 Genevieve Baddoo1†
Genevieve Baddoo1† Adriana Ene1†
Adriana Ene1† Zubia Merchant2†
Zubia Merchant2† Swarnali Banerjee3
Swarnali Banerjee3 Alan J. Wolfe4
Alan J. Wolfe4 Catherine Putonti1,2*
Catherine Putonti1,2*- 1Bioinformatics Program, Loyola University Chicago, Chicago, IL, United States
- 2Department of Biology, Loyola University Chicago, Chicago, IL, United States
- 3Department of Mathematics and Statistics, Loyola University Chicago, Chicago, IL, United States
- 4Department of Microbiology and Immunology, Stritch School of Medicine, Loyola University Chicago, Maywood, IL, United States
Continued efforts to isolate and sequence bacteria of the urinary tract has increased representation of these species in publicly available databases. This in turn has improved taxonomic classifications of the urinary microbiome (urobiome). Short-read sequencing targeting a variable region(s) of the 16S rRNA gene sequence has been fundamental in characterizing the urobiomes of males and females with and without lower urinary tract symptoms, as well as cancers of the urinary tract. Here, we have compiled a data set of full-length or near-full-length 16S rRNA gene sequences for the urobiome. To generate this data set, we first plated 203 isolates from the bladder on differential media and sequenced their full-length 16S rRNA gene sequence. We combined this data set with publicly available genomes from primarily the female urinary tract. The final data set includes 399 sequences representative of 160 different species from 73 genera. We assessed the ability of publicly available databases to correctly predict these sequences based on the V1-V3, V4, and V4-V6 variable regions. As expected, species designations based upon these variable regions is often not possible or incorrect. We also detected incorrect genus-level classifications. This data set can be used to supplement existing databases, by increasing urobiome species variation, and thus improve future studies characterizing urobiomes.
Introduction
High-throughput sequencing technologies were pivotal in first identifying the presence of bacterial DNA in the healthy urinary tract of both males and females (1–4). Improved culture-based techniques, such as the Expanded Quantitative Urinary Culture (EQUC) method (5), enabled researchers to isolate and subsequently sequence more species inhabiting the urinary tract. In the decade that has followed the discovery of the urinary microbiome (urobiome), numerous studies investigating the bacterial fraction of this community have been conducted (see reviews (6, 7)). Whole genome sequencing of strains isolated from the urinary tract have improved representation of urinary taxa in publicly available databases, which has in turn improved bioinformatic assignment of taxa in urobiome studies.
Most urobiome studies have relied on high-throughput sequencing of variable regions of the 16S rRNA gene sequence. Previously targeted variable regions for the urobiome include V1-V2 (8, 9), V1-V3 (1, 3, 10–12), V3-V4 (13–16), V4-V6 (17, 18), V4 (19–30), and V6 (2, 31). General guidelines for 16S surveys of urobiome samples have been recommended (32). Hoffman and colleagues assessed the GreenGenes, SILVA, and NCBI 16S Microbial databases for their ability to assign taxa to the 16S variable regions extracted from the whole genomes of 149 bladder strains, representing 78 species (33). The authors found that the V2-V3 and V1-V3 regions from this in silico experiment, when combined with the NCBI 16S Microbial database, correctly identified most bladder bacterial species. The V4 region, which is widely used, also fared well, correctly classifying two-thirds of the bacterial species. This performance was impacted by limitations in the representation of 16S rRNA gene sequence diversity among urinary microbes in the database; furthermore, prior studies have shown that different species of common urobiome genera can have full-length 16S rRNA gene sequences that are nearly identical (34–36). Additionally, when current bioinformatic tools and databases are applied to older studies, different taxonomic assignments have been identified, highlighting the importance of adding urinary-derived isolates to reference databases (37).
Here, we have focused on developing a reference set of full-length or near-full-length 16S rRNA gene sequences for the urobiome. With this reference set, we assess the resolution possible when the frequently used V1-V3, V4, and V4-V6 regions are targeted. The selection of these three regions was informed by prior assessment of the taxonomic resolution possible for bacterial species of the female urinary tract (33). Based upon our analysis, we identify taxa for which species resolution is possible, taxa for which only genus resolution is possible, and taxa for which the genus cannot be identified. In particular, we focus our discussion on taxa associated with symptoms and “health.” Thus, urobiome researchers can integrate prior studies with limitations in mind.
Methods
Strain sequencing
Bacterial strains were isolated from urine samples collected via transurethral catheterization as part of prior IRB-approved studies (Loyola: 206439, 209545, 206449; University of California, San Diego: 170077AW) that are described in detail elsewhere (25, 38–41). Briefly, the expanded quantitative urine culture (EQUC) process was performed in the Wolfe lab, as described previously (5). Unique colony morphologies were replated and identified via MALDI-TOF prior to storage at -80°C.
For each isolate examined here, the sample was streaked onto either an anaerobe 5% sheep blood (ANA) agar plate, 5% sheep blood agar plate (BAP), or nalidixic acid (CNA) agar plate, depending upon the genus identified by MALDI-TOF. (All three plates were BD BBL prepared plated media [Becton, Dickinson and Co., Sparks, MD].) Each plate was incubated for 48 h in 5% CO2 at 35°C. All colonies were scraped off of the surface of the plate and added to 1 mL of liquid media. The media was selected based upon the genus predicted by MALDI-TOF; these media include: Lysogeny broth (LB), Actinomyces broth (Sigma-Aldrich), Brain Heart Infusion broth (BHI) (BD) + 1% Tween 80 (BHI+Tween), De Man, Rogosa, and Sharpe broth (MRS) (Millipore) + 1% Tween 80 (MRS+Tween), New York City III broth (NYC III), Tryptic Soy broth (TSB) +5% sheep blood, or TSBYE (TSB+0.5% w/v yeast extract). Liquid cultures were incubated for 48 h at 35°C with 5% CO2, after which bacterial glycerol stocks were created using 1 mL liquid culture and 1 mL 50% (v/v) glycerol. Stocks were then frozen at -80°C until further processing.
Each freezer stock was streaked onto 6 different types of 1.7% agar plates (LB, Actinomyces, TSB, NYC III, BHI+Tween, MRS+Tween) and incubated for 48 h in 5% CO2 at 35°C. Morphologically distinct colonies were identified using a light microscope, and a single colony of each morphology was picked from each plate, added to 1 mL of the liquid media of the plate from which it was derived, and incubated for 48 h in 5% CO2 at 37°C.
DNA was extracted from the liquid culture of the colony using the DNeasy UltraClean Microbial Kit (Qiagen, Hilden, Germany), following the manufacturer’s protocol. DNA concentration was quantified using the Qubit fluorometer (ThermoFisher Scientific, Waltham, MA USA). The 16S rRNA gene sequence was amplified using the 63f (5′-CAG GCC TAA CAC ATG CAA GTC-3′) and 1387r (5′-GGG CGG WGT GTA CAA GGC-3′) primers (42). PCR products were purified from the reaction mixture using the E.Z.N.A. Cycle Pure Kit (Omega Bio-tek, Inc., Norcross, GA USA), following the manufacturer’s protocol, and quantified using the Qubit fluorometer. 16S rRNA gene amplicons were sequenced via Sanger sequencing by Genewiz from Azenta Life Sciences (New Brunswick, NJ, USA) with 2x coverage.
Each forward and reverse read was manually trimmed and assembled in Geneious v. 2021.0.3 (Biomatters, Ltd., Auckland, New Zealand). Sequences were filtered out if they contained 30 or more unassigned nucleotides (Ns) and/or gaps in their assembly. The resulting sequence was queried against the NCBI 16S ribosomal RNA sequences database via megablast (as of April 2022). Hits were manually inspected, and taxonomic classification was made. If two or more identical sequences were generated for isolates from the same sample (signifying that the same species was isolated on two or more medias and/or two or more colonies that were perceived to be morphologically distinct), only one was kept for further analysis; the longer of the two was retained. Taxonomic designations were confirmed in December 2023 via the NCBI Taxonomy database. Genera and species listed in the results and supplemental materials reflect the most up to date names.
Creating a unique set of 16S rRNA sequences from bladder bacteria
First, all rna_genomic files for genome assemblies in the BioProject PRJNA316969 were retrieved (as of April 18, 2022). The genomes within this BioProject are predominately from bladder urine, collected via transurethral catheterization, primarily from adult females; non-bladder isolates, determined by referring to metadata records, were removed from the data set. Supplementary Table S1 lists metadata for the strains included in this data set, as well as the strains from our own sequencing efforts. Multiple sequence alignment was performed using MAFFT v7.490 (43) through Geneious Prime v2022.1.1 (Biomatters Ltd., Auckland, NZ). Duplicate sequences were removed, as were sequences that were subsequences of a longer representative sequence.
Variable region simulation
Full-length 16S rRNA gene sequences were used to derive sequences representative of sequencing the V1-V3, V4, and V4-V6 regions. Table 1 lists the primers used for these regions. The same procedure used by Hoffman et al. (33) was employed here by using their script (https://github.com/lakarstens/BladderBacteriaSpecies/tree/master/split_to_vr). For downstream analyses, the fasta files were converted to fastq files using seqtk seq tools (https://github.com/lh3/seqtk). Fastq files were analyzed using DADA2 (44). The assignTaxonomy function from DADA2 was used to assign taxonomy with the SILVA database (v138.1) (45). Full-length 16S rRNA gene sequences were retrieved from SILVA, which includes relevant updates from the GTDB taxonomy.

Table 1 Primer sequences used to simulate short read sequencing of bladder bacteria.
Phylogenetics
Phylogenetic trees were determined as follows. Sequences were imported into Geneious Prime and aligned using the MAFFT (43) plug-in through Geneious Prime. The phylogenetic tree was derived using the FastTree v2.1.12 (46) plug-in with default parameters through Geneious Prime and visualized using iTOL v6 (47).
Results
Of the 203 bladder isolates plated, 155 had growth on at least one of the six media used here (see Methods). DNA was extracted from 1,008 morphologically distinct single colonies from these plates and 16S rRNA gene sequencing was performed resulting in 831 high-quality sequences (Supplementary Data Sheet S1). Taxonomy was assigned via BLAST against NCBI’s 16S ribosomal RNA sequences database. These near-full length sequences range in length from 811 to 1319 bp (average 1250 bp). These 16S rRNA sequences represent 69 different species and 33 genera were identified.
We next built a 16S rRNA gene sequence data set representative of the phylogenetic diversity within the bladder. We supplemented the sequences generated from our 16S rRNA gene sequencing of bladder isolates with publicly available urinary strains were added. Most of these strains were isolated from catheterized urine samples from females. In total, this data set includes 399 unique sequences – 98 from the full-length 16S rRNA gene sequencing performed here. (Other full-length 16S rRNA gene sequences from the plated isolates were better represented by sequences from the publicly available sequences.) These 399 sequences are provided in Supplementary Data Sheet S2 and associated metadata is available in Supplementary Table S1. The 399 sequence data set represents 160 different species from 73 genera (Supplementary Table S2). While these sequences range in size from 1,139 bp to 1,576 bp, all but five exceed 1,200 bp in length. Figure 1 presents the phylogenetic diversity of the species included in this data set. Additionally, the 399 sequence data set includes 5 sequences (all from prior sequencing efforts) that are assigned a genus, but the species has yet to be resolved: Bacillus sp. UMB0728 (Accession No. NZ_PKLA01000016.1), Brachybacterium sp. UMB0905 (Accession No. NZ_PNHL01000013.1), Microbacterium sp. UMB0228 (Accession No. NZ_PNFU01000016.1), Staphylococcus sp. UMB0328 (Accession No. NZ_PNFS01000002.1), and Streptococcus sp. UMB0029 (Accession No. NZ_PNGD01000027.1).
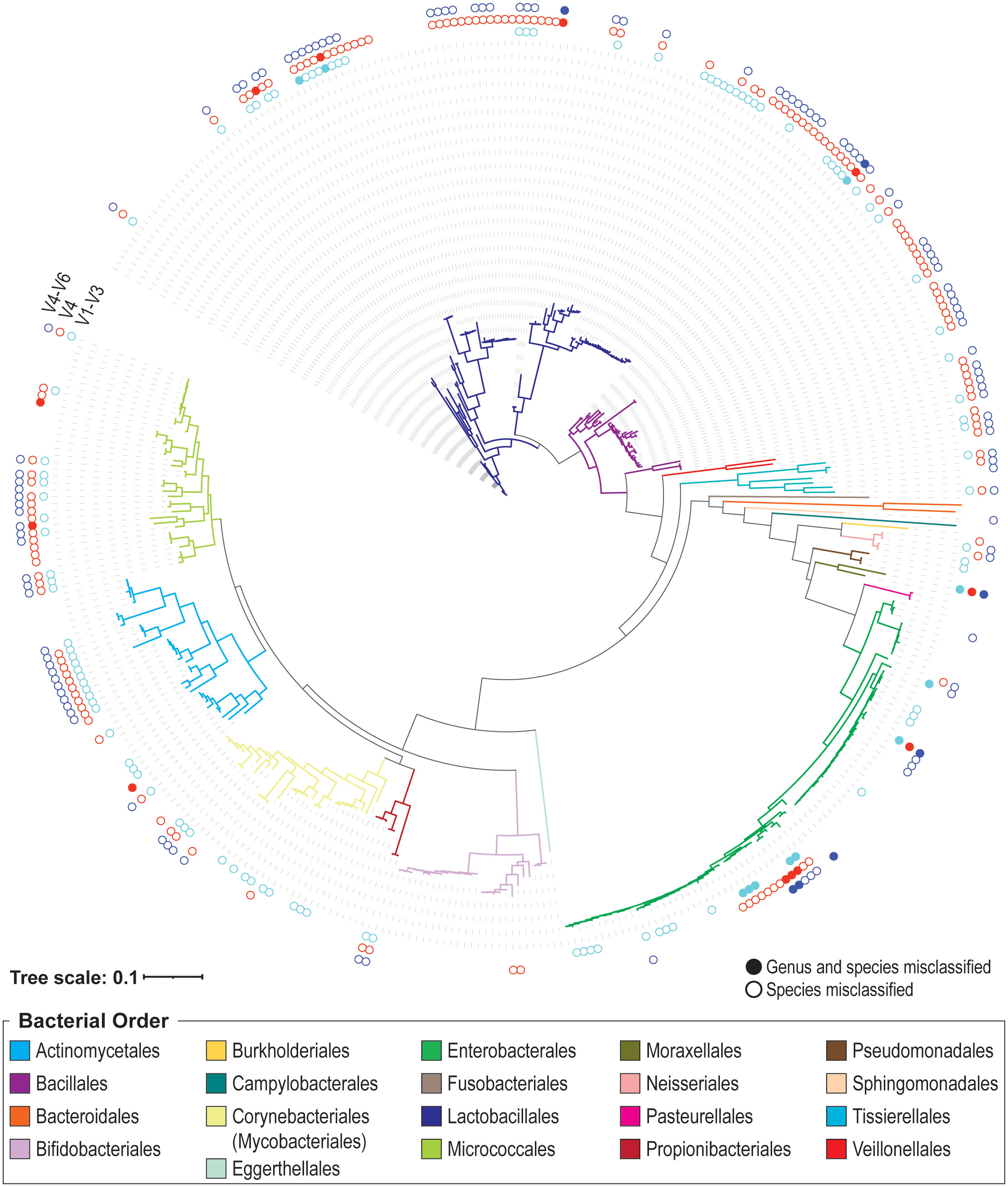
Figure 1 Phylogenetic tree of species included in the 399 urinary 16S rRNA gene sequence data set. Branches are colored according to their bacterial order, as indicated in the legend. The outer rings indicate if the V1-V3 (inner, aqua), V4 (middle, red), and/or V4-V6 (outer, indigo) genus/species predictions are correct. If the variable region identifies the correct genus and species, no dot is drawn. If the genus prediction is correct, but the species is not, the dot is an open circle. If neither the genus nor species prediction is correct, the dot is filled.
As prior short-read 16S rRNA sequencing surveys have cataloged the taxa of the urobiome using either the V1-V3 regions, the V4 region, or the V4-V6 regions, we were interested in ascertaining the variable regions’ ability to correctly identify urinary microbes at the genus and species level. These regions were spliced from the 399 unique 16S rRNA near-full or full-length sequences and classified. Most classifications at the genus-level (often the depth to which short-read 16S rRNA gene sequence surveys report taxa) were correct; however, we identified a few sequences that were incorrectly or unable to be classified at the genus-level (Table 2). For all three variable regions considered (V1-V3, V4, and V4-V6), there were urinary species for which genus-level designations could not be made (Table 2, “Not classified”). Three sequences in our collection, Peribacillus frigoritolerans 462G, Moraxella osloensis UMB0416, and Enterococcus raffinosus UMB9185, were not correctly identified at the genus level by all three regions.
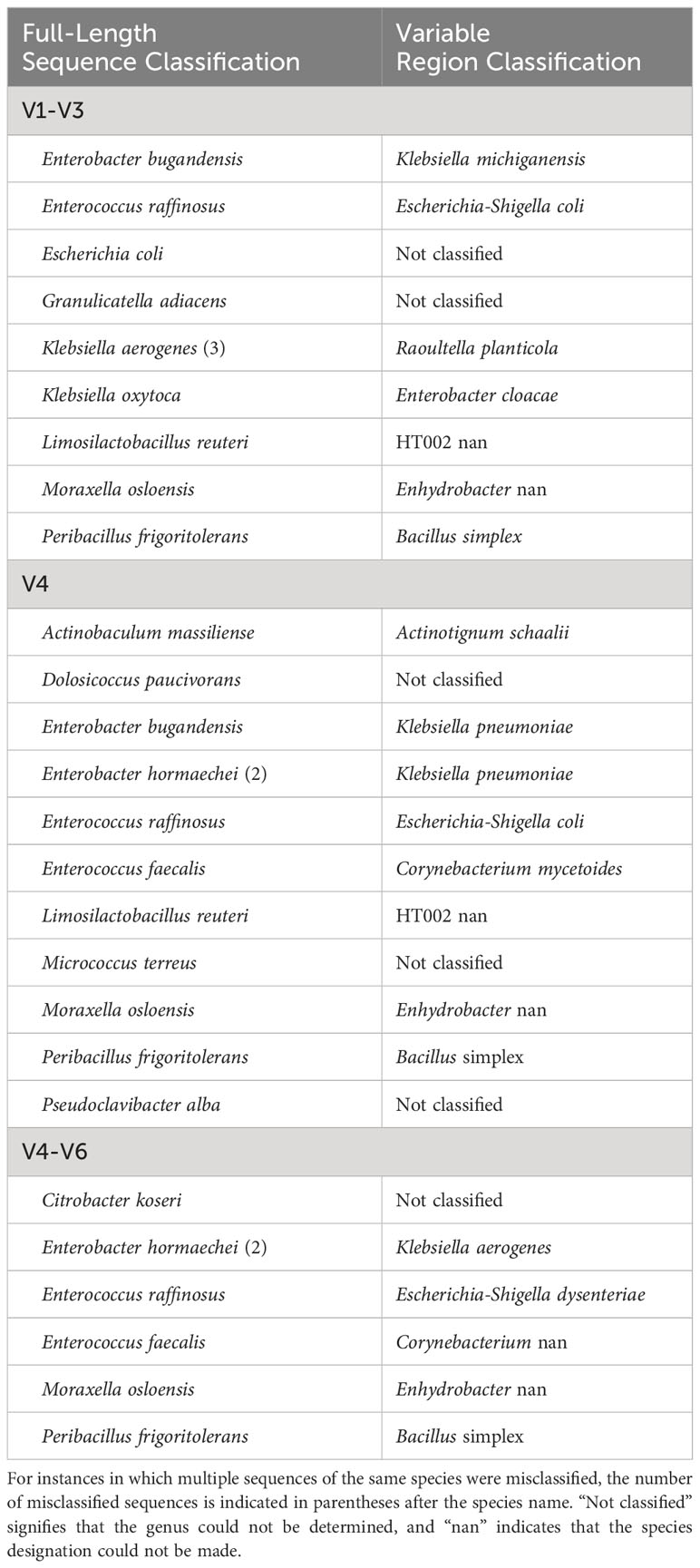
Table 2 Urinary species with a 16S rRNA gene sequence that was misclassified or not classified at the genus-level when considering a variable region(s).
When species predictions were considered (i.e., genus prediction was correct), we found that 100 designations were incorrect for the V1-V3 region (25.06%), 142 for the V4 region (35.59%), and 112 for the V4-V6 region (28.07%) (Figure 1). Many of these sequences were unable to be resolved at the species level. In these cases, the genus prediction was correct, but the species was not predicted. This includes 53 sequences for the V1-V3 region, 47 for the V4 region, and 52 for the V4-V6 region. Supplementary Table S3 lists each of the 399 sequences in the data set, including if their V1-V3, V4, and V4-V6 variable region classification resulted in a genus match (mismatched species), a genus and species match, or a genus and species mismatch. In the case of a mismatch (either genus or species), the predicted taxon is listed. Table 3 summarizes these results for the UTI-associated bacterial species E. coli, Klebsiella pneumoniae, Proteus mirabilis, Morganella morganii, Staphylococcus aureus, and Pseudomonas aeruginosa.
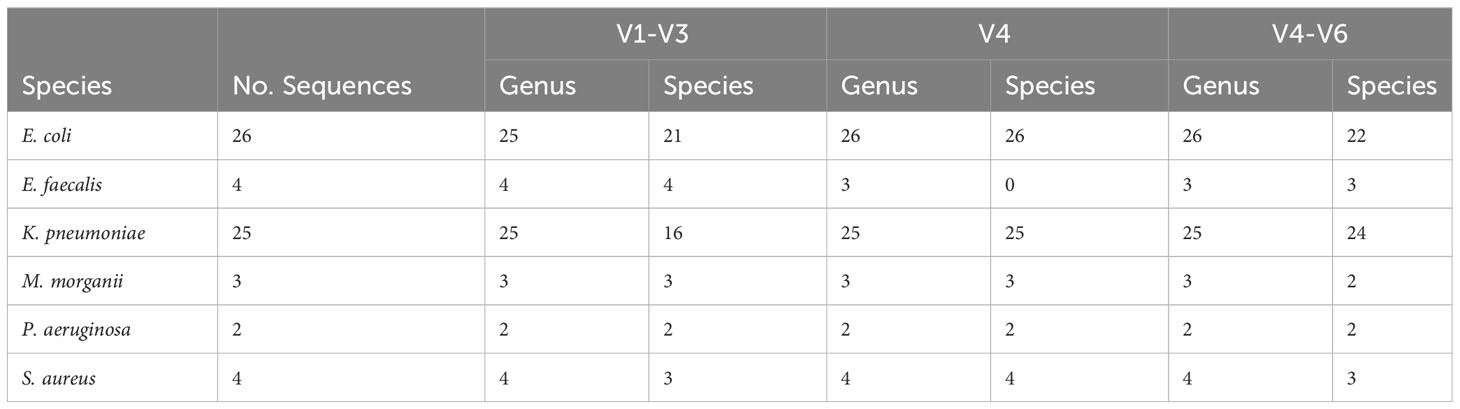
Table 3 Accuracy of prediction of sequences of UTI-associated species at the genus-level and species-level for the V1-V3, V4, and V4-V6 regions.
Discussion
The data set presented here provides a resource for researchers investigating the bacterial diversity of the urinary tract. As most studies of this diversity to date have employed short-read sequencing targeting 16S rRNA gene sequence variable regions, our analyses into the “correctness” of such classifications enables a new interpretation of prior studies of the female urinary microbiome that have targeted the V1-V3, V4, or V4-V6 variable regions. As variable region studies often report taxa at the genus-level, we can first consider those sequences that resulted in a misclassification (or the inability to be classified) at the genus-level. It is important to note that not all of the sequences for the species listed in Table 2 were misclassified/not classified. Nonetheless, this table does include species associated with infection or symptoms or the lack thereof. As more urinary bacteria representatives are included in training models for taxonomic classification, one would anticipate that such instances of misclassification or failed classifications would be reduced. Exceptions, however, would be those species that have nearly identical 16S rRNA gene sequences, e.g., species of lactobacilli, species of Gardnerella (34–36).
16S rRNA gene sequences of the closely related Enterobacter bugandensis (n=1) and Enterobacter hormaechei (n=2) were misclassified as Klebsiella pneumoniae based on the V4 region (Supplementary Table S3). Strains of these closely related Enterobacter species often exhibit multidrug-resistance (MDR) (48, 49). Prior studies have identified MDR strains of E. hormaechei in urine samples from individuals with UTI symptoms (50–52). Strains of K. pneumoniae, a common cause of UTIs due to indwelling catheters, are also frequently MDR (see reviews (53, 54)). The E. bugandensis 16S rRNA gene sequence was also misclassified as Klebsiella michiganensis (V1-V3) and Klebsiella aerogenes (V4-V6); while both of these Klebsiella species similarly have been shown to have MDR, only K. aerogens has been associated with UTIs (55–57). Misclassification of these two Enterobacter species by variable region analysis may underestimate its contribution to UTIs and/or be misleading with regards to the antibiotic resistances present in a sample. While 16S rRNA gene surveys are not a clinical tool, they are frequently used when investigating associations between urobiome constituents and lower urinary tract symptoms (see review (6)).
Enterococcus faecalis and Enterococcus faecium also have been associated with catheter-associated UTIs (CAUTIs) (58). One of the four E. faecalis sequences was misclassified as Corynebacterium based on the V4 and V4-V6 regions. While the other three sequences in the data set were predicted as Enterococcus by their V4 region, they were assigned to the species E. faecium (Figure 1). Recent studies of urinary strains of E. faecalis and E. faecium have found that the species vary in their resistances to antibiotics (58–61). Thus, studies that target the V4 region are limited to a genus-level classification of Enterococcus at best.
As shown in Table 3, most of the other UTI-associated bacterial species are correctly identified at the genus level. Furthermore, false identifications of sequences of M. morganii and P. aeruginosa did not occur for the three regions examined here. Only one sequence – Enterococcus raffinosus UMB918 – was misidentified as E. coli based on the V1-V3 and V4 regions (Supplementary Table S3). In addition to the misidentified E. hormaechi and E. bugandensis sequences previously mentioned, four K. aerogenes and four K. quasipneumoniae sequences were predicted to be K. pneumoniae based on the V4 region; the V1-V3 and V4-V6 sequence species predictions for these other Klebsiella sequences were, however, correct. The V4 sequences of many of the Staphylococcus species, including S. capitis, S. epidermidis, S. haemolyticus, S. hominis, S. lugdunesis, S. pasteuri, and S. warneri, were incorrectly predicted to be S. aureus (Supplementary Table S3). When the expanded V4-V6 region was considered, only three of the four S. haemolyticus sequences were erroneously identified as S. aureus. Therefore, we can conclude that urobiome studies – particularly those considering the V4 region alone – may be overestimating the relative abundance of K. pneumoniae and S. aureus.
Examination of symptom-associated species alone limits our understanding of the urobiome. Numerous studies have associated Lactobacillus species with a lack of lower urinary tract symptoms in females (19, 22, 39, 62, 63). The V1-V3, V4, and V4-V6 regions were unable to correctly assign species designations for many of the L. iners, L. crispatus, and L. gasseri sequences (Supplementary Table S3). These three species are the most frequently detected lactobacilli by the EQUC method among continent females (39). Several L. crispatus strains have shown the ability to inhibit or kill uropathogenic E. coli strains (64–66). In contrast, L. gasseri, L. iners, and L. jensenii are frequently found in the urobiomes of females with urinary incontinence (19, 39). Thus, distinguishing between these Lactobacillus species is key to understanding continence in females. The V1-V3 regions performed best in distinguishing the urinary Lactobacillus species, correctly identifying L. crispatus, L. iners, L. jensenii, L. mulieris, and most L. gasseri (11/12); L. paragasseri strains were predicted to be L. gasseri (Table 2), which is not surprising given the significant sequence similarity of the 16S rRNA gene sequences for these sister taxa (34). In our collection, L. paragasseri and L. gasseri 16S rRNA gene sequences can differ by as few as 1 nucleotide. Thus, urobiome studies that target the V1-V3 region are best for reliably determining Lactobacillus species diversity.
While prior work assessing the capability of variable regions to resolve urinary species did find that NCBI’s database outperformed SILVA, which was used here, this prior assessment was conducted using a previous version of SILVA (v132) (33). A greater precision was observed here, for a larger and more diverse representation of 16S rRNA gene sequences from urinary isolates, than previously reported for this prior version of SILVA (33). Searching the SILVA database used here (v138.1) found that many of the taxa included in our reference data set had numerous representative sequences, highlighting that these databases are only as good as the data publicly available. We will note that after our analyses, we did reinvestigate the taxonomic designations (see Methods) as taxonomies are amended as new information emerges. We chose to use the SILVA database here as most prior 16S rRNA gene sequence surveys of the urobiome have relied on this database, albeit prior versions. It, along with the NCBI collection, are “preferred” databases per the Urobiome Consensus (32). Our data set will contribute to improving these databases. Likewise, additional closed genome sequences for members of the urobiome also will improve representation of genetic diversity of species within this niche.
As our analysis shows, it is possible to reliably characterize the urobiome at the genus level via short read sequencing. However, there are several cases in which distinguishing between two species of the same genera is critical for interpretation. Each of the regions considered here have their own limitations for distinguishing species. Concurring with prior evaluations of the female urobiome characterization by 16S rRNA gene sequence variable regions, we found the V1-V3 region to more accurately resolve taxa than V4-V6 region or V4 region alone (33). However, a similar study of bacteria of the male urobiome identified the V1-V2 amplicon as more precise (67). This highlights the importance of selecting a variable region(s) that can accurately capture the bacterial diversity present in the specific niche being explored. Informed decisions thus necessitate routine assessments of the strengths and limitations of individual variable regions as sequencing platforms, databases, and/or bioinformatic tools improve (37, 68, 69).
Nevertheless, short-read sequencing studies have inherent limitations, which can be overcome by using full-length 16S rRNA gene sequences. With decreasing error rates and costs associated with long-read sequencing technologies, full-length 16S rRNA gene sequencing can reliably resolve species and even strains (70). Full-length 16S rRNA gene sequencing surveys have been instrumental in identifying new 16S rRNA gene variants, e.g., those in the vaginal microbiome (71), bacterial transmission, e.g., mother-to-infant transmission of oral bacteria (72), and temporal dynamics, e.g., gut microbiota post-antibiotic treatment (73). Conducting such full-length 16S rRNA gene sequence surveys of the urobiome have only recently been explored (74). Additional studies are needed to improve the resolution of the bacterial constituents of the urobiome. The 96 sequences generated here have been deposited in GenBank. The data set presented here (Supplementary Data Sheet S2) can be used to supplement existing databases, increasing urobiome species variation for culturable constituents of the urobiome. Full-length 16S rRNA gene sequencing of the urobiome is needed for capturing those members of the urinary community that cannot be grown in the lab.
Data availability statement
The data presented in the study are deposited in GenBank, accession numbers OR975923 through OR976020; accession numbers for all data included in the analyses presented here can be found in Supplementary Table S1.
Author contributions
GB: Formal analysis, Writing – original draft, Writing – review & editing, Data curation, Investigation. AE: Data curation, Formal analysis, Investigation, Writing – review & editing, visualization. ZM: Data curation, Formal analysis, Investigation, Writing – review & editing. SB: Formal analysis, Writing – review & editing. AW: Writing – review & editing, Conceptualization. CP: Conceptualization, Writing – review & editing, Formal analysis, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. GB was supported by a fellowship from Loyola University Chicago. No funding source had a role in the process of writing this manuscript.
Acknowledgments
We acknowledge the clinical and scientific team members of the Loyola Urinary Education and Research Collaborative (LUEREC) who collected the isolates that were part of this work. In particular, the authors thank Thomas Halverson and Jingjie Du.
Conflict of interest
AW discloses advisory board membership for Pathnostics and Urobiome Therapeutics, funding from Pathnostics, the Craig Neilsen Foundation, NIH, and an anonymous donor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) AW declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fruro.2023.1270509/full#supplementary-material
References
1. Dong Q, Nelson DE, Toh E, Diao L, Gao X, Fortenberry JD, et al. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PloS One (2011) 6:e19709. doi: 10.1371/journal.pone.0019709
2. Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol (2011) 11:244. doi: 10.1186/1471-2180-11-244
3. Fouts DE, Pieper R, Szpakowski S, Pohl H, Knoblach S, Suh M-J, et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Trans Med (2012) 10:174. doi: 10.1186/1479-5876-10-174
4. Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, FitzGerald M, et al. Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol (2012) 50:1376–83. doi: 10.1128/JCM.05852-11
5. Hilt EE, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol (2014) 52:871–6. doi: 10.1128/JCM.02876-13
6. Neugent ML, Hulyalkar NV, Nguyen VH, Zimmern PE, De Nisco NJ. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio (2020) 11:e00218-20. doi: 10.1128/mBio.00218-20
7. Perez-Carrasco V, Soriano-Lerma A, Soriano M, Gutiérrez-Fernández J, Garcia-Salcedo JA. Urinary microbiome: yin and yang of the urinary tract. Front Cell Infect Microbiol (2021) 11:617002. doi: 10.3389/fcimb.2021.617002
8. Gottschick C, Deng Z-L, Vital M, Masur C, Abels C, Pieper DH, et al. The urinary microbiota of men and women and its changes in women during bacterial vaginosis and antibiotic treatment. Microbiome (2017) 5, 99. doi: 10.1186/s40168-017-0305-3
9. Anglim B, Phillips C, Shynlova O, Alarab M. The effect of local estrogen therapy on the urinary microbiome composition of postmenopausal women with and without recurrent urinary tract infections. Int Urogynecol J (2022) 33:2107–17. doi: 10.1007/s00192-021-04832-9
10. Lewis DA, Brown R, Williams J, White P, Jacobson SK, Marchesi JR, et al. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front Cell Infection Microbiol (2013) 3:41. doi: 10.3389/fcimb.2013.00041
11. Moustafa A, Li W, Singh H, Moncera KJ, Torralba MG, Yu Y, et al. Microbial metagenome of urinary tract infection. Sci Rep (2018) 8:4333. doi: 10.1038/s41598-018-22660-8
12. Komesu YM, Dinwiddie DL, Richter HE, Lukacz ES, Sung VW, Siddiqui NY, et al. Defining the relationship between vaginal and urinary microbiomes. Am J Obstetrics Gynecology (2020) 222:154.e1–154.e10. doi: 10.1016/j.ajog.2019.08.011
13. Frølund M, Wikström A, Lidbrink P, Abu Al-Soud W, Larsen N, Harder CB, et al. The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PloS One (2018) 13:e0201380. doi: 10.1371/journal.pone.0201380
14. Xu H, Tamrat NE, Gao J, Xu J, Zhou Y, Zhang S, et al. Combined signature of the urinary microbiome and metabolome in patients with interstitial cystitis. Front Cell Infect Microbiol (2021) 11:711746. doi: 10.3389/fcimb.2021.711746
15. Ahn HK, Kim K, Park J, Kim KH. Urinary microbiome profile in men with genitourinary Malignancies. Investig Clin Urol (2022) 63:569–76. doi: 10.4111/icu.20220124
16. Li K, Chen C, Zeng J, Wen Y, Chen W, Zhao J, et al. Interplay between bladder microbiota and overactive bladder symptom severity: a cross-sectional study. BMC Urol (2022) 22:39. doi: 10.1186/s12894-022-00990-0
17. Komesu YM, Richter HE, Carper B, Dinwiddie DL, Lukacz ES, Siddiqui NY, et al. The urinary microbiome in women with mixed urinary incontinence compared to similarly aged controls. Int Urogynecol J (2018) 29:1785–95. doi: 10.1007/s00192-018-3683-6
18. Richter HE, Carnes MU, Komesu YM, Lukacz ES, Arya L, Bradley M, et al. Association between the urogenital microbiome and surgical treatment response in women undergoing midurethral sling operation for mixed urinary incontinence. Am J Obstet Gynecol (2022) 226:93.e1–93.e15. doi: 10.1016/j.ajog.2021.07.008
19. Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. MBio (2014) 5:e01283–01214. doi: 10.1128/mBio.01283-14
20. Pearce MM, Zilliox MJ, Rosenfeld AB, Thomas-White KJ, Richter HE, Nager CW, et al. The female urinary microbiome in urgency urinary incontinence. Am J Obstet Gynecol (2015) 213:347.e1–347.e11. doi: 10.1016/j.ajog.2015.07.009
21. Karstens L, Asquith M, Davin S, Stauffer P, Fair D, Gregory WT, et al. Does the urinary microbiome play a role in urgency urinary incontinence and its severity? Front Cell Infect Microbiol (2016) 6:78. doi: 10.3389/fcimb.2016.00078
22. Thomas-White KJ, Kliethermes S, Rickey L, Lukacz ES, Richter HE, Moalli P, et al. Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. Am J Obstet. Gynecol. (2017) 216:55.e1–55.e16. doi: 10.1016/j.ajog.2016.07.049
23. Wu P, Chen Y, Zhao J, Zhang G, Chen J, Wang J, et al. Urinary microbiome and psychological factors in women with overactive bladder. Front Cell Infect Microbiol (2017) 7:488. doi: 10.3389/fcimb.2017.00488
24. Chen Z, Phan M-D, Bates LJ, Peters KM, Mukerjee C, Moore KH, et al. The urinary microbiome in patients with refractory urge incontinence and recurrent urinary tract infection. Int Urogynecol J (2018) 29:1775–82. doi: 10.1007/s00192-018-3679-2
25. Price TK, Hilt EE, Thomas-White K, Mueller ER, Wolfe AJ, Brubaker L. The urobiome of continent adult women: a cross-sectional study. BJOG (2019) 127:193–201. doi: 10.1111/1471-0528.15920
26. Adebayo AS, Ackermann G, Bowyer RCE, Wells PM, Humphreys G, Knight R, et al. The urinary tract microbiome in older women exhibits host genetic and environmental influences. Cell Host Microbe (2020) 28:298–305.e3. doi: 10.1016/j.chom.2020.06.022
27. Zeng J, Zhang G, Chen C, Li K, Wen Y, Zhao J, et al. Alterations in urobiome in patients with bladder cancer and implications for clinical outcome: A single-institution study. Front Cell Infect Microbiol (2020) 10:555508. doi: 10.3389/fcimb.2020.555508
28. Vaughan MH, Mao J, Karstens LA, Ma L, Amundsen CL, Schmader KE, et al. The urinary microbiome in postmenopausal women with recurrent urinary tract infections. J Urol (2021) 206:1222–31. doi: 10.1097/JU.0000000000001940
29. Nardos R, Leung ET, Dahl EM, Davin S, Asquith M, Gregory WT, et al. Network-based differences in the vaginal and bladder microbial communities between women with and without urgency urinary incontinence. Front Cell Infect Microbiol (2022) 12:759156. doi: 10.3389/fcimb.2022.759156
30. Storm DW, Copp HL, Halverson TM, Du J, Juhr D, Wolfe AJ. A Child’s urine is not sterile: A pilot study evaluating the Pediatric Urinary Microbiome. J Pediatr Urol (2022) 18:383–92. doi: 10.1016/j.jpurol.2022.02.025
31. Siddiqui H, Lagesen K, Nederbragt AJ, Jeansson SL, Jakobsen KS. Alterations of microbiota in urine from women with interstitial cystitis. BMC Microbiol (2012) 12:205. doi: 10.1186/1471-2180-12-205
32. Brubaker L, Gourdine J-PF, Siddiqui NY, Holland A, Halverson T, Limeria R, et al. Forming consensus to advance urobiome research. mSystems (2021) 6:e0137120. doi: 10.1128/mSystems.01371-20
33. Hoffman C, Siddiqui NY, Fields I, Gregory WT, Simon HM, Mooney MA, et al. Species-level resolution of female bladder microbiota from 16S rRNA amplicon sequencing. mSystems (2021) 6:e0051821. doi: 10.1128/mSystems.00518-21
34. Tanizawa Y, Tada I, Kobayashi H, Endo A, Maeno S, Toyoda A, et al. Lactobacillus paragasseri sp. nov., a sister taxon of Lactobacillus gasseri, based on whole-genome sequence analyses. Int J Syst Evol Microbiol (2018) 68:3512–7. doi: 10.1099/ijsem.0.003020
35. Vaneechoutte M, Guschin A, Van Simaey L, Gansemans Y, Van Nieuwerburgh F, Cools P. Emended description of Gardnerella vaginalis and description of Gardnerella leopoldii sp. nov., Gardnerella piotii sp. nov. and Gardnerella swidsinskii sp. nov., with delineation of 13 genomic species within the genus Gardnerella. Int J Syst Evol Microbiol (2019) 69:679–87. doi: 10.1099/ijsem.0.003200
36. Putonti C, Shapiro JW, Ene A, Tsibere O, Wolfe AJ. Comparative Genomic Study of Lactobacillus jensenii and the Newly Defined Lactobacillus mulieris Species Identifies Species-Specific Functionality. mSphere (2020) 5:e00560-20. doi: 10.1128/mSphere.00560-20
37. Siddiqui NY, Ma L, Brubaker L, Mao J, Hoffman C, Dahl EM, et al. Updating urinary microbiome analyses to enhance biologic interpretation. Front Cell Infect Microbiol (2022) 12:789439. doi: 10.3389/fcimb.2022.789439
38. Price TK, Dune T, Hilt EE, Thomas-White KJ, Kliethermes S, Brincat C, et al. The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J Clin Microbiol (2016) 54:1216–22. doi: 10.1128/JCM.00044-16
39. Price TK, Lin H, Gao X, Thomas-White KJ, Hilt EE, Mueller ER, et al. Bladder bacterial diversity differs in continent and incontinent women: a cross-sectional study. Am J Obstet Gynecol (2020) 223:729.e1–729.e10. doi: 10.1016/j.ajog.2020.04.033
40. Burnett LA, Hochstedler BR, Weldon K, Wolfe AJ, Brubaker L. Recurrent urinary tract infection: Association of clinical profiles with urobiome composition in women. Neurourol Urodyn (2021) 40:1479–89. doi: 10.1002/nau.24707
41. Hochstedler BR, Burnett L, Price TK, Jung C, Wolfe AJ, Brubaker L. Urinary microbiota of women with recurrent urinary tract infection: collection and culture methods. Int Urogynecol J (2021) 33:563–570. doi: 10.1007/s00192-021-04780-4
42. Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, et al. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol (1998) 64:795–9. doi: 10.1128/AEM.64.2.795-799.1998
43. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol (2013) 30:772–80. doi: 10.1093/molbev/mst010
44. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods (2016) 13:581–3. doi: 10.1038/nmeth.3869
45. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res (2013) 41:D590–596. doi: 10.1093/nar/gks1219
46. Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PloS One (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
47. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res (2016) 44:W242–5. doi: 10.1093/nar/gkw290
48. Doijad S, Imirzalioglu C, Yao Y, Pati NB, Falgenhauer L, Hain T, et al. Enterobacter bugandensis sp. nov., isolated from neonatal blood. Int J Syst Evol Microbiol (2016) 66:968–74. doi: 10.1099/ijsem.0.000821
49. Urbaniak C, Sielaff AC, Frey KG, Allen JE, Singh N, Jaing C, et al. Detection of antimicrobial resistance genes associated with the International Space Station environmental surfaces. Sci Rep (2018) 8:814. doi: 10.1038/s41598-017-18506-4
50. Kumar MS, Das AP. Molecular identification of multi drug resistant bacteria from urinary tract infected urine samples. Microb Pathog (2016) 98:37–44. doi: 10.1016/j.micpath.2016.06.029
51. Ara B, Urmi UL, Haque TA, Nahar S, Rumnaz A, Ali T, et al. Detection of mobile colistin-resistance gene variants (mcr-1 and mcr-2) in urinary tract pathogens in Bangladesh: the last resort of infectious disease management colistin efficacy is under threat. Expert Rev Clin Pharmacol (2021) 14:513–22. doi: 10.1080/17512433.2021.1901577
52. Perera V, de Silva S, Jayatilleke K, de Silva N, Aydin A, Enne V, et al. Antimicrobial resistance genes, virulence genes, and associated mobile genetic elements of eight multidrug-resistant enterobacterales isolated from hospital-acquired urinary tract infections in Sri Lanka. Microb Drug Resist (2022) 28:882–92. doi: 10.1089/mdr.2022.0003
53. Chung PY. The emerging problems of Klebsiella pneumoniae infections: carbapenem resistance and biofilm formation. FEMS Microbiol Lett (2016) 363:fnw219. doi: 10.1093/femsle/fnw219
54. Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol (2020) 18:344–59. doi: 10.1038/s41579-019-0315-1
55. Otajevwo FD. Urinary tract infection among symptomatic outpatients visiting a tertiary hospital based in midwestern Nigeria. Glob J Health Sci (2013) 5:187–99. doi: 10.5539/gjhs.v5n2p187
56. Li S, Jiang X, Li C, Ju Y, Yue L, Chen F, et al. A bla SIM-1 and mcr-9.2 harboring Klebsiella michiganensis strain reported and genomic characteristics of Klebsiella michiganensis. Front Cell Infect Microbiol (2022) 12:973901. doi: 10.3389/fcimb.2022.973901
57. Rodríguez-Guerrero E, Cabello HR, Expósito-Ruiz M, Navarro-Marí JM, Gutiérrez-Fernández J. Antibiotic resistances of enterobacteriaceae with chromosomal ampc in urine cultures: review and experience of a spanish hospital. Antibiotics (Basel) (2023) 12:730. doi: 10.3390/antibiotics12040730
58. Klein RD, Hultgren SJ. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nat Rev Microbiol (2020) 18:211–26. doi: 10.1038/s41579-020-0324-0
59. Huang L, Huang C, Yan Y, Sun L, Li H. Urinary tract infection etiological profiles and antibiotic resistance patterns varied among different age categories: A retrospective study from a tertiary general hospital during a 12-year period. Front Microbiol (2021) 12:813145. doi: 10.3389/fmicb.2021.813145
60. Li J, Jiang F, Xie A, Jiang Y. Analysis of the distribution and drug resistance of pathogens in patients with urinary tract infection in the eastern chongming area of shanghai from 2018 to 2020. Infect Drug Resist (2022) 15:6413–22. doi: 10.2147/IDR.S384515
61. Carrasco Calzada F, Jairo Aguilera J, Moreno JE, Cuadros González J, Roca Biosca D, Prieto-Pérez L, et al. Differences in virulence factors and antimicrobial susceptibility of uropathogenic enterococcus spp. Strains in a rural area of Uganda and a spanish secondary hospital. Trop Med Infect Dis (2023) 8:282. doi: 10.3390/tropicalmed8050282
62. Abernethy MG, Rosenfeld A, White JR, Mueller MG, Lewicky-Gaupp C, Kenton K. Urinary microbiome and cytokine levels in women with interstitial cystitis. Obstet Gynecol (2017) 129:500–6. doi: 10.1097/AOG.0000000000001892
63. Curtiss N, Balachandran A, Krska L, Peppiatt-Wildman C, Wildman S, Duckett J. A case controlled study examining the bladder microbiome in women with Overactive Bladder (OAB) and healthy controls. Eur J Obstetrics Gynecology Reprod Biol (2017) 214:31–5. doi: 10.1016/j.ejogrb.2017.04.040
64. Butler D, Silvestroni A, Stapleton A. Cytoprotective Effect of Lactobacillus crispatus CTV-05 against Uropathogenic E. coli. Pathogens (2016) 5:27. doi: 10.3390/pathogens5010027
65. Abdul-Rahim O, Wu Q, Price TK, Pistone G, Diebel K, Bugni TS, et al. Phenyl-lactic acid is an active ingredient in bactericidal supernatants of lactobacillus crispatus. J Bacteriol (2021) 203:e0036021. doi: 10.1128/JB.00360-21
66. Song CH, Kim YH, Naskar M, Hayes BW, Abraham MA, Noh JH, et al. Lactobacillus crispatus Limits Bladder Uropathogenic E. coli Infection by Triggering a Host Type I Interferon Response. Proc Natl Acad Sci U.S.A. (2022) 119:e2117904119. doi: 10.1073/pnas.2117904119
67. Heidrich V, Inoue LT, Asprino PF, Bettoni F, Mariotti ACH, Bastos DA, et al. Choice of 16S ribosomal RNA primers impacts male urinary microbiota profiling. Front Cell Infect Microbiol (2022) 12:862338. doi: 10.3389/fcimb.2022.862338
68. Allali I, Arnold JW, Roach J, Cadenas MB, Butz N, Hassan HM, et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol (2017) 17:194. doi: 10.1186/s12866-017-1101-8
69. Onywera H, Meiring TL. Comparative analyses of Ion Torrent V4 and Illumina V3-V4 16S rRNA gene metabarcoding methods for characterization of cervical microbiota: taxonomic and functional profiling. Sci Afr (2020) 7:e00278. doi: 10.1016/j.sciaf.2020.e00278
70. Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun (2019) 10:5029. doi: 10.1038/s41467-019-13036-1
71. Qin H, Jiao JAD, Hua M, Han K, Du H, et al. Single-molecule approach to 16S rRNA for vaginal microbiome signatures in response to metronidazole treatment. Microbiol Spectr (2023) 11:e0170622. doi: 10.1128/spectrum.01706-22
72. Kageyama S, Furuta M, Takeshita T, Ma J, Asakawa M, Yamashita Y. High-level acquisition of maternal oral bacteria in formula-fed infant oral microbiota. mBio (2022) 13:e0345221. doi: 10.1128/mbio.03452-21
73. Graf J, Ledala N, Caimano MJ, Jackson E, Gratalo D, Fasulo D, et al. High-resolution differentiation of enteric bacteria in premature infant fecal microbiomes using a novel rRNA amplicon. mBio (2021) 12:e03656–20. doi: 10.1128/mBio.03656-20
Keywords: urobiome, 16S rRNA gene sequence analysis, female urinary tract, urinary microbiome, 16S rRNA gene sequence
Citation: Baddoo G, Ene A, Merchant Z, Banerjee S, Wolfe AJ and Putonti C (2024) Cataloging variation in 16S rRNA gene sequences of female urobiome bacteria. Front. Urol. 3:1270509. doi: 10.3389/fruro.2023.1270509
Received: 31 July 2023; Accepted: 13 December 2023;
Published: 08 January 2024.
Edited by:
Evann Hilt, University of Minnesota Twin Cities, United StatesReviewed by:
Nazema Siddiqui, Duke University, United StatesHarris Onywera, Africa Centers for Disease Control and Prevention
Mangesh Vasant Suryavanshi, Cleveland Clinic, United States
Copyright © 2024 Baddoo, Ene, Merchant, Banerjee, Wolfe and Putonti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Putonti, Y3B1dG9udGlAbHVjLmVkdQ==
†These authors contributed equally to this work and share first authorship