 Lin Zheng1,2†
Lin Zheng1,2† Ling-Wei Zhu2†Jie Jing2Jia-yao Guan2Ge-Jin Lu2Lin-Hong Xie3Xue Ji2Dong Chu3Yang Sun2
Ling-Wei Zhu2†Jie Jing2Jia-yao Guan2Ge-Jin Lu2Lin-Hong Xie3Xue Ji2Dong Chu3Yang Sun2 Ping Chen1*
Ping Chen1* Xue-Jun Guo2*
Xue-Jun Guo2*- 1School of Food and Engineering, Jilin Agricultural University, Changchun, China
- 2The Key Laboratory of Jilin Province for Zoonosis Prevention and Control, Changchun, China
- 3Wild Animal Sources and Diseases Inspection Station, National Forestry and Grassl and Bureau, Beijing, China
Migratory birds are recently recognized as Vibrio disease vectors, but may be widespread transporters of Vibrio strains. We isolated Vibrio cholerae (V. cholerae) and Vibrio metschnikovii (V. metschnikovii) strains from migratory bird epidemic samples from 2017 to 2018 and isolated V. metschnikovii from migratory bird feces in 2019 from bird samples taken from the Inner Mongolia autonomous region of China. To investigate the evolution of these two Vibrio species, we sequenced the genomes of 40 V. cholerae strains and 34 V. metschnikovii strains isolated from the bird samples and compared these genomes with reference strain genomes. The pan-genome of all V. cholerae and V. metschnikovii genomes was large, with strains exhibiting considerable individual differences. A total of 2,130 and 1,352 core genes were identified in the V. cholerae and V. metschnikovii genomes, respectively, while dispensable genes accounted for 16,180 and 9,178 of all genes for the two strains, respectively. All V. cholerae strains isolated from the migratory birds that encoded T6SS and hlyA were non-O1/O139 serotypes without the ability to produce CTX. These strains also lacked the ability to produce the TCP fimbriae nor the extracellular matrix protein RbmA and could not metabolize trimetlylamine oxide (TMAO). Thus, these characteristics render them unlikely to be pandemic-inducing strains. However, a V. metschnikovii isolate encoding the complete T6SS system was isolated for the first time. These data provide new molecular insights into the diversity of V. cholerae and V. metschnikovii isolates recovered from migratory birds.
Introduction
Vibrio is an abundant bacterial genus in oceans and comprises numerous species including pathogenic types such as Vibrio cholerae, Vibrio parahaemolyticus, and Vibrio metschnikovii. They are often observed in high abundance in marine products. Vibrio are halophilic and naturally found ubiquitously in marine settings, and thus raw seafood naturally harbors these microorganisms and is the main food source responsible for gastroenteritis caused by Vibrio spp. (1). Cholera is caused by V. cholerae that carry the cholera toxin and has resulted in seven pandemics throughout human history. The seventh V. cholerae pandemic continues to present day, and exhibits evolved characteristics compared to previous pandemics, rendering it difficult to treat cholera disease outbreaks (2, 3). One commonality between the sixth and seventh pandemics has been observed to be associated with the CTXΦ bacteriophage that may have introduced the CTX toxin to V. cholerae. Nevertheless, the strains underlying the seventh pandemic did not directly evolve from those underlying the sixth pandemic (2). In contrast to V. cholerae, few reports have described characterizations of V. metschnikovii, with only some studies focused on detection and no reports on their pathogenicity.
Migratory birds have been considered potential vectors of V. cholerae, wherein colonization of their intestines may occur by ingesting water and marine animals infected with V. cholerae (4). Migratory birds travel over long distances and could carry pathogenic microorganisms from one region to another, spreading pathogenic microorganisms by excretion (5, 6). Therefore, migratory birds have been considered as important reservoirs of Vibrio, forming a fecal-food-mouth transmission route (3). Within intestinal environments, Vibrio are induced to evolve into treatment-resistant bacteria or pandemic strains due to stresses they are exposed to. Consequently, increased risk of bacterial spread and difficulty of bacterial source tracking occur due to the long migration distances of vector birds.
Consequently, it is important to analyze the pathogenicity of V. cholerae strains and whether they exhibit the potential to evolve into pandemic strains. The concept of a pan-genome was first proposed in 2005 and is a term that encompasses the sum of all genes of a species, including their core-genome, dispensable genome, and unique genome components (7). Importantly, the population-level evolutionary trends of a species can be evaluated by analyzing their pan-genome.
Migratory birds exhibiting malnutrition, wasting, diarrhea, and high mortality were observed in the Inner Mongolia region of China between 2017 and 2018. Dead birds comprised individuals from several species including Larus ridibundus, Pluvialis squatarola, Tadorna ferrgina, Anas poecilorhyncha, and Aix galericulata, in addition to other migratory birds. Moreover, 40 V. cholerae strains and 34 V. metschnikovii strains were isolated from the migratory bird epidemic materials and water environments. The V. cholerae strains were non-O1/O139 strains without the ctxA/B toxin, based on serotyping and PCR identification. To further investigate this die-off, we conducted a bacteriological examination again on migratory bird feces in Inner Mongolia, China, in 2019. However, V. cholerae strains were again not present in the samples, while only V. metschnikovii was present. In this study, we analyzed the core-/pan- genomes of V. cholerae/V. metschnikovii and compared them with previously sequenced strains to evaluate whether migratory birds harbor Vibrio spp. that have the potential to evolve into pandemic or drug-resistant strains. These analyses thus provide important data to inform the prevention and treatment of V. cholerae and V. metschnikovii infections and/or outbreaks.
Materials and Methods
Sampling
Thirty-six samples (including 10 water samples, 2 aquatic plant samples, 19 epidemic material samples, and 24 feace samples) were taken from birds in 2018 and 2019. DNA was obtained from each fecal sample by stool DNA kit, and molecular method to determine the host source of feaces (8). The visceral organ and intestinal tract samples used for this study were collected aseptically from 19 freshly dead migratory birds (not corrupt). Samples were cultivated via spread plates in triplicate and directly cultivated onto selective thiosulfate citrate bile salts sucrose (TCBS) agar plates and incubated for 24 h at 37°C. Isolates were identified using PCR amplification of ompW, infC, ctxA, hlyA, and chxA genes (9–13). The primers and conditions used for PCR amplification are described in Supplementary Table 1. In addition, V. cholerae isolates were subjected to O1/O139 antigen serotyping using V. cholerae O antisera (Tianjin Biochip Corporation). Isolates were sub-cultured at 37°C in brain heart liquid (Qingdao Haibo, China) or on CHROM agar Vibrio plates (CHROM, Paris, France) unless otherwise specified. Green colonies were again confirmed using serology and PCR assays (see Supplementary Table 1 for primer sequences and PCR conditions). Isolates identified as Vibrio were then stored in a −80°C freezer.
Extraction of Genomic DNA and Library Construction
Genomic DNA was extracted from isolates using the Bacterial Genomic DNA Extraction Kit (Omega). Harvested DNA was then quantified using a Qubit® 2.0 Fluorometer (Thermo Scientific). A total of 1 μg of DNA per sample was used as input material for DNA library preparations. Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following manufacturer's recommendations. In addition, indexing oligonucleotides were added to sequences to identify each sample. Briefly, DNA samples were fragmented by sonication to a size of 350 bp, followed by DNA fragment end-polishing, addition of poly A-tails, and ligation with full-length adaptors for Illumina sequencing, followed by additional PCR amplification. Finally, PCR products were purified (AMPure XP system), and the size distribution of the libraries was analyzed using an Agilent 2100 Bioanalyzer and quantified using real-time PCR. The whole-genomes of strains were sequenced using an Illumina NovaSeq PE150 platform at the Beijing Novogene Bioinformatics Technology Co., Ltd.
Genome Assembly
Reads containing Illumina PCR adapters and low-quality reads were filtered from the dataset using readfq (vision 10). The remaining good quality paired reads were assembled into scaffolds using the SOAP denovo (https://sourceforge.net/projects/soapdenovo2/files/SOAPdenovo2/), SPAdes (http://cab.spbu.ru/software/spades/), and ABySS (http://www.bcgsc.ca/platform/bioinfo/software/abyss) assemblers (14–17). The filtered reads were then used to close gaps in the scaffolds using Readfq with the options: –rq1 input_1.fq, –rq2 input_2.fq,–oq1 out_1.fq,–oq2 out_2.fq,–adp1 adapter_1.lst,–adp2 adapter_2.lst,– Q QUAL, PERCENT,–C QUAL, PERCENT,–N PERCENT,–alen INT,–amis INT,–dup,–gz,–check1 read1.check,–check2 read2.check.
Genome Feature Predictions
Genome component prediction included prediction of coding genes, repetitive sequences, non-coding RNAs, genomic islands, transposons, prophages, and clustered regularly interspaced short palindromic repeat sequences (CRISPRs). The Gene Mark program was used to identify coding genes, while interspersed repetitive sequences were predicted using the Repeat Masker (http://www.repeatmasker.org/). Tandem repeats were identified using the tandem repeats finder (TRF) program. Transfer RNA (tRNA) genes were predicted using tRNA scan-SE (18–21), while ribosomal RNA (rRNA) genes were identified using rRNAmmer (22). Small nuclear RNAs (snRNA) were predicted by BLAST searches against the Rfam database (23, 24). The Island Path-DIOMB program was used to predict genomic islands and the transposon PSI program was used to identify transposons based on the homologous blast method (25). Lastly, PHAST was used to predict prophages (http://phast.wishartlab.com/), and CRISPR Finder was used to identify CRISPRs (26, 27).
Gene Function Prediction
Four databases were used to predict gene functions, including the Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG) (28), and non-redundant (NR) protein databases (29–32). In addition, whole-genome BLAST searches were performed against the above-mentioned four databases using an E-value threshold of <1e-5 and a minimum alignment length percentage >40% as criteria (33). Secretory proteins were predicted using the Signal P database, and the prediction of Type I-VII proteins secreted by pathogenic bacteria was based on the EffectiveT3software program (34, 35). To evaluate pathogenic factors, pathogenicity, and drug resistance analyses were also conducted using the Virulence Factors of Pathogenic Bacteria (VFDB) and Antibiotic Resistance Genes Database (ARDB) databases (36, 37).
Comparative Genomic Analyses
Comparative genomic analyses were conducted including analysis of genomic synteny, the distribution of core- and strain-specific genes, and phylogenetic analysis of gene families. Genomic alignment between the sample and reference genomes was performed using the MUMmer and LASTZ tools while including 41 V. cholerae strains and 4 V. metschnikovii strains as reference genomes (see Supplementary Table 2 for additional details) (38). The analysis of genomic synteny was based on alignment results. Core- and strain-specific genes were identified based on rapid clustering of similar proteins with the CD-HIT program while specifying a threshold of 50% pairwise identity and a 70% length difference cutoff. A Venn diagram was then used to show the relationships of core- and strain-specific genes among samples. The MUSCLE software program was used to align multiple single-copy core-encoded proteins identified by the core-/pan-genome analysis. The aligned sequences were then subjected to phylogenetic analysis using the TreeBeST program, a neighbor-joining tree reconstruction algorithm, and 1,000 bootstrap replicates.
Multilocus gene sequence typing was conducted based on sequence analysis of eight housekeeping genes (adk, gyrB, mdh, metE, pgm, pntA, purM, and pyrC) using previously described PubMLST protocols (https://pubmlst.org/databases.shtml). The nucleotide sequences for each locus were analyzed with the BioNumerics software program (version 7.6; Applied Maths, Belgium) and compared against published sequences on the PubMLST website. Sequence types (STs) were then determined on the basis of the eight locus allelic profiles.
Nucleotide Sequence
Accession Numbers
The 74 sequences of V. cholerae and V. metschnikovii were submitted to FigShare under the public doi: 10.6084/m9.figshare.14417870. And C16-2-29, M13F, M9D, M21D, M28D, and M29D uploaded Genbank database at the same time, accession number GCA_014281135.1, GCA_014305065.1, GCA_014267955.1, GCA_014267965.1, GCA_014305075.1 and GCA_014305185.1, respectively.
Results
Genome Sequencing
A total of 40 V. cholerae and 34 V. Metschnikovii strains were identified in this study using PCR identification and comparison of gene sequences against the non-redundant (NR) protein database. All of the strains lacked the ctxA, tcpA, and chxA genes, and were also non-O1/O139 serotypes by PCR. All isolates were completely turbid after dripping V. cholerae O antisera and no agglutination occurred, the result was the consistent as the PCR, and it was identified as non-O1/O139 V. cholerae (Table 1).
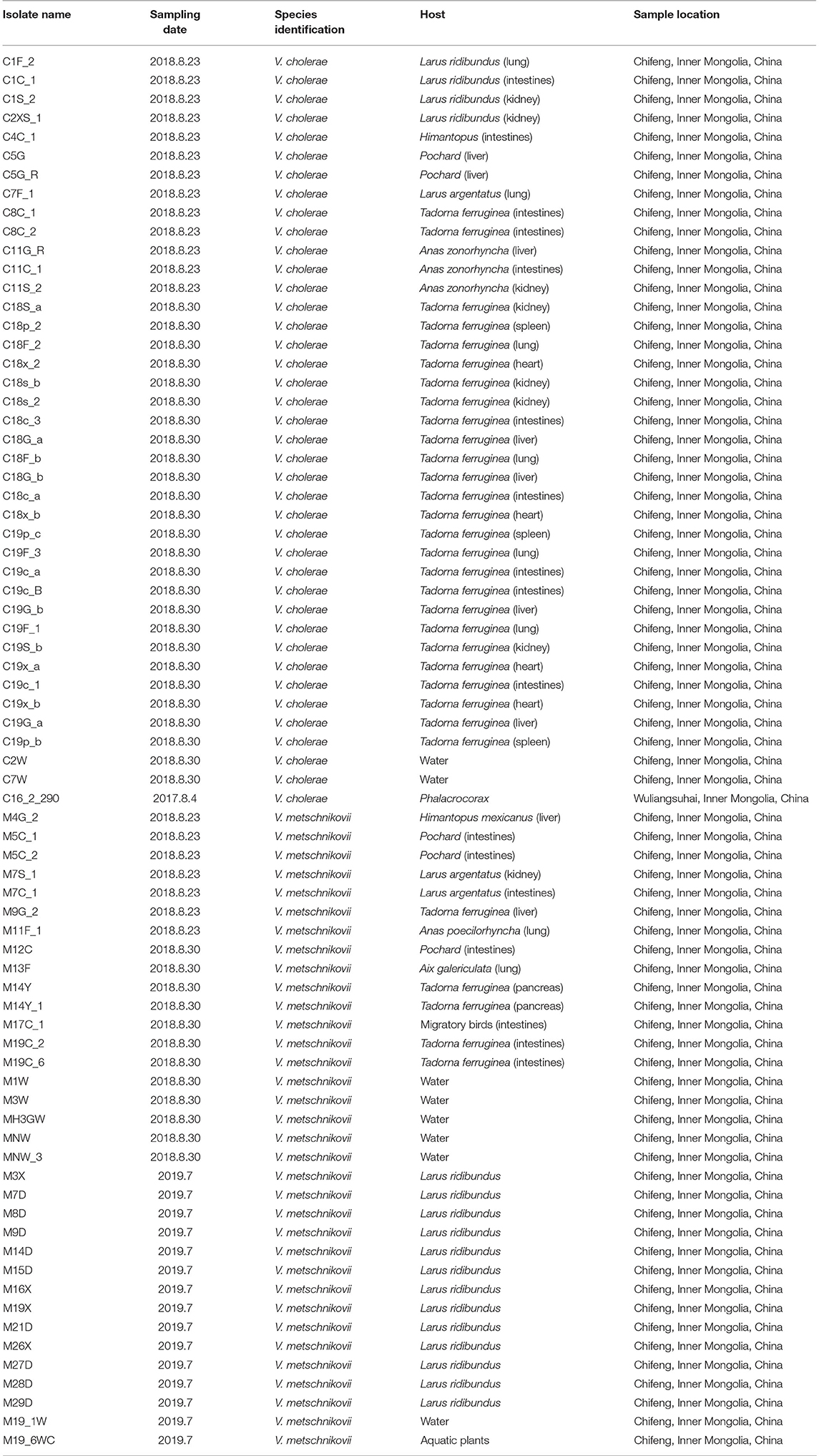
Table 1. Characteristics of Vibrio cholerae and Vibrio metschnikovii isolates identified in migratory birds.
Core- and Pan-Genomic Analysis
The V. cholerae pan-genomes were analyzed by dividing them into core and dispensable genomes. A total of 40 V. cholerae strains isolated from migratory birds were used in comparison against 41 reference V. cholerae genomes retrieved from GenBank. The number of core genes and the pan-genome sizes of the V. cholerae strains are shown in Figure 1A as a function of the number of genes within the genomes. The number of core genes plateaued when plotting the number of core genes against the reciprocal of the number of genomes included in the estimates. In contrast, the pan-genome size steadily increased with the addition of each additional genome, suggesting that V. cholerae exhibited a large pan-genome. Overall, a total of 18,310 pan-genes, 2,130 core-genes, and 16,180 dispensable genes were identified among all V. cholerae strain genomes (Supplementary Table 3). In this study, the dispensable genome was considered to comprise strain-specific genes. A heatmap was constructed to identify the distribution of dispensable genes among different V. cholerae strains. With the exception of the strain TSY216 genome that contained 984 dispensable genes, most other strains in the group C clade contained few dispensable genes.
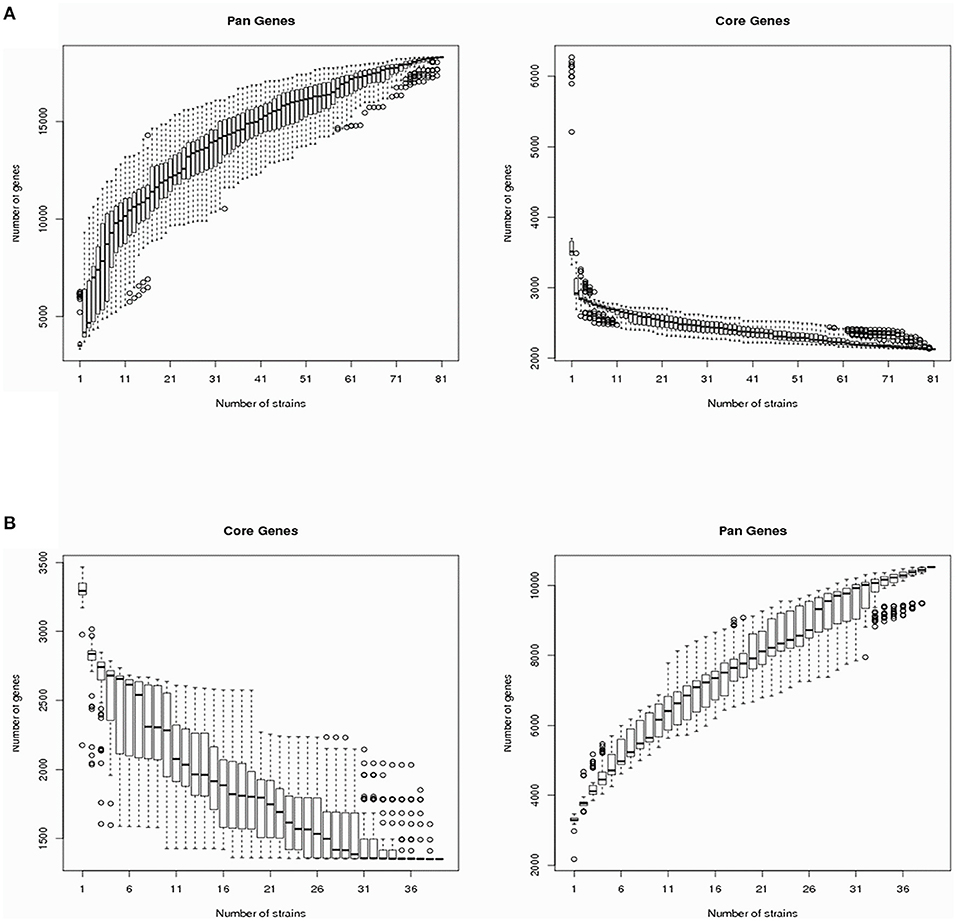
Figure 1. Vibrio cholerae and Vibrio metschnikovii core-/pan-genome diversity curves. (A) The V. cholerae core- and pan-genomes are shown as a function of the number of genomes included in the counts. Boxes represent one standard deviation around the median number of genes within the subsets, while the whiskers indicate two standard deviations from the median. (B) The V. metschnikovii core- and pan-genomes are shown as a function of the number of genomes included in the counts. Boxes represent one standard deviation around the median number of genes within the subsets, while the whiskers indicate two standard deviations from the median.
To analyze the V. metschnikovii pan-genome, 34 V. metschnikovii strain genomes recovered from migratory bird isolates and four reference genomes of V. metschnikovii strains were used for comparative analysis. The size of the pan-genome steadily increased with the addition of each additional genome in the analysis (Figure 1B), suggesting that V. metschnikovii also exhibited a large pan-genome. A total of 10,530 pan-genes, 1,352 core-genes, and 9,178 dispensable genes were identified among all V. metschnikovii strains. Like the genomes of the V. cholerae strains, most V. metschnikovii genomes did not contain many dispensable genes, with the exception of strain M13F, whose genome contained 1,048 dispensable genes.
Phylogenetic Analysis
Phylogenetic analysis indicated the presence of similar branching patterns, wherein the V. cholerae genomes consistently grouped into two major clades. The first clade contained V. cholerae strains from the GenBank reference database and one strain (C16-2-29) from our study. All of the other reference strains, with the exception of strain NCTC30, were present in this clade. In the first clade, strains harboring the cholera toxin (CTX) were divided into group C, while the others in the first clade were considered as group A. All of the strains identified in this study, with the exception of strain C16-2-29 (Wuliangsuhai, 2017), were present in the second clade, which was designated as Group B (Figure 2).
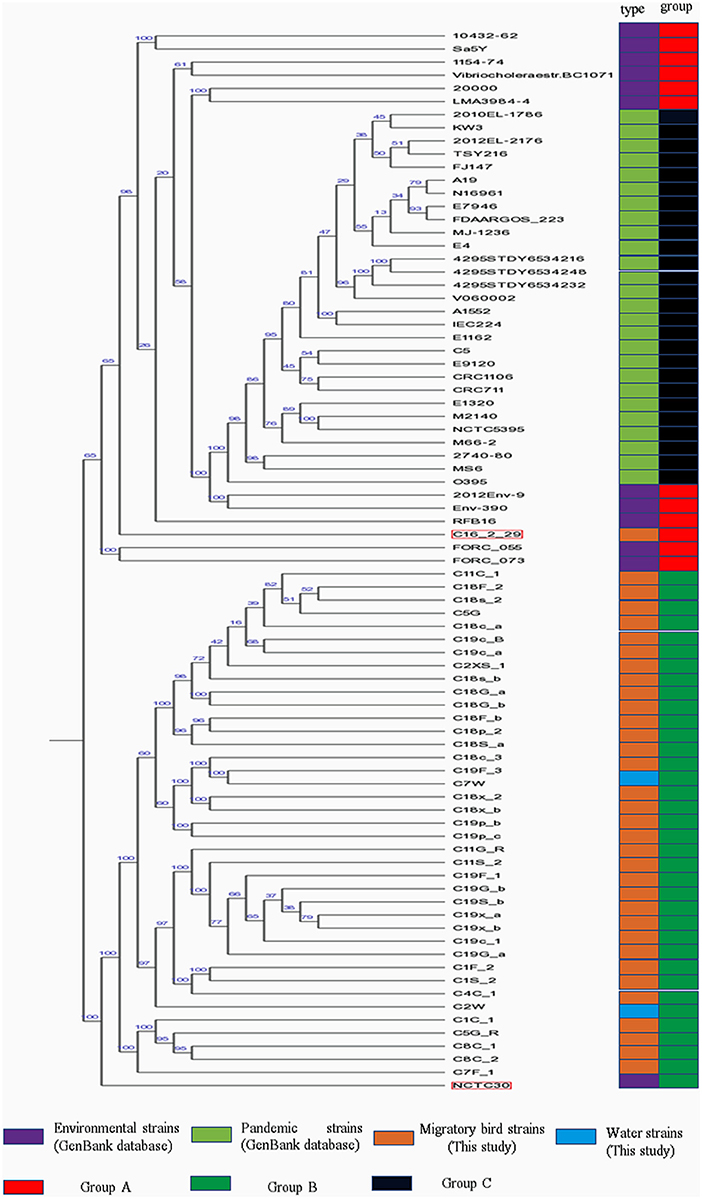
Figure 2. Phylogenetic analysis of V. cholera strains. Different colored blocks are used to indicate types and groups of strains on the left of phylogenetic tree. The color legend is shown below the phylogenetic tree.
MLST analysis of the pandemic V. cholerae strains was conducted using eight different loci (adk, gyrB, mdh, metE, pgm, pntA, purM, and pyrC). However, only seven different loci (adk, gyrB, mdh, metE, pntA, purM, and pyrC) were used for the MLST of the environmental V. cholerae strains (Supplementary Table 3), since pgm was not detected in the environmental strain genomes. With the exception of the C16-2-29 genome, most of the housekeeping genes of V. cholerae strains from migratory birds did not exhibit homology to known genotypes in the pubMLST database. In addition, the pgm gene was detected in the strain C16-2-29 genome, as observed for the pandemic strain genomes. ST69 was the common sequence typing classification for the pandemic strains, while the sixth pandemic strain, O395, exhibited an ST73 sequence type. ST69 was not observed for the V. cholerae strains derived from environments or migratory birds.
Phylognetically, the V. metschnikovii and V. cholerae strains were genetically highly distant. The V. metschnikovii genomes consistently grouped into two major clades, with the first comprising V. metschnikovii strains from different samples and the reference genomes. This first group was designated as Group D. The second clade comprised four strains (M21D, M28D, M9D, and M29D) designated as Group E (Figure 3). A V. metschnikovii MLST database has not been established yet, and thus the V. metschnikovii genomes were not subject to these analyses.
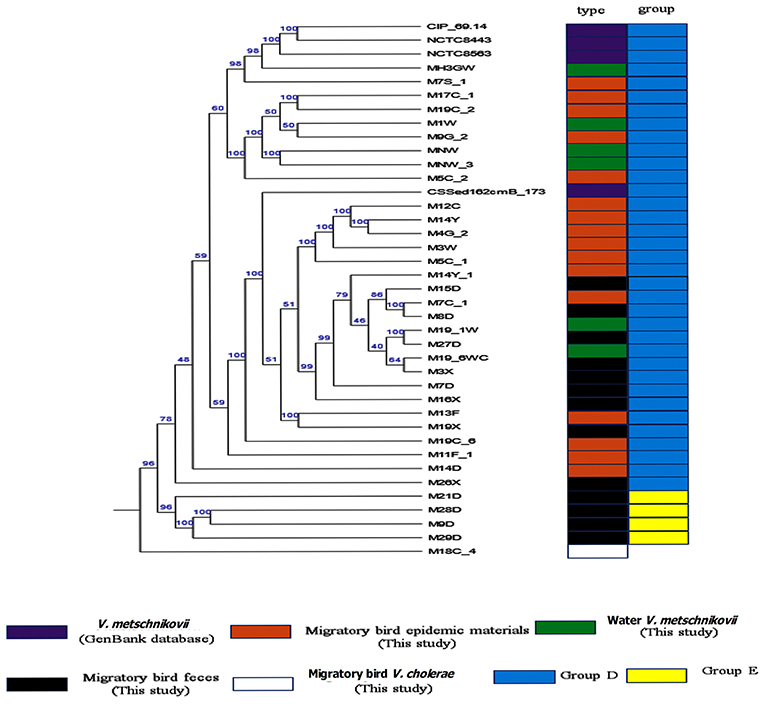
Figure 3. Phylogenetic analysis of V. metschnikovii strain genomes. Different colored blocks are used to indicate types and groups of strains on the left of phylogenetic tree. The color legend is shown below the phylogenetic tree.
Comparison of Gene Functional Enrichment
Gene functions were predicted from the predicted genes and subjected to functional gene enrichment analysis. The V. cholerae core genes were enriched in numerous KEGG pathways including the “E,” amino acid transport and metabolism, and “T,” signal transduction mechanism pathways of the COG database. The numbers of genes within the “T,” signal transduction mechanism pathway, were enriched in group C genomes relative to “E,” amino acid transport and metabolism pathways. However, the other V. cholerae strain genomes exhibited opposite enrichment patterns (Figure 4).
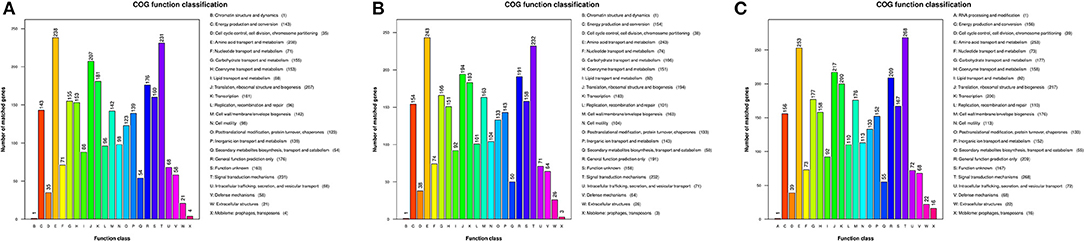
Figure 4. Functional classification of V. cholerae core genome COG functions. (A) COG functional classification of the core genomes of reference environmental V. cholerae strains; (B) COG functional classification of the core genomes of migratory bird V. cholerae strains; (C) COG functional classification of the core genomes of V. cholerae pandemic strains retrieved from the GenBank database.
To further investigate the functional differences encoded by the Vibrio genomes, we analyzed the “metabolism and environmental information processing” functional category encoded by different V. cholerae group genomes. Differences were observed in trimetlylamine oxide (TMAO) metabolism, in addition to two-component systems. Specifically, only group C genomes encoded complete TMAO systems. In addition, the genomes of the other groups lacked TorA, and these isolates are thus unable to metabolize TMAO. Differences between the two groups of V. metschnikovii strain genomes were minimal, although all group genomes were particularly enriched in genes involved in the “E,” amino acid transport and metabolism pathway.
Pathogenicity and Antibiotic-Resistance Potential
The V. cholerae genomes generated in this study only encoded partial RTX protein structures and not rtxA. In addition, all of the groups encoded complete type VI secretory systems (T6SS). The presence of drug resistance genes for V. cholerae in the genomes generated here exhibited low frequencies of resistance genes and the lack of plasmids (Supplementary Table 4). All V. metschnikovii strains lacked CTX, and RTX. However, the genomes of the four strains from migratory birds (M13F, M9D, M29D, and M28D) encoded a T6SS system. Among these, three strains (M9D, M29D, and M28D) lacked other genes involved in secreting proteins (e.g., hcp and vgrG), while only M13F encoded a complete T6SS system V. metschnikovii.
Discussion
Vibrio cholerae acquired cholera toxin (CTX), which is the primary reason for their ability to cause global pandemics, and CTX acquisition occurred over a long evolutionary history (39). CTX was likely introduced to Vibrio by the CTXϕ phage, while the toxin co-regulated pilus (TCP) is also a critical colonization factor of V. cholerae that serves as a receptor for CTXϕ (40). Other studies have indicated that AphA functions in a previously unknown step in the ToxR virulence cascade and helps activate the transcription of tcpPH. TcpP/TcpH, together with ToxR/ToxS, then activate toxT expression, ultimately resulting in the production of virulence factors like the cholera toxin and TCP (41–43). Vibrio strains isolated in this study exhibited unique combinations of virulence factor genes [e.g., ctx(-) tcpA(-) hlyA(+) AphA(+) toxR(+) toxT(-) tcpP(-) tcpH(-)], and thus they did not contain complete virulence factor regulatory networks based on the lack of toxT.
The group C genomes identified in this study also encoded the TMAO metabolic pathway, which differed from other Vibrio groups based on core-/pan-genome analysis. The TorR response regulator mediates TMAO induction of the torCAD operon expression and was intriguingly observed in all of the V. cholerae pandemic strains. With the exception of M66-2, all of the other strains in group C also encoded CTX. The original M66-2 strain did encode CTX, but CTX was knocked out of the strain in a previous study (44). The TMAO metabolic pathway exhibits a unique relationship with CTX production. Specifically, human intestines are anaerobic, and V. cholerae, as a facultative anaerobe, is able to grow by anaerobic respiration. CTX is a major virulence factor of V. cholerae, and its production is highly promoted during anaerobic growth using trimethylamine N-oxide (TMAO) as an alternative electron acceptor (45). The V. cholerae strains from environmental samples lacked the torA operon, which could lead to an inability to conduct TMAO respiration and thus produce CTX. However, the TMAO metabolic signaling pathway was not observed in the migratory bird V. cholerae strain genomes. Thus, it is likely that this metabolic pathway is not present in all V. cholerae strains. However, strain C16-2-29 shared very similar genomic attributes with the reference pandemic strains, unlike the other migratory bird V. cholerae strains. Specifically, the strain harbored the ability to encode a complete TMAO metabolic pathway, despite the apparent inability to produce CTX.
It should be noted that we were not able to exclude geographical factors that can influence bacterial evolution or differences in genomic contents. Strains that were isolated from the same host species but that exhibit differences in two-component systems will respond to external stresses differently. We speculated that conditionally pathogenic V. cholerae would unlike to evolve into a strain with CTX over a short period. Accordingly, the cholera pandemic strains have all been confirmed to be clonally related (2). The genomes of most strains belonging to group C contained fewer dispensable genes based on core-/pan-genomic analysis. A notable exception was the genome of TSY216, which contained more strain-specific genes. In addition to harboring two pairs of chromosomes, strain TSY216 also harbors a giant replicon that could explain its high strain-specificity relative to the others (46).
We additionally searched for other virulence genes in the genomes of V. cholerae from migratory birds by comparing them against the VFDB database. While the genomes contained some of the pathway components to produce the RTX toxin, they lacked the rtxA gene. rtxA deletion can cause a significant reduction in RTX toxin production (47). All strain genomes also encoded the T6SS system, while the V. cholerae genomes from migratory birds also harbored complete T6SS pathways based on KEGG analysis. T6SS was first discovered in V. cholerae in 2006 (48). Several of the V. cholerae groups encoded the T6SS system, and thus T6SS is probably a specific secretion system of V. cholerae. Only one of the three V. metschnikovii strain genomes contained the gene encoding the spike protein for T6SS. The V. metschnikovii strain encoding the spike protein of T6SS was isolated from migratory bird epidemic materials, while the V. metschnikovii strains that did not encode the spike protein of T6SS were isolated from healthy migratory bird feces in the second year. Importantly, T6SS without a spike protein does not confer pathogenic ability. Nevertheless, these results indicate that T6SS systems could have begun to evolve in V. metschnikovii strains. The M19X and M13F strains were closely related in the core genome phylogenetic analysis, although strain M13F did not encode a complete T6SS structure. Furthermore, strain M13F contained the HS I-I system that could activate T6SS, while M19X did not. Thus, different induction environments might activate the expression of HS I-I.
Phylogenetic analysis indicated that the V. cholerae strains from migratory birds (including strain NCTC30) were largely in group B, with the exception of C16-2-29. Strain NCTC30 was isolated from a World War One soldier with diarrhea (49). The strain harbored the T3SS-1 system, conferring it cytotoxicity (50). However, all of the migratory bird V. cholerae strains lacked the T3SS-1 system.
MLST analysis revealed a high degree of diversity among the strains that were evaluated. Indeed, these analyses indicated that the migratory bird strains harbored numerous new V. cholerae alleles and STs that have not been previously observed in databases. A total of seven different loci were used for MLST analysis of the environmental V. cholerae strains (excluding strain C16-2-29), while MLST analysis of the pandemic strains and C16-2-29 was performed using eight different loci (Supplementary Table 2). The combined phylogenetic analyses indicated that C16-2-29 was more closely related to the pandemic strains than to the strains isolated from the migratory birds.
In contrast to other migratory bird V. cholerae strains, C16-2-29 was classified within group A. The primary difference between the genome of this strain and other migratory bird V. cholerae strains was differences in the sequence of the Cbb3-type cytochrome oxidase. Accordingly, the genetic relationship between strain C16-2-29 and strains from group C was low. The COG group, “R,” functional genes were specifically enriched in the C16-2-29 strain relative to the others. In addition, C16-2-29 harbored a complete biosynthesis pathway for siderophore group non-ribosomal peptides based on KEGG annotations, while the other strains did not. The C16-2-29 strain was isolated from the Wuliangsuhai area of the Inner Mongolia autonomous region of China in 2017, while the other migratory bird V. cholerae strains were isolated from the Chifeng area of the Inner Mongolia autonomous region of China in 2018. Consequently, it is unclear whether differences in geographic location influence the evolution of V. cholerae migratory bird strain genomes.
Transmissible elements similar to plasmids were not found integrated into the V. cholerae and V. metschnikovii genomes from migratory bird strains, indicating little chance of transmitting and spreading resistance genes. Rather, inherent resistance mechanisms were identified on the genomic chromosomes. For example, efflux pump-based inhibition is likely to be a viable strategy to overcome antibiotic resistance for V. cholerae and V. metschnikovii strains present in migratory birds. An important pathogenic characteristic to identify is whether V. cholerae can form biofilms based on complete formation pathways. All of the strains analyzed here exhibited this capacity, except for the environmental strains and those from migratory birds. Moreover, the biofilm formation capacity of the V. metschnikovii strains was the same as the environmental strains and V. cholerae strains from the migratory birds, wherein the basement protein-forming gene RmbA was absent. Thus, biofilms formed by these strains might be thinner than those of group C, or they may not develop a three-dimensional structure (51, 52). In addition, CTXϕ phage has been observed in biofilms (53), although it is yet unclear whether a relationship exists between biofilm thickness and the presence of CTXϕ phage.
Overall, our study demonstrates that V. cholerae strains isolated from migratory birds do not exhibit genomic features consistent with an ability to cause pandemics, or otherwise be pathogenic. Specifically, these strains were identified as non-O1/O139 serotypes and only conditional pathogens. In addition, we document the first isolation of V. metschnikovii strains with complete T6SS pathways. Both V. cholerae and V. metschnikovii strains may cause intestinal discomfort in migratory birds through the activities of T6SS and hemolysin. In contrast, all strains with an ability to form biofilms were identified in aquatic plant or animal intestine environments.
Data Availability Statement
The data presented in the study are deposited in the FigShare repository: https://doi.org/10.6084/m9.figshare.14417870.v2.
Ethics Statement
All migratory bird stool samples were collected under the supervision by the Wild Animal Sources and Diseases Inspection Station, National Forestry and Grassland Bureau of China, and did not cause any harm to the animals. All migratory bird epidemic material samples were provided by the local animal disease prevention and control center for bacteriological examination. The experimental protocol was established, according to the ethical guidelines of Helsinki Declaration and was approved by the Laboratory Animal Welfare and Ethics Committee of the Institute of Military Veterinary Science, the Academy of Military Medical Sciences (AMMS - 11 - 2020 - 11).
Author Contributions
PC and X-JG conceived, directed, and carried out the study. L-WZ, DC, L-HX, JJ, YS, and G-JL prepared samples for sequence analysis. XJ, J-yG, and LZ acquired samples and analyzed the data. All authors have read and approved the final manuscript.
Funding
Publication costs are also funded by the National Key Research and Development Programme of China (No. 2016YFD0501305). Funding for study design, data collection, and data generation was provided by the National Science and Technology Major Project of China (Grant agreement 2018ZX10733402).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to members of Dali Nuoer Lake protected area, Chifeng city, Inner Mongolia autonomous region, China, which help for sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.638820/full#supplementary-material
Abbreviations
PCR, Polymerase chain reaction; MLST, Multilocus sequence typing; ARDB, Antibiotic Resistance Genes Database; VFDB, virulence factor database; CARD, Comprehensive Antibiotic Research Database; COG, Cluster of Orthologous Groups of proteins; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; NR, Non-Redundant Protein Database. T6SS, Type VI secretory system; T3SS, Type III secretory system; CTX, cholera toxin.
References
1. Rabbani GH, Greenough WB. Food as a vehicle of transmission of cholera. J Diarrhoeal Dis Res. (1999) 17:1–9.
2. Hu D, Liu B, Feng L, Ding P, Guo X, Wang M, et al. Origins of the current seventh cholera pandemic. Proc Natl Acad Sci USA. (2016) 113:E7730–9. doi: 10.1073/pnas.1608732113
3. Laviad-Shitrit S, Izhaki I, Arakawa E, Halpern M. Wild waterfowl as potential vectors of Vibrio cholerae and Aeromonas species. Trop Med Int Health. (2018) 23:758–64. doi: 10.1111/tmi.13069
4. Miyasaka J, Yahiro S, Arahira Y, Tokunaga H, Katsuki K, Kudo YH. Isolation of Vibrio parahaemolyticus and Vibrio vulnificus from wild aquatic birds in Japan. Epidemiol Infect. (2006) 134:780–5. doi: 10.1017/S0950268805005674
5. Dijk JG, Verhagen JH, Wille M, Waldenström J. Host and virus ecology as determinants of influenza A virus transmission in wild birds Curr Opin Virol. (2018) 28:26–36. doi: 10.1016/j.coviro.2017.10.006
6. Allarda SM, Callahan MT, Bui A, Ferelli AMC, Chopyk J, Sapkota AR, et al. Creek to table: tracking fecal indicator bacteria, bacterial pathogens, and total bacterial communities from irrigation water to kale and radish cropsSci. Total Environ. (2019) 666:461–71. doi: 10.1016/j.scitotenv.2019.02.179
7. Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. (2005) 15:589–94. doi: 10.1016/j.gde.2005.09.006
8. Verma SK, Singh L. Novel universal primers establish identity of an enormous number of animal species for forensic application. Molecular Ecology Notes. (2003) 3:28–31. doi: 10.1046/j.1471-8286.2003.00340.x
9. Cao J, Xu J, Zheng Q, Yan P. Rapid detection of Vibrio metschnikovii in aquatic products by real-time PCR. Folia Microbiol. (2010) 55:607–13. doi: 10.1007/s12223-010-0098-2
10. Awasthi SP, Asakura M, Neogi SB, Hinenoya A, Ramamurthy T, Yamasaki S, et al. Development of a PCR-restriction fragment length polymorphism assay for detection and subtyping of cholix toxin variant genes of Vibrio cholerae. J Med Microbiol. (2014) 63(Pt 5):667–73. doi: 10.1099/jmm.0.070797-0
11. Li F, Du P, Li B, Ke C, Chen A, Chen J, et al. Distribution of virulence-associated genes and genetic relationships in non-O1/O139 Vibrio cholerae aquatic isolates from China. Appl Environ Microbiol. (2014) 80:4987–92. doi: 10.1128/AEM.01021-14
12. Nandi B, Nandy RK, Mukhopadhyay S, Nair GB, Shimada T, Ghose AC. Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein OmpW. J Clin Microbiol. (2000) 38:4145–51. doi: 10.1128/JCM.38.11.4145-4151.2000
13. Alam M, Sultana M, Nair GB, Sack RB, Sack DA, Siddique AK, et al. Toxigenic Vibrio cholerae in the aquatic environment of Mathbaria, Bangladesh. Appl Environ Microbiol. (2006) 72:2849–55. doi: 10.1128/AEM.72.4.2849-2855.2006
14. Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. (2010) 20:265–72. doi: 10.1101/gr.097261.109
15. Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. (2008) 24:713–4. doi: 10.1093/bioinformatics/btn025
16. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Computat Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
17. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. (2009) 19:1117. doi: 10.1101/gr.089532.108
18. Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implicat Finding Sequence Motifs Regulatory Regions Nucleic Acids Res. (2001) 29:2607–18. doi: 10.1093/nar/29.12.2607
19. Saha S, Bridges S, Magbanua ZV, Peterson DG. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. (2008) 36:2284–94. doi: 10.1093/nar/gkn064
20. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research. (1999) 27:573. doi: 10.1093/nar/27.2.573
21. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. (1997) 25:955–64. doi: 10.1093/nar/25.5.955
22. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. (2007) 35:3100–8. doi: 10.1093/nar/gkm160
23. Gardner PP, Daub J, Tate JG, Kolbe DL, Lindgreen S, Wilkinson AC, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. (2009) 37:136–40. doi: 10.1093/nar/gkn766
24. Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. (2009) 25:1335–7. doi: 10.1093/bioinformatics/btp157
25. Hsiao W, Wan I, Jones SJ, Brinkman FS. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics. (2003) 19:418–20. doi: 10.1093/bioinformatics/btg004
26. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucl Acids Res. (2011) 39:347–52. doi: 10.1093/nar/gkr485
27. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. (2007) 35:52–7. doi: 10.1093/nar/gkm360
28. Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. (2015) 43:261–9. doi: 10.1093/nar/gku1223
29. Ashburner M, Ball CA, Blake JA, Botsein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontol ConsortiumNature Genet. (2000) 25:25–9. doi: 10.1038/75556
30. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. (2004) 32:277–80. doi: 10.1093/nar/gkh063
31. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. (2006) 34:354–7. doi: 10.1093/nar/gkj102
32. Li W, Jaroszewski L, Godzik A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics. (2002) 18:77–82. doi: 10.1093/bioinformatics/18.1.77
33. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. (2009) 37:233–8. doi: 10.1093/nar/gkn663
34. Petersen TN, Brunak S, von Heijne GV, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods. (2011) 8:785–6. doi: 10.1038/nmeth.1701
35. Eichinger V, Nussbaumer T, Platzer A, Jehl MA, Arnold R, Rattei T. Effective DB-updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. (2016) 44:669–74. doi: 10.1093/nar/gkv1269
36. Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. (2012) 40:641–5. doi: 10.1093/nar/gkr989
37. Liu B, Pop M. ARDB-antibiotic resistance genes database. Nucleic Acids Res. (2009) 37:443–7. doi: 10.1093/nar/gkn656
38. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. (2004) 5:R12. doi: 10.1186/gb-2004-5-2-r12
39. Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J. Cholera. Lancet. (2017) 390:1539–49. doi: 10.1016/S0140-6736(17)30559-7
40. Kumar P, Thulaseedharan A, Chowdhury G, Ramamurthy T, Thomas S. Characterization of novel alleles of toxin co-regulated pilus A gene (tcpA) from environmental isolates of Vibrio cholerae. Curr Microbiol. (2011) 62:758–63. doi: 10.1007/s00284-010-9774-3
41. Skorupski K, Taylor RK. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol. (1999) 31:763–71. doi: 10.1046/j.1365-2958.1999.01215.x
42. Yu RR, DiRita VJ. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol Microbiol. (2002) 43:119–34. doi: 10.1046/j.1365-2958.2002.02721.x
43. Ghosh C, Nandy RK, Dasgupta SK, Nair GB, Hall RH, Ghose AC. A search for cholera toxin (CT), toxin coregulated pilus (TCP), the regulatory element ToxR and other virulence factors in non-01/non-0139 Vibrio cholerae. Microb Pathog. (1997) 22:199–208. doi: 10.1006/mpat.1996.0105
44. Feng L, Reeves PR, Lan R, Ren Y, Gao C, Zhou Z, et al. A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS ONE. (2008) 3:e4053. doi: 10.1371/journal.pone.0004053
45. Oh YT, Park Y, Yoon MY, Bari W, Go J, Min KB, et al. Cholera toxin production during anaerobic trimethylaminen-oxide respiration is mediated by stringent response in Vibrio cholerae. J Biol Chem. (2014) 289:13232–42. doi: 10.1074/jbc.M113.540088
46. Okada K, Natakuathung W, Na-Ubol M, Roobthaisong A, Wongboot W, Maruyama F, et al. Characterization of 3 megabase-sized circular replicons from Vibrio cholerae. Emerg Infect Dis. (2015) 21:1262–63. doi: 10.3201/eid2107.141055
47. Dolores J, Satchell KJ. Analysis of Vibrio cholerae genome sequences reveals unique rtxA variants in environmental strains and an rtxA-null mutation in recent altered El Tor isolates. mBio. (2013) 4:e00624. doi: 10.1128/mBio.00624-12
48. Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, et al. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. (2006) 103:1528–33. doi: 10.1073/pnas.0510322103
49. Dorman MJ, Kane L, Domman D, Turnbull JD, Cormie C, Fazal MA, et al. The history, genome and biology of NCTC 30: a non-pandemic Vibrio cholerae isolate from World War One. Proc R Soc B. (2019) 286:2018–25. doi: 10.1098/rspb.2018.2025
50. Li S, Zhang Z, Pace L, Lillehoj H, Zhang S. Functions exerted by the virulence-associated type-three secretion systems during Salmonella enterica serovar Enteritidis invasion into and survival within chicken oviduct epithelial cells and macrophages. Avian Pathol. (2009) 38:97–106. doi: 10.1080/03079450902737771
51. Mattei MR, Frunzo L, D'Acunto B, Pechaud Y, Pirozzi F, Esposito G. Continuum and discrete approach in modeling biofilm development and structure: a review. J Math Biol. (2018) 76:945–1003. doi: 10.1007/s00285-017-1165-y
52. Tolker-Nielsen T. Biofilm development. Microbiol Spectr. (2015) 3:MB-001–2014. doi: 10.1128/microbiolspec.MB-0001-2014
Keywords: Vibrio cholerae, Vibrio metschnikovii, comparative genomics, migratory bird, pathogenic
Citation: Zheng L, Zhu L-W, Jing J, Guan J-y, Lu G-J, Xie L-H, Ji X, Chu D, Sun Y, Chen P and Guo X-J (2021) Pan-Genome Analysis of Vibrio cholerae and Vibrio metschnikovii Strains Isolated From Migratory Birds at Dali Nouer Lake in Chifeng, China. Front. Vet. Sci. 8:638820. doi: 10.3389/fvets.2021.638820
Received: 07 December 2020; Accepted: 04 May 2021;
Published: 31 May 2021.
Edited by:
Anuwat Wiratsudakul, Mahidol University, ThailandReviewed by:
Bita Bakhshi, Tarbiat Modares University, IranDuochun Wang, Chinese Center for Disease Control and Prevention (China CDC), China
Bo Pang, National Institute for Communicable Disease Control and Prevention, China
Copyright © 2021 Zheng, Zhu, Jing, Guan, Lu, Xie, Ji, Chu, Sun, Chen and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Chen, MTI0MzM3NzUyM0BxcS5jb20=; Xue-Jun Guo, eHVlanVuZ0B5YWhvby5jb20=
†These authors have contributed equally to this work