartwig AE, Bowen RA. Infection, replication, and transmission of middle east respiratory syndrome coronavirus in alpacas. Emerg Infect Dis. (2016) 22:1031–7. doi: 10.3201/eid2206.160192
111. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45.e1039. doi: 10.1016/j.cell.2020.04.026
112. Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat Microbiol. (2019) 4:789–99. doi: 10.1038/s41564-019-0371-3
113. Gorbunova V, Seluanov A, Kennedy BK. The world goes bats: Living longer and tolerating viruses. Cell Metab. (2020) 32:31–43. doi: 10.1016/j.cmet.2020.06.013
114. Amman BR, Carroll SA, Reed ZD, Sealy TK, Balinandi S, Swanepoel R, et al. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog. (2012) 8:e1002877. doi: 10.1371/journal.ppat.1002877
115. Sheikh A, Al-Taher A, Al-Nazawi M, Al-Mubarak AI, Kandeel M. Analysis of preferred codon usage in the coronavirus N genes and their implications for genome evolution and vaccine design. J Virol Methods. (2020) 277:113806. doi: 10.1016/j.jviromet.2019.113806
116. Simmonds P. Rampant C→ U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere. (2020) 5:e00408–20. doi: 10.1128/mSphere.00408-20
117. Schäfer A, Baric RS. Epigenetic landscape during coronavirus infection. Pathogens. (2017) 6:8. doi: 10.3390/pathogens6010008
118. Bakkers MJ, Lang Y, Feitsma LJ, Hulswit RJ, de Poot SA, van Vliet AL, et al. Betacoronavirus adaptation to humans involved progressive loss of hemagglutinin-esterase lectin activity. Cell Host Microbe. (2017) 21:356–66. doi: 10.1016/j.chom.2017.02.008
119. Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg Infect Dis. (2007) 13:1526–32. doi: 10.3201/eid1310.070448
120. Gessain A. Mechanisms of viral emergence and interspecies transmission: the exemple of simian foamy viruses in Central Africa. Bull Acad Natl Med. (2013) 197:1655–67. doi: 10.1016/S0001-4079(19)31387-1
121. Streicker DG. From persistence to cross-species emergence of a viral zoonosis. Science. (2013) 342:1185–6. doi: 10.1126/science.1247566
122. Kin N, Miszczak F, Diancourt L, Caro V, Moutou F, Vabret A, et al. Comparative molecular epidemiology of two closely related coronaviruses, bovine coronavirus (BCoV) and human coronavirus OC43 (HCoV-OC43), reveals a different evolutionary pattern. Infect Genet Evol. (2016) 40:186–91. doi: 10.1016/j.meegid.2016.03.006
123. Li Z, He W, Lan Y, Zhao K, Lv X, Lu H, et al. The evidence of porcine hemagglutinating encephalomyelitis virus induced nonsuppurative encephalitis as the cause of death in piglets. PeerJ. (2016) 4:e2443. doi: 10.7717/peerj.2443
124. Han HJ, Wen HL, Zhao L, Liu JW, Luo LM, Zhou CM, et al. Novel coronaviruses, astroviruses, adenoviruses and circoviruses in insectivorous bats from northern China. Zoo Public Health. (2017) 64:636–46. doi: 10.1111/zph.12358
125. Oong XY, Ng KT, Takebe Y, Ng LJ, Chan KG, Chook JB, et al. Identification and evolutionary dynamics of two novel human coronavirus OC43 genotypes associated with acute respiratory infections: phylogenetic, spatiotemporal and transmission network analyses. Emerg Microbes Infect. (2017) 6:e3. doi: 10.1038/emi.2016.132
126. Guo J, Fang L, Ye X, Chen J, Xu S, Zhu X, et al. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound Emerg Dis. (2019) 66:111–8. doi: 10.1111/tbed.12991
127. De Wilde AH, Snijder EJ, Kikkert M, Van Hemert MJ. Host factors in coronavirus replication. Curr Top Microbiol Immunol. (2018) 419:1–42. doi: 10.1007/82_2017_25
128. Kaden R. Early phylogenetic diversification of SARS-CoV-2: determination of variants and the effect on epidemiology, immunology, and diagnostics. J Clin Med. (2020) 9:1615. doi: 10.3390/jcm9061615
130. Holmes KV, Enjuanes L. The SARS coronavirus: A postgenomic era. Science. (2003) 300:1377–8. doi: 10.1126/science.1086418
131. Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. (2007) 17:67–91. doi: 10.1002/rmv.520
132. Lvov DK, Alkhovsky SV. Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (betacoronavirus: coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus). Vopr Virusol. (2020) 65:62–70. doi: 10.36233/0507-4088-2020-65-2-62-70
133. Gray RR, Salemi M. Integrative molecular phylogeography in the context of infectious diseases on the human-animal interface. Parasitol. (2012) 139:1939–51. doi: 10.1017/S0031182012001102
134. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. (1992) 56:152–79. doi: 10.1128/MR.56.1.152-179.1992
135. Antonovics J, Hood M, Partain J. The ecology and genetics of a host shift: microbotryum as a model system. Am Nat. (2002) 160:S40–53. doi: 10.1086/342143
136. Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, et al. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. (2006) 80:7481–90. doi: 10.1128/JVI.00697-06
137. Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. (2014) 101:45–56. doi: 10.1016/j.antiviral.2013.10.013
138. Slingenbergh J. Animal virus ecology and evolution are shaped by the virus host-body infiltration and colonization pattern. Pathogens. (2019) 8:72. doi: 10.3390/pathogens8020072
139. French RK, Holmes EC. An ecosystems perspective on virus evolution and emergence. Trends Microbiol. (2020) 28:165–75. doi: 10.1016/j.tim.2019.10.010
140. Rodhain F. Bats and viruses: complex relationships. Bull Soc Path Exot. (2015) 108:272–89. doi: 10.1007/s13149-015-0448-z
141. Morens DM, Daszak P, Taubenberger JK. Escaping pandora's box — another novel coronavirus. NEJM. (2020) 382:1293–5. doi: 10.1056/NEJMp2002106
142. Letko M, Seifert SN, Olival KJ, Plowright RK, Munster VJ. Bat-borne virus diversity, spillover and emergence. Nat Rev Microbiol. (2020) 18:461–71. doi: 10.1038/s41579-020-0394-z
143. Dos Santos Bezerra R, Valença IN, De Cassia Ruy P, Ximenez JPB, Da Silva Junior WA, Covas DT, et al. The novel coronavirus SARS-CoV-2: from a zoonotic infection to coronavirus disease 2019. J Med Virol. (2020) 92:2607–15. doi: 10.1002/jmv.26072
144. Joffrin L, Goodman SM, Wilkinson DA, Ramasindrazana B, Lagadec E, Gomard Y, et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci Rep. (2020) 10:6873. doi: 10.1038/s41598-020-63799-7
145. Kunz TH, Braun De Torrez E, Bauer D, Lobova T, Fleming TH. Ecosystem services provided by bats. Ann N Y Acad Sci. (2011) 1223:1–38. doi: 10.1111/j.1749-6632.2011.06004.x
146. Gossner C, Danielson N, Gervelmeyer A, Berthe F, Faye B, Kaasik Aaslav K, et al. Human-dromedary camel interactions and the risk of acquiring zoonotic Middle East Respiratory Syndrome coronavirus infection. Zoo Publ Hlth. (2016) 63:1–9. doi: 10.1111/zph.12171
147. Nishiura H, Ejima K, Mizumoto K. Missing information in animal surveillance of MERS-CoV. Lancet Infect Dis. (2014) 14:70699–6. doi: 10.1016/S1473-3099(13)70699-6
148. Wang M, Yan M, Xu H, Liang W, Kan B, Zheng B, et al. SARS-CoV infection in a restaurant from palm civet. Emerg Infect Dis. (2005) 11:1860–5. doi: 10.3201/eid1112.041293
149. Oreshkova N, Molenaar RJ, Vreman S, Harders F, Oude Munnink BB, Hakze-Van Der Honing RW, et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Euro Surveill. (2020) 25:2001005. doi: 10.2807/1560-7917.ES.2020.25.23.2001005
150. Schillaci MA, Jones-Engel L, Engel GA, Paramastri Y, Iskandar E, Wilson B, et al. Prevalence of enzootic simian viruses among urban performance monkeys in Indonesia. Trop Med Int Health. (2005) 10:1305–14. doi: 10.1111/j.1365-3156.2005.01524.x
151. Yeşilbag K, Alpay G, Karakuzulu H. A serologic survey of viral infections in captive ungulates in Turkish zoos. J Zoo Wildl Med. (2011) 42:44–8. doi: 10.1638/2010-0009.1
152. Ampel NM. Plagues–what's past is present: thoughts on the origin and history of new infectious diseases. Rev Infect Dis. (1991) 13:658–65. doi: 10.1093/clinids/13.4.658
153. Baric RS, Sullivan E, Hensley L, Yount B, Chen W. Persistent infection promotes cross-species transmissibility of mouse hepatitis virus. J Virol. (1999) 73:638–49. doi: 10.1128/JVI.73.1.638-649.1999
154. Baric RS. SARS-CoV: lessons for global health. Virus Res. (2008) 133:1–3. doi: 10.1016/j.virusres.2007.03.024
155. Dobson AP. Virology. What links bats to emerging infectious diseases? Science. (2005) 310:628–9. doi: 10.1126/science.1120872
156. Medley GF, Nokes DJ. Epidemiology. Does viral diversity matter? Science. (2009) 325:274–5. doi: 10.1126/science.1177475
157. Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, et al. Global patterns in coronavirus diversity. Virus Evol. (2017) 3:vex012. doi: 10.1093/ve/vex012
158. O'shea TJ, Cryan PM, Cunningham AA, Fooks AR, Hayman DTS, Luis AD, et al. Bat flight and zoonotic viruses. Emerg Infect Dis. (2014) 20:741–5. doi: 10.3201/eid2005.130539
159. Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. (2012) 86:3995–4008. doi: 10.1128/JVI.06540-11
160. Verhagen JH, Herfst S, Fouchier RA. Infectious disease. How a virus travels the world. Science. (2015) 347:616–7. doi: 10.1126/science.aaa6724
161. Hepojoki S, Lindh E, Vapalahti O, Huovilainen A. Prevalence and genetic diversity of coronaviruses in wild birds, Finland. Infect Ecol Epidemiol. (2017) 7:1408360. doi: 10.1080/20008686.2017.1408360
162. Kreuder JC, Hitchens PL, Smiley ET, Goldstein T, Thomas K, Clements A, et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci Rep. (2015) 5:14830. doi: 10.1038/srep14830
163. Hu B, Zeng LP, Yang XL, Ge Y, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. (2017) 13:e1006698. doi: 10.1371/journal.ppat.1006698
164. Cho M, Son HS. Prediction of cross-species infection propensities of viruses with receptor similarity. Infect Genet Evol. (2019) 73:71–80. doi: 10.1016/j.meegid.2019.04.016
165. Satterfield DA, Marra PP, Sillett TS, Altizer S. Responses of migratory species and their pathogens to supplemental feeding. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170094. doi: 10.1098/rstb.2017.0094
166. Seltmann A, Corman VM, Rasche A, Drosten C, Czirják G, Bernard H, et al. Seasonal fluctuations of astrovirus, but not coronavirus shedding in bats inhabiting human-modified tropical forests. Ecohealth. (2017) 14:272–84. doi: 10.1007/s10393-017-1245-x
167. Lens L, Van Dongen S, Matthysen E. Fluctuating asymmetry as an early warning system in the critically endangered Taita thrush. Conserv Biol. (2002) 16:479–87. doi: 10.1046/j.1523-1739.2002.00516.x
168. Melaun C, Werblow A, Busch MW, Liston A, Klimpel S. Bats as potential reservoir hosts for vector-borne diseases. In: Klimpel S, Mehlhorn H, editors. Bats (Chiroptera) as Vectors of Diseases and Parasites. Parasitology Research Monographs, Vol. 5. Berlin; Heidelberg: Springer (2014). p. 25–61. doi: 10.1007/978-3-642-39333-4_3
170. Guan Y, Zheng B, He Y, Liu X, Zhuang Z, Cheung C, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. (2003) 302:276–8. doi: 10.1126/science.1087139
171. Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA. (2005) 102:14040–5. doi: 10.1073/pnas.0506735102
172. Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. (2005) 310:676–9. doi: 10.1126/science.1118391
173. Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, et al. Identification of a novel coronavirus in bats. J Virol. (2005) 79:2001–9. doi: 10.1128/JVI.79.4.2001-2009.2005
174. Holmes EC. Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. (2003) 11:543–6. doi: 10.1016/j.tim.2003.10.006
175. Boni MF, Lemey P, Jiang X, Lam TT, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. (2020) 5:1408–17. doi: 10.1038/s41564-020-0771-4
176. Flores-Alanis A, Sandner-Miranda L, Delgado G, Cravioto A, Morales-Espinosa R. The receptor binding domain of SARS-CoV-2 spike protein is the result of an ancestral recombination between the bat-CoV RaTG13 and the pangolin-CoV MP789. BMC Res Notes. (2020) 13:398. doi: 10.1186/s13104-020-05242-8
177. Bermingham A, Chand MA, Brown CS, Aarons E, Tong C, Langrish C, et al. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill. (2012) 17:20290. doi: 10.2807/ese.17.40.20290-en
178. Müller MA, Raj VS, Muth D, Meyer B, Kallies S, Smits SL, et al. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio. (2012) 3:e00515–00512. doi: 10.1128/mBio.00515-12
179. Chan RWY, Poon LLM. The emergence of human coronavirus EMC: How scared should we be? mBio. (2013) 4:e00191–00113. doi: 10.1128/mBio.00191-13
180. Kindler E, Jónsdóttir HR, Muth D, Hamming OJ, Hartmann R, Rodriguez R, et al. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio. (2013) 4:e00611–2. doi: 10.1128/mBio.00611-12
181. Ramadan N, Shaib H. Middle East respiratory syndrome coronavirus (MERS-CoV): a review. Germs. (2019) 9:35–42. doi: 10.18683/germs.2019.1155
182. Cotten M, Watson SJ, Kellam P, Al-Rabeeah AA, Makhdoom HQ, Assiri A, et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. (2013) 382:1993–2002. doi: 10.1016/S0140-6736(13)61887-5
183. Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. (2013) 19:1697–9. doi: 10.3201/eid1910.130946
184. Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, et al. Spread, circulation, and evolution of the Middle East Respiratory Syndrome Coronavirus. mBio. (2014) 5:e01062-13. doi: 10.1128/mBio.01062-13
185. Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. (2013) 19:1819–23. doi: 10.3201/eid1911.131172
186. Sharmin R, Islam AB. Conserved antigenic sites between MERS-CoV and Bat-coronavirus are revealed through sequence analysis. Source Code Biol Med. (2016) 11:3. doi: 10.1186/s13029-016-0049-7
187. Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, et al. Further evidence for bats as the evolutionary source of Middle East Respiratory Syndrome Coronavirus. mBio. (2017) 8:e00373–00317. doi: 10.1128/mBio.00373-17
188. Song MH, Lee J, Cho SP, Lee KJ, Yoo SK. Support vector machine based arrhythmia classification using reduced features. Int J Control Automat Syst. (2005) 3:571–9. doi: 10.1016/j.artmed.2008.04.007
189. Reusken CB, Haagmans BL, Müller MA, Gutierrez C, Godeke GJ, Meyer B, et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis. (2013) 13:859–66. doi: 10.1016/S1473-3099(13)70164-6
190. Zhang W, Zheng XS, Agwanda B, Ommeh S, Zhao K, Lichoti J, et al. Serological evidence of MERS-CoV and HKU8-related CoV co-infection in Kenyan camels. Emerg Microb Infect. (2019) 8:1528–34. doi: 10.1080/22221751.2019.1679610
191. Adney DR, Letko M, Ragan IK, Scott D, Van Doremalen N, Bowen RA, et al. Bactrian camels shed large quantities of Middle East respiratory syndrome coronavirus (MERS-CoV) after experimental infection(). Emerg Microbes Infect. (2019) 8:717–23. doi: 10.1080/22221751.2019.1618687
192. Hui DS. Tracking the transmission and evolution of MERS-CoV. Lancet Digital Hlth. (2013) 382:1962–4. doi: 10.1016/S0140-6736(13)61955-8
193. Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, Hashem AM, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med. (2014) 370:2499–505. doi: 10.1056/NEJMoa1401505
194. Chu DK, Poon LL, Gomaa MM, Shehata MM, Perera RA, Abu Zeid D, et al. MERS coronaviruses in dromedary camels, Egypt. Emerg Infect Dis. (2014) 20:1049–53. doi: 10.3201/eid2006.140299
195. Liljander A, Meyer B, Jores J, Müller MA, Lattwein E, Njeru I, et al. MERS-CoV antibodies in humans, Africa, 2013-2014. Emerg Infect Dis. (2016) 22:1086–9. doi: 10.3201/eid2206.160064
196. Group FOWMTW. MERS: Progress on the global response, remaining challenges and the way forward. Antiviral Res. (2018) 159:35–44. doi: 10.1016/j.antiviral.2018.09.002
197. Abbad A, Perera RA, Anga L, Faouzi A, Minh NNT, Malik MMR, et al. Middle East respiratory syndrome coronavirus (MERS-CoV) neutralising antibodies in a high-risk human population, Morocco, November 2017 to January 2018. Euro Surveillance. (2019) 24:1900244. doi: 10.2807/1560-7917.ES.2019.24.48.1900244
198. AlBalwi MA, Khan A, AlDrees M, Gk U, Manie B, Arabi Y, et al. Evolving sequence mutations in the Middle East Respiratory Syndrome Coronavirus (MERS-CoV). J Infect Public Health. (2020) 13:1544–50. doi: 10.1016/j.jiph.2020.06.030
199. Forni D, Cagliani R, Mozzi A, Pozzoli U, Al-Daghri N, Clerici M, et al. Extensive positive selection drives the evolution of nonstructural proteins in lineage C betacoronaviruses. J Virol. (2016) 90:3627–39. doi: 10.1128/JVI.02988-15
200. El-Kafrawy SA, Corman VM, Tolah AM, Al Masaudi SB, Hassan AM, Müller MA, et al. Enzootic patterns of Middle East respiratory syndrome coronavirus in imported African and local Arabian dromedary camels: a prospective genomic study. Lancet Planetary Hlth. (2019) 3:e521–8. doi: 10.1016/S2542-5196(19)30243-8
201. Al-Shomrani BM, Manee MM, Alharbi SN, Altammami MA, Alshehri MA, Nassar MS, et al. Genomic sequencing and analysis of eight camel-derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) isolates in Saudi Arabia. Viruses. (2020) 12:611. doi: 10.3390/v12060611
202. Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. (2013) 500:227–31. doi: 10.1038/nature12328
203. Borucki MK, Lao V, Hwang M, Gardner S, Adney D, Munster V, et al. Middle East respiratory syndrome coronavirus intra-host populations are characterized by numerous high frequency variants. PLoS ONE. (2016) 11:e0146251. doi: 10.1371/journal.pone.0146251
204. Raj VS, Mou H, Smits SL, Dekkers DH, Müller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. (2013) 495:251–4. doi: 10.1038/nature12005
205. Te N, Vergara-Alert J, Lehmbecker A, Pérez M, Haagmans BL, Baumgärtner W, et al. Co-localization of Middle East respiratory syndrome coronavirus (MERS-CoV) and dipeptidyl peptidase-4 in the respiratory tract and lymphoid tissues of pigs and llamas. Transbound Emerg Dis. (2019) 66:831–41. doi: 10.1111/tbed.13092
206. Alnaeem A, Kasem S, Qasim I, Al-Doweriej A, Refaat M, Al-Shabebi A, et al. The dipeptidyl peptidase-4 expression in some MERS-CoV naturally infected dromedary camels in Saudi Arabia 2018-2019. Virus Dis. (2020) 31:1–4. doi: 10.1007/s13337-020-00586-y
207. Lau SKP, Zhang L, Luk HKH, Xiong L, Peng X, Li KSM, et al. Receptor usage of a novel bat lineage c betacoronavirus reveals evolution of Middle East respiratory syndrome-related coronavirus spike proteins for human dipeptidyl peptidase 4 binding. J Infect Dis. (2018) 218:197–207. doi: 10.1093/infdis/jiy018
208. Letko M, Miazgowicz K, Mcminn R, Seifert SN, Sola I, Enjuanes L, et al. Adaptive evolution of MERS-CoV to species variation in DPP4. Cell Rep. (2018) 24:1730–7. doi: 10.1016/j.celrep.2018.07.045
209. Naeem A, Hamed ME, Alghoribi MF, Aljabr W, Alsaran H, Enani MA, et al. Molecular evolution and structural mapping of n-terminal domain in spike gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Viruses. (2020) 12:502. doi: 10.3390/v12050502
210. Algaissi A, Agrawal AS, Han S, Peng BH, Luo C, Li F, et al. Elevated human dipeptidyl peptidase 4 expression reduces the susceptibility of hDPP4 transgenic mice to Middle East Respiratory Syndrome Coronavirus infection and disease. J Infect Dis. (2019) 219:829–35. doi: 10.1093/infdis/jiy574
211. Tang XC, Agnihothram SS, Jiao Y, Stanhope J, Graham RL, Peterson EC, et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc Natl Acad Sci USA. (2014) 111:E2018–26. doi: 10.1073/pnas.1402074111
212. Zhang XW, Yap YL, Danchin A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch Virol. (2005) 150:1–20. doi: 10.1007/s00705-004-0413-9
213. Watabe T, Kishino H. Structural considerations in the fitness landscape of a virus. Mol Biol Evol. (2010) 27:1782–91. doi: 10.1093/molbev/msq056
214. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. (2003) 300:1394–9. doi: 10.1126/science.1085952
215. Liò P, Goldman N. Phylogenomics and bioinformatics of SARS-CoV. Trends Microbiol. (2004) 12:106–11. doi: 10.1016/j.tim.2004.01.005
216. Holmes KV. Structural biology. Adaptation of SARS coronavirus to humans. Science. (2005) 309:1822–3. doi: 10.1126/science.1118817
217. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. (2005) 309:1864–8. doi: 10.1126/science.1116480
218. Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Res. (2013) 100:246–54. doi: 10.1016/j.antiviral.2013.08.014
219. Wang LF, Shi Z, Zhang S, Field H, Daszak P, Eaton BT. Review of bats and SARS. Emerg Infect Dis. (2006) 12:1834. doi: 10.3201/eid1212.060401
220. Feng HP. Crossing the species barrier. Nat Struct Mol Biol. (2005) 12:831. doi: 10.1038/nsmb1005-831
221. Wu K, Peng G, Wilken M, Geraghty RJ, Li F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J Biol Chem. (2012) 287:8904–11. doi: 10.1074/jbc.M111.325803
222. Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. (2005) 102:2430–5. doi: 10.1073/pnas.0409608102
223. Li F. Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol. (2008) 82:6984–91. doi: 10.1128/JVI.00442-08
224. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. (2013) 503:535–8. doi: 10.1038/nature12711
225. Zheng M, Zhao X, Zheng S, Chen D, Du P, Li X, et al. Bat SARS-Like WIV1 coronavirus uses the ACE2 of multiple animal species as receptor and evades IFITM3 restriction via TMPRSS2 activation of membrane fusion. Emerg Microb Infect. (2020) 9:1567–79. doi: 10.1080/22221751.2020.1787797
226. Yang XL, Hu B, Wang B, Wang MN, Zhang Q, Zhang W, et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J Virol. (2015) 90:3253–6. doi: 10.1128/JVI.02582-15
227. Li W, Wong SK, Li F, Kuhn JH, Huang IC, Choe H, et al. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol. (2006) 80:4211–9. doi: 10.1128/JVI.80.9.4211-4219.2006
228. Hou Y, Peng C, Yu M, Li Y, Han Z, Li F, et al. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch Virol. (2010) 155:1563–9. doi: 10.1007/s00705-010-0729-6
229. Hernández M, Abad D, Eiros JM, Rodríguez-Lázaro D. Are animals a neglected transmission route of SARS-CoV-2? Pathogens. (2020) 9:480. doi: 10.3390/pathogens9060480
230. Wang J, Xu X, Zhou X, Chen P, Liang H, Li X, et al. Molecular simulation of SARS-CoV-2 spike protein binding to pangolin ACE2 or human ACE2 natural variants reveals altered susceptibility to infection. J Gen Virol. (2020) 101:921–4. doi: 10.1099/jgv.0.001452
231. Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. (2010) 84:11336–49. doi: 10.1128/JVI.00650-10
232. Cárdenas-Conejo Y, Liñan-Rico A, García-Rodríguez DA, Centeno-Leija S, Serrano-Posada H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human-pathogenic coronavirus (SARS-CoV-2). J Med Virol. (2020) 92:688–92. doi: 10.1002/jmv.25758
233. Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol. (2020) 92:602–11. doi: 10.1002/jmv.25731
234. Abdel-Moneim AS, Abdelwhab EM. Evidence for SARS-CoV-2 infection of animal hosts. Pathogens. (2020) 9:529. doi: 10.3390/pathogens9070529
235. Lau SK, Feng Y, Chen H, Luk HK, Yang WH, et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J Virol. (2015) 89:10532–47. doi: 10.1128/JVI.01048-15
236. Menachery VD, Yount BL, Sims AC, Debbink K, Agnihothram SS, Gralinski LE, et al. SARS-like WIV1-CoV poised for human emergence. Proc Natl Acad Sci USA. (2016) 113:3048–53. doi: 10.1073/pnas.1517719113
237. Menachery VD, Mitchell HD, Cockrell AS, Gralinski LE, Yount B. L, Graham RL, et al. MERS-CoV accessory ORFs play key role for infection and pathogenesis. mBio. (2017) 8:e00665–00617. doi: 10.1128/mBio.00665-17
238. Han Y, Du J, Su H, Zhang J, Zhu G, Zhang S, et al. Identification of diverse bat alphacoronaviruses and betacoronaviruses in china provides new insights into the evolution and origin of coronavirus-related diseases. Front Microbiol. (2019) 10:1900. doi: 10.3389/fmicb.2019.01900
239. Tang JW, Toovey OT, Harvey KN, Hui DD. Introduction of the South African SARS-CoV-2 variant 501Y. V2 into the UK. J Infect. (2021) 82:e8–10. doi: 10.1016/j.jinf.2021.01.007
240. Omotoso OE. Contributory role of SARS-CoV-2 genomic variations and life expectancy in COVID-19 transmission and low fatality rate in Africa. Egypt J Med Human Genet. (2020) 21:1–6. doi: 10.1186/s43042-020-00116-x
241. Heinzelman P, Romero PA. Discovery of human ACE2 variants with altered recognition by the SARS-CoV-2 spike protein. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.09.17.301861
242. Chaw SM, Tai JH, Chen SL, Hsieh CH, Chang SY, Yeh SH, et al. The origin and underlying driving forces of the SARS-CoV-2 outbreak. J Biomed Sci. (2020) 27:73. doi: 10.1186/s12929-020-00665-8
243. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the Covid-19 outbreak. Curr Biol. (2020) 30:1346–51.e1342. doi: 10.1016/j.cub.2020.03.063
244. Liu P, Chen W, Chen JP. Viral metagenomics revealed sendai virus and coronavirus infection of Malayan Pangolins (Manis javanica). Viruses. (2019) 11:979. doi: 10.3390/v11110979
245. Liu P, Jiang JZ, Wan XF, Hua Y, Li L, Zhou J, et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. (2020) 16:e1008421. doi: 10.1371/journal.ppat.1008421
246. Rehman SU, Shafique L, Ihsan A, Liu Q. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens. (2020) 9:240. doi: 10.3390/pathogens9030240
247. Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou J, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. (2020) 583:286–89. doi: 10.1038/s41586-020-2313-x
248. Friend T, Stebbing J. What is the intermediate host species of SARS-CoV-2? Fut Virol. (2021). doi: 10.2217/fvl-2020-0390
249. Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat Commun. (2021) 12:1–9. doi: 10.1038/s41467-021-21768-2
250. Ji W, Li X. Response to comments on “Cross-species transmission of the newly identified coronavirus 2019-nCoV” and “Codon bias analysis may be insufficient for identifying host(s) of a novel virus.” J Med Virol. (2020) 92:1440. doi: 10.1002/jmv.26048
251. Ji W, Wang W, Zhao X, Zai J, Li X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J Med Virol. (2020) 92:433–40. doi: 10.1002/jmv.25682
252. Liu Z, Xiao X, Wei X, Li J, Yang J, Tan H, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. (2020) 92:595–601. doi: 10.1002/jmv.25726
253. Lopes LR, De Mattos Cardillo G, Paiva PB. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Brazilian J Microbiol. (2020) 51:1593–9. doi: 10.1007/s42770-020-00321-1
254. Sheahan T, Rockx B, Donaldson E, Sims A, Pickles R, Corti D, et al. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J Virol. (2008) 82:2274–85. doi: 10.1128/JVI.02041-07
255. Lakdawala SS, Menachery VD. The search for a COVID-19 animal model. Science. (2020) 368:942–3. doi: 10.1126/science.abc6141
256. Zhai X, Sun J, Yan Z, Zhang J, Zhao J, Zhao Z, et al. Comparison of severe acute respiratory syndrome coronavirus 2 spike protein binding to ACE2 receptors from human, pets, farm animals, and putative intermediate hosts. J Virol. (2020) 94:e00831-20. doi: 10.1128/JVI.00831-20
257. Hammer AS, Quaade ML, Rasmussen TB, Fonager J, Rasmussen M, Mundbjerg K, et al. SARS-CoV-2 transmission between Mink (Neovison vison) and humans, Denmark. Emerg Infect Dis. (2020) 27:547–51. doi: 10.3201/eid2702.203794
258. Oude Munnink BB, Sikkema RS, Nieuwenhuijse DF, Molenaar RJ, Munger E, Molenkamp R, et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science. (2020) 371:172–7. doi: 10.1126/science.abe5901
259. Gollakner R, Capua I. Is COVID-19 the first pandemic that evolves into a panzootic? Vet Ital. (2020) 56:7–8. doi: 10.12834/VetIt.2246.12523.1
260. Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science. (2020) 368:1016–20. doi: 10.1126/science.abb7015
261. Villar M, Fernández De Mera IG, Artigas-Jerónimo S, Contreras M, Gortázar C, De La Fuente J. Coronavirus in cat flea: findings and questions regarding COVID-19. Parasites Vect. (2020) 13:409. doi: 10.1186/s13071-020-04292-y
262. Dhama K, Patel SK, Sharun K, Pathak M, Tiwari R, Yatoo MI, et al. SARS-CoV-2 jumping the species barrier: zoonotic lessons from SARS, MERS and recent advances to combat this pandemic virus. Travel Med Infect Dis. (2020) 37:101830. doi: 10.1016/j.tmaid.2020.101830
263. Opriessnig T, Huang YW. Coronavirus disease 2019 (COVID-19) outbreak: Could pigs be vectors for human infections? Xenotransplantation. (2020) 27:e12591. doi: 10.1111/xen.12591
264. Cao Y, Sun Y, Tian X, Bai Z, Gong Y, Qi J, et al. Analysis of ACE2 gene-encoded proteins across mammalian species. Front Vet Sci. (2020) 7:457. doi: 10.3389/fvets.2020.00457
265. Sajini Aa AA, Mubaraki FA. The recombination potential between SARS-CoV-2 and MERS-CoV from cross-species spill-over infections. J Epidemiol Global Health. (2020) 2210:6006. doi: 10.2991/jegh.k.201105.001
266. Bruyndonckx N, Dubey S, Ruedi M, Christe P. Molecular cophylogenetic relationships between European bats and their ectoparasitic mites (Acari, Spinturnicidae). Mol Phylogenet Evol. (2009) 51:227–37. doi: 10.1016/j.ympev.2009.02.005
267. Lau SKP, Woo PCY, Yip CCY, Fan RYY, Huang Y, Wang M, et al. Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J Virol. (2012) 86:5481–96. doi: 10.1128/JVI.06927-11
268. Damas J, Hughes GM, Keough KC, Painter CA, Persky NS, Corbo M, et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc Natl Acad Sci USA. (2020) 117:22311–22. doi: 10.1073/pnas.2010146117
269. Goumenou M, Spandidos DA, Tsatsakis A. Possibility of transmission through dogs being a contributing factor to the extreme Covid-19 outbreak in North Italy. Mol Med Rep. (2020) 21:2293–5. doi: 10.3892/mmr.2020.11037
270. Hossain MG, Javed A, Akter S, Saha S. SARS-CoV-2 host diversity: an update of natural infections and experimental evidence. J Microbiol Immunol Infect. (2021) 54:175–181. doi: 10.1016/j.jmii.2020.06.006
271. Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature. (2020) 583:834–8. doi: 10.1038/s41586-020-2342-5
272. Parrish CR, Holmes EC, Morens DM, Park EC, Burke DS, Calisher CH, et al. Cross-Species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev. (2008) 72:457–70. doi: 10.1128/MMBR.00004-08
273. Halliday JE, Allan KJ, Ekwem D, Cleaveland S, Kazwala RR, Crump JA. Endemic zoonoses in the tropics: a public health problem hiding in plain sight. Vet Rec. (2015) 176:220–5. doi: 10.1136/vr.h798
274. Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol. (2020) 92:418–23. doi: 10.1002/jmv.26234
275. Guth S, Visher E, Boots M, Brook CE. Host phylogenetic distance drives trends in virus virulence and transmissibility across the animal-human interface. Philos Trans R Soc Lond B Biol Sci. (2019) 374:20190296. doi: 10.1098/rstb.2019.0296
276. Poulin R, Mouillot D. Combining phylogenetic and ecological information into a new index of host specificity. J Parasitol. (2005) 91:511–4. doi: 10.1645/GE-398R
277. Qu XX, Hao P, Song XJ, Jiang SM, Liu YX, Wang PG, et al. Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy. J Biol Chem. (2005) 280:29588–95. doi: 10.1074/jbc.M500662200
278. Decaro N, Desario C, Elia G, Mari V, Lucente MS, Cordioli P, et al. Serological and molecular evidence that canine respiratory coronavirus is circulating in Italy. Vet Microbiol. (2007) 121:225–30. doi: 10.1016/j.vetmic.2006.12.001
279. Tang X, Li G, Vasilakis N, Zhang Y, Shi Z, Zhong Y, et al. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evolution Biol. (2009) 9:52. doi: 10.1186/1471-2148-9-52
280. An DJ, Jeoung HY, Jeong W, Park JY, Lee MH, Park BK, et al. Prevalence of Korean cats with natural feline coronavirus infections. Virol J. (2011) 8:455. doi: 10.1186/1743-422X-8-455
281. Richard M, Fouchier RAM. Influenza A virus transmission via respiratory aerosols or droplets as it relates to pandemic potential. FEMS Microbiol Rev. (2016) 40:68–85. doi: 10.1093/femsre/fuv039
282. Hemida MG, Chu DKW, Perera R, Ko RLW, So RTY, Ng BCY, et al. Coronavirus infections in horses in Saudi Arabia and Oman. Transbound Emerg Dis. (2017) 64:2093–103. doi: 10.1111/tbed.12630
283. Mira F, Purpari G, Lorusso E, Di Bella S, Gucciardi F, Desario C, et al. Introduction of Asian canine parvovirus in Europe through dog importation. Transbound Emerg Dis. (2018) 65:16–21. doi: 10.1111/tbed.12747
284. Hiebl A, Auer A, Bagrinovschi G, Stejskal M, Hirt R, Rümenapf HT, et al. Detection of selected viral pathogens in dogs with canine infectious respiratory disease in Austria. J Small Anim Pract. (2019) 60:594–600. doi: 10.1111/jsap.13051
285. Lun ZR, Qu LH. Animal-to-human SARS-associated coronavirus transmission? Emerg Infect Dis. (2004) 10:959. doi: 10.3201/eid1005.040022
286. Krueger WS, Heil GL, Gray GC. No serologic evidence for zoonotic canine respiratory coronavirus infections among immunocompetent adults. Zoo Publ Hlth. (2013) 60:349–54. doi: 10.1111/zph.12005
287. Hofmann H, Pyrc K, Van Der Hoek L, Geier M, Berkhout B, Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA. (2005) 102:7988–93. doi: 10.1073/pnas.0409465102
288. Milewska A, Nowak P, Owczarek K, Szczepanski A, Zarebski M, Hoang A, et al. Entry of human coronavirus NL63 into the cell. J Virol. (2018) 92:e01933–e01917. doi: 10.1128/JVI.01933-17
289. Rastogi YR, Sharma A, Nagraik R, Aygün A, Sen F. The novel coronavirus 2019-nCoV: its evolution and transmission into humans causing global COVID-19 pandemic. Int J Environ Sci Technol. (2020) 26:1–8. doi: 10.1007/s13762-020-02781-2
290. Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. (2012) 4:1011–33. doi: 10.3390/v4061011
291. Yang Y, Du L, Liu C, Wang L, Ma C, Tang J, et al. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc Nat Acad Sci USA. (2014) 111:12516–21. doi: 10.1073/pnas.1405889111
292. Hammond RG, Tan X, Johnson MA. SARS-unique fold in the Rousettus bat coronavirus HKU9. Protein Sci. (2017) 26:1726–37. doi: 10.1002/pro.3208
293. Luo CM, Wang N, Yang XL, Liu HZ, Zhang W, Li B, et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East respiratory syndrome coronavirus. J Virol. (2018) 92:e00116-18. doi: 10.1128/JVI.00116-18
294. Menachery VD, Yount BL, Debbink K, Agnihothram S, Gralinski LE, Plante JA, et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. (2015) 21:1508–13. doi: 10.1038/nm.3985
295. Huang C, Qi J, Lu G, Wang Q, Yuan Y, Wu Y, et al. Putative receptor binding domain of bat-derived coronavirus HKU9 spike protein: evolution of betacoronavirus receptor binding motifs. Biochem. (2016) 55:5977–88. doi: 10.1021/acs.biochem.6b00790
296. Mylne AQ, Pigott DM, Longbottom J, Shearer F, Duda KA, Messina JP, et al. Mapping the zoonotic niche of Lassa fever in Africa. Trans R Soc Trop Med Hyg. (2015) 109:483–92. doi: 10.1093/trstmh/trv047
297. Pigott DM, Golding N, Mylne A, Huang Z, Weiss DJ, Brady OJ, et al. Mapping the zoonotic niche of Marburg virus disease in Africa. Trans R Soc Trop Med Hyg. (2015) 109:366–78. doi: 10.1093/trstmh/trv024
298. Ramshaw RE, Letourneau ID, Hong AY, Hon J, Morgan JD, Osborne JCP, et al. A database of geopositioned Middle East respiratory syndrome coronavirus occurrences. Sci Data. (2019) 6:318. doi: 10.1038/s41597-019-0330-0
299. Pigott DM, Golding N, Mylne A, Huang Z, Henry AJ, Weiss DJ, et al. Mapping the zoonotic niche of Ebola virus disease in Africa. Elife. (2014) 3:e04395. doi: 10.7554/eLife.04395.017
300. Hasan S, Ahmad SA, Masood R, Saeed S. Ebola virus: A global public health menace: A narrative review. J Family Med Prim Care. (2019) 8:2189–201. doi: 10.4103/jfmpc.jfmpc_297_19
301. Gao S, Luan J, Cui H, Zhang L. ACE2 isoform diversity predicts the host susceptibility of SARS-CoV-2. Transbound Emerg Dis. (2020). doi: 10.1111/tbed.13773
302. Konda M, Dodda B, Konala VM, Naramala S, Adapa S. Potential zoonotic origins of SARS-CoV-2 and insights for preventing future pandemics through one health approach. Cureus. (2020) 12:e8932. doi: 10.7759/cureus.8932
303. Woo PC, Lau SK, Lam CS, Tsang AK, Hui SW, Fan RY, et al. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in gammacoronavirus. J Virol. (2014) 88:1318–31. doi: 10.1128/JVI.02351-13
304. Stern A, Te Yeh M, Zinger T, Smith M, Wright C, Ling G, et al. The evolutionary pathway to virulence of an RNA virus. Cell. (2017) 169:35–46.e19. doi: 10.1016/j.cell.2017.03.013
305. Miłek J, Blicharz-Domańska K. Coronaviruses in avian species - review with focus on epidemiology and diagnosis in wild birds. J Vet Res. (2018) 62:249–55. doi: 10.2478/jvetres-2018-0035
306. Lee S, Lee DU, Noh YH, Lee SC, Choi HW, Yang HS, et al. Molecular characteristics and pathogenic assessment of porcine epidemic diarrhoea virus isolates from the 2018 endemic outbreaks on Jeju Island, South Korea. Transbound Emerg Dis. (2019) 66:1894–909. doi: 10.1111/tbed.13219
307. He WT, Ji X, He W, Dellicour S, Wang S, Li G, et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol Biol Evol. (2020) 37:2641–54. doi: 10.1093/molbev/msaa117
Keywords: zoonoses, coronavirus, SARS-CoV-2, zoonotic transmission, ecology, evolution, reservoir species
Citation: Jelinek HF, Mousa M, Alefishat E, Osman W, Spence I, Bu D, Feng SF, Byrd J, Magni PA, Sahibzada S, Tay GK and Alsafar HS (2021) Evolution, Ecology, and Zoonotic Transmission of Betacoronaviruses: A Review. Front. Vet. Sci. 8:644414. doi: 10.3389/fvets.2021.644414
Received: 21 December 2020; Accepted: 25 March 2021;
Published: 20 May 2021.
Edited by:
Dirk Werling, Royal Veterinary College (RVC), United KingdomReviewed by:
Francisco Rivera-Benítez, Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias (INIFAP), MexicoAmy Yomiko Vittor, University of Florida, United States
Reviewed by:
Francisco Rivera-Benítez, Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias (INIFAP), MexicoAmy Yomiko Vittor, University of Florida, United States
Copyright © 2021 Jelinek, Mousa, Alefishat, Osman, Spence, Bu, Feng, Byrd, Magni, Sahibzada, Tay and Alsafar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Habiba S. Alsafar, aGFiaWJhLmFsc2FmYXJAa3UuYWMuYWU=