
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 26 August 2020
Sec. Veterinary Pharmacology and Toxicology
Volume 7 - 2020 | https://doi.org/10.3389/fvets.2020.00580
 Diyala S. Shihadih1Todd R. Harris1Sean D. Kodani1Sung-Hee Hwang1
Diyala S. Shihadih1Todd R. Harris1Sean D. Kodani1Sung-Hee Hwang1 Kin Sing Stephen Lee1,2
Kin Sing Stephen Lee1,2 Vengai Mavangira3Briana Hamamoto4
Vengai Mavangira3Briana Hamamoto4 Alonso Guedes4,5
Alonso Guedes4,5 Bruce D. Hammock1
Bruce D. Hammock1 Christophe Morisseau1*
Christophe Morisseau1*The veterinary pharmacopeia available to treat pain and inflammation is limited in number, target of action and efficacy. Inhibitors of soluble epoxide hydrolase (sEH) are a new class of anti-inflammatory, pro-resolving and analgesic drugs being tested in humans that have demonstrated efficacy in laboratory animals. They block the hydrolysis, and thus, increase endogenous concentrations of analgesic and anti-inflammatory signaling molecules called epoxy-fatty acids. Here, we screened a library of 2,300 inhibitors of the sEH human against partially purified feline, canine and equine hepatic sEH to identify inhibitors that are broadly potent among species. Six very potent sEH inhibitors (IC50 < 1 nM for each enzyme tested) were identified. Their microsomal stability was then measured in hepatic extracts from cat, dog and horse, as well as their solubility in solvents suitable for the formulation of drugs. The trans-4-{4-[3-(4-trifluoromethoxy-phenyl)-ureido]-cyclohexyloxy}-benzoic acid (t-TUCB, 1,728) appears to be the best compromise between stability and potency across species. Thus, it was selected for further testing in veterinary clinical trials of pain and inflammation in animals.
Pain management is central to the effective treatment of both human and veterinary patients. Pain control improves both the outcomes of disease and the quality of life (1). Currently, the main class of veterinary drugs used to reduce pain and inflammation in animals, including horses, cats, and dogs, are non-steroidal anti-inflammatory drugs (NSAIDs), cyclooxygenase inhibitors (COXIBs) and corticosteroids. As well as being marginally effective in some species, these categories of pharmacological drugs have dose dependent deleterious side effects that limits their use, especially in the management of chronic pain (2). For example, chronic laminitis in horses is an extremely painful condition that too often leads to euthanasia due to the inability of currently available analgesics to adequately control pain (3). Osteoarthritis is another chronic painful condition, especially in elderly cats and dogs, that can be difficult to manage with currently available analgesics. It cannot be treated with NSAIDs or COXIBs chronically because of adverse side effects, such as liver toxicity and gastric ulceration (4, 5). These challenges underline the urgent need for novel pain medication in veterinary medicine.
In vertebrates, the arachidonic acid (AA) cascade plays a major role in the regulation of multiple biological processes, including pain and inflammation (6). While the cyclooxygenase (COX) and lipoxygenase (LOX) branches of the cascade are targets of many pharmaceuticals on the market, including NSAIDs and COXIBs, the CYP450 branch of the cascade has yet to be targeted in the same manner. Within the P450 branch, epoxide metabolites of AA, called epoxy-eicosatrienoic acids (EETs), have anti-inflammatory and analgesic effects (7). However, the endogenous hydrolysis of the EETs to their corresponding diols by soluble epoxide hydrolase (sEH) reduces their biological activity (8). Both in vitro and in vivo studies have demonstrated that the modulation of chronic inflammation and neuronal pain by EETs is inversely dependent on the extent of their hydrolysis by sEH (7–9). Thus, the use of sEH inhibitors to maintain high in vivo levels of EETs is a promising pharmacological approach to treat pain and inflammation (10). Consequently, sEH inhibitors are in development for the treatment of neuropathic pain in human patients (9). In addition to their use in humans, sEH inhibitors may become suitable analgesics for use in veterinary medicine (11–14).
Preliminary data obtained with compounds optimized for the human sEH show that sEH inhibition has the potential to reduce pain in dogs with osteoarthritis (14), and horses with chronic laminitis (12). However, sEH inhibitors optimized for use in primates and rodents are typically not sufficiently potent inhibitors or are metabolized too rapidly for chronic use in other mammals (15). Therefore, a library of 2,300 sEH inhibitors developed for the human enzyme was screened with partially purified feline, canine and equine hepatic sEH to identify inhibitors that are broadly potent among species. In addition, microsomal stability of potent compounds was tested, as well as the solubility of the selected inhibitor in solvents typically used in the formulation of drugs.
(3-Phenyl-oxiranyl)-acetic acid cyano-(6-methoxy-naphthalen -2-yl)-methyl ester (PHOME), cyano(6-methoxynaphthalen-2-yl)methyl phenyl butyrate (CMNPB), cyano(6-methoxynaphthalen-2-yl)methyl ((3-phenyloxiran-2-yl)methyl) carbonate (CMNPC), and 4-((4-(3-(4-(trifluoromethoxy)phenyl)ureido)cyclohexyl)oxy)benzoic acid (t-TUCB) were synthesized previously in the laboratory (16, 17). Paraoxon and other chemicals were obtained from Sigma or Fisher Scientific and used without further purification. The library of human sEH inhibitors was previously prepared in the laboratory (18). The library was prepared in 2-mL deep-well polypropylene 96-well assay plates. Every compound was prepared at 10 mM in dimethyl sulfoxide (DMSO). Only compounds completely soluble at 10 mM in DMSO were included in the library. In each plate, the wells in the first column contained only DMSO to serve as controls. In the remainder of the plate, we dispensed one compound per well, with 88 compounds total per plate. Twenty-six plates were created with different chemicals for a total of 2,288 compounds. The plates were tightly sealed with EVA copolymer sealing mats. The plates were then sealed in a 2-mil. thick plastic bag, to avoid condensation, and stored at −20°C until use. Upon usage, the plates were allowed to warm-up to room temperature overnight before removal from the plastic bag. Using a robotic pipetting station (Quadra 96 – 96 well-automated pipettor; Tomtec, Hamden, CT), each well was first mixed and the compound solutions were diluted 10-fold in DMSO (down to 1 mM) and then further diluted in the appropriate buffer and transferred to black 96-well plates.
Fresh liver tissues samples (5–30 g) were collected promptly after death from healthy adult cat, dog and horse euthanized for reasons unrelated to this study, and whose bodies were given to science by the owners in line with federal regulations and protocol approved by the Institutional Animal Care and Use Committee at University of California Davis. Similarly, tissues from the other animals tested herein were collected from healthy animals immediately after being slaughter at the university abattoir. Because no animals were directly used or killed for this study, sample collections were exempted from approval by the Institutional Animal Care and Use Committee of the University of California Davis and Michigan State University. Samples were taken from livers that upon gross evaluation did not display any sign of clinical disease. The tissues were flash-frozen in liquid nitrogen following removal, and then kept at −80°C until used. After thawing on ice, the samples were homogenized in chilled sodium phosphate buffer (20 mM pH 7.4) containing 5 mM EDTA, 1 mM DTT and 1 mM PMSF (tissue weight:buffer volume ratio 1:4). These homogenates were centrifuged at 100,000 g for 60 min at 4°C. The supernatant (the cytosolic fraction) was separated from the pellet, aliquoted and flash frozen in liquid nitrogen, then kept frozen at −80°C until usage.
The sEH activity from the cat, dog and horse liver was further purified to eliminate contaminating esterase activity that could interfere with the screening fluorescent assay (16). The cytosolic fraction was loaded on a Q-Sepharose column (100 mL) equilibrated with sodium phosphate buffer (20 mM pH 7.4). After washing of the bound proteins, the sEH activity was eluted with a step gradient of NaCl (0–1 M). The fractions containing the sEH activity were pooled and concentrated using a Centriprep-30. The concentrates were aliquoted and flash frozen, then kept frozen at −80°C until usage. To ensure total elimination of residual esterase activity, the purified enzymes were further treated with 100 μM of Paraoxon, a non-selective pan-esterase inhibitor (19) that does not inhibit sEH (18). Protein concentration was quantified using the Pierce BCA assay (Pierce, Rockford, IL), using Fraction V bovine serum albumin (BSA) as the calibrating standard.
The sEH activity was measured using [3H]-labeled trans-diphenyl propene oxide ([3H]t-DPPO) as substrate (20), while the esterase activity was measured using CMNPB as substrate (21). For both assays, the cellular extracts were diluted in sodium phosphate buffer (0.1 M pH 7.4) containing 0.1 mg/mL of BSA. The activities were measured with a final substrates concentration of 50 μM, for 5–10 min at 37°C).
Primary screening of the library was performed with an inhibitor concentration of 100 nM using PHOME as substrate (22). To black 96-well plates containing 20 μL of 1 μM of the test-compound solutions, 150 μL of sodium phosphate buffer (0.1 M pH 7.4) containing 0.1 mg/mL of BSA were added in wells A1 to D1 (these four wells served as background control), and 150 μL of the enzyme diluted in the same buffer ([protein]final for horse sEH: 17 μg/mL; cat sEH: 99 μg/mL; dog sEH: 208 μg/mL) were added to the rest of the plate (wells E1 to H1 served as full activity control). The plates were then mixed and incubated at 30°C for 5 min. Across each plate, 30 μL of the working substrate solution (270 μL of 5 mM PHOME solution in DMSO diluted with 3,680 μL of buffer) were added quickly to yield [S]final: 50 μM. Each plate was further incubated at room temperature (horse sEH: 60 min; cat sEH: 45 min; dog sEH: 30 min). The formation of the fluorescent product, 6-methoxynaphthaldehyde (λexcitation = 330 nm, λemission = 465 nm) was measured with a Spectramax M2 spectrophotometer (Molecular Devices, Sunnyvale, CA). To ensure the validity of the high throughput screen (HTS), results from the background control wells (A1 to D1) and full activity control wells (E1 to H1) were used to calculate the signal to noise ratio and Z′ factor (23).
Compounds that gave more than 90% inhibition for one of the three enzymes tested during the primary screen were collected and distributed in five new 96-well plates (88 compounds per plate, 390 compounds in total). The library of selected compounds was screened with [I] = 100 nM using PHOME as substrate, in conditions similar to the primary screening with the exception of the plate reading to identify possible false positive. Following substrate addition, the activity was immediately measured at 30°C kinetically for 15 min by measuring the fluorescent signal (λexcitation = 330 nm, λemission = 465 nm, 30°C) every 30 s.
The IC50 is the concentration of an inhibitor that reduces the sEH activity by 50%. The IC50 values reported herein were determined using either a fluorescent based assay (CMNPC as substrate) for purified enzyme preparations or a radioactive based assay ([3H]t-DPPO as substrate) for crude enzyme preparations (20). The fluorescent assay was used with the purified proteins. The enzymes ([E]final for horse sEH: 17 μg/mL; cat sEH: 99 μg/mL; dog sEH: 208 μg/mL) were incubated at 30°C with the inhibitors ([I]final = 0.4–1,000 nM) for 5 min in 100 mM sodium phosphate buffer (200 μL pH 7.4) containing 0.1 mg/mL of BSA and 1% of DMSO. The substrate (CMNPC) was then added ([S]final = 5 μM). Activity was assessed by measuring the appearance of the fluorescent 6-methoxynaphthaldehyde product (λex = 330 nm, λem = 465 nm) every 30 s for 10 min at 30°C. The radioactive fluorescent assay was used with the liver crude extracts. Extracts were incubated with inhibitors ([I]final = 1–1,000 nM) at 30° C for 5 min in sodium phosphate buffer (pH 7.4) containing 0.1 mg/mL of BSA and 1% of DMSO, prior to substrate introduction ([3H]t-DPPO [S]final: 50 μM; ~12,000 dpm/assay). The enzymes were incubated at 30°C for 10 min, and the reaction was quenched by the addition of 60 μL methanol and 200 μL isooctane, which extracts the remaining epoxide from the aqueous phase. The activity was followed by measuring the quantity of radioactive diol formed in the aqueous phase using a liquid scintillation counter (Tri-Carb 2810TR, Perkin Elmer, Waltham, MA). Under the conditions used, rates were linear with both time and enzyme concentration and resulted in at least 5% but no more than 30% hydrolysis of the substrate. Assays were performed in triplicate. The IC50 values were calculated from at least five different concentrations, each in triplicate, with at least two on either side of 50% activity mark.
The stability of the sEH inhibitors was determined using liver microsomes as described (24). The inhibitors ([I]final = 1 μM) were added to a suspension of liver microsomes ([protein]final= 1 mg/mL) in potassium phosphate buffer (0.1M pH7.4) containing 3 mM MgCl2 and 1 mM EDTA. After 5 min incubation at 37°C, the reaction was started by the addition of an NADPH generating system (buffer was added in the control tubes) (24). After 30 min at 37°C, the reaction was stopped by the addition of one volume of methanol containing CUDA (200 nM) as surrogate. The amount of remaining inhibitor was determined by LC/MS/MS. The stability is reported as a percentage of compound remaining after 30 min in these conditions. Results are triplicate average ± standard deviation.
Mass spectrometry analyses were performed using a Waters Quattro Premier triple quadrupole tandem mass spectrometer (Micromass, Manchester, UK) interfaced to an electrospray ionization (ESI) source. The MS was coupled with a Waters Acquity UPLC (Waters, Milford, MA, USA). A Varian Pursuit5 C18 RP HPLC column (150 mm × 2.1 mm, particle size 5 μm) was used to separate the analytes. The ESI was performed following HPLC in the positive mode at 2.51 kV capillary voltage. The source and the desolvation temperatures were set at 120 and 300°C, respectively. Cone gas (N2) and desolvation gas (N2) were maintained at flow rates of 10 and 700 L/h, respectively. Dwell time was set to 0.1 s. A regression curve for each compound was obtained from at least six different concentrations of standard stock solutions (R2 > 0.99).
Solubility was determined by shaking vigorously excess t-TUCB (0.25–2 grams) in 10 mL of 11 different solvents at 30 or 40°C for 24 h in glass vials. The un-dissolved compounds were filtered through a 0.22 μm centrifuge filter at 30 or 40°C and the supernatant further diluted 10-fold with methanol. The concentration of t-TUCB in the samples was determined by LC/MS/MS analysis as previously described (15).
Liver represents an excellent source of sEH because it is a large organ and the sEH activity is the highest in this organ across species (25). Liver cytosolic fractions are commonly used to measure sEH activity with the radioactive substrate [3H]t-DPPO (20). However, this substrate is not appropriate for an HTS of a library of compounds due to the low throughput of the technique. Unfortunately, the sEH fluorescent substrate developed for the HTS, PHOME, is not suitable for usage with crude lysate since it can also be hydrolyzed rapidly by esterases and can react chemically and enzymatically with glutathione (22). The glutathione and these enzymes are highly abundant in the liver, thus yielding a very high background that cannot be eliminated totally with chemical treatment. Esterase activity was measured using CMNPB, a fluorescent substrate that is structurally similar to the EH substrate but does not contain the epoxide moiety. To eliminate the glutathione cofactor and to reduce the esterase activity, a partial purification of the liver sEH was performed. A simple gravity anionic exchange column allows the removal of all the glutathione and over 90% of the unwanted esterase activity while retaining more than 67% of the targeted sEH activity, resulting in a 6.5- to 9.9-fold increase in specific activity (Table 1). The remaining small amount of esterase activity was eliminated totally with treatment of the partially purified preparation with paraoxon, a non-selective serine esterase inhibitor (19) that does not inhibit sEH. These preparations were used for the screening of the library of compounds.
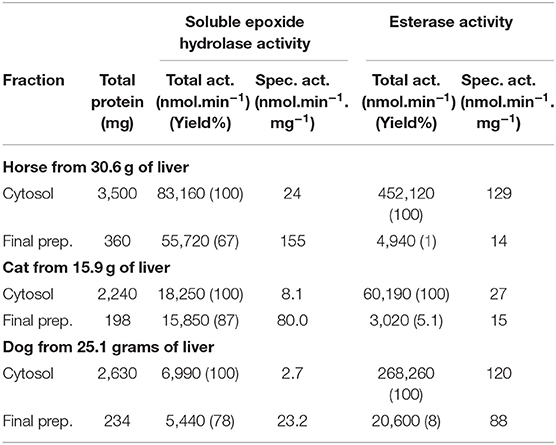
Table 1. Partial purification of sEH activity from horse, cat and dog liver to remove esterase activity.
The compound library was initially screened using an endpoint assay. For all three species, the amount of enzyme and time of incubation were optimized to ensure linear substrate hydrolysis over the period tested, while limiting the total substrate conversion to under 20% as recommended in the literature (22). Using the optimized conditions described in the method section, a 26-plate library (2,288 compounds in total) was screened at a final inhibitor concentration of 100 nM. To determine the appropriateness and robustness of the assays for HTS, the Z′ factor and S/N were calculated (Table 2) (23). For all three enzymes, Z' values were between 0.7 and 0.8 and the S/N value > 80. This demonstrated that the assay has a large window between samples and blank signals and thereby confirms that it was suitable for HTS. For each enzyme tested, between 4 and 15% of compounds in the targeted library were positive hits (> 90% inhibition). In total, 390 compounds were found to strongly inhibit sEH of at least one of the animal species tested. Occasionally, false positives can occur when the tested inhibitor quenches the fluorescence of the enzymatic product. Thus, these positive hits were reorganized in five plates, and the potency of this subset of compounds was tested against the three enzymes at 100 nM. The plates were read in a kinetic mode to eliminate false positives. Again, both the Z' and S/N factors were high for all three enzymes (Table 2), underlying the robustness of the assay. For most of the compounds tested, the potency of the primary screening was confirmed in the secondary screening (Table S1) indicating the accuracy of the primary screening.
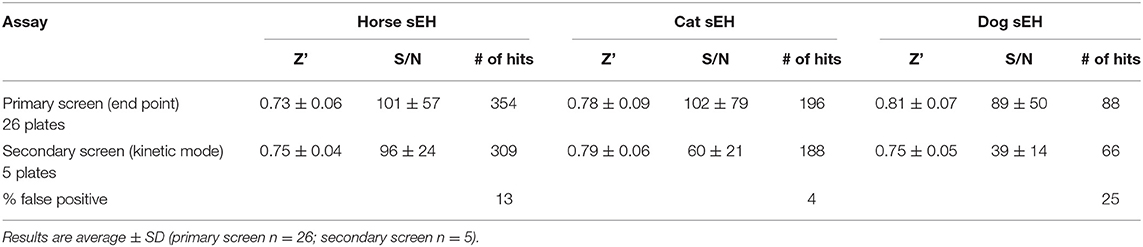
Table 2. Screening assay performances.
Narrowing the focus to compounds that inhibit sEH in all three species, inhibitors yielding at least 80% inhibition for each enzyme were selected, and their inhibitory potencies (IC50) were determined (Table S2). Consistent with previous observations, the most potent inhibitors across species (Table S2) generally contained a central urea pharmacophore (17). This is due to the relatively strong H-bonding interaction between this pharmacophore and the enzyme active site residues that are conserved across species. On one side of the urea functional group, the presence of an adamantyl or substituted phenyl group yielded a potent inhibitor for all three enzymes. On the opposite side of the urea, alkyl or cyclohexyl were preferred, while phenyl or piperidine groups did not yield potent inhibitors of any of the enzymes tested. The six most potent inhibitors for all three animal species were selected for further testing (Table 3).
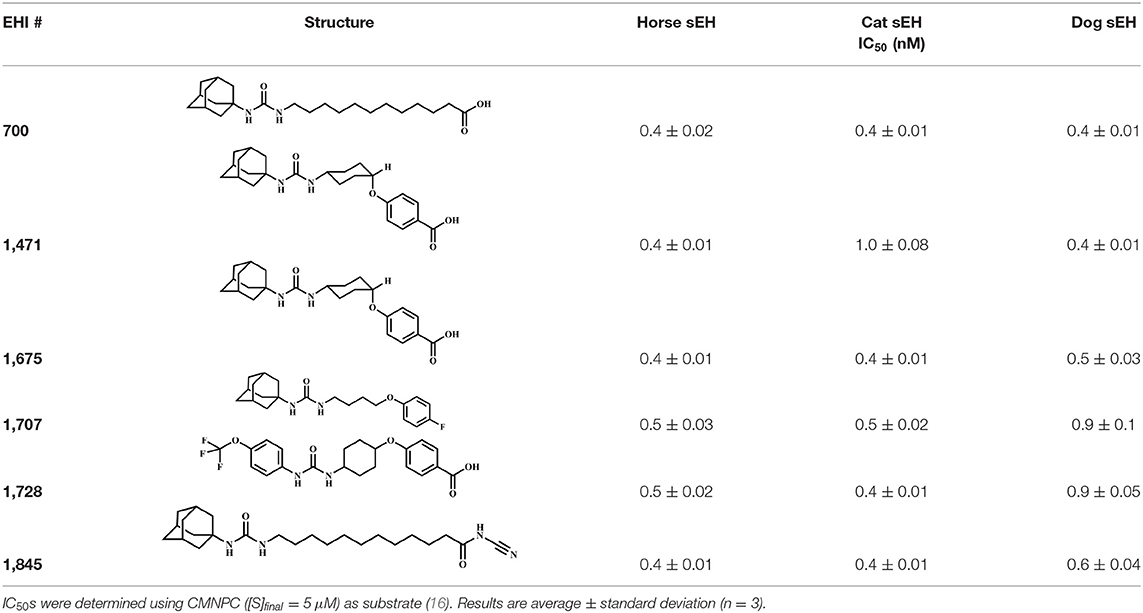
Table 3. Inhibition potency of selected compounds for the horse, cat, and dog sEH.
Since sEH inhibitors could also be beneficial to treat animals other than horses, cats and dogs, especially farm animals, the potency of the selected six inhibitors was tested against the sEH activity of several common animals (Table 4) and compared to 1,770 (a.k.a. TPPU) a compound with demonstrated efficacy in rodent models. Except for 1,845, the selected compounds were potent inhibitors of the sEH from the other tested species and so are potentially useful in the treatment of diverse mammalian species.
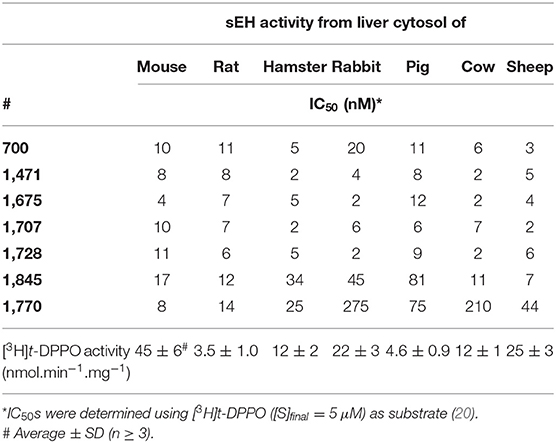
Table 4. Inhibition potency of the selected compounds for the sEH from other animals of veterinary medicine importance.
In addition to their on-target potency, inhibitors also need to be soluble and metabolically stable to be clinically useful. cLogP, a measure of both solubility and bioavailability, and microsomal stability, an in vitro measure of metabolism, were previously shown to influence greatly the in vivo potency of sEH inhibitors in rodents (26). The cLogP, and microsomal stability were determined for all six selected compounds (Table 5). The cLogPs were calculated using ChemBioDraw (v.16.0, CambridgeSoft). In their neutral form, all inhibitors have a cLogP above 4.5, suggesting limited solubility and possibly bioavailability (27). For 700 (a.k.a. AUDA), 1,471 (a.k.a. t-AUCB), 1,675, and 1,728 (a.k.a. t-TUCB), the charged form (a.k.a. base form) of the acid function yields a significantly lower cLogP. This suggests that water solubility could be greatly increased in neutral to basic solutions.
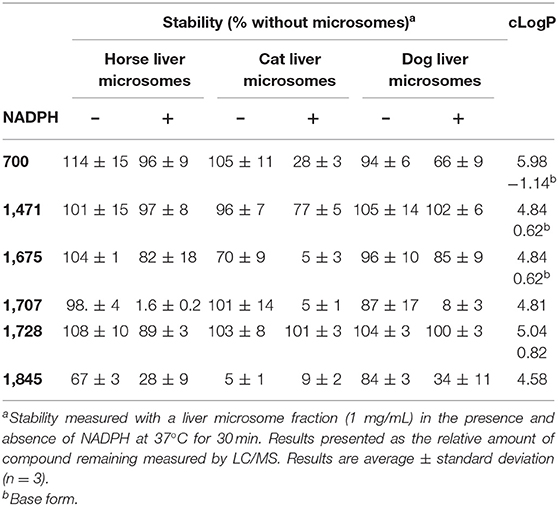
Table 5. cLogP and stability of selected compounds.
The stability of the six inhibitors (1 μM) in equine, feline and canine liver microsomes (1 mg/mL) was measured in the presence and absence of NADPH. After a 30-min incubation at 37°C, the amount of remaining inhibitor was quantified using mass spectrometry (Table 5) (28). The stability of the compounds varied widely based on structure and species. Even in the absence of NADPH, 1,845 was not stable, suggesting a non-P450 metabolism. It was metabolized faster in the cat than in the horse and dog. Because 1,845 contains an amide, its rapid metabolism probably resulted from the action of amidases that are well-known to be present in liver microsomes (29, 30). In dog and horse, 1,845 compound was further metabolized in the presence of NADPH underlying its overall instability, thus limiting its potential usage in vivo. While the other compounds were stable in absence of NADPH, their stability varied when it was added to the microsomes. While 1,707 was metabolized rapidly by the P450 s of all three species, 700, and 1,675 were quickly transformed by the cat enzymes only, while 1,471 metabolism was slower. 1,728 was the most stable compound for all three species tested.
Besides having adequate potency, availability and stability, a good drug must also be amenable to formulation in a relatively non-toxic vehicle that can be administered to the patient. Urea derivatives have generally low solubility (17), which could be in part mitigated by high potency and slow metabolism, butt formulation is still a challenge. Because 1,728 has such low solubility in water, an organic co-solvent will likely be needed to solubilize it. Thus, the solubility of 1,728 was measured in various solvents commonly used for drug formulation (Table 6) (31). While barely soluble in protic solvents, 1,728 displayed a good solubility (> 20 mg/mL) in numerous organic liquids used in the formulation of drugs such as Carbitol and polyethylene glycol (PEG). As expected, increasing the temperature from 30 to 40°C significantly increased the amount of compound in solution.
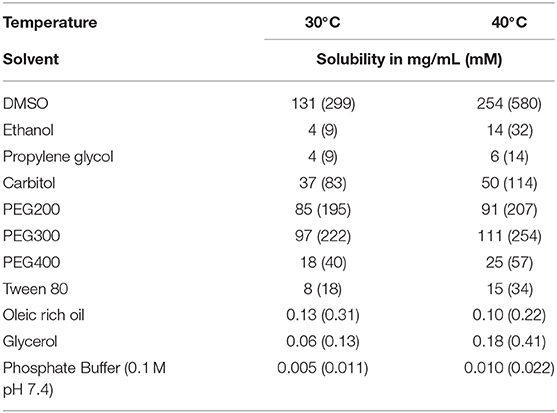
Table 6. Formulation ease: solubility of 1,728 in various solvents classically used for formulation.
From our screen of 2,300 sEH inhibitors, 1,728 is overall the best compound (high potency, good solubility, and stability) for all three species. While less stable, 1,471 is a good alternative for veterinary usage. Although the results obtained herein are only in vitro, they suggest that 1,728 will be relatively easy to formulate for dosing in animals. This compound could provide a new approach to relieve pain and inflammation in companion and farm animals through the inhibition of sEH.
Inflammation is necessary for the body to combat and survive infections and damages. However, uncontrolled inflammatory response could lead to cell death, organ damages and contribute to chronic disease such as cancer, heart disease and diabetes. Additionally, both acute and chronic inflammation cause pain as a secondary indication that can reduce quality of life. Acute inflammation causes localized pain that resolves with the injury, while chronic inflammation can result in organ damages and neuropathic pain that is difficult to treat. Inhibition of sEH has been shown to decrease inflammation as well as the inflammatory and neuropathic pain associated with it in animal models (7–10). Recent results show that sEH inhibition reduce pain in horses with chronic laminitis (11–13). The positive effect of the sEH inhibitor used in these studies, 1,728, was observed either as an additive to the standard-of-care therapy, which was marginally effective (11, 12), or by itself in horses with lipopolysaccharide (LPS)-induced radiocarpal synovitis (13). Upon dosing (0.1–1 mg/kg i.v.), there was a significant and rapid reduction in pain without observed adverse effects (11–13). In a separated study, 1,728 was also able to reduce pain perception in dogs with osteoarthritis (14). Additional promising results have been obtained in an in vitro model. Treating chondrocytes, the key cell type that maintain joint function, in vitro with EETs, the lipid signaling molecules increased through sEH inhibition in vivo, reduced inflammatory markers and was cytoprotective (14). Thus, sEH inhibition, by enhancing concentrations of EETs in vivo, could reduce synovial inflammation and subsequent pain leading to reduce synovial inflammation (14). These results indicate that the compound identified here as a potential sEH inhibitor for veterinary medicine application could be efficient in vivo.
Beside laminitis in horses and osteoarthritis in dogs, there are many possible therapeutic applications for sEH inhibitors in veterinary medicine. Mirroring human applications (10), along with providing a potential therapy for inflammation and painful diseases in animals, sEH inhibition could also have applications in the treatment of cardiovascular and respiratory diseases. For example, in cattle, the respiratory disease syndrome in feedlots and mastitis of dairy cows commonly due to bacterial infection, are characterized by severe inflammation leading to significant costs to producers in part due to antimicrobial drugs use but also loss of market shares (32). Because epoxy-fatty acids seem to play a role in cattle during mastitis (33), sEH inhibition may provide an alternative therapy for inflammation that may result in improved treatment outcomes and the reduction in the use of antimicrobials in animal agriculture. The selection of a compound, t-TUCB (1,728), an sEH inhibitor that that is potent across species and displays good physical properties, will allow researchers and veterinarians to easily study the effect of sEH inhibition in pets and farm animals.
Put together, 1,728 is overall the best sEH inhibitor (high potency, good solubility and stability) for all the animal species tested herein. This compound through the inhibition of sEH and stabilization of epoxy-fatty acids could provide a new approach to relieve pain and inflammation in companion and farm animals. Toward this end, 1,728 pharmacological properties are currently measured in several animal species toward testing it in animal clinical trials soon.
All datasets generated for this study are included in the article/Supplementary Material.
VM, BH, SK, S-HH, and AG: provide key materials. DS, TH, SK, KL, and CM: run experiments and acquire the data. TH, KL, and CM: data analysis. DS, BDH, and CM: study design. TH, SK, S-HH, KL, VM, AG, BDH, and CM: manuscript writing and editing. All authors contributed to the article and approved the submitted version.
This study was supported, in part by a grant from the National Institute of Environmental Health Sciences (NIEHS) Grant R35ES030443 and NIEHS Superfund Research Program P42 ES004699. KL is also partially supported by NIEHS R00 ES024806 and National Science Foundation DMS-1761320.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The University of California holds patents on the sEH inhibitors used in this study as well as their use to treat inflammation, inflammatory pain, and neuropathic pain. AG, BDH, and CM are inventors on some of these patents. BDH is cofounder of EicOsis L.L.C., a startup company advancing sEH inhibitors as potential therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2020.00580/full#supplementary-material
1. Epstein M, Rodan I, Griffenhagen G, Kadrlik J, Petty M, Robertson S, et al. 2015 AAHA/AAFP pain management guidelines for dogs and cats. J Am Anim Hosp Assoc. (2015) 51:67–84. doi: 10.5326/JAAHA-MS-7331
2. Cooper C, Chapurlat R, Al-Daghri N, Herrero-Beaumont G, Bruyère O, Rannou F, et al. Safety of oral non-selective non-steroidal anti-inflammatory drugs in osteoarthritis: what does the literature say? Drugs Aging. (2019) 36:15–24. doi: 10.1007/s40266-019-00660-1
3. Driessen B, Bauquier SH, Zarucco L. Neuropathic pain management in chronic laminitis. Vet Clin North Am Equine Pract. (2010) 26:315–37. doi: 10.1016/j.cveq.2010.04.002
4. McLean MK, Khan SA. Toxicology of frequently encountered nonsteroidal anti-inflammatory drugs in dogs and cats: an update. Vet Clin North Am Small Anim Pract. (2018) 48:969–84. doi: 10.1016/j.cvsm.2018.06.003
5. Bergh MS, Budsberg SC. The coxib NSAIDs: potential clinical and pharmacologic importance in veterinary medicine. J Vet Intern Med. (2005) 19:633–43. doi: 10.1892/0891-6640(2005)19[633:tcnpca]2.0.co;2
6. Sala A, Proschak E, Steinhilber D, Rovati GE. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem Pharmacol. (2018) 158:161–73. doi: 10.1016/j.bcp.2018.10.007
7. Wagner K, Vito S, Inceoglu B, Hammock BD. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. (2014) 113–5:2–12. doi: 10.1016/j.prostaglandins.2014.09.001
8. Kodani SD, Morisseau C. Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie. (2019) 159:59–65. doi: 10.1016/j.biochi.2019.01.020
9. Wagner KM, McReynolds CB, Schmidt WK, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther. (2017) 180:62–76. doi: 10.1016/j.pharmthera.2017.06.006
10. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. (2013) 53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244
11. Guedes AG, Morisseau C, Sole A, Soares JH, Ulu A, Dong H, et al. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg. (2013) 40:440–8. doi: 10.1111/vaa.12030
12. Guedes A, Galuppo L, Hood D, Hwang SH, Morisseau C, Hammock BD. Soluble epoxide hydrolase activity and pharmacologic inhibition in horses with chronic severe laminitis. Equine Vet J. (2017) 49:345–51. doi: 10.1111/evj.12603
13. Guedes AGP, Aristizabal F, Sole A, Adedeji A, Brosnan R, Knych H, et al. Pharmacokinetics and antinociceptive effects of the soluble epoxide hydrolase inhibitor t-TUCB in horses with experimentally induced radiocarpal synovitis. J Vet Pharmacol Ther. (2018) 41:230–8. doi: 10.1111/jvp.12463
14. McReynolds CB, Hwang SH, Yang J, Wan D, Wagner K, Morisseau C, et al. Pharmaceutical effects of inhibiting the soluble epoxide hydrolase in canine osteoarthritis. Front Pharmacol. (2019) 10:533. doi: 10.3389/fphar.2019.00533
15. Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, et al. Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci. (2010) 40:222–38. doi: 10.1016/j.ejps.2010.03.018
16. Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. (2005) 343:66–75. doi: 10.1016/j.ab.2005.03.041
17. Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. (2007) 50:3825–40. doi: 10.1021/jm070270t
18. Shihadih DS, Harris TR, Yang J, Merzlikin O, Lee KS, Hammock BD, et al. Identification of potent inhibitors of the chicken soluble epoxide hydrolase. Bioorg Med Chem Lett. (2015) 25:276–9. doi: 10.1016/j.bmcl.2014.11.053
19. Crow JA, Bittles V, Herring KL, Borazjani A, Potter PM, Ross MK. Inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by chlorpyrifos oxon, paraoxon and methyl paraoxon. Toxicol Appl Pharmacol. (2012) 258:145–50. doi: 10.1016/j.taap.2011.10.017
20. Morisseau C, Hammock BD. Measurement of soluble epoxide hydrolase (sEH) activity. Curr Protoc Toxicol. (2007) 4:4.23. doi: 10.1002/0471140856.tx0423s33
21. Shan G, Hammock BD. Development of sensitive esterase assays based on alpha-cyano-containing esters. Anal Biochem. (2001) 299:54–62. doi: 10.1006/abio.2001.5388
22. Wolf NM, Morisseau C, Jones PD, Hock B, Hammock BD. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal Biochem. (2006) 355:71–80. doi: 10.1016/j.ab.2006.04.045
23. Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. (1999) 4:67–73. doi: 10.1177/108705719900400206
24. Pecic S, Zeki AA, Xu X, Jin GY, Zhang S, Kodani S, et al. Novel piperidine-derived amide sEH inhibitors as mediators of lipid metabolism with improved stability. Prostaglandins Other Lipid Mediat. (2018) 136:90–5. doi: 10.1016/j.prostaglandins.2018.02.004
25. Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. (2005) 44:1–51. doi: 10.1016/j.plipres.2004.10.001
26. Liu JY, Lin YP, Qiu H, Morisseau C, Rose TE, Hwang SH, et al. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci. (2013) 48:619–27. doi: 10.1016/j.ejps.2012.12.013
27. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. (2001) 46:3–26. doi: 10.1016/s0169-409x(00)00129-0
28. Burmistrov V, Morisseau C, Lee KS, Shihadih DS, Harris TR, Butov GM, et al. Symmetric adamantyl-diureas as soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. (2014) 24:2193–7. doi: 10.1016/j.bmcl.2014.03.016
29. Biernacki M, Skrzydlewska E. Metabolism of endocannabinoids. Postepy Hig Med Dosw. (2016) 70:830–43. doi: 10.5604/17322693.1213898
30. Di L. The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr Drug Metab. (2019) 20:91–102. doi: 10.2174/1389200219666180821094502
32. Trevisi E, Zecconi A, Cogrossi S, Razzuoli E, Grossi P, Amadori M. Strategies for reduced antibiotic usage in dairy cattle farms. Res Vet Sci. (2014) 96:229–33. doi: 10.1016/j.rvsc.2014.01.001
Keywords: epoxyeicosatrienoic acids, dog, cat, horse, pain
Citation: Shihadih DS, Harris TR, Kodani SD, Hwang S-H, Lee KSS, Mavangira V, Hamamoto B, Guedes A, Hammock BD and Morisseau C (2020) Selection of Potent Inhibitors of Soluble Epoxide Hydrolase for Usage in Veterinary Medicine. Front. Vet. Sci. 7:580. doi: 10.3389/fvets.2020.00580
Received: 28 May 2020; Accepted: 20 July 2020;
Published: 26 August 2020.
Edited by:
Lloyd Reeve-Johnson, University of the Sunshine Coast, AustraliaCopyright © 2020 Shihadih, Harris, Kodani, Hwang, Lee, Mavangira, Hamamoto, Guedes, Hammock and Morisseau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Morisseau, Y2htb3Jpc3NlYXVAdWNkYXZpcy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.